
Abstract
Rheumatoid arthritis (RA) is one of the inflammatory joint diseases in a heterogeneous group of disorders that share features of destruction of the extracellular matrices of articular cartilage and bone. The underlying disturbance in immune regulation that is responsible for the localized joint pathology results in the release of inflammatory mediators in the synovial fluid and synovium that directly and indirectly influence cartilage homeostasis. Analysis of the breakdown products of the matrix components of joint cartilage in body fluids and quantitative imaging techniques have been used to assess the effects of the inflammatory joint disease on the local remodeling of joint structures. The role of the chondrocyte itself in cartilage destruction in the human rheumatoid joint has been difficult to address but has been inferred from studies in vitro and in animal models. This review covers current knowledge about the specific cellular and biochemical mechanisms that account for the disruption of the integrity of the cartilage matrix in RA.
Similar content being viewed by others


Rheumatoid arthritis
Rheumatoid arthritis (RA) is an inflammatory joint disease that most frequently affects the anatomical components of articular and juxta-articular tissues of diarthrodial joints. The diarthrodial joints join two opposing bone surfaces that are covered by a specialized hyaline cartilage providing a low-friction, articulating interface. The synovium lines the joint cavity and is the site of production of synovial fluid, which provides the nutrition for the articular cartilage and lubricates the cartilage surfaces. In RA, the synovial lining of diarthrodial joints is the site of the initial inflammatory process [1, 2]. This lesion is characterized by proliferation of the synovial lining cells, increased vascularization, and infiltration of the tissue by inflammatory cells, including lymphocytes, plasma cells, and activated macrophages [3–5]. With the growth and expansion of the synovial lining, there is eventual extension of the inflammatory tissue mass to the adjacent articular cartilage with progressive overgrowth of the articular surface and formation of the so-called pannus, which is derived from the Latin word meaning 'cloth' and the Greek word meaning 'web'. At the interface between the RA synovium and articular cartilage, tongues of proliferating cells can be seen penetrating the extracellular matrix of the cartilage. Similarly, at the interface between the inflamed synovium and adjacent subchondral bone, there is evidence of local activation of bone resorption with destruction of the mineralized bone matrix, accompanied by cells expressing phenotypic features of osteoclasts, including calcitonin receptor mRNA, cathepsin K, and tartrate-resistant acid phosphatase (TRAP) [6, 7]. RA synovium produces a broad spectrum of factors possessing the capacity to stimulate cartilage matrix destruction and bone erosion [3, 4]. Although there is an association between inflammation and the development of joint damage, the destruction may progress in spite of attenuated inflammatory activity, and cartilage and bone erosions may develop in the absence of overt clinical signs of inflammation [8–11]. Recent evidence from human and animal studies indicates that although the specific cellular mechanisms of cartilage and bone destruction are different, TNF-α, IL-1, and additional proinflammatory cytokines and mediators can drive elements of both processes [10, 12]. The recent development of assays for specific biological markers that reflect quantitative and dynamic changes in the synthetic and degradation products of cartilage and bone matrix components has offered the possibility of identifying patients at risk for rapid joint damage and also the possibility of early monitoring of the efficacy of disease-modifying anti-rheumatic therapies [13–15]. This review will focus on the unique ways in which the chondrocyte responds to the inflammatory milieu and contributes to the disease process in the cartilage.
The chondrocyte in adult articular cartilage
Adult human articular cartilage, which covers the articulating surfaces of long bones, is populated exclusively by chondrocytes that are somewhat unique to this tissue. The collagen network of the interterritorial cartilage matrix is composed of types II, IX, and XI collagens, which provide tensile strength and promote the retention of proteoglycans. Type XI collagen is part of the type II collagen fibril, and type IX integrates with the surface of the fibril with the non-collagen domain projecting outward, permitting association with other matrix components. The other major component, the large aggregating proteoglycan aggrecan, which is attached to hyaluronic acid polymers via link protein, bestows compressive resistance. A large number of other non-collagen molecules are present in the interterritorial matrix; these molecules include several small proteoglycans such as biglycan, decorin, fibromodulin, the matrilins, and cartilage oligomeric matrix protein (COMP). The chondrocytes are surrounded by a pericellular matrix composed of type VI collagen microfibrils that interact with hyaluronic acid, biglycan, and decorin and maintain chondrocyte attachment, but little or no fibrillar collagen. Under physiological conditions, the chondrocytes maintain a stable equilibrium between the synthesis and the degradation of matrix components, with a half-life of more than 100 years for type II collagen [16] and a half-life for aggrecan core protein in the range 3 to 24 years [17]. The glycosaminoglycan components of aggrecan and other cartilage matrix constituents also are synthesized by chondrocytes under conditions of low turnover, and the matrix turnover may be more rapid in the immediate pericellular zones.
Under normal conditions, chondrocyte proliferation is limited, and penetration of other cell types from the joint space or subchondral bone is restricted. In the absence of a vascular supply, the chondrocyte must rely on diffusion from the articular surface or subchondral bone for the exchange of nutrients and metabolites. Glucose serves both as the major energy source for the chondrocytes and as an essential precursor for glycosaminoglycan synthesis. Facilitated glucose transport in chondrocytes is mediated by several distinct glucose transporter proteins (GLUTs) that are either expressed constitutively (GLUT3 and GLUT8) or inducible by cytokines (GLUT1 and GLUT6) [18, 19]. Chondrocytes do not contain abundant mitochondria, but they maintain active membrane transport systems for exchange of cations, including Na+, K+, Ca2+, and H+, whose intracellular concentrations fluctuate with charge, biomechanical forces, and alterations in the composition of the cartilage matrix [20]. Furthermore, chondrocyte metabolism operates at low oxygen tension, ranging from 10% at the surface to less than 1% in the deep zones of the cartilage. Chondrocytes adapt to low oxygen tensions by upregulating hypoxia-inducible factor (HIF)-1α, which can stimulate the expression of GLUTs [19] and angiogenic factors such as vascular endothelial growth factor (VEGF) [21, 22], as well as ascorbate transport [23] and several genes associated with cartilage anabolism and chondrocyte differentiation, including Sox9 and type II collagen [24]. By modulating the intracellular expression of survival factors such as HIF-1α, chondrocytes have a high capacity to survive in the avascular cartilage matrix and to respond to environmental changes.
Joint inflammation and cartilage remodeling in RA
Cartilage destruction in RA occurs primarily in areas contiguous with the proliferating synovial pannus [25, 26]. In the cartilage–pannus junction, there is evidence of attachment of both fibroblast-like and macrophage-like synovial cell types, which can release proteinases capable of digesting the cartilage matrix components [27]. A distinctive fibroblast-like cell type, the so-called 'pannocyte', present in RA synovium exhibits anchorage-independent growth and can invade cartilage in the absence of an inflammatory environment [2]. Nevertheless, there is evidence of loss of proteoglycan throughout the cartilage matrix, particularly in the superficial zone in contact with the synovial fluid at sites not directly associated with the pannus [28, 29]. This has been attributed to the release of inflammatory mediators and degradative enzymes released by polymorphonuclear leuko-cytes and other inflammatory cells in the synovial fluid. In early RA, however, the loss of proteoglycan occurs throughout the cartilage matrix, and selective damage to type II collagen fibrils can be observed in middle and deep zones [30, 31], suggesting that the chondrocyte may also participate in degrading its own matrix by releasing autocrine–paracrine factors.
Of the matrix metalloproteinases (MMPs) involved in the degradation of cartilage collagens and proteoglycans in RA, the MMPs of the collagenase and stromelysin families have been given greatest attention because they specifically degrade native collagens and proteoglycans. Active stromelysin also serves as an activator of latent collagenases [32]. MMPs are localized at sites of degradation in cartilage derived from patients with RA [33]. Collagenases 1, 2, and 3 (MMP-1, MMP-8, and MMP-13, respectively), gelatinases (MMP-2 and MMP-9), stromelysin-1 (MMP-3), and membrane type I MMP (MT1-MMP; MMP–14) are present in active RA synovium [34, 35]. Although elevated levels of MMPs in the synovial fluid probably originate from the synovium, intrinsic chondrocyte-derived chondrolytic activity is present at the cartilage–pannus junction as well as in deeper zones of cartilage matrix in some RA specimens [36]. For example, MMP-1 does not derive from the RA synovial pannus but is produced by chondrocytes [37]. MMP-10, similarly to MMP-3, activates procollagenases and is produced by both the synovium and chondrocytes in response to inflammatory cytokines [38]. In contrast, MMP-14, produced principally by the synovial tissue, is important for synovial invasiveness, and inhibition of the expression of this membrane proteinase by antisense mRNA has been shown to reduce cartilage destruction [39].
Other MMPs, including MMP-16 and MMP-28 [40, 41], and a large number of members of the reprolysin-related proteinases of the ADAM (a disintegrin and metalloproteinase) family, including ADAM-17/TACE (TNF-α converting enzyme) [42], are expressed in cartilage, but their roles in cartilage damage in RA have yet to be defined [32, 43, 44]. Although several of the MMPs, including MMP-3, MMP-8, and MMP-14, are capable of degrading proteoglycans, ADAMTS (ADAM with thrombospondin-1 domains)-4 and ADAMTS-5 are now regarded as the principal mediators of aggrecan degradation [45, 46]. ADAMTS-4 is expressed constitutively, whereas ADAMTS-5 is more prominently regulated by inflammatory cytokines. However, the activities of MMPs and aggrecanases are complementary [47]. Of the aggrecanases, so far only aggrecanase-2, ADAMTS5, seems to be associated with increased susceptibility to osteoarthritis, as shown in Adamts5-deficient mice [48, 49]. Tissue inhibitor of metallo-proteinases (TIMP)-3, but not TIMP-1, TIMP-2, or TIMP-4, is a potent inhibitor of ADAMTS-4 and ADAMTS-5 in vitro [50]. That capacity of transforming growth factor (TGF)-β to increase TIMP gene expression may partly account for its protective effects against cartilage breakdown mediated by MMP and by ADAMTS [51, 52].
Other proteinases, including the urokinase-type plasminogen activator and the cathepsins B, L, and D, which degrade various cartilage matrix components and may be produced by the chondrocytes themselves, also contribute to breakdown of the cartilage matrix [53, 54]. Cathepsin K is expressed in synovial fibroblasts on the cartilage surface at the cartilage–pannus junction and is upregulated by inflammatory cytokines [55]. Among the known cathepsins, cathepsin K is the only proteinase that is capable of hydrolyzing types I and II collagens at multiple sites within the triple-helical regions, and its requirement for acidic pH may be provided by the micro-environment between the synovial pannus and the cartilage [56].
Degraded cartilage matrix components are to be considered both diagnostic markers of cartilage damage and potential autoantigens in the induction and maintenance of RA synovial inflammation [13, 15]. Molecules originating from the articular cartilage, including aggrecan fragments, which contain chondroitin sulfate and keratan sulfate, type II collagen fragments, collagen pyridinoline cross-links, and COMP, are usually released as degradation products as a result of catabolic processes. Specific antibodies that detect either synthetic or cleavage epitopes have been developed to study biological markers of cartilage metabolism in RA body fluids (reviewed in [14]). These include the C2C antibody (previously known as Col2-3/4CLong mono), which has been used to detect cleavage of the triple helix of type II collagen in experimental models of RA and in RA cartilage [57]. Similarly, the degradation of aggrecan in cartilage has been characterized by using antibodies 846, 3B3- and 7D4 (which detect chondroitin sulfate neoepitopes), 5D4 (which detects keratan sulfate epitopes), and the VIDIPEN and NITEGE antibodies (which recognize aggrecanase and MMP cleavage sites, respectively), within the interglobular G1 domain of aggrecan [45, 54].
