
Abstract
Background
The purpose of this research is to study the relationship between superoxide dismutase (SOD) and lung redox state in an animal model of sepsis.
Methods
Sepsis was induced in rats by the cecal ligation and perforation model (CLP). After 3, 6, and 12 h, CLP protein content and expression of SOD1, SOD2, and SOD3 were evaluated, and SOD activity was assessed. Oxidative damage was determined by quantifying nitrotyrosine content. Lung localization of SOD3 was performed by immunohistochemistry. The protective effect of a SOD mimetic on oxidative damage, inflammation, and lung permeability was assessed 12 and 24 h after sepsis induction.
Results
Lung levels of SOD1 decreased 3 and 12 h after sepsis, but SOD2 and SOD3 increased, as well as SOD activity. These alterations were not associated with changes in sod gene expression. Nitrotyrosine levels increased 3 and 12 h after sepsis. The administration of a SOD mimetic decreased nitrotyrosine and proinflammatory cytokine levels and improved lung permeability.
Conclusions
SOD2 and SOD3 increased after sepsis induction, but this was insufficient to protect the lung. Treatments based on SOD mimetics could have a role in lung injury associated with sepsis.
Similar content being viewed by others


Background
Several molecular mechanisms of inflammation and cellular damage have been implicated in the pathogenesis of sepsis including excessive reactive oxygen species (ROS) generation [1]. Main sources of ROS in the lung during sepsis are inflammatory cells and mitochondria [2, 3]. Production of ROS leads to lipid, protein, and extracellular matrix damage, which increases pulmonary inflammation [4, 5].
Antioxidants are known to counteract the deleterious effects of ROS. Superoxide dismutase (SOD) is a component of antioxidant response and catalyzes the conversion of superoxide anions to hydrogen peroxide [6]. Three SODs are found in mammals and regulate the concentration of superoxide: a cytosolic (SOD1), a mitochondrial (SOD2), and an extracellular (SOD3), which bind to both cell surfaces and extracellular matrices [7]. sod3 gene is highly expressed in the lung, where it plays a major protective role by controlling oxidative stress and inflammation and regulating redox homeostasis of the airways [8]. Localization of SOD3 in the lung depends on its ability to bind to the extracellular matrix by a heparan sulfate domain, which can be fragmented by oxidative damage [5, 9]. Furthermore, extracellular matrix fragments stimulate inflammatory cell migration, which is of concern since matrix components are widely distributed throughout the interstitium [9]. Proteolytic cleavage of SOD3's anchorage domain alters its tissue distribution and, consequently, the oxidant/antioxidant balance [10].
In addition, bioactivity of nitric oxide (NO) partially depends on its interaction with ROS, particularly superoxide anions [11]. NO reacts with superoxide to form peroxynitrite (ONOO−), which induces protein oxidation and DNA damage [12]. SOD3 prevents the inactivation of NO by superoxide; therefore, an increase in sod3 expression in blood vessels preserves endothelial function by overcoming oxidative stresses [13]. Furthermore, several evidences suggest that SOD3 may have a protective role against inflammation [10, 14].
Thus, it was hypothesized that endogenous SOD3 could have a major role in lung defenses against oxidative stress during sepsis development. The aims of this study are (1) to determine if there is a relationship between the expression and activity of SOD and the occurrence of oxidative stress and (2) to determine if the administration of a SOD mimetic is able to prevent lung damage in an animal model of sepsis.
Methods
Animals
Three-month-old male Wistar rats (350 to 400 g) were obtained from our breeding colony. The rats were caged in groups of five, had free access to food and water, and were maintained on a 12-h light–dark cycle (600 to 1800 hours) in a temperature-controlled colony room (22°C ± 1°C). The research protocol was approved by the Ethical Committee for Animal Experimentation of Universidade do Extremo Sul Catarinense under protocol number 21/2011.
