
Abstract
Partial duplication and partial deletion of the short arm of chromosome 9 have each been reported in the literature as clinically recognizable syndromes. We present clinical, cytogenetic, and molecular findings on a five-week-old female infant with concomitant duplication and terminal deletion of the short arm of chromosome 9. To our knowledge ten such cases have previously been reported. Conventional cytogenetic analysis identified additional material on chromosome 9 at band p23. FISH analysis aided in determining the additional material consisted of an inverted duplication with a terminal deletion of the short arm. Microarray analysis confirmed this interpretation and further characterized the abnormality as a duplication of about 32.7 Mb, from 9p23 to 9p11.2, and a terminal deletion of about 11.5 Mb, from 9p24.3 to 9p23. The infant displayed characteristic features of Duplication 9p Syndrome (hypotonia, bulbous nose, single transverse palmar crease, cranial anomalies), as well as features associated with Deletion 9p Syndrome (flat nasal bridge, long philtrum, cardiac anomalies) despite the deletion being distal to the reported critical region for this syndrome. This case suggests that there are genes or regulatory elements that lie outside of the reported critical region responsible for certain phenotypic features associated with Deletion 9p Syndrome. It also underscores the importance of utilizing array technology to precisely define abnormalities involving the short arm of 9p in order to further refine genotype/phenotype associations and to identify additional cases of duplication/deletion.
Similar content being viewed by others
Background
Rearrangements involving the distal region of 9p, particularly p22-p24, have been well described in the literature and are associated with either Duplication 9p Syndrome or Deletion 9p Syndrome (OMIM 158170) [1–5]. Few cases, however, have been reported with concurrent duplication and terminal deletion of 9p [5–14]. Here we present another such case and compare our clinical and cytogenetic findings with those in the literature.
Clinical report
The patient is the third child of a healthy 19-year-old woman of African-American and Hispanic descent. She was born preterm at 36 weeks gestation. At birth, she weighed 2.7 kg; her head circumference was 33.5 cm and length was 48 cm (all around the 10th percentile). She was evaluated by pediatric cardiology at three days of life because of a heart murmur. An echocardiogram, at that time, revealed a small perimembraneous ventricular septal defect, moderate patent ductus arteriosus, and a moderate to large secundum atrial septal defect. She initially had hypothermia and hypoglycemia, however, these conditions resolved quickly. She also exhibited hypotonia and intermittent tongue thrusting.
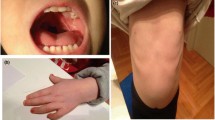
Notable findings on physical examination at five weeks of age included wide-open anterior fontanelle and sagittal suture, prominent nose, flat nasal bridge, long grooved philtrum, thin lips, extra skin at the back of the neck, and a single transverse palmar crease on the left hand. External genitalia were that of a normal female.
Results
Cytogenetics and fluorescence in situ hybridization (FISH)
Cytogenetic analysis identified additional material on one of the homologs of chromosome 9 at band p23 (Figure 1A). FISH analysis using whole chromosome 9 painting probe, WCP 9, revealed the extra material was comprised entirely of material from chromosome 9 (Figure 1B). Additionally, FISH identified a terminal deletion of 9p using the 9p subtelomeric probe, 305J7T7 (Figure 1C). Based on both conventional cytogenetics and FISH analysis, the derivative chromosome 9 was determined to have an inverted duplication of the region between bands p11.2 and p23 as well as a concurrent deletion of the terminal pter-p23 segment. We were unable to determine whether this abnormality was inherited or de novo as parental blood was unavailable.

Identification and characterization of the 9p abnormality by conventional cytogenetics and FISH analysis. (A) G-banded chromosomes 9 with der(9)(pter→p24.3::p11.2→p23::p23→qter). Ideogram showing the region that is duplicated, p11.2-p23 (blue bar), and inverted (red bar). (B) FISH analysis using WCP 9 and (C) subtelomeric probes 305J7T7 for 9p (green) and LSI 9q34 for 9q (aqua). Arrow indicates terminal deletion of 9p.
Microarray analysis
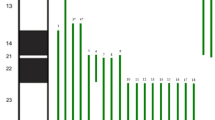
Affymetrix Genome-Wide Human SNP Array 6.0 confirmed the apparent interstitial duplication and distal deletion on the short arm of chromosome 9 (Figure 2). The duplication was found to span approximately 32.7 Mb, from 11,576,113 bp (9p23), to 44,244,868 (9p11.2). The proximal deletion breakpoint occurred between genomic probe positions 11,575,785 and 11,576,113 (9p23), with the deletion encompassing the region distal to the breakpoint, indicating that genes including and distal to PTPRD are deleted and those proximal to this break on the p arm are duplicated. The final karyotype after molecular characterization was: 46,XX,der(9)(pter→p24.3::p11.2→p23::p23→qter).arrcgh9p24.3p23(37,746-11,575,785)x1,9p23p11.2(11,576,113-44,244,868)x3.