Several studies have shown that COMP levels reflect processes in cartilage that are distinct from inflammatory aspects of the disease and serve as a general indicator of cartilage turnover [58]. YKL-40/HC-gp39, also known as chitinase 3-like protein 1 (CH3L1), is a specific histological marker in inflamed RA synovium that forms immune complexes with HLA-DR4 [59]. The immune response to YKL-40, which is biased toward the regulatory, suppressor T-cell phenotype in healthy individuals, is shifted from an anti-inflammatory to a proinflammatory phenotype in patients with RA [60]. In cartilage, CH3L1 is induced by inflammatory cytokines. It inhibits cytokine-induced cellular responses and may function as a feedback regulator [61, 62]. A related member of the chitinase family, YKL-39, may be a more specific serum marker as a cartilage-derived autoantigen [63, 64]. Another novel molecule is the cartilage-derived retinoic-acid-sensitive protein (CD-RAP), also known as melanoma inhibitory activity, which is found at high levels in synovial fluids from patients with mild RA and decreases with disease progression [65].
Mediators of cartilage degradation in RA
There is evidence that the chondrocytes may not only participate in the destruction of the cartilage matrix by responding to the proinflammatory cytokines released from the synovium but may themselves also be the source of pro-inflammatory cytokines that, by means of autocrine or paracrine mechanisms, increase tissue catabolism and suppress anabolic repair processes. The resultant disequilibrium in remodeling probably contributes to the rapid loss of cartilage matrix components characteristic of the RA joint lesion. Our understanding of basic cellular mechanisms regulating chondrocyte responses to inflammatory cytokines has been inferred from numerous studies in vitro with cultures of cartilage fragments or isolated chondrocytes and is supported by studies in experimental models of inflammatory arthritis such as collagen-induced arthritis (CIA) and antigen-induced arthritis (AIA) in mice. Less information has been derived from direct analysis of cartilage or chondrocytes obtained from patients with RA in whom cartilage damage is extensive.
Inflammatory cytokines
Alterations in products of cartilage matrix turnover and levels of matrix-degrading proteinases and inhibitors described above are accompanied by changes in the levels of various cytokines in the rheumatoid synovial fluids (Fig. 1). Numerous studies in vitro and in vivo indicate that IL-1 and TNF-α are the predominant catabolic cytokines involved in the destruction of the articular cartilage in RA [10, 66, 67]. The first recognition of IL-1 as a regulator of chondrocyte function stems largely from work in culture models showing that activities derived from synovium or monocyte-macrophages induce the production of cartilage-degrading proteinases (reviewed in [66]). IL-1 has the capacity to stimulate the production of most, if not all, of the proteinases involved in cartilage destruction and it colocalizes with TNF-α, MMP-1, MMP-3, MMP-8, and MMP-13, and type II collagen cleavage epitopes in regions of matrix depletion in RA cartilage [34, 57]. Originally known as cachectin, TNF-α produces many effects on chondrocytes in vitro that are similar to those of IL-1, including stimulation of the production of matrix-degrading proteinases and suppression of cartilage matrix synthesis. IL-1 is 100-fold to 1,000-fold more potent on a molar basis than TNF-α, but strong synergistic effects occur at low concentrations of the two cytokines together [10].

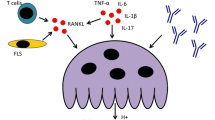
Cytokine networks and cellular interactions in cartilage destruction in rheumatoid arthritis. This scheme represents the progressive destruction of the cartilage associated with the invading synovial pannus in rheumatoid arthritis. As a result of immune cell interactions involving T and B lymphocytes, monocyte/macrophages, and dendritic cells, several different cytokines are produced in the inflamed synovium as a result of the influx of inflammatory cells from the circulation and synovial cell hyperplasia. The upregulation of proinflammatory cytokines produced primarily in the synovium, but also by chondrocytes, results in the upregulation of cartilage-degrading enzymes, of the matrix metalloproteinase (MMP) and ADAM with thrombospondin-1 domains (ADAMTS) families, at the cartilage–pannus junction. Chemokines, nitric oxide (NO), and prostaglandins (PGs) also contribute to the inflammation and tissue catabolism. SDF, stromal cell-derived factor 1; TNF, tumor necrosis factor; TGF, transforming growth factor; IFN, interferon; Treg, regulatory T lymphocytes; Th, T helper cells.
The concept that TNF-α drives acute inflammation, whereas IL-1 has a pivotal role in sustaining both inflammation and cartilage erosion, has been derived from work in transgenic or knockout mouse models [67]. For example, the spontaneous development of a chronic destructive arthritis in mice deficient in IL-1 receptor antagonist (IL-1Ra) established the importance of IL-1 in arthritis [68]. In the original study showing that transgenic or dysregulated overexpression of the TNF-α in causes polyarthritis in mice, chondrocytes were found to express the human transgene [69]. When backcrossed with arthritis-susceptible DBA/1 mice, a more severe, erosive arthritis developed during successive generations [70]. Because few chondrocytes remained in older mice with advanced arthritis and the extracellular matrix of the cartilage was relatively preserved, it was proposed that the chondrocytes may die early in the life of the mice by TNF-α-driven apoptosis before significant proteoglycan degradation can occur [70]. The higher potency of IL-1 compared with TNF-α in driving cartilage erosion is supported by studies showing that blockade of IL-1 is more effective than TNF-α neutralization in CIA mice [71] and that IL-1 is a secondary mediator in TNF-α transgenic mice [72]. Later studies in the human RA/SCID (severe combined immunodeficiency) mouse chimera indicated that TNF-α is a key molecule in the inflammatory changes that occur in the rheumatoid synovium, whereas cartilage damage occurs independently of this cytokine [73]. Despite these findings in animal models, anti-TNF therapy in patients with RA has been more successful in preventing cartilage and bone destruction. This could be related to the pharmacokinetic properties of IL-1Ra. It has been suggested that alternative approaches for targeting IL-1, including the use of soluble receptors and neutralizing antibodies, need to be tested [67, 74]. Supporting the concept that IL-1 drives cartilage destruction are the findings of a recent study by Schett's group in which crossing the arthritic human TNF transgenic (hTNFtg) mice with mice deficient in IL-1α and IL-1β protected against cartilage erosion without affecting synovial inflammation [75].
Cytokine networks
IL-1 and TNF-α can also induce chondrocytes to produce several other proinflammatory cytokines, including IL-6, leukemia inhibitory factor (LIF), IL-17, and IL-18, and chemokines [76, 77] (Fig. 1). IL-6 seems to perform a dual function by increasing products that downregulate inflammation such as IL-1Ra, soluble TNF receptor (sTNFR), and TIMPs, while also enhancing immune cell function and inflammation [41, 78]. The inhibition of proteoglycan synthesis and other chondro-cyte responses in vitro require the soluble IL-6 receptor α (sIL-6Rα), which permits the synergistic stimulation of MMP expression by IL-1 and IL-6 [79]. IL-6 blockade is under current investigation in animal models and clinical trials [80, 81]. The use of the IL-6 gene promoter as an inducible adenoviral gene delivery system proposed for the local treatment of arthritis would presumably target cartilage destruction as well as inflammation [82]. Other members of the IL-6 family that act through receptors that heterodimerize with gp130 may also modulate chondrocyte function. IL-11 shares several actions of IL-6, including the stimulation of TIMP production without affecting MMP production [79] and may actually inhibit cartilage destruction [83]. Leukemia inhibitory factor (LIF), similarly to the other chondrocyte-derived autocrine factors described above, may participate in a positive feedback loop by increasing the production of IL-6 by chondrocytes. Oncostatin M (OSM), which is a product of macrophages and activated T cells, can act alone or synergistically with IL-1 to stimulate the production of MMPs and aggrecanases by chondrocytes [38, 79, 84]. Direct evidence supporting a role for OSM in contributing to cartilage loss in inflammatory arthritis is provided by studies in animal models [85, 86].
IL-17A, one of at least six family members, is primarily a product of T helper type 17 (Th17) cells, a newly described subset of T cells, which is a potent inducer of catabolic responses in chondrocytes by itself or in synergy with other cytokines [87, 88]. IL-17 can drive T-cell-dependent erosive arthritis in the TNF-deficient and IL-1Ra knockout mice, and treatment of mice with CIA or AIA with neutralizing IL-17 antibody effectively inhibits cartilage destruction in those models of RA [89–92].
The IL-1R/Toll-like receptor (TLR) superfamily of receptors has a key role in innate immunity and inflammation. Studies in arthritis induced with streptococcal cell wall showed that joint inflammation and cartilage proteoglycan loss is predominantly dependent on TLR-2 signaling [93]. Human articular chondrocytes can express TLR-1, TLR-2, and TLR-4, and activation of TLR-2 by IL-1, TNF-α, peptidoglycans, lipopolysaccharide, or fibronectin fragments increases the production of MMPs, nitric oxide (NO), prostaglandin E (PGE), and VEGF [94–96]. In arthritis mediated by immune complex, TLR-4 regulates early-onset inflammation and cartilage destruction by IL-10-mediated upregulation of Fcγ receptor expression and enhanced production of cytokines [97]. Because the IL-18 receptor shares homology with IL-1RI and has a TLR signaling domain, therapeutic strategies similar to those for targeting IL-1 signaling have been explored [78, 98]. In animal models, IL-18, by means of TLR-2, promotes joint inflammation in a partly TNF-α-dependent manner and induces IL-1-driven cartilage destruction [99]. IL-18 has effects similar to IL-1 in human chondrocytes, and stimulates chondrocyte apoptosis, although studies do not suggest a pivotal role in cartilage destruction in RA [100–102]. Of the other members of the IL-1 family recently identified by DNA database searches, IL-1F8 seems to be capable of stimulating the production of IL-6, IL-8, and NO by human chondrocytes, but at 100-fold to 1,000-fold higher concentrations than that of IL-1 [103]. IL-32, a recently discovered cytokine that induces TNF-α, IL-1β, IL-6, and chemokines and is expressed in the synovia of patients with RA, contributes to TNF-α-dependent inflammation and a loss of cartilage proteoglycan [104].
IL-4, IL-10, and IL-13 are generally classified as inhibitory or modulatory cytokines because they are able to inhibit many of the cartilage catabolic processes induced by proinflammatory cytokines [105]. Their therapeutic application has been proposed to restore the cytokine balance in RA [106, 107]. The efficacy of IL-4, IL-10, and IL-13 in retarding cartilage damage may be related, in part, to their stimulatory effects on IL-1Ra production [108, 109]. Despite the capacity of IL-4 to inhibit the effects of proinflammatory cytokines on chondrocyte function [110, 111], differential effects have been observed in mice, depending on the model used [112, 113]. Gene transfer of IL-10 in combination with IL-1Ra inhibits cartilage destruction by a mechanism involving activin, a TGF-β family member [114]. IL-10 is part of the response induced by immunomodulatory neuropeptides that have recently been shown to inhibit inflammation and cartilage and bone destruction by downregulating the Th1-driven immune response and upregulating IL-10/TGF-β-producing regulatory T (Treg) lymphocytes [115]. IL-13 decreases the breakdown of collagen and proteoglycans by inhibiting IL-1- and OSM-induced MMP-3 and MMP-13 expression [116]. Local gene transfer of IL-13 inhibits chondrocyte death and MMP-mediated cartilage degradation despite enhanced inflammation in the immune-complex arthritis model [117].