Cecal ligation and perforation surgery
The animals were subjected to cecal ligation and perforation (CLP) as previously described [15]. Briefly, the rats were anesthetized with ketamine (80 mg/kg). Under aseptic conditions, a 3-cm midline laparotomy was performed to allow exposure of the cecum with the adjoining intestine. The cecum was tightly ligated with a 3.0 silk suture at its base (below the ileocecal valve) and was then perforated a single time with a 14-gauge needle. The cecum was gently squeezed to extrude a small amount of feces from the perforation site into the peritoneal cavity. The animals were resuscitated with normal saline (50 mL/kg, subcutaneous) immediately and 12 h after CLP. The sham-operated group was submitted to all surgical procedures, but the cecum was neither ligated nor perforated. The animals were killed by decapitation at 3, 6, and 12 h after surgery, and the lung was removed for subsequent analyses. The number of animals in each group per experiment was ten. In some experiments, a SOD mimetic was administered once by intra-tracheal instillation immediately after CLP induction (50 mg/kg), and the lung was removed 24 h after for subsequent analyses. This mimetic has been previously described by Horn et al. [16].
Total SOD activity
The SOD activity was measured by inhibition of adrenaline auto-oxidation followed by spectrophotometry as previously described [17].
Immunoblotting
The lung samples were lysed in Laemmli sample buffer (62.5 mM Tris–HCl, pH 6.8, 1% (w/v) SDS, 10% v/v) glycerol). Protein (30 μg) was fractionated by SDS-polyacrylamide gel electrophoresis and then electroblotted onto nitrocellulose membranes. Protein loading and electroblotting efficiencies were verified by Ponceau S staining. The membrane was blocked in Tween-Tris-buffered saline (TTBS, 100 mM Tris–HCl, pH 7.5, containing 0.9% NaCl and 0.1% Tween-20) containing 5% albumin. The membranes were incubated overnight at 4°C with rabbit polyclonal antibody, targeting SOD1 (Santa Cruz Biotechnology, CA, USA) (dilution range 1:400), SOD2 (Santa Cruz Biotechnology) (dilution range 1:400), SOD3 (Santa Cruz Biotechnology) (dilution range 1:750), iNOS (Santa Cruz Biotechnology) (dilution range 1:400) or anti-β-actin 1:2000, in the presence of 5% milk. Thereafter, the membranes were washed with TTBS. Anti-rabbit immunoglobulin G (IgG) peroxidase-linked secondary antibody was incubated (1:10,000 dilution range), and the immunoreactivity was detected by enhanced chemiluminescence using ECL Plus kit (Thermo Fisher Scientific Inc., Pittsburgh, PA, USA). Densitometric analyses of the films were performed with ImageQuant software (GE Healthcare Life Sciences, Billerica, MA, USA). The blots were developed such that the signals are linear and non-saturating which are required for densitometry. All results were expressed as a relative ratio comparing the immunocontent of SOD1, SOD2, SOD3, and iNOS with that of the β-actin internal control [18].
Enzyme-linked immunosorbent assay to 3-nitrotyrosine contents
An indirect enzyme-linked immunosorbent assay (ELISA) was performed to analyze the changes in 3-nitrotyrosine content. Briefly, an anti-3-nitrotyrosine polyclonal rabbit antibody (Santa Cruz Biotechnology) was diluted 2,000-fold in PBS with 5% albumin according to the manufacturer's instructions. Then, microtiter plates (96 wells, with flat bottom) were coated for 24 h with the samples that had been diluted 1:2 in PBS with 5% albumin. The plates were washed four times with wash buffer (PBS with 0.05% Tween-20), and the antibody was added to each plate for 2 h at room temperature. After washing, a second incubation with anti-rabbit antibody peroxidase conjugate (diluted 1:1,000) was performed for 1 h at room temperature. After the addition of substrates, the samples were read in a plate spectrophotometer at 450 nm. The results are expressed as changes in the percentage among the groups [19].
