
Abstract
Background
Inherited retinal dystrophies, including Retinitis Pigmentosa and Leber Congenital Amaurosis among others, are a group of genetically heterogeneous disorders that lead to variable degrees of visual deficits. They can be caused by mutations in over 100 genes and there is evidence for the presence of as yet unidentified genes in a significant proportion of patients. We aimed at identifying a novel gene for an autosomal recessive form of early onset severe retinal dystrophy in a patient carrying no previously described mutations in known genes.
Methods
An integrated strategy including homozygosity mapping and whole exome sequencing was used to identify the responsible mutation. Functional tests were performed in the medaka fish (Oryzias latipes) model organism to gain further insight into the pathogenic role of the ADAMTS18 gene in eye and central nervous system (CNS) dysfunction.
Results
This study identified, in the analyzed patient, a homozygous missense mutation in the ADAMTS18 gene, which was recently linked to Knobloch syndrome, a rare developmental disorder that affects the eye and the occipital skull. In vivo gene knockdown performed in medaka fish confirmed both that the mutation has a pathogenic role and that the inactivation of this gene has a deleterious effect on photoreceptor cell function.
Conclusion
This study reveals that mutations in the ADAMTS18 gene can cause a broad phenotypic spectrum of eye disorders and contribute to shed further light on the complexity of retinal diseases.
Similar content being viewed by others

Background
Inherited retinal dystrophies (IRD) are a genetically heterogeneous group of disorders that represent the most frequent causes of genetic blindness in the western world [1, 2]. They include Retinitis Pigmentosa (RP) and Leber Congenital Amaurosis (LCA), among the others. RP is the most frequent form of retinal dystrophies with an approximate incidence ranging between 1 in 3000 and 1 in 5000 individuals [1, 3, 4]. LCA is characterized by a severe visual impairment that starts in the first years of life [5]. Both RP and LCA are generally characterized by a large extent of genetic heterogeneity. Over the last few years, about 230 genes causing inherited retinal diseases have been mapped to chromosomal locations (see RETnet web site: http://www.sph.uth.tmc.edu/RetNet/) and about 190 responsible genes have been identified. It is currently possible to determine the molecular defect underlying IRDs in up to 50% of patients [6], which strongly indicates the existence of additional genes responsible for these conditions.
We recently analyzed a cohort of over 400 Italian patients with autosomal recessive retinal dystrophies, including Retinitis Pigmentosa (ARRP) and LCA ([7] and unpublished data). Patients were screened for the presence of previously described mutations in genes with a known pathogenic role in ARRP and LCA using genotyping microchips based on the allele-specific primer extension (APEX) technique [8]. About 70% of the analyzed patients did not harbor any previously described mutation in known LCA/ARRP genes. We reasoned that this subset of patients could be a valuable resource to identify novel retinal dystrophy genes. Therefore, we selected a subset of the latter patients, preferentially those with some evidence of belonging to consanguineous families, for homozygosity mapping genotyping followed by whole exome sequencing analysis, which has proven to be an effective strategy for the identification of novel autosomal-recessive retinal disease genes [9, 10].
Methods
Human subjects
We used standard methods to isolate genomic DNA from peripheral blood of the patients and their family members. Informed consent was obtained from all participating patients and families according to the Declaration of Helsinki and the studies were approved by the Research Ethics Committee of the Second University of Naples.
Ophthalmologic examination including best corrected visual acuity using Snellen charts or Teller Acuity Cards, measurement of objective refractive error after cycloplegia, slit-lamp biomicroscopy, dilated fundus examination, bilateral full-field ERGs and optical coherence tomography (OCT) recordings, was carried out as previously described [11].
SNP genotyping
Genotyping was performed on SNP microarray (GeneChip Genome-Wide Human SNP Array 5.0, Affymetrix, Santa Clara, CA). Array experiments were performed according to protocols provided by the manufacturer. Genotypes were called with the Genotype Console program, ver. 2.1 (Affymetrix) and regions of homozygosity were identified using PLINK [12], with a sliding window of 50 SNPs and allowing 2 heterozygous SNPs (miscalls) and 10 missing SNPs (no calls) per window. Regions containing more than 250 consecutive homozygous SNPs were considered to be significant homozygous regions, on average corresponding to a genomic size of 1 Megabase (Mb).