Mediators and mechanisms in the responses of chondrocytes to inflammatory cytokines
In addition to inducing the synthesis of MMPs and other proteinases by chondrocytes, IL-1 and TNF-α upregulate the production of NO by means of inducible nitric oxide synthetase (iNOS, or NOS2), and that of PGE2 by stimulating the expression or activities of cyclooxygenase (COX)-2, microsomal PGE synthase-1 (mPGES-1), and soluble phospholipase A2 (sPLA2). Although PGE2 and NO have been well characterized as proinflammatory mediators, there is evidence of crosstalk between them in the regulation of chondrocyte function (reviewed in [118]). COX-2 is also involved in the chondrocyte response to high shear stress, associated with decreased antioxidant capacity and increased apoptosis [119]. In the production of prosta-glandins, mPGES-1, which is induced by IL-1 in chondro-cytes, is a major player [120, 121]. In addition to opposing the induction of COX-2, iNOS, and MMPs and the suppression of aggrecan synthesis by IL-1, activators of the peroxisome proliferator-activated receptor γ (PPAR-γ), including the endogenous ligand 15-deoxy-Δ12,14-prostaglandin J2 (PGJ2), inhibit the IL-1-induced expression of mPGES-1 [122, 123]. Recent evidence indicates that PPAR-α agonists may protect chondrocytes against IL-1-induced responses by increasing the expression of IL-1Ra [124].
Adipokines, which were originally identified as products of adipocytes, have recently been shown to have roles in cartilage metabolism [125]. White adipose tissue has been proposed as a major source of both proinflammatory and anti-inflammatory cytokines, including IL-Ra and IL-10 [126]. Leptin expression is enhanced during acute inflammation, correlating negatively with inflammatory markers in RA sera [127], and has been proposed to serve as a link between the neuroendocrine and immune systems [128]. The elevated expression of leptin in OA cartilage and in osteophytes, and its capacity to stimulate insulin-like growth factor (IGF)-1 and TGF-β1 synthesis, suggest a role for this adipokine in anabolic responses of chondrocytes [129]. Leptin synergizes with IL-1 or interferon-γ to increase NO production in chondrocytes [130], and leptin deficiency attenuates inflammatory processes in experimental arthritis [131]. It has been proposed that the dysregulated balance between leptin and other adipokines, such as adiponectin, promotes destructive inflammatory processes [132].
Several additional mediators that affect chondrocyte metabolism have been described. The IL-1-induced SOCS3 (suppressor of cytokine signaling 3) acts as a negative feedback regulator during desensitization toward IGF-1 in the absence of NO by inhibiting the phosphorylation of insulin receptor substrate (IRS)-1 [133]. Recent evidence indicates that RAGE, the receptor for advanced glycation end products (AGEs), interacts preferentially with S100A4, a member of the S100 family of calcium-binding proteins, in chondrocytes and stimulates MMP-13 production through the phosphorylation of Pyk2, mitogen-activated protein kinases, and NF-κB [134]. The fibroblast activation protein α (FAP-α), a membrane serine proteinase, which colocalizes in synovium with MMP-1 and MMP-13 and is induced by IL-1 and OSM in chondrocytes, may have a role in collagen degradation [135, 136]. Many of these proteins may be activated during the chondrocyte response to abnormal stimuli and may serve as endogenous mediators of cellular responses to stress and inflammation.
Signaling mechanisms, gene transcription, and genome analyses
Signal transduction molecules and transcription factors activated by inflammatory mediators in chondrocytes and synovial cells have been studied to identify potential therapeutic targets. For example, NF-κB is a 'master switch' of the inflammatory cascade [137], and the signaling intermediates in the p38 and JNK pathways have also been targeted for future therapeutic development [138]. In addition to NF-κB, members of the CCAAT-enhancer-binding protein (C/EBP), Ets, and activator protein (AP)-1 families are important for the regulation of gene expression by IL-1 and TNF-α [43, 139–142] and have been localized in rheumatoid tissues [143, 144]. The JAK/Stat3 signaling pathway is important for signaling by gp130 cytokines [145]. Cytokine-induced transcription factors also suppress the expression of several genes associated with the differentiated chondrocyte phenotype, including type II collagen (COL2A1), aggrecan, and CD-RAP [146–148]. Chondrocyte-specific transcription factors, including Sox9 (which regulates cartilage formation during development [139]), have not been studied in the context of cartilage metabolism in RA. Genomic and proteomic analyses that have been performed in cytokine-treated chondrocytes, in cartilage from patients with osteoarthritis, and in rheumatoid synovium have provided some insights into novel mechanisms that might govern chondrocyte responses in RA [149–154]. So far, more than 1,000 differentially expressed transcripts have been identified in cartilage derived from patients with arthritis [155].
Chemokines
The role of chemokines in RA synovium, where they are involved in neutrophil activation, chemotaxis, and angio-genesis, is well established, but their potential contribution to cartilage metabolism has been recognized only recently [156–159]. IL-8, probably the most potent and abundant chemotactic agent in RA synovial fluids, and other chemo-kines, such as monocyte chemoattractant protein (MCP)-1 and RANTES, are produced primarily by the synovium and serve as indicators of synovitis. Chondrocytes, when activated by IL-1 and TNF-α, express several chemokines, including IL-8, MCP-1, and MCP-4, macrophage inflammatory protein (MIP)-1α, MIP-1β, RANTES, and GROα, as well as receptors that enable responses to some of these chemokines, and may feedback regulate synovial cell responses [160, 161]. High levels of stromal cell-derived factor 1 (SDF-1) are detected in RA synovial fluids, and its receptor, CXCR4, is expressed by chondrocytes but not synovial fibroblasts, suggesting a direct influence of this chemokine on cartilage damage [162]. Microarray studies have elucidated several chemokines that are inducible in chondrocytes by fibronectin fragments and cytokines [154].
Adhesion molecules and angiogenesis
In addition to the requirement of chemokines for the recruitment of T lymphocytes and other inflammatory cells to the subsynovial lining, adhesion receptors must be available on synovial blood vessels for binding the circulating leukocytes and other cell types with which they interact in the inflamed tissue, including macrophages, dendritic cells, and fibro-blasts. The principal families of adhesion molecules involved are the selectins, the integrins, the cadherins, and variants of the immunoglobulin supergene family. Although these molecules are common to different inflammatory sites, many of the prominent adhesion proteins expressed in the inflamed rheumatoid synovium are also expressed in cartilage. For example, vascular cell adhesion molecule (VCAM)-1 and intercellular adhesion molecule (ICAM)-1, which are members of the immunoglobulin family, are expressed by human articular chondrocytes as well as synovial and endothelial cells, although their function on chondrocytes may not be significant unless damage to the matrix permits cell-cell interactions [163]. VCAM-1, as well as VEGF, fibroblast growth factor (FGF), and TNF-α, contributes to angiogenesis during synovitis and to the activation of chondrocytes during cartilage degradation [164, 165]. VEGF expression is upregulated by inflammatory cytokines in both chondrocytes and synovial cells and by hypoxia [166, 167], and Vegfb knockout mice are protected against synovial angiogenesis in the CIA and AIA models [168].
Several members of the integrin family are expressed by chondrocytes. The α 1β1 and α 5β1 integrins function as receptors for fragments of collagen and fibronectin, respectively. The stimulation of α 5β1 integrin by integrin-activating antibodies or fibronectin fragments results in increased MMP production and requires reactive oxygen species [169]. In contrast, the discoidin domain receptor-2 specifically increases MMP-13 production by recognizing intact type II collagen fibrils that have been denuded by proteoglycans, as occurs in osteoarthritis [170, 171], but its role in RA has not been determined. Specific roles for the hyaluronan receptor, CD44, in cell–matrix interactions in cartilage have also been identified [172]. CD44 expression is upregulated on chondrocytes in articular cartilage and synoviocytes from patients with RA [173, 174]. Hyaluronan binding to CD44 increases MMP-13 and NO production by chondrocytes [175]. Furthermore, induction of MMP-specific cleavage of type II collagen and NO production by the heparin-binding fragment of fibronectin is mediated by CD44 [176].
Cadherins are adhesion molecules that mediate cell–cell adhesion by binding a cadherin of the same cell type on an adjacent cell. The recent identification of cadherin-11 as a key adhesion molecule, which regulates the formation of the synovial lining during development and the synoviocyte function postnatally, has provided the opportunity to examine its role in inflammatory joint disease [177]. Cadherin-11 deficiency, or treatment with cadherin-11 antibody or a cadherin-11 fusion protein, decreased synovial inflammation and decreased cartilage erosion in an animal model of arthritis. Furthermore, cadherin-11 facilitated synoviocyte invasion into cartilage-like extracellular matrix in an in vitro model, suggesting that this molecule could serve as a specific target for therapy against cartilage destruction in inflammatory arthritis [178].
Bone-related factors
The potent induction by IL-17 of receptor activator of NF-κB ligand (RANKL), which is produced by synoviocytes and T cells in RA synovium [179] and mediates osteoclast differentiation and activity, may partly account for the capacity of IL-17 to induce bone destruction in an IL-1-independent manner and bypass the requirement for TNF in the development of inflammatory arthritis [88]. Both RANKL and its receptor RANK, a member of the TNF receptor family, are expressed in adult articular chondrocytes [180], but a direct action in cartilage has not yet been identified. Although RANKL deficiency blocks bone destruction without direct effects on cartilage destruction in inflammatory models, it is possible that indirect cartilage-protective effects may occur through interference with the degradation of subchondral bone [179, 181, 182].
Wnt signaling, through the canonical β-catenin pathway and activation of T-cell factor (TCF)/Lef transcription factors, functions in a cell-autonomous manner to induce osteoblast differentiation and suppress chondrocyte differentiation in early osteo-chondroprogenitors [183]. During chondrogenesis, Wnt/β-catenin acts at two stages, at low levels to promote chondroprogenitor differentiation and later at high levels to promote chondrocyte hypertrophic differentiation and subsequent endochondral ossification [183, 184]. Because ectopic Wnt/β-catenin signaling leads to enhanced ossification and the suppression of cartilage formation during skeletal development, the disruption of Wnt signaling in adult cartilage would be expected to have pathological consequences. For example, activation of β-catenin in mature cartilage cells stimulates hypertrophy, matrix mineralization, and expression of VEGF, ADAMTS5, MMP-13, and several other MMPs [184]. A recent study showed limited expression of β-catenin in joint tissues of patients with RA, but high expression of the inhibitor of Wnt/β-catenin signaling, DKK-1, in the inflamed synovium, especially in the synoviocytes and synovial microvessels, and in cartilage adjacent to inflammatory tissue [185]. This study also showed expression of DKK-1 in a TNF-α-dependent manner in TNF transgenic mice and blockade of RANKL-dependent bone resorption by the administration of DKK-1 antibody, as a result of upregulation of the RANKL inhibitor osteoprotegerin [185] (reviewed in [186]).