Characterization of the breakpoints involved in the duplication and deletion of 9p using the Affymetrix Genome-Wide Human SNP Array 6.0. The plot shows chromosome 9 log2 test over reference ratio and allelic difference (Y-axis) plotted against the Mb position from pter to the centromere (X-axis). A significant terminal loss of approximately 11.5 Mb, from 9pter to 9p23, and an adjacent duplication of approximately 32.7 Mb, from 9p23 to 9p11.2, were identified.
Discussion
Partial duplication of 9p is one of the most commonly detected autosomal structural abnormalities in live-borns [15, 16]. Despite variability in size of the duplicated segment, patients with partial duplication of 9p display considerable phenotypic similarity. Characteristic features of this syndrome, which include microcephaly, hypertelorism, upslanting palpebral fissures, broad nasal root with bulbous nasal tip, downturned corners of the mouth, anomalous ears, single palmar crease, skeletal malformations, hypotonia, and developmental delay, are thought to result from duplication of genes lying within 9p22 [15, 16]. The current case has a duplication of approximately 32.7 Mb, from 9p23 to 9p11.2, and displays some of the cardinal features of Duplication 9p Syndrome (Figure 3).

Comparison of deletion and duplication in the present case with the previously reported 2 Mb terminal deletion[10], the proposed critical region in Deletion 9p Syndrome, and the region associated with Duplication 9p Syndrome. Clinical features associated with each region are indicated. Features in the present case that correspond to the deleted or duplicated regions are shown in bold.
Deletion 9p syndrome is a well described clinical entity with over 80 cases reported in the literature. Phenotypic features include mental retardation, trigonocephaly, midface hypoplasia, flat nasal bridge, long philtrum, microstomia, micro/retrognathia, cardiac anomalies, and genital anomalies in males. Work from several groups has narrowed down the critical region to p22.2-p23, a 3.5 Mb region between 11.4-14.9 Mb from the 9p telomere [4, 9, 17, 18]. More recently, Swinkels et al. proposed refining the critical region to approximately 300 kb within band 9p22.3 [5].
The deletion in the present case lies distal to the critical region proposed by these groups. Nonetheless, our patient displays features associated with Deletion 9p Syndrome such as long philtrum, flat nasal bridge, and heart murmurs/defects. Similar reports of patients with distal terminal deletions exhibiting features of Deletion 9p Syndrome have also been reported [14, 19]. Recently, Hauge et al. identified a minimal 9p terminal deletion of 2 Mb, distal to the previously described critical region, in 5 patients who manifested features common to Deletion 9p Syndrome [10]. Figure 3 depicts the proposed critical region for Deletion 9p Syndrome, the 2 Mb minimally deleted region [10], the deleted region in the present case, and the clinical features associated with each. Our report, in addition to similar reports [10, 14, 19], submits that deletion of genes more distal to the previously described critical region contributes to features commonly associated with Deletion 9p Syndrome (Figure 3) and that further refinement to the genotype/phenotype correlation for this syndrome is needed.
One of the major features of Deletion 9p Syndrome, trigonocephaly, was absent in the present case. Previous reports have mapped trigonocephaly to a region in p22.3 between 14.7 and 15.1 Mb. Two genes that lie within this region, FREM1 and CER1, have been implicated as candidate genes [18, 20]. FREM1 mutations exhibit variable penetrance and expressivity, suggesting that other factors, possibly mutations or deletions of additional genes or regulatory elements, contribute to the phenotype. Features of trigonocephaly have also been reported in patients with deletions that lie distal to FREM1[4, 10, 19], further suggesting that other genes may be involved.
In contrast to trigonocephaly, the patient in the present study exhibited delayed closure of the anterior fontanelle and sagittal suture. This feature is occasionally seen in patients with Duplication 9p Syndrome. Interestingly, the duplicated region in our case coincides with the deleted region commonly associated with trigonocephaly. It is possible that a dosage sensitive gene (or genes), possibly FREM1 and/or CER1, is involved in the development and/or closure of skeletal plates.