Whole exome sequencing
Whole exome enrichment was carried out using the SureSelect All Exon kit v.1 (Agilent Technologies, Santa Clara, CA, USA) according to the manufacturer instructions. Whole exome sequencing was carried out on a SOLiD 3 Plus System (Life Technologies, Carlsbad, CA, USA). Sequencing reads were mapped to the reference genome (UCSC, hg19 build) using the software BioScope v1.3 (Life Technologies, Carlsbad, CA, USA). Single nucleotide variations (SNV) and in-del mutation calling analyses were carried out using the diBayes algorithm with medium stringency settings and the SOLiD Small Indel Fragment Tool, respectively.
RNA in situ hybridization and immunohistochemistry
RNA in situ hybridization on mouse sections and immunohistochemistry experiments on medaka eyes were performed according to previously published protocols [13, 14].
Medaka stocks and mRNA injections
The Cab-strain of wild type medaka fish were kept and staged as described [15]. A morpholino (Mo; Gene Tools LLC, Oregon, USA) was designed against the ATG initiation codon and the 5′ untranslated region of the medaka ortholog of the ADAMTS18 gene (olAdamts18) whereas a control Mo carrying five mismatches (mmMo-Adamts18) was used as a control (see sequences in Additional file 1: Table S2). The specificity and inhibitory efficiencies of Mo-Adamts18 were determined as described in [16]. Mo-Adamts18 was injected at a 90 μM concentration into one blastomere at the two-cell stage. In vitro synthesis of the human wild type ADAMTS18 mRNAs as well as of the c.T3235 > C mutated form was performed as described [14]. Morpholino and mRNA injections were carried out as previously described [14, 17, 18].
The light-induced photoreceptor cell degeneration assay was performed as previously described [19]. Medaka embryos were incubated with phenyl thiourea (PTU) to prevent pigmentation [18]. Fish were sacrificed and analyzed after 5 days of constant light.
Results and discussion
Clinical features of patient A24
Among the selected patients, subject A24, a 30-year old male, displayed a severe early-onset retinal dystrophy accompanied by an autistic disorder. He is the second and last son of two parents that were born in the same small community in Southern Italy. Delivery was at full term without any complication and birth weight was 3750 g. At the age of 2.5, he was diagnosed with autism characterized by emotional indifference and poor/no response to environmental stimuli. A brain CT scan performed at that age was normal. At 2 years of age, the patient started to display exotropia and nystagmus and visual impairment, namely night blindness and reduced peripheral vision. At the age of 5, following a full-field electroretinogram recorded under sedation that showed extinguished rod and cone responses, the patient received a diagnosis of Early-Onset Severe Retinal Dystrophy [20–22]. At the age of 15, he underwent a more detailed ophthalmological evaluation that revealed a hypermetropic refraction (Right Eye: + 2; Left Eye: + 3) and a non-measurable visual acuity due to poor cooperation. Ocular motility testing revealed the presence of exotropia and nystagmus. No signs of fibrillar vitreous condensation were observed by slit lamp analysis. Fundus examination showed attenuated retinal vessels, macular dystrophy, and diffuse mid-peripheral RPE mottling with myriads of tiny white and black dots (Figure 1A). During the follow-up examination at the age of 21, his best corrected visual acuity was 20/1000 and full-field electroretinogram showed extinguished rod and cone responses. At the age of 30, an incipient posterior subcapsular cataract in the right eye was also observed, while all other ocular defects were unchanged. He underwent an OCT (Optical Coherence Tomography) test that demonstrated an absence of foveal depression due to the presence of macular pucker associated with intraretinal cystoid spaces and partial vitreous attachment to the optic disc (data not shown). Complete physical evaluation failed to reveal involvement of other organs and systems.