Conclusion
Significant advances have been made in recent years that have contributed to our understanding of the cellular interactions in the RA joint involving macrophages, T and B lymphocytes, and synovial fibroblasts. Laboratory investigations in vitro and in vivo have resulted in new findings about the role of the chondrocyte in remodeling the cartilage matrix in the RA joint. Although the mediators involved in immunomodulation and synovial cell function, including cytokines, chemokines, and adhesion molecules, have primary roles in the inflammatory and catabolic processes in the joint, they may also promote cartilage damage, directly or indirectly. Despite the clinical success of anti-TNF therapy for RA, there is still a need for therapeutic strategies that prevent the extensive cartilage and bone loss. Recent work that has identified novel molecules and mechanisms, as well as providing new understanding of the contributions of known mediators, offers the possibility of developing new therapies for targeting cartilage destruction in inflammatory joint disease.
Note
This review is part of a series on Cells of the synovium in rheumatoid arthritis edited by Gary Firestein.
Other articles in this series can be found at http://arthritis-research.com/articles/review-series.asp?series=ar_Cells
Abbreviations
- AIA:
-
= antigen-induced arthritis
- ADAM:
-
= a disintegrin and metalloproteinase
- ADAMTS:
-
= ADAM with thrombospondin-1 domains
- CD-RAP:
-
= cartilage-derived retinoic-acid-sensitive protein
- CH3L1:
-
= chitinase 3-like protein 1
- CIA:
-
= collagen-induced arthritis
- COMP:
-
= cartilage oligomeric matrix protein
- COX:
-
= cyclooxygenase
- GLUT:
-
= glucose transporter protein
- HIF:
-
= hypoxia-inducible factor
- IGF:
-
= insulin-like growth factor
- IL:
-
= interleukin
- IL-1Ra:
-
= IL-1 receptor antagonist
- iNOS:
-
= inducible nitric oxide synthetase
- MCP:
-
= monocyte chemoattractant protein
- MIP:
-
= macrophage inflammatory protein
- MMP:
-
= matrix metalloproteinase
- mPGES-1:
-
= microsomal PGE synthase-1
- NF:
-
= nuclear factor
- OSM:
-
= onco-statin M
- PGE:
-
= prostaglandin E
- PPAR:
-
= peroxisome proliferator-activated receptor
- RA:
-
= rheumatoid arthritis
- RANKL:
-
= receptor activator of NF-κB ligand
- TGF:
-
= transforming growth factor
- Th:
-
= T helper
- TIMP:
-
= tissue inhibitor of metalloproteinases
- TLR:
-
= Toll-like receptor
- TNF:
-
= tumor necrosis factor
- TRAP:
-
= tartrate-resistant acid phosphatase
- VCAM:
-
= vascular cell adhesion molecule
- VEGF:
-
= vascular endothelial growth factor.
References
Tak PP, Bresnihan B: The pathogenesis and prevention of joint damage in rheumatoid arthritis: advances from synovial biopsy and tissue analysis. Arthritis Rheum. 2000, 43: 2619-2633. 10.1002/1529-0131(200012)43:12<2619::AID-ANR1>3.0.CO;2-V.
Firestein GS: Evolving concepts of rheumatoid arthritis. Nature. 2003, 423: 356-361. 10.1038/nature01661.
Meyer LH, Franssen L, Pap T: The role of mesenchymal cells in the pathophysiology of inflammatory arthritis. Best Pract Res Clin Rheumatol. 2006, 20: 969-981. 10.1016/j.berh.2006.06.005.
Knedla A, Neumann E, Muller-Ladner U: Developments in the synovial biology field 2006. Arthritis Res Ther. 2007, 9: 209-10.1186/ar2140.
Szekanecz Z, Koch AE: Macrophages and their products in rheumatoid arthritis. Curr Opin Rheumatol. 2007, 19: 289-295. 10.1097/BOR.0b013e32805e87ae.
Gravallese EM, Harada Y, Wang J-T, Gorn A, Thornhill T, Goldring SR: Identification of cell types responsible for bone resorption in rheumatoid arthritis and juvenile rheumatoid arthritis. Am J Pathol. 1998, 152: 943-951.
Pettit AR, Walsh NC, Manning C, Goldring SR, Gravallese EM: RANKL protein is expressed at the pannus-bone interface at sites of articular bone erosion in rheumatoid arthritis. Rheumatology (Oxford). 2006, 45: 1068-1076. 10.1093/rheumatology/kel045.
Mulherin D, Fitzgerald O, Bresnihan B: Clinical improvement and radiological deterioration in rheumatoid arthritis: evidence that the pathogenesis of synovial inflammation and articular erosion may differ. Br J Rheumatol. 1996, 35: 1263-1268. 10.1093/rheumatology/35.12.1263.
Kirwan J, Byron M, Watt I: The relationship between soft tissue swelling, joint space narrowing and erosive damage in hand X-rays of patients with rheumatoid arthritis. Rheumatology (Oxford). 2001, 40: 297-301. 10.1093/rheumatology/40.3.297.
van den Berg WB, van Riel PL: Uncoupling of inflammation and destruction in rheumatoid arthritis: myth or reality?. Arthritis Rheum. 2005, 52: 995-999. 10.1002/art.20981.
Graudal N: The natural history and prognosis of rheumatoid arthritis: association of radiographic outcome with process variables, joint motion and immune proteins. Scand J Rheumatol Suppl. 2004, 1-38. 10.1080/03009740310004847.
van Lent PL, Grevers L, Lubberts E, de Vries TJ, Nabbe KC, Verbeek S, Oppers B, Sloetjes A, Blom AB, van den Berg WB: Fcγ receptors directly mediate cartilage, but not bone, destruction in murine antigen-induced arthritis: uncoupling of cartilage damage from bone erosion and joint inflammation. Arthritis Rheum. 2006, 54: 3868-3877. 10.1002/art.22253.
Verstappen SM, Poole AR, Ionescu M, King LE, Abrahamowicz M, Hofman DM, Bijlsma JW, Lafeber FP: Radiographic joint damage in rheumatoid arthritis is associated with differences in cartilage turnover and can be predicted by serum biomarkers: an evaluation from 1 to 4 years after diagnosis. Arthritis Res Ther. 2006, 8: R31-10.1186/ar1882.
Goldring SR, Goldring MB: Rheumatoid arthritis and other inflammatory joint pathologies. Dynamics of Bone and Cartilage Metabolism. Edited by: Seibel MJ, Robins SP, Bilezikian JP. 2006, San Diego: Academic Press, 843-869. 2
Charni-Ben Tabassi N, Garnero P: Monitoring cartilage turnover. Curr Rheumatol Rep. 2007, 9: 16-24. 10.1007/s11926-007-0017-y.
Verzijl N, DeGroot J, Thorpe SR, Bank RA, Shaw JN, Lyons TJ, Bijlsma JW, Lafeber FP, Baynes JW, TeKoppele JM: Effect of collagen turnover on the accumulation of advanced glycation end products. J Biol Chem. 2000, 275: 39027-39031. 10.1074/jbc.M006700200.
Maroudas A, Bayliss MT, Uchitel-Kaushansky N, Schneiderman R, Gilav E: Aggrecan turnover in human articular cartilage: use of aspartic acid racemization as a marker of molecular age. Arch Biochem Biophys. 1998, 350: 61-71. 10.1006/abbi.1997.0492.
Shikhman AR, Brinson DC, Lotz MK: Distinct pathways regulate facilitated glucose transport in human articular chondrocytes during anabolic and catabolic responses. Am J Physiol Endocrinol Metab. 2004, 286: E980-E985. 10.1152/ajpendo.00243.2003.
Mobasheri A, Richardson S, Mobasheri R, Shakibaei M, Hoyland JA: Hypoxia inducible factor-1 and facilitative glucose transporters GLUT1 and GLUT3: putative molecular components of the oxygen and glucose sensing apparatus in articular chondrocytes. Histol Histopathol. 2005, 20: 1327-1338.
Wilkins RJ, Browning JA, Ellory JC: Surviving in a matrix: membrane transport in articular chondrocytes. J Membr Biol. 2000, 177: 95-108. 10.1007/s002320001103.
Pufe T, Kurz B, Petersen W, Varoga D, Mentlein R, Kulow S, Lemke A, Tillmann B: The influence of biomechanical parameters on the expression of VEGF and endostatin in the bone and joint system. Ann Anat. 2005, 187: 461-472.
Lin C, McGough R, Aswad B, Block JA, Terek R: Hypoxia induces HIF-1α and VEGF expression in chondrosarcoma cells and chondrocytes. J Orthop Res. 2004, 22: 1175-1181. 10.1016/j.orthres.2004.03.002.
McNulty AL, Stabler TV, Vail TP, McDaniel GE, Kraus VB: Dehydroascorbate transport in human chondrocytes is regulated by hypoxia and is a physiologically relevant source of ascorbic acid in the joint. Arthritis Rheum. 2005, 52: 2676-2685. 10.1002/art.21254.
Robins JC, Akeno N, Mukherjee A, Dalal RR, Aronow BJ, Koopman P, Clemens TL: Hypoxia induces chondrocyte-specific gene expression in mesenchymal cells in association with transcriptional activation of Sox9. Bone. 2005, 37: 313-322. 10.1016/j.bone.2005.04.040.
Kobayashi I, Ziff M: Electron microscopic studies of the cartilage–pannus junction in rheumatoid arthritis. Arthritis Rheum. 1975, 18: 475-483. 10.1002/art.1780180507.
Woolley DE, Crossley MJ, Evanson JM: Collagenase at sites of cartilage erosion in the rheumatoid joint. Arthritis Rheum. 1977, 20: 1231-1239. 10.1002/art.1780200612.
Edwards JCW: Fibroblast biology. Development and differentiation of synovial fibroblasts in arthritis. Arthritis Res. 2000, 2: 344-347. 10.1186/ar110.
Kimura H, Tateishi HJ, Ziff M: Surface ultrastructure of rheumatoid articular cartilage. Arthritis Rheum. 1977, 20: 1085-1098. 10.1002/art.1780200508.
Dodge GR, Poole AR: Immunohistochemical detection and immunochemical analysis of type II collagen degradation in human normal, rheumatoid, and osteoarthritic articular cartilages and in explants of bovine articular cartilage cultured with interleukin 1. J Clin Invest. 1989, 83: 647-661.
Mitchell NS, Shepard N: Changes in proteoglycan and collagen in cartilage in rheumatoid arthritis. J Bone Joint Surg AM. 1978, 60: 342-348.
Dodge GR, Pidoux I, Poole AR: The degradation of type II collagen in rheumatoid arthritis: an immunoelectron microscopic study. Matrix. 1991, 11: 330-338.
Murphy G, Lee MH: What are the roles of metalloproteinases in cartilage and bone damage?. Ann Rheum Dis. 2005, 64 (Suppl 4): iv44-iv47. 10.1136/ard.2005.042465.
Hembry RM, Bagga MR, Reynolds JJ, Hamblen DL: Immunolocalisation studies on six matrix metalloproteinases and their inhibitors, TIMP-1 and TIMP-2, in synovia from patients with osteo- and rheumatoid arthritis. Ann Rheum Dis. 1995, 54: 25-32.
Tetlow LC, Woolley DE: Comparative immunolocalization studies of collagenase 1 and collagenase 3 production in the rheumatoid lesion, and by human chondrocytes and synovio-cytes in vitro. Br J Rheumatol. 1998, 37: 64-70. 10.1093/rheumatology/37.1.64.
