
Abstract
The phosphatidylinositol 3-kinase (PI3K) pathway is commonly deregulated in cancer. In recent years, the results of the first phase I clinical trials with PI3K inhibitors have become available. In comparison to other targeted agents such v-raf murine sarcoma viral oncogene homolog B1 (BRAF) inhibitors in melanoma or crizotinib in anaplastic lymphoma receptor tyrosine kinase (ALK) translocated tumors, the number of objective responses to PI3K inhibitors is less dramatic. In this review we propose possible strategies to optimize the clinical development of PI3K inhibitors: by exploring the potential role of PI3K isoform-specific inhibitors in improving the therapeutic index, molecular characterization as a basis for patient selection, and the relevance of performing serial tumor biopsies to understand the associated mechanisms of drug resistance. The main focus of this review will be on PI3K isoform-specific inhibitors by describing the functions of different PI3K isoforms, the preclinical activity of selective PI3K isoform-specific inhibitors and the early clinical data of these compounds.
Similar content being viewed by others


Introduction
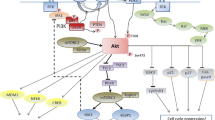
Phosphatidylinositol 3-kinases (PI3Ks) represent a family of lipid kinases that plays a key role in signal transduction, cell metabolism and survival [1, 2]. The PI3K family is divided into three classes, I, II and III, based on their substrate specificity and structure. Among them, class I PI3K seems to be the most relevant in cancer. Class I PI3K has a catalytic subunit (p110) and a regulatory subunit (p85) that stabilizes p110 and inactivates its kinase activity at basal state. Physiologically, PI3K transduces signals received from activated tyrosine kinase receptors (RTK), G protein-coupled receptors (GPCR) or from activated RAS. Upon receipt of such signals, the p85 regulatory subunit interacts with the phosphorylated tyrosine residues of activated RTKs. This engagement then causes release of the p85-mediated inhibition of p110, such that p110 can interact with the lipid membranes to phosphorylate phosphatidylinositol 4,5-bisphosphate (PIP2) to phosphatidylinositol 3,4,5-trisphosphate (PIP3). This reaction triggers a signaling cascade through the activation of AKT and its downstream effectors. The amount of PIP3 generated and resultant PI3K pathway activation are tightly regulated by the tumor suppressor protein, phosphatase and tensin homologue deleted on chromosome 10 (PTEN). PTEN can inactivate the PI3K pathway by converting PIP3 into PIP2 (Figure 1). The PI3K pathway can be activated not only via RTKs, but also by RAS and GPCR. RAS can activate the PI3K pathway by its direct interaction with p110α, p110γ, and p110δ subunits, while GPCRs can interact with p110β and p110γ subunits [2].

Phosphatidylinositol 3-kinase (PI3K) pathway activation. Tyrosine kinase receptors (TKR) can interact with several PI3K isoforms. RAS proteins can activate PI3Kα and γ isoforms. In addition certain RAS proteins can activate PI3Kδ isoform. G protein-coupled receptors (GPCR) preferentially interact with the PI3Kβ or γ isoforms. Once activated by any of these mechanisms, PI3K interacts with the lipid membrane phosphorylating phosphatidylinositol 4,5-bisphosphate (PIP2) generating phosphatidylinositol 3,4,5-trisphosphate (PIP3). PTEN (phosphatase and tensin homologue deleted on chromosome 10) converts PIP3 into PIP2, regulating the final amount of PIP3 generated. PIP3 triggers a signaling cascade through the activation of AKT, phosphoinositide-dependent protein kinase 1 (PDK1) and their downstream effectors. G = G protein G; p110 = PI3K catalytic subunit; p85 = class IA PI3K regulatory subunit; p87 = class IB PI3K regulatory subunit; p101 = class IB regulatory subunit.
The PI3K pathway is commonly deregulated in cancer, with the most common events being mutation or increased gene copy numbers of PIK3CA or other PI3K isoforms, loss of expression of the pathway suppressors (for example, PTEN), or hyperactivation of RTKs through receptor overexpression or activating mutations (Table 1). The first results of several early phase I clinical trials investigating different PI3K inhibitors (Table 2) have been presented in recent years (Table 3). Other targeted agents evaluated in specific oncogenically addicted patient populations in the early trial setting, such as vemurafenib [3] or dabrafenib [4] in v-raf murine sarcoma viral oncogene homolog B1 (BRAF) V600E mutant melanoma, or crizotinib in echinoderm microtubule-associated protein-like 4-anaplastic lymphoma kinase (EML4-ALK) translocated non-small cell lung cancer [5], have demonstrated dramatic antitumor activity. In contrast, the objective responses observed thus far with PI3K inhibitors have been more modest and in many cases of short duration. Several strategies may be considered to optimize the development of PI3K inhibitors in clinical trials.