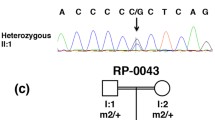
The ADAMTS18 gene is mutated in a patient with Early Onset Severe Retinal Dystrophy. (A) Fundus examination of patient A24 revealed attenuated retinal vessels, macular dystrophy, and diffuse mid-peripheral RPE mottling with myriads of tiny white and black dots. (B) Schematic representation, visualized with the Integrative Genomics Viewer (IGV) browser, of the mapped exome sequencing reads around the ADAMTS18 c.T3235 > C sequence variant in patient A24. The upper part shows the wild-type sequence and the coverage per base. The c.T3235 > C homozygous variant is shown in brown. Please note that the sequence variant shown in red corresponds to a described SNP (rs35478105). (C) Validation by Sanger sequencing of the c.T3235 > C in patient A24 while his parents and his unaffected brother are heterozygous for the variation (red arrows); the rs35478105 SNP is labeled by an asterisk. (D) The top diagram shows a schematic representation of the ADAMTS18 protein. The c.T3235 > C (p.C1079R) variant affects a cysteine residue localized within the third TSP type 1 motifs, which is highly conserved across evolution (red asterisk, bottom panel). (E) RNA in situ hybridization experiment showing that the murine Adamts18 is expressed in the adult mouse retina (see text for further details). ON, optic nerve.
Identification of a mutation in the ADAMTS18 gene in patient A24
We carried out SNP genotyping analysis on a genomic DNA sample from patient A24. This analysis revealed the presence of four large homozygous regions in the genome of the patient, namely on chromosomes 8, 4, 3 and 16 for a total of 15.5 Mb (Additional file 2: Table S1).
We then performed whole exome sequencing analysis on patient A24. Over 65 million sequencing reads, corresponding to about 3.3 Gigabases (Gb) of mappable sequences were obtained using a SOLiD 3 Plus System (Life Technologies, Carlsbad, CA, USA). In total, 86.12% reads could be mapped to the reference genome (UCSC, hg19 build). About 77% of the uniquely mapped reads were aligned on the targeted exome after duplicate reads removal, with 70% of the targeted exons covered at > 20x depth. Overall, we identified 13.388 exonic variants including 6.213 non-synonymous SNVs, 152 small insertions or deletions, 71 stop-gain or stop-loss variants and 40 variants putatively affecting splice sites. Further filtering based on the exclusion of all known dbSNP variants (using dbSNP130) reduced the total number to 760 and the application of an autosomal-recessive model of inheritance for the disease left 110 sequence variants. By limiting the analysis to the regions of homozygosity and by only considering genes with reported evidence of significant expression in the eye [23, 24], we were left with only a single homozygous missense variation, namely c.T3235 > C (p.C1079R) in the ADAMTS18 gene (NM_199355) that is localized to the long arm of chromosome 16 (Figure 1B). Intriguingly, Aldahmesh et al. recently reported a homozygous missense variation [25] in this gene in a Saudi Arabian family with Knobloch syndrome, a rare autosomal recessive developmental disorder that affects the eye and the occipital region of the skull and the brain (On-line Mendelian Inheritance in Man (OMIM) #267750). They therefore proposed ADAMTS18 as the gene responsible for Knobloch syndrome even if they did not provide any functional evidence in support of their hypothesis [25]. Due to the above-mentioned observations, we regarded this gene as a good candidate for a pathogenic role in the phenotype of patient A24 and therefore selected it for further analysis. ADAMTS18 is a member of the ADAMTS protein family, a family of metalloproteinases similar to the ADAM proteins (A Disintegrin-like And Metalloproteinase), but distinct by the additional presence of ThromboSpondin (TSP) motifs in the C-terminus and the lack of transmembrane domains [26]. The c.T3235 > C mutation causes the substitution of a highly conserved cysteine residue with an arginine in one of the four C-terminal TSP type 1 motifs of the protein (Figure 1D), which are known to be important in modulating ADAMTS-mediated proteolysis [27] and influence protein recognition and matrix localization [28].
Sanger sequencing confirmed that the c.T3235 > C sequence variation was present in homozygosity in patient A24 (Figure 1C). The patient’s parents and his healthy brother were found to be heterozygous carriers of the mutation, which is in line with the autosomal recessive inheritance of the disorder in the family (Figure 1C). The variation was not present in neither the Exome Variant Server (http://evs.gs.washington.edu/EVS/) nor in over 350 Italian control DNA samples. Finally, the c.T3235 > C variation was predicted to be “disease causing” by MutationTaster (http://www.mutationtaster.org/) with high probability and “probably damaging” by Polyphen2 (http://genetics.bwh.harvard.edu/pph2/index.shtml), further supporting its putative pathogenic role.