Cunnane G, FitzGerald O, Hummel KM, Youssef PP, Gay RE, Gay S, Bresnihan B: Synovial tissue protease gene expression and joint erosions in early rheumatoid arthritis. Arthritis Rheum. 2001, 44: 1744-1753. 10.1002/1529-0131(200108)44:8<1744::AID-ART309>3.0.CO;2-K.
Woolley DE, Tetlow LC: Observations on the microenvironmental nature of cartilage degradation in rheumatoid arthritis. Ann Rheum Dis. 1997, 56: 151-161.
Ainola MM, Mandelin JA, Liljestrom MP, Li TF, Hukkanen MV, Konttinen YT: Pannus invasion and cartilage degradation in rheumatoid arthritis: involvement of MMP-3 and interleukin-1β. Clin Exp Rheumatol. 2005, 23: 644-650.
Barksby HE, Milner JM, Patterson AM, Peake NJ, Hui W, Robson T, Lakey R, Middleton J, Cawston TE, Richards CD, et al: Matrix metalloproteinase 10 promotion of collagenolysis via procollagenase activation: implications for cartilage degradation in arthritis. Arthritis Rheum. 2006, 54: 3244-3253. 10.1002/art.22167.
Rutkauskaite E, Volkmer D, Shigeyama Y, Schedel J, Pap G, Muller-Ladner U, Meinecke I, Alexander D, Gay RE, Drynda S, et al: Retroviral gene transfer of an antisense construct against membrane type 1 matrix metalloproteinase reduces the invasiveness of rheumatoid arthritis synovial fibroblasts. Arthritis Rheum. 2005, 52: 2010-2014. 10.1002/art.21156.
Kevorkian L, Young DA, Darrah C, Donell ST, Shepstone L, Porter S, Brockbank SM, Edwards DR, Parker AE, Clark IM: Expression profiling of metalloproteinases and their inhibitors in cartilage. Arthritis Rheum. 2004, 50: 131-141. 10.1002/art.11433.
Cawston TE, Wilson AJ: Understanding the role of tissue degrading enzymes and their inhibitors in development and disease. Best Pract Res Clin Rheumatol. 2006, 20: 983-1002. 10.1016/j.berh.2006.06.007.
Patel IR, Attur MG, Patel RN, Stuchin SA, Abagyan RA, Abramson SB, Amin AR: TNF-α convertase enzyme from human arthritis-affected cartilage: isolation of cDNA by differential display, expression of the active enzyme, and regulation of TNF-α. J Immunol. 1998, 160: 4570-4579.
Burrage PS, Huntington JT, Sporn MB, Brinckerhoff CE: Regulation of matrix metalloproteinase gene expression by a retinoid X receptor-specific ligand. Arthritis Rheum. 2007, 56: 892-904. 10.1002/art.22417.
Overall CM, Blobel CP: In search of partners: linking extracellular proteases to substrates. Nat Rev Mol Cell Biol. 2007, 8: 245-257. 10.1038/nrm2120.
Arner EC: Aggrecanase-mediated cartilage degradation. Curr Opin Pharmacol. 2002, 2: 322-329. 10.1016/S1471-4892(02)00148-0.
Plaas A, Osborn B, Yoshihara Y, Bai Y, Bloom T, Nelson F, Mikecz K, Sandy JD: Aggrecanolysis in human osteoarthritis: confocal localization and biochemical characterization of ADAMTS5–hyaluronan complexes in articular cartilages. Osteoarthritis Cartilage. 2007, 15: 719-734. 10.1016/j.joca.2006.12.008.
Sandy JD: A contentious issue finds some clarity: on the independent and complementary roles of aggrecanase activity and MMP activity in human joint aggrecanolysis. Osteoarthritis Cartilage. 2006, 14: 95-100. 10.1016/j.joca.2005.09.004.
Stanton H, Rogerson FM, East CJ, Golub SB, Lawlor KE, Meeker CT, Little CB, Last K, Farmer PJ, Campbell IK, et al: ADAMTS5 is the major aggrecanase in mouse cartilage in vivo and in vitro. Nature. 2005, 434: 648-652. 10.1038/nature03417.
Glasson SS, Askew R, Sheppard B, Carito B, Blanchet T, Ma HL, Flannery CR, Peluso D, Kanki K, Yang Z, et al: Deletion of active ADAMTS5 prevents cartilage degradation in a murine model of osteoarthritis. Nature. 2005, 434: 644-648. 10.1038/nature03369.
Kashiwagi M, Tortorella M, Nagase H, Brew K: TIMP-3 is a potent inhibitor of aggrecanase 1 (ADAM-TS4) and aggrecanase 2 (ADAM-TS5). J Biol Chem. 2001, 276: 12501-12504. 10.1074/jbc.C000848200.
Hui W, Cawston T, Rowan AD: Transforming growth factor β1 and insulin-like growth factor 1 block collagen degradation induced by oncostatin M in combination with tumour necrosis factor α from bovine cartilage. Ann Rheum Dis. 2003, 62: 172-174. 10.1136/ard.62.2.172.
El Mabrouk M, Qureshi HY, Li WQ, Sylvester J, Zafarullah M: Interleukin-4 antagonizes oncostatin M and transforming growth factor β-induced responses in articular chondrocytes. J Cell Biochem. 2007, [epub ahead of print, June 1]
Yamanishi Y, Firestein GS: Pathogenesis of rheumatoid arthritis: the role of synoviocytes. Rheum Dis Clin North Am. 2001, 27: 355-371. 10.1016/S0889-857X(05)70206-4.
Nagase H, Kashiwagi M: Aggrecanases and cartilage matrix degradation. Arthritis Res Ther. 2003, 5: 94-103. 10.1186/ar630.
Hou WS, Li W, Keyszer G, Weber E, Levy R, Klein MJ, Gravallese EM, Goldring SR, Bromme D: Comparison of cathepsins K and S expression within the rheumatoid and osteoarthritic synovium. Arthritis Rheum. 2002, 46: 663-674. 10.1002/art.10114.
Salminen-Mankonen HJ, Morko J, Vuorio E: Role of cathepsin K in normal joints and in the development of arthritis. Curr Drug Targets. 2007, 8: 315-323. 10.2174/138945007779940188.
Fraser A, Fearon U, Billinghurst RC, Ionescu M, Reece R, Barwick T, Emery P, Poole AR, Veale DJ: Turnover of type II collagen and aggrecan in cartilage matrix at the onset of inflammatory arthritis in humans: relationship to mediators of systemic and local inflammation. Arthritis Rheum. 2003, 48: 3085-3095. 10.1002/art.11331.
Lindqvist E, Eberhardt K, Bendtzen K, Heinegard D, Saxne T: Prognostic laboratory markers of joint damage in rheumatoid arthritis. Ann Rheum Dis. 2005, 64: 196-201. 10.1136/ard.2003.019992.
Baeten D, Steenbakkers PG, Rijnders AM, Boots AM, Veys EM, De Keyser F: Detection of major histocompatibility complex/ human cartilage gp-39 complexes in rheumatoid arthritis synovitis as a specific and independent histologic marker. Arthritis Rheum. 2004, 50: 444-451. 10.1002/art.20012.
van Bilsen JH, van Dongen H, Lard LR, van der Voort EI, Elferink DG, Bakker AM, Miltenburg AM, Huizinga TW, de Vries RR, Toes RE: Functional regulatory immune responses against human cartilage glycoprotein-39 in health vs. proinflammatory responses in rheumatoid arthritis. Proc Natl Acad Sci USA. 2004, 101: 17180-17185. 10.1073/pnas.0407704101.
Ling H, Recklies AD: The chitinase 3-like protein human cartilage glycoprotein 39 inhibits cellular responses to the inflammatory cytokines interleukin-1 and tumour necrosis factor-α. Biochem J. 2004, 380: 651-659. 10.1042/BJ20040099.
Recklies AD, Ling H, White C, Bernier SM: Inflammatory cytokines induce production of CHI3L1 by articular chondrocytes. J Biol Chem. 2005, 280: 41213-41221. 10.1074/jbc.M510146200.
Sekine T, Masuko-Hongo K, Matsui T, Asahara H, Takigawa M, Nishioka K, Kato T: Recognition of YKL-39, a human cartilage related protein, as a target antigen in patients with rheumatoid arthritis. Ann Rheum Dis. 2001, 60: 49-54. 10.1136/ard.60.1.49.
Knorr T, Obermayr F, Bartnik E, Zien A, Aigner T: YKL-39 (chitinase 3-like protein 2), but not YKL-40 (chitinase 3-like protein 1), is up regulated in osteoarthritic chondrocytes. Ann Rheum Dis. 2003, 62: 995-998. 10.1136/ard.62.10.995.
Saito S, Kondo S, Mishima S, Ishiguro N, Hasegawa Y, Sandell LJ, Iwata H: Analysis of cartilage-derived retinoic-acid-sensitive protein (CD-RAP) in synovial fluid from patients with osteoarthritis and rheumatoid arthritis. J Bone Joint Surg Br. 2002, 84: 1066-1069. 10.1302/0301-620X.84B7.12177.
Dayer JM: The process of identifying and understanding cytokines: from basic studies to treating rheumatic diseases. Best Pract Res Clin Rheumatol. 2004, 18: 31-45. 10.1016/j.berh.2003.09.009.
van den Berg WB: Animal models of arthritis. What have we learned?. J Rheumatol Suppl. 2005, 72: 7-9.
Horai R, Saijo S, Tanioka J, Nakae S, Sudo K, Okahara A, Ikuse T, Asano M, Iwakura Y: Development of chronic inflammatory arthropathy resembling rheumatoid arthritis in interleukin 1 receptor antagonist-deficient mice. J Exp Med. 2000, 191: 313-320. 10.1084/jem.191.2.313.
Keffer J, Probert L, Cazlaris H, Georgopoulos S, Kaslaris E, Kioussis D, Kollias G: Transgenic mice expressing human tumour necrosis factor: a predictive genetic model of arthritis. EMBO J. 1991, 10: 4025-4031.
Butler DM, Malfait AM, Mason LJ, Warden PJ, Kollias G, Maini RN, Feldmann M, Brennan FM: DBA/1 mice expressing the human TNF-α transgene develop a severe, erosive arthritis: characterization of the cytokine cascade and cellular composition. J Immunol. 1997, 159: 2867-2876.
Joosten LA, Helsen MM, van de Loo FA, van den Berg WB: Anti-cytokine treatment of established type II collagen-induced arthritis in DBA/1 mice. A comparative study using anti-TNFα, anti-IL-1α/β, and IL-1Ra. Arthritis Rheum. 1996, 39: 797-809. 10.1002/art.1780390513.
Probert L, Plows D, Kontogeorgos G, Kollias G: The type I interleukin-1 receptor acts in series with tumor necrosis factor (TNF) to induce arthritis in TNF-transgenic mice. Eur J Immunol. 1995, 25: 1794-1797. 10.1002/eji.1830250647.
Matsuno H, Yudoh K, Katayama R, Nakazawa F, Uzuki M, Sawai T, Yonezawa T, Saeki Y, Panayi GS, Pitzalis C, et al: The role of TNF-α in the pathogenesis of inflammation and joint destruction in rheumatoid arthritis (RA): a study using a human RA/ SCID mouse chimera. Rheumatology (Oxford). 2002, 41: 329-337. 10.1093/rheumatology/41.3.329.