Strategies to optimize the development of PI3K inhibitors
The development of PI3K inhibitors is rapidly evolving with newer and more potent compounds entering clinical trials. Of particular interest are the isoform-specific PI3K inhibitors, which offer the potential of achieving greater selective target blockade while minimizing off-target effects due to inhibition of other isoforms as in the case of pan-PI3K inhibitors (Table 2). Whether these compounds may be superior to pan-PI3K inhibitors in safety and efficacy, and which patient populations may benefit the most from their use, are questions yet to be addressed. In addition, first-in-human studies of different PI3K inhibitors [6–27] have used variable approaches in patient inclusion ranging from unselected populations to restriction of patients with PI3K pathway alterations (Table 3). The results of these studies may help guide the design of future clinical trials. Patient selection can be enhanced through an improved understanding of the biological significance of PI3K pathway alterations in each tumor type and, even more specifically, in each patient. Lastly, the translation of antitumor activity observed in preclinical models to the clinical setting has been largely disappointing for PI3K inhibitors. As in the case of many other anticancer agents whereby proof of target inhibition in phase I trials is not straightforward, it is often uncertain if the dose ranges delivered in early trials of PI3K inhibitors can induce such effects at the tumoral level. Thus, there is a continued need whenever feasible to obtain tumor tissues during treatment for mechanistic proof of pathway engagement. Such pharmacodynamic data, together with relevant pharmacokinetic results, may help guide optimal dosing schedules. Tumor biopsy at disease progression among initial responders is also highly encouraged, in order to appreciate the underlying mechanisms of resistance and enable selection of the most appropriate therapy to overcome them.
PI3K isoform-specific inhibitors
The PI3Ks are grouped into three classes (I, II and III) based on their structural characteristics and substrate specificity [2]. Class I PI3Ks are further divided into class IA enzymes, which include p110α, p110β and p110δ, while p110γ constitutes class IB [2]. In mammals, p110α and p110β are ubiquitous while p110γ and p110δ are expressed preferentially in leukocytes [28, 29]. This distribution justifies the most relevant role of p110γ and p110δ in inflammatory diseases and the implication of p110δ in hematological malignancies. Class II PI3Ks seem to be implicated in exocytosis, cell migration, smooth muscle cell contraction, glucose metabolism and apoptosis [30]. Class III PI3Ks regulate cellular trafficking of vesicles and proteins [2]. Class I PI3Ks are involved in cell growth, survival and metabolism, therefore represent one of the most sought after targets in cancer therapeutics.
PI3Kα-specific inhibitors
In addition to its effects on cell growth, proliferation and survival, class IA PI3K regulates glucose metabolism through insulin signaling [31–33]. It is commonly deregulated in cancer through mutations or amplifications of the PIK3CA gene or through alterations in the function of upstream tumor suppressors such as PTEN (Table 1). About 80% of the mutations of the PIK3CA gene are clustered in three hotspots in the p110α gene that encodes the catalytic subunit: two in the helical domain (E542K and E545K) and one in the kinase domain (H1047R) [34]. PIK3CA mutations are oncogenic per se, as they can induce the generation of tumors in several preclinical models without other molecular aberrations [35–37].
In addition to experiments in genetically engineered mice [31, 32], the first generation of PI3Kα-specific inhibitors, while less isoform selective than the more recent compounds, have been instrumental in defining the biologic role of different PI3K isoforms in normal and cancer cells [33, 38, 39]. However, these agents have provided only inconclusive data on their antitumor activity in cell lines harboring PIK3CA mutations compared to those that are PIK3CA wild-type [40, 41]. One of the main reasons is the limited number of cell lines in which these compounds have been evaluated. Cell lines without PIK3CA mutations often harbor alterations in oncogenic tyrosine kinase receptors, such as ERBB2 amplification, which preferentially uses the p110α isoform for signal transduction [32]. However, some of the cell lines harboring PIK3CA mutations had additional molecular aberrations, some of which are known mechanisms of resistance [41].
The new PI3Kα-isoform specific inhibitors have shown promising activity in cell lines harboring PIK3CA mutations [42, 43]. In addition, the screening of one of these compounds, BYL719, in a large genomically characterized cell line panel, has revealed that besides PIK3CA mutations, the presence of PIK3CA amplification or ERBB2 amplification correlated with higher drug sensitivity. Conversely, BRAF and PTEN mutations were correlated with resistance. v-Ki-ras2 Kirsten rat sarcoma viral oncogene homolog (KRAS) mutation by itself was not associated with either sensitivity or resistance, although the coexistence of KRAS and PIK3CA mutations was usually associated with a lack of response [42].