This is the second report that describes a putative pathogenic role of the ADAMTS18 gene in human genetic diseases that affect the eye and the central nervous system (CNS). Due to the recent description of another ADAMTS18 homozygous missense mutation, i.e., the p.S179L, in a family with Knobloch syndrome [25], we carefully revised the clinical history and phenotype of patient A24 to detect a possible overlap with the Knobloch phenotype. However, the lack of any detectable occipital defect as well as the lack of myopia (the patient is actually hypermetropic) and of the classical signs of vitreoretinal degeneration [29] prompted us to exclude the diagnosis of Knobloch syndrome in patient A24 (Figure 1A and data not shown) thus suggesting that the ADAMTS18 gene is also responsible for non-Knobloch forms of retinal diseases.
To determine whether mutations in ADAMTS18 are more widely involved in retinal dystrophies, we analyzed 450 unrelated individuals who had ARRP, LCA or autosomal recessive Cone Rod dystrophy and who had significant homozygous regions (see Methods). In 9 families, ADAMTS18 was located in significantly large homozygous regions: sequence analysis of probands from these families, however, did not reveal the presence of additional mutation in ADAMTS18.
ADAMTS18 is expressed in the adult eye
ADAMTS18 was previously reported to be expressed in the developing mouse eye [25]. We aimed at determining whether ADAMTS18 is also expressed in the adult eye, and mainly in the retina, i.e., the main target of the phenotype present in patient A24. To that purpose, we performed RT-PCR experiments on human retina cDNA (Clontech). As a result, we detected the expected size products (data not shown) using oligonucleotide primers specific for the ADAMTS18 mRNA sequence (Additional file 1: Table S2) and we confirmed their identity by Sanger sequencing. To define the sites of expression of this gene at the cellular level, we performed RNA in situ hybridization experiments on murine adult eye section collected at P60. This experiment revealed the murine Adamts18 gene to be strongly expressed in the retinal ganglion cell layer (GCL), in the inner part of the inner nuclear layer (INL) and in the retinal pigment epithelium (RPE) (Figure 1E).
In vivo analysis in medaka fish
To further prove the pathogenic effect of the ADAMTS18 c.T3235 > C variation in patient A24, we carried out an in vivo functional test in the Oryzias latipes (medaka fish) model system by means of gene knock-down and rescue assays, as already described [17]. We designed a specific morpholino oligonucleotide against the ATG initiation codon and the 5′ untranslated region of the medaka ortholog of the ADAMTS18 gene (olAdamts18) (Additional file 1: Table S2) and we injected it into fertilized one-cell embryos. We observed an aberrant CNS phenotype at stage (st)40 in morphant embryos (79 ± 3% of 1,300 injected embryos), particularly in the telencephalic and mesencephalic areas (Figure 2A-B”) of the brain. In particular, we observed an altered morphology of the dorsolateral part of the telencephalon (DLT in Figure 2B’) and a size reduction of the telencephalic ventricles (TV in Figure 2B’). Furthermore, we observed an expansion of the optic tectum in the mesencephalon (OT in Figure 2B”). Interestingly, the areas of the medaka brain affected by the olAdamts18 knockdown correspond to the most posterior areas of the human brain, which can be altered in Knobloch syndrome patients (OMIM #267750). The above-described aberrant medaka phenotypes were significantly rescued when the olAdamts18 morpholino was co-injected with the full-length coding human ADAMTS18 synthetic mRNA (93 ± 2% of 600 injected embryos; Figure 2C-C”) but not with the human mRNA carrying the c.T3235 > C mutation (Figure 2D-D”). These data provide in vivo evidence that the c.T3235 > C missense variation in the ADAMTS18 gene has a pathogenic effect. We believe that the in vivo gene knockdown/rescue assays in medaka can be used as a rapid and efficient tool to screen the possible pathogenic role of sequence variants identified in the course of whole exome or whole genome sequencing efforts in patients with genetic diseases, particularly in those characterized by a developmental phenotype.