Burger D, Dayer JM, Palmer G, Gabay C: Is IL-1 a good therapeutic target in the treatment of arthritis?. Best Pract Res Clin Rheumatol. 2006, 20: 879-896. 10.1016/j.berh.2006.06.004.
Zwerina J, Redlich K, Polzer K, Joosten L, Kroenke G, Distler J, Hess A, Pundt N, Pap T, Hoffmann O, et al: TNF-induced structural joint damage is mediated by IL-1. Proc Natl Acad Sci USA. 2007, 104: 11742-11747. 10.1073/pnas.0610812104.
Goldring SR, Goldring MB: The role of cytokines in cartilage matrix degeneration in osteoarthritis. Clin Orthop Relat Res. 2004, 427 (Suppl): S27-S36. 10.1097/01.blo.0000144854.66565.8f.
Aida Y, Maeno M, Suzuki N, Namba A, Motohashi M, Matsumoto M, Makimura M, Matsumura H: The effect of IL-1β on the expression of inflammatory cytokines and their receptors in human chondrocytes. Life Sci. 2006, 79: 764-771. 10.1016/j.lfs.2006.02.038.
Connell L, McInnes IB: New cytokine targets in inflammatory rheumatic diseases. Best Pract Res Clin Rheumatol. 2006, 20: 865-878. 10.1016/j.berh.2006.05.011.
Rowan AD, Koshy PJ, Shingleton WD, Degnan BA, Heath JK, Vernallis AB, Spaull JR, Life PF, Hudson K, Cawston TE: Synergistic effects of glycoprotein 130 binding cytokines in combination with interleukin-1 on cartilage collagen breakdown. Arthritis Rheum. 2001, 44: 1620-1632. 10.1002/1529-0131(200107)44:7<1620::AID-ART285>3.0.CO;2-B.
Choy E: Clinical experience with inhibition of interleukin-6. Rheum Dis Clin North Am. 2004, 30: 405-415. 10.1016/j.rdc.2004.01.008.
Nishimoto N, Kishimoto T: Interleukin 6: from bench to bedside. Nat Clin Pract Rheumatol. 2006, 2: 619-626. 10.1038/ncprheum0338.
van de Loo FA, de Hooge AS, Smeets RL, Bakker AC, Bennink MB, Arntz OJ, Joosten LA, van Beuningen HM, van der Kraan PK, Varley AW, et al: An inflammation-inducible adenoviral expression system for local treatment of the arthritic joint. Gene Ther. 2004, 11: 581-590. 10.1038/sj.gt.3302182.
Sack U, Sehm B, Kahlenberg F, Murr A, Lehmann J, Tannapfel A, Uberla K, Moessner A, Dietrich A, Emmrich F, et al: Investigation of arthritic joint destruction by a novel fibroblast-based model. Ann NY Acad Sci. 2005, 1051: 291-298. 10.1196/annals.1361.070.
Koshy PJ, Lundy CJ, Rowan AD, Porter S, Edwards DR, Hogan A, Clark IM, Cawston TE: The modulation of matrix metalloproteinase and ADAM gene expression in human chondrocytes by interleukin-1 and oncostatin M: a time-course study using real-time quantitative reverse transcription-polymerase chain reaction. Arthritis Rheum. 2002, 46: 961-967. 10.1002/art.10212.
Plater-Zyberk C, Buckton J, Thompson S, Spaull J, Zanders E, Papworth J, Life PF: Amelioration of arthritis in two murine models using antibodies to oncostatin M. Arthritis Rheum. 2001, 44: 2697-2702. 10.1002/1529-0131(200111)44:11<2697::AID-ART450>3.0.CO;2-#.
Rowan AD, Hui W, Cawston TE, Richards CD: Adenoviral gene transfer of interleukin-1 in combination with oncostatin M induces significant joint damage in a murine model. Am J Pathol. 2003, 162: 1975-1984.
Koshy PJ, Henderson N, Logan C, Life PF, Cawston TE, Rowan AD: Interleukin 17 induces cartilage collagen breakdown: novel synergistic effects in combination with proinflammatory cytokines. Ann Rheum Dis. 2002, 61: 704-713. 10.1136/ard.61.8.704.
Lubberts E, Koenders MI, van den Berg WB: The role of T cell interleukin-17 in conducting destructive arthritis: lessons from animal models. Arthritis Res Ther. 2005, 7: 29-37. 10.1186/ar1478.
Lubberts E, Koenders MI, Oppers-Walgreen B, van den Bersselaar L, Coenen-de Roo CJ, Joosten LA, van den Berg WB: Treatment with a neutralizing anti-murine interleukin-17 antibody after the onset of collagen-induced arthritis reduces joint inflammation, cartilage destruction, and bone erosion. Arthritis Rheum. 2004, 50: 650-659. 10.1002/art.20001.
Koenders MI, Lubberts E, Oppers-Walgreen B, van den Bersselaar L, Helsen MM, Di Padova FE, Boots AM, Gram H, Joosten LA, van den Berg WB: Blocking of interleukin-17 during reactivation of experimental arthritis prevents joint inflammation and bone erosion by decreasing RANKL and interleukin-1. Am J Pathol. 2005, 167: 141-149.
Koenders MI, Joosten LA, van den Berg WB: Potential new targets in arthritis therapy: interleukin (IL)-17 and its relation to tumour necrosis factor and IL-1 in experimental arthritis. Ann Rheum Dis. 2006, 65 (Suppl 3): iii29-iii33. 10.1136/ard.2006.058529.
Koenders MI, Lubberts E, van de Loo FA, Oppers-Walgreen B, van den Bersselaar L, Helsen MM, Kolls JK, Di Padova FE, Joosten LA, van den Berg WB: Interleukin-17 acts independently of TNF-α under arthritic conditions. J Immunol. 2006, 176: 6262-6269.
Joosten LA, Koenders MI, Smeets RL, Heuvelmans-Jacobs M, Helsen MM, Takeda K, Akira S, Lubberts E, van de Loo FA, van den Berg WB: Toll-like receptor 2 pathway drives streptococcal cell wall-induced joint inflammation: critical role of myeloid differentiation factor 88. J Immunol. 2003, 171: 6145-6153.
Su SL, Tsai CD, Lee CH, Salter DM, Lee HS: Expression and regulation of Toll-like receptor 2 by IL-1β and fibronectin fragments in human articular chondrocytes. Osteoarthritis Cartilage. 2005, 13: 879-886. 10.1016/j.joca.2005.04.017.
Kim HA, Cho ML, Choi HY, Yoon CS, Jhun JY, Oh HJ, Kim HY: The catabolic pathway mediated by Toll-like receptors in human osteoarthritic chondrocytes. Arthritis Rheum. 2006, 54: 2152-2163. 10.1002/art.21951.
Varoga D, Paulsen F, Mentlein R, Fay J, Kurz B, Schutz R, Wruck C, Goldring MB, Pufe T: TLR-2-mediated induction of vascular endothelial growth factor (VEGF) in cartilage in septic joint disease. J Pathol. 2006, 210: 315-324. 10.1002/path.2059.
van Lent PL, Blom AB, Grevers L, Sloetjes A, van den Berg WB: Toll-like receptor 4 induced FcγR expression potentiates early onset of joint inflammation and cartilage destruction during immune complex arthritis: Toll-like receptor 4 largely regulates FcγR expression by interleukin 10. Ann Rheum Dis. 2007, 66: 334-340. 10.1136/ard.2006.057471.
Asquith DL, McInnes IB: Emerging cytokine targets in rheumatoid arthritis. Curr Opin Rheumatol. 2007, 19: 246-251. 10.1097/BOR.0b013e3280eec78c.
Joosten LA, Smeets RL, Koenders MI, van den Bersselaar LA, Helsen MM, Oppers-Walgreen B, Lubberts E, Iwakura Y, van de Loo FA, van den Berg WB: Interleukin-18 promotes joint inflammation and induces interleukin-1-driven cartilage destruction. Am J Pathol. 2004, 165: 959-967.
Matsui K, Tsutsui H, Nakanishi K: Pathophysiological roles for IL-18 in inflammatory arthritis. Expert Opin Ther Targets. 2003, 7: 701-724. 10.1517/14728222.7.6.701.
Dai SM, Shan ZZ, Nishioka K, Yudoh K: Implication of interleukin 18 in production of matrix metalloproteinases in articular chondrocytes in arthritis: direct effect on chondrocytes may not be pivotal. Ann Rheum Dis. 2005, 64: 735-742. 10.1136/ard.2004.026088.
John T, Kohl B, Mobasheri A, Ertel W, Shakibaei M: Interleukin-18 induces apoptosis in human articular chondrocytes. Histol Histopathol. 2007, 22: 469-482.
Magne D, Palmer G, Barton JL, Mezin F, Talabot-Ayer D, Bas S, Duffy T, Noger M, Guerne PA, Nicklin MJ, et al: The new IL-1 family member IL-1F8 stimulates production of inflammatory mediators by synovial fibroblasts and articular chondrocytes. Arthritis Res Ther. 2006, 8: R80-10.1186/ar1946.
Joosten LA, Netea MG, Kim SH, Yoon DY, Oppers-Walgreen B, Radstake TR, Barrera P, van de Loo FA, Dinarello CA, van den Berg WB: IL-32, a proinflammatory cytokine in rheumatoid arthritis. Proc Natl Acad Sci USA. 2006, 103: 3298-3303. 10.1073/pnas.0511233103.
Bessis N, Boissier MC: Novel pro-inflammatory interleukins: potential therapeutic targets in rheumatoid arthritis. Joint Bone Spine. 2001, 68: 477-481. 10.1016/S1297-319X(01)00310-4.
van de Loo FA, Smeets RL, van den Berg WB: Gene therapy in animal models of rheumatoid arthritis: are we ready for the patients?. Arthritis Res Ther. 2004, 6: 183-196. 10.1186/ar1214.
van de Loo FA, Geurts J, van den Berg WB: Gene therapy works in animal models of rheumatoid arthritis ... so what!. Curr Rheumatol Rep. 2006, 8: 386-393. 10.1007/s11926-006-0070-y.
Palmer G, Guerne PA, Mezin F, Maret M, Guicheux J, Goldring MB, Gabay C: Production of interleukin-1 receptor antagonist by human articular chondrocytes. Arthritis Res. 2002, 4: 226-231. 10.1186/ar411.
Fernandes JC, Martel-Pelletier J, Pelletier JP: The role of cytokines in osteoarthritis pathophysiology. Biorheology. 2002, 39: 237-246.
Chowdhury TT, Bader DL, Lee DA: Anti-inflammatory effects of IL-4 and dynamic compression in IL-1β stimulated chondro-cytes. Biochem Biophys Res Commun. 2006, 339: 241-247. 10.1016/j.bbrc.2005.11.016.
Schuerwegh AJ, Dombrecht EJ, Stevens WJ, Van Offel JF, Bridts CH, De Clerck LS: Influence of pro-inflammatory (IL-1α, IL-6, TNF-α, IFN-γ) and anti-inflammatory (IL-4) cytokines on chondrocyte function. Osteoarthritis Cartilage. 2003, 11: 681-687. 10.1016/S1063-4584(03)00156-0.