Several new generation PI3Kα-selective inhibitors are currently being evaluated in phase I clinical trials, including BYL719 (NCT01219699), INK-1114 (NCT01449370) and GDC-0032 (NCT01296555). The clinical results of the dose escalation part of the phase I trial investigating BYL719 have recently been presented [25]. Trial enrollment was restricted to patients with solid tumors harboring PIK3CA mutations or amplifications. This population was selected based on the higher antitumor activity observed in preclinical models with PIK3CA mutations or amplifications using the Cancer Cell Line Encyclopedia [42]. This was the first reported study of a PI3K inhibitor in which molecular prescreening was undertaken starting from the dose escalation part. A total of 35 patients have been enrolled thus far and the maximum tolerated dose has been determined as 400 mg orally on a continuous once daily schedule. Three patients, all of whom received doses ≥ 270 mg/day, have achieved a partial response. The tumor types of these responders were estrogen receptor positive breast cancer, cervical cancer and KRAS-mutant colon cancer, and PIK3CA mutations were detected in all three cases (E542K/V, E545K and R88Q respectively). In addition, prolonged disease stabilization, defined as that lasting for ≥ 4 months, has been observed in ten patients with primary tumor sites from oral cavity, salivary gland, colon, and estrogen receptor positive breast. Among them, five patients have remained on study treatment for more than 6 months [25]. The clinical response observed in the colon cancer patient with coexistent KRAS and PIK3CA mutations contrasts with the preclinical finding in which such coexpression generally conferred resistance to BYL719. Tumor heterogeneity may partly explain the clinical results, if for instance, these mutations are not coexistent in all geographic areas, or if the two mutations have different tumor-driving functions. Furthermore, this case illustrates the molecular complexities in human malignancies that often cannot be reliably reflected by preclinical models.
From a safety perspective, the most commonly observed adverse effects associated with BYL719 were hyperglycemia, nausea, fatigue, rash and gastrointestinal toxicities [25], all of which are also frequently encountered with the pan-PI3K inhibitors. Although the spectrum of toxicities encountered between BYL719 and the pan-PI3K inhibitors are similar, hyperglycemia represents the most frequent and dose-limiting adverse event with BYL719. Given the interaction between PI3K pathway inhibition and insulin signaling, occurrence of this on-target toxicity supports proof-of-mechanism. A relevant question is whether an isoform-selective PI3K inhibitor is able to achieve greater target inhibition than the pan-PI3K inhibitors while producing a similar degree and extent of side effects. At present, there is a paucity of published preclinical data comparing any of the PI3Kα-selective inhibitors currently in clinical development with pan-isoform PI3K inhibitors. While early results from the phase I trial of BYL719 appear encouraging, direct comparison of the preliminary efficacy results achieved with this agent against those reported with the pan-isoform PI3K inhibitors would be invalid, as none of the early phase trials involving pan-PI3K inhibitors have been specifically designed to evaluate only the PIK3CA mutant population. Even among those cases which utilized an enrichment strategy in the expansion cohort to select for patients with molecular alteration in the PI3K pathway, a variety of alterations such as PIK3CA mutation or amplification, PTEN mutation or loss of PTEN expression have been included (Table 3).
PI3Kδ-specific inhibitors
In contrast to the ubiquitously expressed p110α and p110β isoforms, p110δ is mainly expressed in leukocytes [28, 29]. Its overexpression has been observed in a wide range of lymphoproliferative disorders including chronic lymphocytic leukemia (CLL) [44], multiple myeloma [45], diffuse large B-cell lymphoma [46], B-cell acute lymphoblastic leukemia [46], follicular lymphoma [46], mantle cell lymphoma [47, 48], and Hodgkin's lymphoma [49].
Currently, two PI3Kδ-specific inhibitors are in clinical development: GS-1101, previously known as CAL-101, and AMG 319. GS-1101 has shown preclinical activity as a single agent against different lymphoid malignancies including CLL [44, 46], multiple myeloma [45], mantle cell lymphoma [47], Hodgkin's lymphoma [49] and B-cell acute lymphoblastic leukemia [46]. GS-1101 has been shown to partially revert stroma-induced resistance to conventional cytotoxic drugs in CLL [50, 51]. In addition, synergy with targeted therapies such as the mammalian target of rapamycin (mTOR) inhibitor everolimus [47] or the proteasome inhibitor bortezomib [45], has been described in mantle cell lymphoma and multiple myeloma, respectively. AMG 319 has shown activity against several cell lines derived from B-cell malignancies [52], and synergy with vincristine in diffuse B-cell lymphoma has been observed [52, 53].