Semi-quantitative reverse transcription polymerase chain reaction
All transcriptional analyses were performed in the samples in which prior Western blotting experiments revealed differences in the immunocontent. The goal was to evaluate the contribution of each gene to transcriptional changes in the immunocontent of each enzyme. Total RNA was isolated from the rat lung using TRIzol® reagent (Invitrogen, Carlsbad, CA, USA), according to the manufacturer's instructions. Using a spectrophotometer, the purity of the RNA was quantified by calculating the ratio between absorbance values at 260 and 280 nm, and its integrity was confirmed by electrophoresis using a 1.0% agarose gel. Afterward, cDNA species were synthesized using ImProm-II™ Reverse Transcription System (Promega®, Madison, WI, USA), as described by the supplier's instruction. The cDNA products (1 μL) were used as a template for each polymerase chain reaction (PCR) amplification. The PCR parameters were first optimized. Thereafter, the reactions were performed, such that product detection could be performed within the linear phase of messenger ribonucleic acid (mRNA) transcript amplification for each primer pair (Table 1). PCR for the β-actin gene was performed in a total volume of 20 μL using 0.1 μM of each primer, 0.2 μM dNTP, 1.6 mM MgCl2, and 0.2 U Taq platinum DNA polymerase (Invitrogen). For PCR amplification of sod1, sod2, and sod3, the reaction was performed in a total volume of 25 μL using 0.2 μM of each primer, 0.2 μM dNTP, 1.6 mM MgCl2, and 0.25 U Taq platinum DNA polymerase (Invitrogen). The conditions for sod1, sod2, and sod3 PCRs were as follows: initial 1-min denaturation step at 94°C, another 1-min denaturation step at 94°C, 1-min annealing step at 60°C, 1-min extension step at 72°C for 30 cycles, and a final 10-min extension at 72°C. The conditions for the β-actin PCR were as follows: initial 1-min denaturation step at 94°C, another 1-min denaturation step at 94°C, 1-min annealing step at 54°C, 1-min extension step at 72°C for 35 cycles, and a final 10-min extension at 72°C. For each PCR set, a negative control was included. The PCR products were then analyzed on a 1% agarose gel containing GelRed® (Biotium, Hayward, CA, USA) and visualized with ultraviolet light. The Low DNA Mass Ladder (Invitrogen) was used as a molecular marker, and the samples were normalized against the constitutively expressed β-actin gene. The band intensities were measured by optical densitometry analysis, and the enzyme/β-actin mRNA ratios were established for each treatment using the freeware Image J 1.37. Each experiment was repeated at least four times using RNA isolated from independent extractions.
Immunohistochemistry
Lung sections (sham 0 and 12 and CLP 0 and 12, n = 3 in each group) were deparaffinized and hydrated. To examine their histological features, the lungs were stained with hematoxylin and eosin (H&E). After blocking of endogenous peroxidase, antigen retrieval was performed in a high-temperature Tris-citrate buffer (pH 7.2). The rabbit polyclonal anti-SOD3 (Santa Cruz Biotechnology) (diluted 1:1,600) was used as the primary antibody. The Vectastin ABC Kit (Vector Laboratories, Burlingame, CA, USA) was used as the secondary antibody, and 3,3-diaminobenzidine (DAB, Sigma, St. Louis, MO, USA) was used as the chromogen. Thereafter, the sections were counterstained with Harris hematoxylin (Merck, Darmstadt, Germany). In the negative controls, the first antibody was omitted from the procedure, and the tissues were incubated with bovine serum albumin (BSA) instead.
Cytokine determination
Lung levels of tumor necrosis factor (TNF)-α, interleukin (IL)-1β, IL-6, and IL-10 were determined by ELISA according to the manufacturer's instructions (PrepoTech, Ribeirão Preto, SP, Brazil). All samples were assayed in duplicate.