Nandakumar KS, Holmdahl R: Arthritis induced with cartilage-specific antibodies is IL-4-dependent. Eur J Immunol. 2006, 36: 1608-1618. 10.1002/eji.200535633.
Ho SH, Hahn W, Lee HJ, Kim DS, Jeong JG, Kim S, Yu SS, Jeon ES, Kim JM: Protection against collagen-induced arthritis by electrotransfer of an expression plasmid for the interleukin-4. Biochem Biophys Res Commun. 2004, 321: 759-766. 10.1016/j.bbrc.2004.07.028.
Neumann E, Judex M, Kullmann F, Grifka J, Robbins PD, Pap T, Gay RE, Evans CH, Gay S, Scholmerich J, et al: Inhibition of cartilage destruction by double gene transfer of IL-1Ra and IL-10 involves the activin pathway. Gene Ther. 2002, 9: 1508-1519. 10.1038/sj.gt.3301811.
Gonzalez-Rey E, Chorny A, Varela N, O'Valle F, Delgado M: Therapeutic effect of urocortin on collagen-induced arthritis by down-regulation of inflammatory and Th1 responses and induction of regulatory T cells. Arthritis Rheum. 2007, 56: 531-543. 10.1002/art.22394.
Cleaver CS, Rowan AD, Cawston TE: Interleukin 13 blocks the release of collagen from bovine nasal cartilage treated with proinflammatory cytokines. Ann Rheum Dis. 2001, 60: 150-157. 10.1136/ard.60.2.150.
Nabbe KC, van Lent PL, Holthuysen AE, Sloetjes AW, Koch AE, Radstake TR, van den Berg WB: Local IL-13 gene transfer prior to immune-complex arthritis inhibits chondrocyte death and matrix-metalloproteinase-mediated cartilage matrix degradation despite enhanced joint inflammation. Arthritis Res Ther. 2005, 7: R392-R401. 10.1186/ar1502.
Goldring MB, Berenbaum F: The regulation of chondrocyte function by proinflammatory mediators: prostaglandins and nitric oxide. Clin Orthop Relat Res. 2004, 427 (Suppl): S37-S46. 10.1097/01.blo.0000144484.69656.e4.
Healy ZR, Lee NH, Gao X, Goldring MB, Talalay P, Kensler TW, Konstantopoulos K: Divergent responses of chondrocytes and endothelial cells to shear stress: cross-talk among COX-2, the phase 2 response, and apoptosis. Proc Natl Acad Sci USA. 2005, 102: 14010-14015. 10.1073/pnas.0506620102.
Masuko-Hongo K, Berenbaum F, Humbert L, Salvat C, Goldring MB, Thirion S: Up-regulation of microsomal prostaglandin E synthase 1 in osteoarthritic human cartilage: critical roles of the ERK-1/2 and p38 signaling pathways. Arthritis Rheum. 2004, 50: 2829-2838. 10.1002/art.20437.
Whiteman M, Spencer JP, Zhu YZ, Armstrong JS, Schantz JT: Peroxynitrite-modified collagen-II induces p38/ERK andNF-κB-dependent synthesis of prostaglandin E2 and nitric oxide in chondrogenically differentiated mesenchymal progenitor cells. Osteoarthritis Cartilage. 2006, 14: 460-470. 10.1016/j.joca.2005.11.002.
Li X, Afif H, Cheng S, Martel-Pelletier J, Pelletier JP, Ranger P, Fahmi H: Expression and regulation of microsomal prostaglandin E synthase-1 in human osteoarthritic cartilage and chondrocytes. J Rheumatol. 2005, 32: 887-895.
Cheng S, Afif H, Martel-Pelletier J, Pelletier JP, Li X, Farrajota K, Lavigne M, Fahmi H: Activation of peroxisome proliferator-activated receptor γ inhibits interleukin-1β-induced membrane-associated prostaglandin E2 synthase-1 expression in human synovial fibroblasts by interfering with Egr-1. J Biol Chem. 2004, 279: 22057-22065. 10.1074/jbc.M402828200.
Francois M, Richette P, Tsagris L, Fitting C, Lemay C, Benallaoua M, Tahiri K, Corvol MT: Activation of the peroxisome proliferator-activated receptor α pathway potentiates interleukin-1 receptor antagonist production in cytokine-treated chondro-cytes. Arthritis Rheum. 2006, 54: 1233-1245. 10.1002/art.21728.
Loeser RF: Systemic and local regulation of articular cartilage metabolism: where does leptin fit in the puzzle?. Arthritis Rheum. 2003, 48: 3009-3012. 10.1002/art.11315.
Dayer JM, Chicheportiche R, Juge-Aubry C, Meier C: Adipose tissue has anti-inflammatory properties: focus on IL-1 receptor antagonist (IL-1Ra). Ann N Y Acad Sci. 2006, 1069: 444-453. 10.1196/annals.1351.043.
Popa C, Netea MG, Radstake TR, van Riel PL, Barrera P, van der Meer JW: Markers of inflammation are negatively correlated with serum leptin in rheumatoid arthritis. Ann Rheum Dis. 2005, 64: 1195-1198. 10.1136/ard.2004.032243.
Otero M, Lago R, Gomez R, Dieguez C, Lago F, Gomez-Reino J, Gualillo O: Towards a pro-inflammatory and immunomodulatory emerging role of leptin. Rheumatology (Oxford). 2006, 45: 944-950. 10.1093/rheumatology/kel157.
Dumond H, Presle N, Terlain B, Mainard D, Loeuille D, Netter P, Pottie P: Evidence for a key role of leptin in osteoarthritis. Arthritis Rheum. 2003, 48: 3118-3129. 10.1002/art.11303.
Otero M, Lago R, Lago F, Reino JJ, Gualillo O: Signalling pathway involved in nitric oxide synthase type II activation in chondrocytes: synergistic effect of leptin with interleukin-1. Arthritis Res Ther. 2005, 7: R581-R591. 10.1186/ar1708.
Palmer G, Aurrand-Lions M, Contassot E, Talabot-Ayer D, Ducrest-Gay D, Vesin C, Chobaz-Peclat V, Busso N, Gabay C: Indirect effects of leptin receptor deficiency on lymphocyte populations and immune response in db/db mice. J Immunol. 2006, 177: 2899-2907.
Lago F, Dieguez C, Gomez-Reino JJ, Gualillo O: The emerging role of adipokines as mediators of inflammation and immune responses. Cytokine Growth Factor Rev. 2007, 18: 313-325. 10.1016/j.cytogfr.2007.04.007.
Smeets RL, Veenbergen S, Arntz OJ, Bennink MB, Joosten LA, van den Berg WB, van de Loo FA: A novel role for suppressor of cytokine signaling 3 in cartilage destruction via induction of chondrocyte desensitization toward insulin-like growth factor. Arthritis Rheum. 2006, 54: 1518-1528. 10.1002/art.21752.
Yammani RR, Carlson CS, Bresnick AR, Loeser RF: Increase in production of matrix metalloproteinase 13 by human articular chondrocytes due to stimulation with S100A4: role of the receptor for advanced glycation end products. Arthritis Rheum. 2006, 54: 2901-2911. 10.1002/art.22042.
Bauer S, Jendro MC, Wadle A, Kleber S, Stenner F, Dinser R, Reich A, Faccin E, Godde S, Dinges H, et al: Fibroblast activation protein is expressed by rheumatoid myofibroblast-like synoviocytes. Arthritis Res Ther. 2006, 8: R171-10.1186/ar2080.
Milner JM, Kevorkian L, Young DA, Jones D, Wait R, Donell ST, Barksby E, Patterson AM, Middleton J, Cravatt BF, et al: Fibroblast activation protein α is expressed by chondrocytes following a pro-inflammatory stimulus and is elevated in osteoarthritis. Arthritis Res Ther. 2006, 8: R23-10.1186/ar1877.
Firestein GS: NF-κB: Holy Grail for rheumatoid arthritis?. Arthritis Rheum. 2004, 50: 2381-2386. 10.1002/art.20468.
Vincenti MP, Brinckerhoff CE: Transcriptional regulation of collagenase (MMP-1, MMP-13) genes in arthritis: integration of complex signaling pathways for the recruitment of gene-specific transcription factors. Arthritis Res. 2002, 4: 157-164. 10.1186/ar401.
Imamura T, Imamura C, Iwamoto Y, Sandell LJ: Transcriptional co-activators CREB-binding protein/p300 increase chondrocyte Cd-rap gene expression by multiple mechanisms including sequestration of the repressor CCAAT/enhancer-binding protein. J Biol Chem. 2005, 280: 16625-16634. 10.1074/jbc.M411469200.
Grall FT, Prall WC, Wei W, Gu X, Cho JY, Choy BK, Zerbini LF, Inan MS, Goldring SR, Gravallese EM, et al: The Ets transcription factor ESE-1 mediates induction of the COX-2 gene by LPS in monocytes. FEBS J. 2005, 272: 1676-1687. 10.1111/j.1742-4658.2005.04592.x.
Raymond L, Eck S, Mollmark J, Hays E, Tomek I, Kantor S, Elliott S, Vincenti M: Interleukin-1β induction of matrix metalloproteinase-1 transcription in chondrocytes requires ERK-dependent activation of CCAAT enhancer-binding protein-β. J Cell Physiol. 2006, 207: 683-688. 10.1002/jcp.20608.
Rannou F, Francois M, Corvol MT, Berenbaum F: Cartilage breakdown in rheumatoid arthritis. Joint Bone Spine. 2006, 73: 29-36. 10.1016/j.jbspin.2004.12.013.
Grall F, Gu X, Tan L, Cho J-Y, Inan MS, Pettit A, Thamrongsak U, Choy BK, Manning C, Akbarali Y, et al: Responses to the pro-inflammatory cytokines interleukin-1 and tumor necrosis factor α in cells derived from rheumatoid synovium and other joint tissues involve NFκB-mediated induction of the Ets transcription factor ESE-1. Arthritis Rheum. 2003, 48: 1249-1260. 10.1002/art.10942.
Benito MJ, Murphy E, Murphy EP, van den Berg WB, FitzGerald O, Bresnihan B: Increased synovial tissue NF-κB1 expression at sites adjacent to the cartilage–pannus junction in rheumatoid arthritis. Arthritis Rheum. 2004, 50: 1781-1787. 10.1002/art.20260.
Katoh M: STAT3-induced WNT5A signaling loop in embryonic stem cells, adult normal tissues, chronic persistent inflammation, rheumatoid arthritis and cancer [review]. Int J Mol Med. 2007, 19: 273-278.
Okazaki K, Li J, Yu H, Fukui N, Sandell LJ: CCAAT/Enhancer-binding proteins β and δ mediate the repression of gene transcription of cartilage-derived retinoic acid-sensitive protein induced by interleukin-1β. J Biol Chem. 2002, 277: 31526-31533. 10.1074/jbc.M202815200.
Tan L, Peng H, Osaki M, Choy BK, Auron PE, Sandell LJ, Goldring MB: Egr-1 mediates transcriptional repression of COL2A1 promoter activity by interleukin-1β. J Biol Chem. 2003, 278: 17688-17700. 10.1074/jbc.M301676200.
Seguin CA, Bernier SM: TNFα suppresses link protein and type II collagen expression in chondrocytes: role of MEK1/2 and NF-κB signaling pathways. J Cell Physiol. 2003, 197: 356-369. 10.1002/jcp.10371.