Of the genes that fall within the deleted region in our patient (Additional file 1: Table S1), several are associated with a particular phenotype. Deletion of FOXD4 is associated with speech and language delays [10], and mutations in a related forkhead box gene, FOXP2, cause linguistic impairment and difficulties with motor coordination [21]. Haploinsufficiency of DMRT1 2, and 3 may result in ambiguous external genitalia, particularly in karyotypic males, as well as gonadal dysgenesis [7, 22, 23]. Disruption of DOCK8 was identified in patients with mental retardation and/or seizures [10, 24]. Similarly, mutations in VLDR1 are associated with cognitive impairment, cerebellar ataxia, and seizures [25]. Our patient exhibited intermittent tongue thrusting, however, whether it is associated with mental retardation or epilepsy is difficult to assess due to her age. Deletions within the paternal KANK1 gene have been identified in several children with cerebral palsy [26]. The significance of the KANK1 deletion in our patient is unknown as the parental origin of the 9p deletion/duplication was not able to be determined.
To our knowledge, 10 cases with both a proximal duplication and a terminal deletion of 9p have previously been reported. Comparison of the deleted and duplicated regions in the current case and reported cases for which microarray analysis had been performed [5, 9–12, 14] is given in Figure 4. Deletions are placed into three groups: Group 1 (two cases) with terminal deletions of less than 2 Mb, Group 2 (three cases) with terminal deletions of 3.7-11.5 Mb, encompassing the 2 Mb minimally deleted terminal region of 9p identified by Hauge et al. [10], and Group 3 (two cases) with larger deletions of 14.8-17.3 Mb, which include the Deletion 9p Syndrome critical region. The size of the duplication varied between cases with most encompassing the p22 region. Clinical features varied among cases (Table 1), owing to the different sizes and locations of the deletion and duplication; nevertheless, they could be divided into 3 categories: those associated with a 2 Mb terminal deletion of 9p, those related to Deletion 9p Syndrome, and those common to Duplication 9p Syndrome.

Comparison of all reported cases of deletion and duplication of 9p for which microarray analysis was performed. Shaded boxes represent the 2 Mb terminally deleted region (green), the proposed critical region for Deletion 9p Syndrome (blue), and the region associated with Duplication 9p Syndrome (red). Group 1 includes cases with deletions below 2 Mb, Group 2 comprises cases with deletions covering the 2 Mb terminal region, and Group 3 includes cases with deletions encompassing the proposed critical region for Deletion 9p Syndrome (Group 3).
We report the eleventh case involving partial duplication of 9p with a concomitant cryptic terminal deletion. Similar cases have likely been missed in the past due to the sole use of conventional cytogenetics which has undoubtedly confounded genotype/phenotype correlations, underscoring the importance of using molecular techniques to identify these often small, however, clinically significant terminal deletions. The use of microarray technology is critical for the proper identification and precise molecular characterization of these abnormalities and will aid in better defining critical regions, facilitating more accurate genotype/phenotype correlations, and potentially describing new clinical syndromes, such as 9p Deletion/Duplication Syndrome.
Materials and methods
Cytogenetics and FISH
Peripheral blood from the patient was cultured for 72 hr in the presence of phytohemagglutinin. Metaphase spread preparations and GTG-banding were performed according to standard methods. FISH analysis was performed using WCP 9 and subtelomeric probes, 305J7T7 for 9p (green) and LSI 9q34 for 9q (aqua) (Vysis/Abbott Molecular Inc., Des Plaines, IL).
Molecular karyotyping
SNP array was performed using the Affymetrix Genome-Wide Human SNP Array 6.0, which contains 906,600 single nucleotide polymorphism probes (SNPs) and over 946,000 probes for copy number (Affymetrix, Santa Clara, CA, USA). Manufacturer-provided protocols for experimentation (http://media.affymetrix.com/support/downloads/manuals/) and analysis (Chromosome Analysis Suite (ChAS) software package, version 1.2) were followed. Genomic positions are given as mapped to the GRCh37/hg19 genome build.
Consent
Written informed consent for this cytogenetic study was obtained from a parent of the patient. A copy of the written consent is available for review by the Editor-in-Chief of this journal.
References
Rethoré MO, Larget-Piet L, Abonyi D, Boeswillwad M, Berger R, Carpentier S, Cruveiller J, Dutrillau B, Lafourcade J, Penneau M, Lejeune J: Cases of trisomy for the short arm of chromosome 9. Individualization of a new morbid entity. Ann Genet 1970, 13: 217–232.
Alfi O, Donnell GN, Crandall BF, Derencsenyi A, Menon R: Deletion of the short arm of chromosome no.9 (46,9p-): a new deletion syndrome. Ann Genet 1973, 16: 17–22.