Lung permeability assay
Permeability changes were measured by Evan's blue dye (EBD) leakage from the blood into the airways. EBD (20 mg/kg) was administered by femoral vein injection 1 h before the end of the experiments. One hour later, the mice were bled by cardiac puncture, and the pulmonary vasculature was flushed by right ventricle puncture. The pulmonary vessels were perfused with normal saline to remove EBD from the vascular spaces. The lungs were removed en bloc and dried at 60°C for 24 h. EBD was extracted in formamide at 37°C for 24 h and quantitated by its fluorescence intensity. The extravasated EBD concentration in lung homogenate was calculated against a standard curve.
Statistical analysis
Data are expressed as mean ± standard deviation. The means for the different groups were compared by t test or one-way or two-way ANOVA followed by Tukey test, depending on the number of experimental groups. Statistical significance was assigned to p < 0.05.
Results
Levels of SOD were determined in the lung 3 to 12 h after sepsis. There was a decrease in the immunocontent of SOD1 at 3 and 12 hours, but not at 6 h, after sepsis induction (Figure 1A,B,C). Furthermore, an increase in the immunocontent of SOD2 was observed at all times (Figure 2A,B,C), and there was an increase in the immunocontent of SOD3 at 6 and 12 h after sepsis induction (Figure 3A,B,C). The SOD activity increased at 6 and 12 h (6 h, 6.03 ± 2.1 vs. 21.1 ± 5.6, p = 0.02; 12 h, 4.9 ± 1.7 vs. 22.3 ± 7.6 U/mg protein, p = 0.01) when compared to the sham and CLP animals, respectively. To determine if these changes of protein content were associated with alterations in gene expression, semi-quantitative reverse transcription polymerase chain reaction (RT-PCR) was performed. There was no consistent pattern of gene expression; while sod1 and sod2 gene expressions increased 12 h after sepsis (Figure 1D,E and Figure 2D,E,F), sod3 gene expression decreased at this time point (Figure 3B).

Cytosolic superoxide dismutase (SOD1) alterations early after sepsis induction. The animals were submitted to sepsis or sham-operated, and 3 h (A), 6 h (B), and 12 h (C) after surgery, the immunocontent of SOD1 was evaluated. The sod1 gene expression was evaluated at 3 h (D) and 12 h (E) after surgery. The data are expressed as mean ± SD (n = ten animals/group). p value < 0.05 was considered significant when compared to the control. * denotes p < 0.05.

Mitochondrial superoxide dismutase (SOD2) alterations early after sepsis induction. The animals were submitted to sepsis or sham-operated, and 3 h (A), 6 h (B), and 12 h (C) after surgery, the immunocontent of SOD2 was evaluated. The sod2 gene expression was evaluated at 3 h (D), 6 h (E), and 12 h (F) after surgery. The data are expressed as mean ± SD (n = ten animals/group). p value < 0.05 was considered significant when compared to the control.

Extracellular superoxide dismutase (SOD3) alterations early after sepsis induction. The animals were submitted to sepsis or sham-operated, and 3 h (A), 6 h (B), and 12 h (C) after surgery, the immunocontent of SOD3 was evaluated. The sod3 gene expression was evaluated at 3 h (D) and 12 h (E) after surgery. The data are expressed as mean ± SD (n = ten animals/group). p value < 0.05 was considered significant when compared to the control.
Nitrotyrosine levels increased 3 and 12 hours after CLP induction (Figure 4), suggesting that despite the increase of SOD3 content the reaction between superoxide and NO was occurring. Lung content of iNOS did not increase in the lung of septic animals (Figure 5A-C), but it was increased in the pulmonary artery (Figure 5D-E).

Nitrotyrosine content early after sepsis induction. The animals were submitted to sepsis or sham-operated, and 3, 6, and 12 h after surgery, the content of nitrotyrosine was evaluated. The data are expressed as mean ± SD (n = ten animals/group). p value < 0.05 was considered significant when compared to the control.

Inducible nitric oxide synthase (iNOS) alterations early after sepsis induction. The animals were submitted to sepsis or sham-operated, and 3 h (A), 6 h (B), and 12 h (C) after surgery, the immunocontent of iNOS was evaluated in the lung and in the pulmonary artery (3 h (D), 6 h (E), 12 h (F)). The data are expressed as mean ± SD (n = ten animals/group). p value < 0.05 was considered significant when compared to the control.