The c.T3235 > C ADAMTS18 mutation has a deleterious effect in vivo . Bright-field stereomicroscopy images of dorsal views of control (A), Mo-AdamTS18- (B), MO-AdamTS18/ADAMTS18wt- (C), and MO-AdamTS18/ADAMTS18mut- (D) injected medaka embryos. (A’,A”,B’,B”,C’,C”, D’,D”) Frontal cryostat sections of St40 medaka embryos stained with DAPI (white). Panels A’-D’ are frontal sections through the telencephalon (the section plan is marked by the blue line), whereas panels A”-C” are frontal sections through the mesencephalon (the section plan is marked by the red line). (B’) In Mo-AdamTS18-injected embryos, both the dorsolateral part of the telencephalon (DLT) and the size of the telencephalic ventricles (TV) are altered with respect to control embryos (A’). (B”) Expansion of the optic tectum (OT) in the mesencephalon is present in Mo-AdamTS18-injected embryos. (C’-C”) Wild-type human ADAMTS18 mRNA co-injection with Mo-AdamTS18 restores the correct pattern of both telencephalic and mesencephalic tissue. (D’-D”) Co-injection of the human ADAMTS18 mRNA carrying the c.T3235 > C mutation with the Mo-AdamTS18 does not rescue the telencephalic and mesencephalic phenotypes.
We could not detect any gross morphological abnormality of the eye in morphant embryos until st40. Therefore, in order to gain further insight into the involvement of ADAMTS18 in retinal function in vertebrates, we decided to perform a functional test in medaka fish. In particular, we decided to use the light-induced photoreceptor degeneration model. In teleost fish, including medaka fish, exposure of the retina to intense light is a noninvasive method to elicit the selective degeneration of photoreceptors [30]. We carried out this test in st40 olAdamts18 knockdown embryos and we analyzed their retinas 5 days later (post-hatching stage, n = 20 eyes). We observed a notable increase of light-induced rod photoreceptor damage in olAdamts18 knockdown vs. wild type fish, as assessed by immunofluorescence analysis with an anti-Rhodopsin antibody (Figure 3). In particular, we detected a reduction of about 50% of the Rhodopsin-positive retinal areas in Adamts18-deficient eyes as compared with wild-type eyes, following light damage. These data strongly indicate that ADAMTS18 is required for proper photoreceptor function and further corroborate the pathogenic role of this gene in inherited retinal dystrophies. The evidence that this gene does not seem to be expressed in photoreceptors (Figure 1E) is not in contrast with the above findings. ADAMTS18 is expressed in the RPE, a tissue that plays a pathogenic role in many forms of retinal disorders [31, 32].

Exposure to intense light induces a severe retinal degeneration in Mo-AdamTS18-injected medaka fish. Representative frontal eye sections, immunostained with an anti-Rhodopsin antibody (green), from P5 control (A) and Mo-AdamTS18–injected (B) medaka fish after 5 days of constant light exposure. Sections are counterstained with DAPI (blue). We observed a significant reduction in both the thickness of the photoreceptor outer segment (OS) and in the intensity of rhodopsin staining in Mo-AdamTS18–injected medaka fish as compared with control animals following constant and intense light exposure. A’ and B’ are higher magnifications of the areas marked by the white dashed boxes in A and B, respectively. Other abbreviations: ONL, Outer Nuclear Layer; INL, Inner Nuclear Layer. Scale bars, red lines: 20 μm.
Conclusions
We determined, by using a multidisciplinary strategy involving the use of advanced genomic procedures and in vivo functional analysis, that mutations in the ADAMTS18 gene, recently proposed to cause Knobloch syndrome, can also be responsible, although with a relatively low frequency, for early-onset severe retinal dystrophy possibly accompanied by other CNS features, such as autism and neurodevelopmental delay. Importantly, the results of our ADAMTS18 knockdown experiment in medaka provide for the first time in vivo functional support to the pathogenic role of this gene in Knobloch syndrome as well. Our data indicate that different mutations in the ADAMTS18 can be linked to the pathogenesis of different eye disorders and contribute to shed further light on the molecular mechanisms underlying the complexity of inherited retinal dystrophies.