Centerwall WR, Beatty-DeSana JW: The trisomy 9p syndrome. Pediatrics 1975, 56: 748–755.
Huret JL, Leonard C, Forestier B, Rethore MO, Lejeune J: Eleven new cases of del(9p) and features from 80 cases. J Med Genet 1988, 25: 741–749. 10.1136/jmg.25.11.741
Swinkels ME, Simons A, Smeets DF, Vissers LE, Veltman JA, Pfundt R, de Vries BB, Faas BH, Schrander-Stumpel CT, McCann E, Sweeney E, May P, Draaisma JM, Knoers NV, van Kessel AG, van Ravenswaaij-Arts CM: Clinical and cytogenetic characterization of 13 Dutch patients with deletion 9p syndrome: Delineation of the critical region for a consensus phenotype. Am J Med Genet A 2008, 146A: 1430–1438. 10.1002/ajmg.a.32310
Teebi AS, Gibson L, McGrath J, Meyn MS, Breg WR, Yang-Feng TL: Molecular and cytogenetic characterization of 9p- abnormalities. Am J Med Genet 1993, 46: 288–292. 10.1002/ajmg.1320460310
Muroya K, Okuyama T, Goishi K, Ogiso Y, Fukuda S, Kameyama J, Sato H, Suzuki Y, Terasaki H, Gomyo H, Wakui K, Fukushima Y, Ogata T: Sex-determining gene(s) on distal 9p: clinical and molecular studies in six cases. J Clin Endocrinol Metab 2000, 85: 3094–3100. 10.1210/jc.85.9.3094
Krepischi-Santos AC, Vianna-Morgante AM: Disclosing the mechanisms of origin of de novo short-arm duplications of chromosome 9. Am J Med Genet A 2003, 117A: 41–46. 10.1002/ajmg.a.10634
Faas BH, de Leeuw N, Mieloo H, Bruinenberg J, de Vries BB: Further refinement of the candidate region for monosomy 9p syndrome. Am J Med Genet A 2007, 143A: 2353–2356. 10.1002/ajmg.a.31961
Hauge X, Raca G, Cooper S, May K, Spiro R, Adam M, Martin CL: Detailed characterization of, and clinical correlations in, 10 patients with distal deletions of chromosome 9p. Genet Med 2008, 10: 599–611. 10.1097/GIM.0b013e31817e2bde
Mosca AL, Callier P, Faivre L, Marle N, Mejean N, Thauvin-Robinet C, Masurel-Paulet A, Madinier N, Durand C, Couillaud G, Ragot S, Huet F, Teyssier JR, Mugneret F: Polymicrogyria in a child with inv dup del(9p) and 22q11.2 microduplication. Am J Med Genet A 2009, 149A: 475–481. 10.1002/ajmg.a.32665
Hulick PJ, Noonan KM, Kulkarni S, Donovan DJ, Listewnik M, Ihm C, Stoler JM, Weremowicz S: Cytogenetic and array-CGH characterization of a complex de novo rearrangement involving duplication and deletion of 9p and clinical findings in a 4-month-old female. Cytogenet Genome Res 2009, 126: 305–312. 10.1159/000251966
Al Achkar W, Wafa A, Moassass F, Liehr T: Partial trisomy 9p22 to 9p24.2 in combination with partial monosomy 9pter in a Syrian girl. Mol Cytogenet 2010, 3: 18. 10.1186/1755-8166-3-18
Chen CP, Su YN, Chern SR, Hsu CY, Tsai FJ, Wu PC, Lee CC, Chen YT, Lee MS, Wang W: Inv dup del(9p): prenatal diagnosis and molecular cytogenetic characterization by fluorescence in situ hybridization and array comparative genomic hybridization. Taiwan J Obstet Gynecol 2011, 50: 67–73. 10.1016/j.tjog.2011.01.038
Haddad BR, Lin AE, Wyandt H, Milunsky A: Molecular cytogenetic characterisation of the first familial case of partial 9p duplication (p22p24). J Med Genet 1996, 33: 1045–1047. 10.1136/jmg.33.12.1045
Fujimoto A, Lin MS, Schwartz S: Direct duplication of 9p22→p24 in a child with duplication 9p syndrome. Am J Med Genet 1998, 77: 268–271. 10.1002/(SICI)1096-8628(19980526)77:4<268::AID-AJMG3>3.0.CO;2-J
Christ LA, Crowe CA, Micale MA, Conroy JM, Schwartz S: Chromosome breakage hotspots and delineation of the critical region for the 9p-deletion syndrome. Am J Hum Genet 1999, 65: 1387–1395. 10.1086/302606
Kawara H, Yamamoto T, Harada N, Yoshiura K, Niikawa N, Nishimura A, Mizuguchi T, Matsumoto N: Narrowing candidate region for monosomy 9p syndrome to a 4.7-Mb segment at 9p22.2–p23. Am J Med Genet A 2006, 140: 373–377.