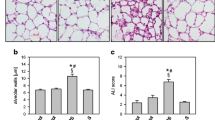
These alterations were associated with capillary congestion and infiltration of neutrophils into the alveolar septa (Figure 6). It was possible that the increase in SOD3 protein content did not protect the lung from oxidant injury due to a disorganized distribution resulting from proteolytic cleavage of its heparin-binding domain, but SOD was adequately distributed across the lung (Figure 6). It was expressed mainly in the bronchial and alveolar epithelia of the CLP animals and to a minor extent in the leukocytes and endothelial cells (Figure 6).

Distribution of extracellular superoxide dismutase (SOD3) in the lung early after sepsis. The animals were submitted to sepsis or sham-operated, and 12 h after surgery, the distribution of lung SOD3 was determined by immunohistochemistry. Representative photographs of septic (A) and sham (B) animals. Original magnification is × 6,200.
It was expected that SOD3 protected the lung from oxidative damage and inflammation, but this was not supported by the presented data. Thus, it was administered a SOD mimetic, and it decreased the lung nitrotyrosine and cytokine levels, as well as preserved lung permeability (Figure 7A,B,C).

Effects of the treatment with a SOD mimetic on lung injury induced by sepsis. The animals were submitted to sepsis or sham-operated, and immediately after surgery, a SOD mimetic was administered once by intra-tracheal instillation. Twenty-four hours after surgery, the content of nitrotyrosine (A), lung permeability (B), interleukin-6 (C), and tumor necrosis factor (D) was determined. The data are expressed as mean ± SD (n = ten animals/group). p value < 0.05 was considered significant when compared to the control.
Discussion
Here, in a CLP model, we demonstrated that during sepsis development, the levels of SOD2 and SOD3 increased in the lung, but these did not prevent nitrosative damage and inflammation. Furthermore, we showed that superoxide-derived lung inflammation could be attenuated by the administration of an exogenous SOD mimetic.
An increase in the levels of SOD2 and SOD3 was observed; thus, it was believed that the lung was protected from superoxide-derived oxidative damage. SOD3 is a potent inhibitor of inflammation in lung injury models [20], and mice lacking the sod3 gene are more sensitive to lethal levels of hyperoxia [14]. Furthermore, SOD3 protects against endothelial dysfunction in mice treated with endotoxin [21]. In a murine model of emphysema, the mice that overexpress the sod3 gene or are treated with a SOD mimetic have improved lung compliance, decreased neutrophil influx, release of proinflammatory mediators, and oxidative damage [22]. Here, we show that despite the increase in SOD content, there is an increase in lung nitrotyrosine content.
In general, inflammatory conditions (including lipopolysaccharide, hypoxia, asbestos exposure, bleomycin, and hyperoxia) induce a decrease in SOD3 content. This observation is most likely a result of proteolysis of its heparin-binding domain rather than alterations in its gene expression [20, 23]. In contrast, at earlier time points after sepsis induction, we demonstrate an increase in SOD3 levels that are properly distributed throughout the lung parenchyma, but this is not sufficient to prevent the formation of peroxynitrite. Increased oxidative damage, despite the presence of higher levels of SOD3, was described in a model of cerebral ischemia [24].
The reaction of NO with superoxide may lead to an increase in ONOO−, which mediates nitration of tyrosine residues in certain enzymes, including SODs. This modification leads to a reduction in their activity and, as a result, further increase in superoxide levels [25]. Specifically, ONOO− is known to inactivate SOD1 and SOD2 [26, 27]. Due to the structural similarity between SOD1 and SOD3, we speculate that the activity of SOD3 may similarly be decreased by ONOO−. Here, we demonstrate that septic animals have increased levels of nitrotyrosine. This occurs even in the presence of higher SOD3 levels and total SOD activity. This pattern is not expected since in different models, an increase in SOD3 activity is usually associated with a decrease in ONOO−[28]. However, when we used an exogenous SOD mimetic, it is observed that there is a decrease in the nitrotyrosine levels. These observations suggest that the endogenous SOD system is not able to prevent oxidative damage in the CLP model. In addition, a SOD mimetic decreased the markers of lung inflammation and improved lung permeability. Moreover, these findings raise the possibility that SOD mimetics could be used to treat sepsis-related lung injury.