References
Berson EL: Retinitis pigmentosa. The Friedenwald lecture. Invest Ophtalmol Vis Sci. 1993, 34: 1659-1676.
Dryja TP, Li T: Molecular genetics of retinitis pigmentosa. Hum Mol Genet. 1995, 4: 1739-1743. Spec No
Rivolta C, Sharon D, DeAngelis MM, Dryja TP: Retinitis pigmentosa and allied diseases: numerous diseases, genes, and inheritance patterns. Hum Mol Genet. 2002, 11: 1219-1227. 10.1093/hmg/11.10.1219.
Saleem RA, Walter MA: The complexities of ocular genetics. Clin Genet. 2002, 61: 79-88. 10.1034/j.1399-0004.2002.610201.x.
Cremers FP, van den Hurk JA, den Hollander AI: Molecular genetics of Leber congenital amaurosis. Hum Mol Genet. 2002, 11: 1169-1176. 10.1093/hmg/11.10.1169.
Neveling K, Collin RW, Gilissen C, van Huet RA, Visser L, Kwint MP, Gijsen SJ, Zonneveld MN, Wieskamp N, de Ligt J, et al: Next-generation genetic testing for retinitis pigmentosa. Hum Mutat. 2012, 33: 963-972. 10.1002/humu.22045.
Simonelli F, Ziviello C, Testa F, Rossi S, Fazzi E, Bianchi PE, Fossarello M, Signorini S, Bertone C, Galantuomo S, et al: Clinical and molecular genetics of Leber’s congenital amaurosis: a multicenter study of Italian patients. Invest Ophthalmol Vis Sci. 2007, 48: 4284-4290. 10.1167/iovs.07-0068.
Zernant J, Kulm M, Dharmaraj S, den Hollander AI, Perrault I, Preising MN, Lorenz B, Kaplan J, Cremers FP, Maumenee I, et al: Genotyping microarray (disease chip) for Leber congenital amaurosis: detection of modifier alleles. Invest Ophthalmol Vis Sci. 2005, 46: 3052-3059. 10.1167/iovs.05-0111.
Estrada-Cuzcano A, Neveling K, Kohl S, Banin E, Rotenstreich Y, Sharon D, Falik-Zaccai TC, Hipp S, Roepman R, Wissinger B, et al: Mutations in C8orf37, encoding a ciliary protein, are associated with autosomal-recessive retinal dystrophies with early macular involvement. Am J Hum Genet. 2012, 90: 102-109. 10.1016/j.ajhg.2011.11.015.
Littink KW, den Hollander AI, Cremers FP, Collin RW: The power of homozygosity mapping: discovery of new genetic defects in patients with retinal dystrophy. Adv Exp Med Biol. 2012, 723: 345-351. 10.1007/978-1-4614-0631-0_45.
Testa F, Surace EM, Rossi S, Marrocco E, Gargiulo A, Di Iorio V, Ziviello C, Nesti A, Fecarotta S, Bacci ML, et al: Evaluation of Italian patients with leber congenital amaurosis due to AIPL1 mutations highlights the potential applicability of gene therapy. Invest Ophthalmol Vis Sci. 2011, 52: 5618-5624. 10.1167/iovs.10-6543.
Purcell S, Neale B, Todd-Brown K, Thomas L, Ferreira MA, Bender D, Maller J, Sklar P, de Bakker PI, Daly MJ, Sham PC: PLINK: a tool set for whole-genome association and population-based linkage analyses. Am J Hum Genet. 2007, 81: 559-575. 10.1086/519795.
Conte I, Lestingi M, den Hollander A, Miano MG, Alfano G, Circolo D, Pugliese M, Testa F, Simonelli F, Rinaldi E, et al: Characterization of MPP4, a gene highly expressed in photoreceptor cells, and mutation analysis in retinitis pigmentosa. Gene. 2002, 297: 33-38. 10.1016/S0378-1119(02)00872-7.
Alfano G, Conte I, Caramico T, Avellino R, Arno B, Pizzo MT, Tanimoto N, Beck SC, Huber G, Dolle P, et al: Vax2 regulates retinoic acid distribution and cone opsin expression in the vertebrate eye. Development. 2011, 138: 261-271. 10.1242/dev.051037.