Barbaro M, Balsamo A, Anderlid BM, Myhre AG, Gennari M, Nicoletti A, Pittalis MC, Oscarson M, Wedell A: Characterization of deletions at 9p affecting the candidate regions for sex reversal and deletion 9p syndrome by MLPA. Eur J Hum Genet 2009, 17: 1439–1447. 10.1038/ejhg.2009.70
Vissers LE, Cox TC, Maga AM, Short KM, Wiradjaja F, Janssen IM, Jehee F, Bertola D, Liu J, Yagnik G, Sekiguchi K, Kiyozumi D, van Bokhoven H, Marcelis C, Cunningham ML, Anderson PJ, Boyadjiev SA, Passos-Bueno MR, Veltman JA, Smyth I, Buckley MF, Roscioli T: Heterozygous mutations of FREM1 are associated with an increased risk of isolated metopic craniosynostosis in humans and mice. PLoS Genet 2011, 7: e1002278. 10.1371/journal.pgen.1002278
Fisher SE, Vargha-Khadem F, Watkins KE, Monaco AP, Pembrey ME: Localisation of a gene implicated in a severe speech and language disorder. Nature Genet 1998, 18: 168–170. 10.1038/ng0298-168
Shan Z, Zabel B, Trautmann U, Hillig U, Ottolenghi C, Wan Y, Haaf T: FISH mapping of the sex-reversal region on human chromosome 9p in two XY females and in primates. Eur J Hum Genet 2000, 8: 167–173. 10.1038/sj.ejhg.5200431
Livadas S, Mavrou A, Sofocleous C, van Vliet-Constantinidou C, Dracopoulou M, Dacou-Voutetakis C: Gonadoblastoma in a patient with del(9)(p22) and sex reversal: report of a case and review of the literature. Cancer Genet Cytogenet 2003, 143: 174–177. 10.1016/S0165-4608(02)00849-X
Griggs BL, Ladd S, Saul RA, DuPont BR, Srivastava AK: Dedicator of cytokinesis 8 is disrupted in two patients with mental retardation and developmental disabilities. Genomics 2008, 91: 195–202. 10.1016/j.ygeno.2007.10.011
Boycott KM, Flavelle S, Bureau A, Glass HC, Fujiwara TM, Wirrell E, Davey K, Chudley AE, Scott JN, McLeod DR, Parboosingh JS: Homozygous deletion of the very low density lipoprotein receptor gene causes autosomal recessive cerebellar hypoplasia with cerebral gyral simplification. Am J Hum Genet 2005, 77: 477–483. 10.1086/444400
Lerer I, Sagi M, Meiner V, Cohen T, Zlotogora J, Abeliovich D: Deletion of the ANKRD15 gene at 9p24.3 causes parent-of-origin-dependent inheritance of familial cerebral palsy. Hum Mol Genet 2005, 14: 3911–3920. 10.1093/hmg/ddi415
Author information
Authors and Affiliations
Corresponding author
Additional information
Competing interests
The authors declare they have no competing interests.
Authors’ contributions
DD analyzed conventional and molecular cytogenetic findings, collected relevant data, and drafted the paper. ME, RO, and TS performed the microarray studies and analysis. FG performed clinical examination of the patient and provided insight into the manuscript. VP and SM supervised the conventional and molecular cytogenetic analysis. SM provided insight into the manuscript. All of the authors read and approved the final manuscript.
Electronic supplementary material
Authors’ original submitted files for images
Below are the links to the authors’ original submitted files for images.
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Rights and permissions
This article is published under license to BioMed Central Ltd. This is an Open Access article distributed under the terms of the Creative Commons Attribution License (http://creativecommons.org/licenses/by/2.0), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
About this article
Cite this article
Di Bartolo, D.L., El Naggar, M., Owen, R. et al. Characterization of a complex rearrangement involving duplication and deletion of 9p in an infant with craniofacial dysmorphism and cardiac anomalies. Mol Cytogenet 5, 31 (2012). https://doi.org/10.1186/1755-8166-5-31
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/1755-8166-5-31