There are some limitations to our study. Firstly, SOD mimetic was administered immediately after CLP, when the septic response was not fully developed and animals did not presented clinical signs of severe infection. Thus, this design did not reflect the clinical scenario, but can be relevant to understand the mechanisms associated with sepsis development. Secondly, nitrotyrosine was not measured by gold-standard techniques, such as tandem mass spectrometry or high-performance liquid chromatography. Since the increase of nitrotyrosine is classically found in sepsis models, the use of semi-quantitative ELISA as a marker of oxidative damage seems to be adequate to the study aim.
Conclusions
In conclusion, this study demonstrated an increase in SOD3 levels following sepsis; however, this was not able to prevent oxidative stress and inflammation associated with ONOO− production. Nonetheless, the administration of a SOD mimetic had a positive impact, which suggests that administration of exogenous SOD may improve lung function in sepsis.
References
Salvemini D, Cuzzocrea S: Oxidative stress in septic shock and disseminated intravascular coagulation. Free Radic Biol Med 2002, 33(9):1173–1185. 10.1016/S0891-5849(02)00961-9
Powell CS, Jackson RM: Mitochondrial complex I, aconitase, and succinate dehydrogenase during hypoxia-reoxygenation: modulation of enzyme activities by MnSOD. Am J Physiol Lung Cell Mol Physiol 2003, 285: 189–198. doi:10.1152/ajplung.00253.2002
Kurosaki M, Li Calzi M, Scanziani E, Garattini E, Terao M: Tissue- and cell-specific expression of mouse xanthine oxidoreductase gene in vivo: regulation by bacterial lipopolysaccharide. Biochem J 1995, 306(1):225–234.
Dahl M, Bauer AK, Arredouani M, Soininen R, Tryggvason K, Kleeberger SR, Kobzik L: Protection against inhaled oxidants through scavenging of oxidized lipids by macrophage receptors MARCO and SR-AI/II. J Clin Invest 2007, 117: 757–764. doi:10.1172/JCI29968 10.1172/JCI29968
Kliment CR, Tobolewski JM, Manni ML, Tan RJ, Enghild J, Oury TD: Extracellular superoxide dismutase protects against matrix degradation of heparan sulfate in the lung. Antioxid Redox Signal 2008, 10: 261–268. doi:10.1089/ars.2007.1906 10.1089/ars.2007.1906
Kinnula VL, Crapo JD: Superoxide dismutases in the lung and human lung diseases. Am J Respir Crit Care Med 2003, 167: 1600–1619. doi:10.1164/rccm.200212–1479SO 10.1164/rccm.200212-1479SO
Clarke MB, Wright R, Irwin D, Bose S, Van Rheen Z, Birari R, Stenmark KR, McCord JM, Nozik-Grayck E: Sustained lung activity of a novel chimeric protein, SOD2/3, after intratracheal administration. Free Radic Biol Med 2010, 49: 2032–2039. doi:10.1016/j.freeradbiomed.2010.09.028 10.1016/j.freeradbiomed.2010.09.028
Gongora MC, Lob HE, Landmesser U, Guzik TJ, Martin WD, Ozumi K, Wall SM, Wilson DS, Murthy N, Gravanis M, Fukai T, Harrison DG: Loss of extracellular superoxide dismutase leads to acute lung damage in the presence of ambient air: a potential mechanism underlying adult respiratory distress syndrome. Am J Pathol 2008, 173(4):915–926. doi:10.2353/ajpath.2008.080119 10.2353/ajpath.2008.080119
Dahl M, Bowler RP, Juul K, Crapo JD, Levy S, Nordestgaard BG: Superoxide dismutase 3 polymorphism associated with reduced lung function in two large populations. Am J Respir Crit Care Med 2008, 178: 906–912. doi:10.1164/rccm.200804–549OC 10.1164/rccm.200804-549OC
Gao F, Kinnula VL, Myllärniemi M, Oury TD: Extracellular superoxide dismutase in pulmonary fibrosis. Antioxid Redox Signal 2008, 10(2):343–354. doi:10.1089/ars.2007.1908 10.1089/ars.2007.1908
Oury TD, Day BJ, Crapo JD: Extracellular superoxide dismutase: a regulator of nitric oxide bioavailability. Lab Invest 1996, 75: 617–636.