Conte I, Bovolenta P: Comprehensive characterization of the cis-regulatory code responsible for the spatio-temporal expression of olSix3.2 in the developing medaka forebrain. Genome Biol. 2007, 8: R137-10.1186/gb-2007-8-7-r137.
Conte I, Marco-Ferreres R, Beccari L, Cisneros E, Ruiz JM, Tabanera N, Bovolenta P: Proper differentiation of photoreceptors and amacrine cells depends on a regulatory loop between NeuroD and Six6. Development. 2010, 137: 2307-2317. 10.1242/dev.045294.
Indrieri A, van Rahden VA, Tiranti V, Morleo M, Iaconis D, Tammaro R, D’Amato I, Conte I, Maystadt I, Demuth S, et al: Mutations in COX7B Cause Microphthalmia with Linear Skin Lesions, an Unconventional Mitochondrial Disease. Am J Hum Genet. 2012, 91: 942-949. 10.1016/j.ajhg.2012.09.016.
Conte I, Carrella S, Avellino R, Karali M, Marco-Ferreres R, Bovolenta P, Banfi S: miR-204 is required for lens and retinal development via Meis2 targeting. Proc Natl Acad Sci USA. 2010, 107: 15491-15496. 10.1073/pnas.0914785107.
Vihtelic TS, Hyde DR: Light-induced rod and cone cell death and regeneration in the adult albino zebrafish (Danio rerio) retina. J Neurobiol. 2000, 44: 289-307. 10.1002/1097-4695(20000905)44:3<289::AID-NEU1>3.0.CO;2-H.
Lewis CA, Batlle IR, Batlle KG, Banerjee P, Cideciyan AV, Huang J, Aleman TS, Huang Y, Ott J, Gilliam TC, et al: Tubby-like protein 1 homozygous splice-site mutation causes early-onset severe retinal degeneration. Invest Ophthalmol Vis Sci. 1999, 40: 2106-2114.
Thompson DA, Li Y, McHenry CL, Carlson TJ, Ding X, Sieving PA, Apfelstedt-Sylla E, Gal A: Mutations in the gene encoding lecithin retinol acyltransferase are associated with early-onset severe retinal dystrophy. Nat Genet. 2001, 28: 123-124. 10.1038/88828.
Lorenz B, Gyurus P, Preising M, Bremser D, Gu S, Andrassi M, Gerth C, Gal A: Early-onset severe rod-cone dystrophy in young children with RPE65 mutations. Invest Ophthalmol Vis Sci. 2000, 41: 2735-2742.
Diez-Roux G, Banfi S, Sultan M, Geffers L, Anand S, Rozado D, Magen A, Canidio E, Pagani M, Peluso I, et al: A high-resolution anatomical atlas of the transcriptome in the mouse embryo. PLoS Biol. 2011, 9: e1000582-10.1371/journal.pbio.1000582.
Wu C, Orozco C, Boyer J, Leglise M, Goodale J, Batalov S, Hodge CL, Haase J, Janes J, Huss JW, Su AI: BioGPS: an extensible and customizable portal for querying and organizing gene annotation resources. Genome Biol. 2009, 10: R130-10.1186/gb-2009-10-11-r130.
Aldahmesh MA, Khan AO, Mohamed JY, Alkuraya H, Ahmed H, Bobis S, Al-Mesfer S, Alkuraya FS: Identification of ADAMTS18 as a gene mutated in Knobloch syndrome. J Med Genet. 2011, 48: 597-601. 10.1136/jmedgenet-2011-100306.
Kuno K, Kanada N, Nakashima E, Fujiki F, Ichimura F, Matsushima K: Molecular cloning of a gene encoding a new type of metalloproteinase-disintegrin family protein with thrombospondin motifs as an inflammation associated gene. J Biol Chem. 1997, 272: 556-562. 10.1074/jbc.272.1.556.