Heller RA, Schena M, Chai A, Shalon D, Bedilion T, Gilmore J, Woolley DE, Davis RW: Discovery and analysis of inflammatory disease-related genes using cDNA microarrays. Proc Natl Acad Sci USA. 1997, 94: 2150-2155. 10.1073/pnas.94.6.2150.
Vincenti MP, Brinckerhoff CE: Early response genes induced in chondrocytes stimulated with the inflammatory cytokine interleukin-1β. Arthritis Res. 2001, 3: 381-388. 10.1186/ar331.
Barksby HE, Hui W, Wappler I, Peters HH, Milner JM, Richards CD, Cawston TE, Rowan AD: Interleukin-1 in combination with oncostatin M up-regulates multiple genes in chondrocytes: implications for cartilage destruction and repair. Arthritis Rheum. 2006, 54: 540-550. 10.1002/art.21574.
Aigner T, Fundel K, Saas J, Gebhard PM, Haag J, Weiss T, Zien A, Obermayr F, Zimmer R, Bartnik E: Large-scale gene expression profiling reveals major pathogenetic pathways of cartilage degeneration in osteoarthritis. Arthritis Rheum. 2006, 54: 3533-3544. 10.1002/art.22174.
Sato T, Konomi K, Yamasaki S, Aratani S, Tsuchimochi K, Yokouchi M, Masuko-Hongo K, Yagishita N, Nakamura H, Komiya S, et al: Comparative analysis of gene expression profiles in intact and damaged regions of human osteoarthritic cartilage. Arthritis Rheum. 2006, 54: 808-817. 10.1002/art.21638.
Pulai JI, Chen H, Im HJ, Kumar S, Hanning C, Hegde PS, Loeser RF: NF-κB mediates the stimulation of cytokine and chemokine expression by human articular chondrocytes in response to fibronectin fragments. J Immunol. 2005, 174: 5781-5788.
Attur MG, Dave MN, Akamatsu M, Katoh M, Amin AR: A system biology approach to bioinformatics and functional genomics in complex human diseases: arthritis. Curr Issues Mol Biol. 2002, 4: 129-146.
Goldring MB, Goldring SR: Role of cytokines and chemokines in cartilage and bone destruction in arthritis. Curr Opin Orthopaed. 2002, 13: 351-362. 10.1097/00001433-200210000-00005.
Koch AE: Chemokines and their receptors in rheumatoid arthritis: future targets?. Arthritis Rheum. 2005, 52: 710-721. 10.1002/art.20932.
Vergunst CE, van de Sande MG, Lebre MC, Tak PP: The role of chemokines in rheumatoid arthritis and osteoarthritis. Scand J Rheumatol. 2005, 34: 415-425. 10.1080/03009740500439159.
Nakamura H, Masuko K, Yudoh K, Kato T, Nishioka K: Effects of celecoxib on human chondrocytes – enhanced production of chemokines. Clin Exp Rheumatol. 2007, 25: 11-16.
Borzi RM, Mazzetti I, Marcu KB, Facchini A: Chemokines in cartilage degradation. Clin Orthop Relat Res. 2004, 427 (Suppl): S53-S61. 10.1097/01.blo.0000143805.64755.4f.
Iwamoto T, Okamoto H, Iikuni N, Takeuchi M, Toyama Y, Tomatsu T, Kamatani N, Momohara S: Monocyte chemoattractant protein-4 (MCP-4)/CCL13 is highly expressed in cartilage from patients with rheumatoid arthritis. Rheumatology (Oxford). 2006, 45: 421-424. 10.1093/rheumatology/kei209.
Kanbe K, Takemura T, Takeuchi K, Chen Q, Takagishi K, Inoue K: Synovectomy reduces stromal-cell-derived factor-1 (SDF-1) which is involved in the destruction of cartilage in osteoarthritis and rheumatoid arthritis. J Bone Joint Surg Br. 2004, 86: 296-300. 10.1302/0301-620X.86B2.14474.
Green DM, Noble PC, Ahuero JS, Birdsall HH: Cellular events leading to chondrocyte death after cartilage impact injury. Arthritis Rheum. 2006, 54: 1509-1517. 10.1002/art.21812.
Fearon U, Griosios K, Fraser A, Reece R, Emery P, Jones PF, Veale DJ: Angiopoietins, growth factors, and vascular morphology in early arthritis. J Rheumatol. 2003, 30: 260-268.
Szekanecz Z, Gaspar L, Koch AE: Angiogenesis in rheumatoid arthritis. Front Biosci. 2005, 10: 1739-1753. 10.2741/1657.
Honorati MC, Neri S, Cattini L, Facchini A: Interleukin-17, a regulator of angiogenic factor release by synovial fibroblasts. Osteoarthritis Cartilage. 2005, 14: 345-352. 10.1016/j.joca.2005.10.004.
Fearon U, Mullan R, Markham T, Connolly M, Sullivan S, Poole AR, FitzGerald O, Bresnihan B, Veale DJ: Oncostatin M induces angiogenesis and cartilage degradation in rheumatoid arthritis synovial tissue and human cartilage cocultures. Arthritis Rheum. 2006, 54: 3152-3162. 10.1002/art.22161.
Mould AW, Tonks ID, Cahill MM, Pettit AR, Thomas R, Hayward NK, Kay GF: Vegfb gene knockout mice display reduced pathology and synovial angiogenesis in both antigen-induced and collagen-induced models of arthritis. Arthritis Rheum. 2003, 48: 2660-2669. 10.1002/art.11232.
Del Carlo M, Schwartz D, Erickson EA, Loeser RF: Endogenous production of reactive oxygen species is required for stimulation of human articular chondrocyte matrix metalloproteinase production by fibronectin fragments. Free Radic Biol Med. 2007, 42: 1350-1358. 10.1016/j.freeradbiomed.2007.01.035.
Xu L, Peng H, Wu D, Hu K, Goldring MB, Olsen BR, Li Y: Activation of the discoidin domain receptor 2 induces expression of matrix metalloproteinase 13 associated with osteoarthritis in mice. J Biol Chem. 2005, 280: 548-555.
Hu K, Xu L, Cao L, Flahiff CM, Brussiau J, Ho K, Setton LA, Youn I, Guilak F, Olsen BR, et al: Pathogenesis of osteoarthritis-like changes in the joints of mice deficient in type IX collagen. Arthritis Rheum. 2006, 54: 2891-2900. 10.1002/art.22040.
Knudson CB, Knudson W: Hyaluronan and CD44: modulators of chondrocyte metabolism. Clin Orthop Relat Res. 2004, 427 (Suppl): S152-S162. 10.1097/01.blo.0000143804.26638.82.
Takagi T, Okamoto R, Suzuki K, Hayashi T, Sato M, Kurosaka N, Koshino T: Up-regulation of CD44 in rheumatoid chondro-cytes. Scand J Rheumatol. 2001, 30: 110-113. 10.1080/03009740151095420.
Naor D, Nedvetzki S: CD44 in rheumatoid arthritis. Arthritis Res Ther. 2003, 5: 105-115. 10.1186/ar746.
Ohno S, Im HJ, Knudson CB, Knudson W: Hyaluronan oligosac-charides induce matrix metalloproteinase 13 via transcriptional activation of NFκB and p38 MAP kinase in articular chondrocytes. J Biol Chem. 2006, 281: 17952-17960. 10.1074/jbc.M602750200.
Yasuda T, Kakinuma T, Julovi SM, Yoshida M, Hiramitsu T, Akiyoshi M, Nakamura T: COOH-terminal heparin-binding fibronectin fragment induces nitric oxide production in rheumatoid cartilage through CD44. Rheumatology (Oxford). 2004, 43: 1116-1120. 10.1093/rheumatology/keh274.
Valencia X, Higgins JM, Kiener HP, Lee DM, Podrebarac TA, Dascher CC, Watts GF, Mizoguchi E, Simmons B, Patel DD, et al: Cadherin-11 provides specific cellular adhesion between fibroblast-like synoviocytes. J Exp Med. 2004, 200: 1673-1679. 10.1084/jem.20041545.
Lee DM, Kiener HP, Agarwal SK, Noss EH, Watts GF, Chisaka O, Takeichi M, Brenner MB: Cadherin-11 in synovial lining formation and pathology in arthritis. Science. 2007, 315: 1006-1010. 10.1126/science.1137306.
Kong YY, Yoshida H, Sarosi I, Tan HL, Timms E, Capparelli C, Morony S, Oliveira-dos-Santos AJ, Van G, Itie A, et al: OPGL is a key regulator of osteoclastogenesis, lymphocyte development and lymph-node organogenesis. Nature. 1999, 397: 315-323. 10.1038/16852.
Komuro H, Olee T, Kuhn K, Quach J, Brinson DC, Shikhman A, Valbracht J, Creighton-Achermann L, Lotz M: The osteoprotegerin/receptor activator of nuclear factor κB/receptor activator of nuclear factor κB ligand system in cartilage. Arthritis Rheum. 2001, 44: 2768-2776. 10.1002/1529-0131(200112)44:12<2768::AID-ART464>3.0.CO;2-I.
Pettit AR, Ji H, von Stechow D, Muller R, Goldring SR, Choi Y, Benoist C, Gravallese EM: TRANCE/RANKL knockout mice are protected from bone erosion in a serum transfer model of arthritis. Am J Pathol. 2001, 159: 1689-1699.
Zwerina J, Hayer S, Redlich K, Bobacz K, Kollias G, Smolen JS, Schett G: Activation of p38 MAPK is a key step in tumor necrosis factor-mediated inflammatory bone destruction. Arthritis Rheum. 2006, 54: 463-472. 10.1002/art.21626.
Day TF, Guo X, Garrett-Beal L, Yang Y: Wnt/β-catenin signaling in mesenchymal progenitors controls osteoblast and chondrocyte differentiation during vertebrate skeletogenesis. Dev Cell. 2005, 8: 739-750. 10.1016/j.devcel.2005.03.016.
Tamamura Y, Otani T, Kanatani N, Koyama E, Kitagaki J, Komori T, Yamada Y, Costantini F, Wakisaka S, Pacifici M, et al: Developmental regulation of Wnt/β-catenin signals is required for growth plate assembly, cartilage integrity, and endochondral ossification. J Biol Chem. 2005, 280: 19185-19195. 10.1074/jbc.M414275200.
Diarra D, Stolina M, Polzer K, Zwerina J, Ominsky MS, Dwyer D, Korb A, Smolen J, Hoffmann M, Scheinecker C, et al: Dickkopf-1 is a master regulator of joint remodeling. Nat Med. 2007, 13: 156-163. 10.1038/nm1538.
Goldring SR, Goldring MB: Eating bone or adding it: the Wnt pathway decides. Nat Med. 2007, 13: 133-134. 10.1038/nm0207-133.
Author information
Authors and Affiliations
Corresponding author
Additional information
Competing interests
The authors declare that they have no competing interests.
An erratum to this article is available at http://dx.doi.org/10.1186/ar2351.
Authors’ original submitted files for images
Below are the links to the authors’ original submitted files for images.
Rights and permissions
About this article
Cite this article
Otero, M., Goldring, M.B. Cells of the synovium in rheumatoid arthritis. Chondrocytes. Arthritis Res Ther 9, 220 (2007). https://doi.org/10.1186/ar2292
Published:
DOI: https://doi.org/10.1186/ar2292