Hayreh SS, Piegors DJ, Heistad DD: Serotonin-induced constriction in ocular arteries in atherosclerotic monkeys: implications for ischemic disorders of the retina and optic nerve head. Arch Ophthalmol 1997, 115: 220–228. 10.1001/archopht.1997.01100150222012
Heistad DD, Paul M, Endothelial function in the time of the giants: Vanhoutte Lecture. J Cardiovasc Pharmacol 2008, 52(5):385–392. doi:10.1097/FJC.0b013e31818a403b 10.1097/FJC.0b013e31818a403b
Carlsson LM, Jonsson J, Edlund T, Marklund SL: Mice lacking extracellular superoxide dismutase are more sensitive to hyperoxia. Proc Natl Acad Sci USA 1995, 92: 6264–6268. 10.1073/pnas.92.14.6264
Ritter C, Andrades M, Frota Júnior ML, Bonatto F, Pinho RA, Polydoro M, Klamt F, Pinheiro CT, Menna-Barreto SS, Moreira JC, Dal-Pizzol F: Oxidative parameters and mortality in sepsis induced by cecal ligation and perforation. Intensive Care Med 2003, 29(10):1782–1789. doi:10.1007/s00134–003–1861–5 10.1007/s00134-003-1789-9
Horn A Jr, Parrilha GL, Melo KV, Fernandes C, Horner M, Visentin Ldo C, Santos JA, Santos MS, Eleutherio EC, Pereira MD: An iron-based cytosolic catalase and superoxide dismutase mimic complex. Inorg Chem 2010, 49(4):1274–1276. doi:10.1021/ic901904b 10.1021/ic901904b
Bannister JV, Calaberese L: Assays for SOD. Methods Biochem Anal 1987, 32: 279–312.
Pasquali MAB, Gelain DP, Oliveira MR, Behr GA, Motta LL, Rocha RF, Klamt F, Moreira JC: Vitamin A supplementation induces oxidative stress and decreases the immunocontent of catalase and superoxide dismutase in rat lungs. Exp Lung Res 2009, 35: 427–438. 10.1080/01902140902747436
Oliveira MR, Lorenzi R, Schnorr CE, Morrone M, Moreira JC: Increased 3-nitrotyrosine levels in mitochondrial membranes and impaired respiratory chain activity in brain regions of adult female rats submitted to daily vitamin A supplementation for 2 months. Brain Res Bull 2011, 86: 246–253. doi:1016/j.brainresbull.2011.08.006 10.1016/j.brainresbull.2011.08.006
Bowler RP, Nicks M, Tran K, Tanner G, Chang LY, Young SK, Worthen GS: Extracellular superoxide dismutase attenuates lipopolysaccharide-induced neutrophilic inflammation. Am J Respir Cell Mol Biol 2004, 31(4):432–439. doi:10.1165/rcmb.2004–0057OC 10.1165/rcmb.2004-0057OC
Lund DD, Chu Y, Brooks RM, Faraci FM, Heistad DD: Effects of a common human gene variant of ECSOD on endothelial function after endotoxin in mice. J Physiol 2007, 584(2):583–590. doi:10.1113/jphysiol.2007.140830 10.1113/jphysiol.2007.140830
Yao H, Arunachalam G, Hwang J, Chung S, Sundar IK, Kinnula VL, Crapo JD, Rahman I: Extracellular superoxide dismutase protects against pulmonary emphysema by attenuating oxidative fragmentation of ECM. Proc Nat Acad Sci USA 2010, 107(35):15571–15576. doi:10.1073/pnas.1007625107 10.1073/pnas.1007625107
Oury TD, Schaefer LM, Fattman CL, Choi A, Weck KE, Watkins SC: Depletion of pulmonary EC-SOD after exposure to hyperoxia. Am J Physiol Lung Cell Mol Physiol 2002, 283: L777-L784. doi:10.1152/ajplung.00011.2002