Zeng W, Corcoran C, Collins-Racie LA, Lavallie ER, Morris EA, Flannery CR: Glycosaminoglycan-binding properties and aggrecanase activities of truncated ADAMTSs: comparative analyses with ADAMTS-5, -9, -16 and −18. Biochim Biophys Acta. 2006, 1760: 517-524. 10.1016/j.bbagen.2006.01.013.
Tortorella MD, Malfait F, Barve RA, Shieh HS, Malfait AM: A review of the ADAMTS family, pharmaceutical targets of the future. Curr Pharm Des. 2009, 15: 2359-2374. 10.2174/138161209788682433.
Khan AO, Aldahmesh MA, Mohamed JY, Al-Mesfer S, Alkuraya FS: The distinct ophthalmic phenotype of Knobloch syndrome in children. Br J Ophthalmol. 2012, 96: 890-895. 10.1136/bjophthalmol-2011-301396.
Taylor S, Chen J, Luo J, Hitchcock P: Light-induced photoreceptor degeneration in the retina of the zebrafish. Methods Mol Biol. 2012, 884: 247-254. 10.1007/978-1-61779-848-1_17.
Davidson AE, Millar ID, Urquhart JE, Burgess-Mullan R, Shweikh Y, Parry N, O’Sullivan J, Maher GJ, McKibbin M, Downes SM, et al: Missense mutations in a retinal pigment epithelium protein, bestrophin-1, cause retinitis pigmentosa. Am J Hum Genet. 2009, 85: 581-592. 10.1016/j.ajhg.2009.09.015.
Hamel CP, Tsilou E, Pfeffer BA, Hooks JJ, Detrick B, Redmond TM: Molecular cloning and expression of RPE65, a novel retinal pigment epithelium-specific microsomal protein that is post-transcriptionally regulated in vitro. J Biol Chem. 1993, 268: 15751-15757.
Acknowledgements
We thank the participating patients and their families. We are grateful to B. Franco and G. Diez-Roux for critical reading of the manuscript. We also thank C. Gilissen and J.A. Veltman for helpful discussion; S. Crispi and the High Throughput Sequencing IGB facility for support in exome sequencing; R. Rispoli and the TIGEM Bioinformatics Core for support in exome data analysis and F. Salierno for technical support. The members of the European Retinal Disease Consortium involved in this study are Elfride De Baere, Robert K. Koenekoop, Bart P. Leroy, Frans P. Cremers, Susanne Kohl, Christian Hamel, Carmen Ayuso, Bernd Wissinger, Chris Inglehearn, Carmel Toomes and Anneke den Hollander. This work was supported by grants from the Retina Italia, RP-Liguria and the Italian Telethon Foundations (to S.B.) and by the European Union, EU FP7/2007-2013 under grant agreement no. 223143 (project acronym: TECHGENE).
Ethics approval for the work described was provided by the Second University of Naples Medical Ethical Committee.
Author information
Authors and Affiliations
Consortia
Corresponding author
Additional information
Competing interests
All authors have non-financial interests that may be relevant to the submitted work.
Authors’ contributions
IP, MP and CZ performed the genotyping and sequencing analysis. FT, AMB, MABM and FS recruited the family described herein and collected the clinical data. RWJ, GD and MM performed the statistical interpretation of linkage and sequencing analyses. analyses. NM performed the gene expression analysis. The members of the European Retinal Disease Consortium involved in this study contributed to the mutation analysis on a large dataset of retinal dystrophy patients. Sara B and IC carried out the in vivo functional studies. IP, IC, VN, FS and SB oversaw all aspects of the research. SB initiated, planned and coordinated the study. IP, IC, FS and SB wrote the manuscript. All authors read, edited and approved the final version of the manuscript.
Authors’ original submitted files for images
Below are the links to the authors’ original submitted files for images.
Rights and permissions
Open Access This article is published under license to BioMed Central Ltd. This is an Open Access article is distributed under the terms of the Creative Commons Attribution License ( https://creativecommons.org/licenses/by/2.0 ), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
About this article
Cite this article
Peluso, I., Conte, I., Testa, F. et al. The ADAMTS18 gene is responsible for autosomal recessive early onset severe retinal dystrophy. Orphanet J Rare Dis 8, 16 (2013). https://doi.org/10.1186/1750-1172-8-16
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/1750-1172-8-16