Fukui S, Ookawara T, Nawashiro H, Suzuki K, Shima K: Post-ischemic transcriptional and translational responses of EC-SOD in mouse brain and serum. Free Radic Biol Med 2002, 32(3):289–298. doi:10.1016/S0891–5849(01)00804–8 10.1016/S0891-5849(01)00804-8
Van der Loo B, Labugger R, Skepper JN, Bachschmid M, Kilo J, Powell JM, Palacios-Callender M, Erusalimsky JD, Quaschning T, Malinski T, Gygi D, Ullrich V, Lüscher TF: Enhanced peroxynitrite formation is associated with vascular aging. J Exp Med 2000, 192: 1731–1744. doi:10.1084/jem.192.12.1731 10.1084/jem.192.12.1731
Alvarez B, Demicheli V, Duran R, Trujillo M, Cerveñansky C, Freeman BA, Radi R: Inactivation of human Cu, Zn superoxide dismutase by peroxynitrite and formation of histidinyl radical. Free Radic Biol Med 2004, 37: 813–822. doi:10.1016/j.freeradbiomed.2004.06.006 10.1016/j.freeradbiomed.2004.06.006
Sun D, Huang A, Yan EH, Wu Z, Yan C, Kaminski PM, Oury TD, Wolin MS, Kaley G: Reduced release of nitric oxide to shear stress in mesenteric arteries of aged rats. Am J Physiol Heart Circ Physiol 2004, 286: H2249-H2256. doi:10.1152/ajpheart.00854.2003 10.1152/ajpheart.00854.2003
Iida S, Chu Y, Weiss RM, Kang YM, Faraci FM, Heistad DD: Vascular effects of a common gene variant of extracellular superoxide dismutase in heart failure. Am J Physiol Heart Circ Physiol 2006, 291(2):H914-H920. doi:10.1152/ajpheart.00080.2006 10.1152/ajpheart.00080.2006
Acknowledgements
This research was supported by grants from CAPES, FAPESC, UNESC, and CNPq-Instituto Nacional de Ciência e Tecnologia Translacional em Medicina (INCT-TM). Further acknowledgment is expressed to all authors who contributed to this study.
Author information
Authors and Affiliations
Corresponding author
Additional information
Competing interests
The authors declare that they have no competing interests.
Authors’ contributions
LC, MRB, JCFM, AH, TM, CR, and FD-P conceived this study, participated in the design of the study, and drafted the manuscript. RCG, VRG, CDT, FV, LWK, GMTO, MABP, KVM, and CF participated in the design of the study and in the collection of data. All authors read and approved the final manuscript.
Authors’ original submitted files for images
Below are the links to the authors’ original submitted files for images.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made.
The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder.
To view a copy of this licence, visit https://creativecommons.org/licenses/by/4.0/.
About this article

{kind=link}
{kind=link}
{kind=link}
{kind=link}
Cite this article
Constantino, L., Gonçalves, R.C., Giombelli, V.R. et al. Regulation of lung oxidative damage by endogenous superoxide dismutase in sepsis. ICMx 2, 17 (2014). https://doi.org/10.1186/2197-425X-2-17
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/2197-425X-2-17