Among the PI3Kδ-specific inhibitors, clinical data have been published thus far only with GS-1101. Early signs of antitumor activity were found in the phase I clinical trial in selected relapsed or refractory hematologic malignancies including patients with CLL and non-Hodgkin's lymphoma [54, 55]. The most recent report has shown that in 80% of the 54 patients with CLL enrolled in the phase I trial, ≥ 50% lymphadenopathy shrinkage was observed and the overall intention-to-treat response rate by the 2008 International Workshop on Chronic Lymphocytic Leukemia (IWCLL) response criteria [56] was 26%. The most relevant grade 3 or higher adverse events were pneumonia, neutropenia (7% of patients developed febrile neutropenia), thrombocytopenia, anemia, and transaminase elevation [26]. In patients with non-Hodgkin's lymphoma, 15 out of 24 patients with indolent non-Hodgkin's lymphoma and 10 out of 16 patients with mantle cell lymphoma achieved a partial response. However, none of the nine patients with diffuse large B-cell lymphoma had a partial response. The observed serious toxicities (grade ≥ 3) were similar to those reported in the CLL arm, which included hematological toxicities and transaminase elevation [27]. Based on the striking monotherapy activity observed in these two population groups, GS-1101 is being evaluated in a phase I clinical trial in combination with several compounds active in hematological malignancies. The data of the combination arms of GS-1101 with rituximab with or without bendamustine [57, 58] and in combination with ofatumumab [59] have been recently presented. Substantial antitumor activity has been described with both regimens with the expected toxicities based on the single agent toxicity profile. Results from the fludarabine, chlorambucil, everolimus and bortezomib arms have not been presented yet (NCT01088048). There are ongoing phase III clinical trials for patients with CLL investigating the combination of GS-1101 with rituximab (NCT01539512), and with rituximab and bendamustine (NCT01569295). Besides GS-1101, AMG 319 is another PI3Kδ-specific inhibitor in clinical development, a phase I clinical trial of this agent in patients with relapsed or refractory lymphoid malignancies is ongoing (NCT01300026).
The question of whether PI3Kδ-specific or pan-isoform PI3K inhibition constitutes a more optimal therapeutic strategy in patients with lymphoid malignancies is still under debate. Preclinically, some pan-isoform PI3K inhibitors have shown signs of activity in selected lymphomas and CLL [48, 60–63]. In certain lymphoma subtypes, the activity of the pan-isoform PI3K inhibitors GDC-0941 and SF-1126 could potentially be superior [48, 62, 63].
In the clinical setting, the pan-isoform PI3K inhibitor SAR245408 (XL147) [64] and the pan-isoform PI3K and mTOR inhibitor SAR245409 (XL765) [15] have been evaluated in patients with lymphoma, as an expansion cohort of the respective phase I clinical trials. Observed grade 3 or higher adverse events with SAR245408 have been primarily hematological toxicities including neutropenia and thrombocytopenia, as well as hyperglycemia [64]. Hyperglycemia was not commonly reported with SAR245409, but grade 3 transaminase elevation was observed in 2 out of 15 patients [15]. The antitumor activity of SAR245408 has not yet been reported [64]. Among the 13 patients with lymphoma treated in the phase I clinical trial with SAR245409, 3 patients (mantle cell lymphoma, transformed lymphoma and diffuse large B-cell lymphoma) achieved a partial response [15]. SAR245409 is currently being evaluated as a single agent in a phase II clinical trial in patients with selected types of lymphoma or leukemia (NCT01403636) and in a phase I trial in combination with bendamustine with or without rituximab (NCT01410513).
PI3Kβ-specific inhibitors: the role of PTEN alteration
The signaling of the PI3Kβ isoform is mediated via GPCR [65–71] while the PI3Kα isoform preferentially mediates via RTK, however, platelet-derived growth factor receptor is able to sustain its signaling through the PI3Kβ isoform in the absence of the PI3Kα isoform [32].
The PI3Kβ isoform is oncogenic when deregulated [65]. There are no PIK3CB mutations described in cancer so far. The most common event that leads to PI3Kβ-isoform signaling deregulation is PTEN deficiency, although PIK3CB amplification has been described in breast cancer [72]. PTEN is a lipid phosphatase that dephosphorylates the 3-phosphoinositide products of PI3K [73]. PTEN deficiency is a frequent event in cancer [74] (Table 1), which can occur through several mechanisms including PTEN mutation, PTEN deletion, epigenetic changes [75–79], miRNA-mediated regulation [80–82] or post-translational modifications [83, 84].
In preclinical models, it has been demonstrated that PTEN-deficient tumors depend on the PI3Kβ isoform for pathway activation, growth and survival [65, 85]. The preclinical activity of several PI3Kβ-specific inhibitors in PTEN-deficient cell lines and xenograft models has been recently communicated [86–88]. In the clinical setting, a phase I clinical trial with the selective PI3Kβ-selective inhibitor GSK2636771 in patients with advanced solid tumors with PTEN deficiency is currently ongoing (NCT01458067), and a phase I clinical trial with the PI3Kβ-selective inhibitor (SAR260301) in solid tumors as a single agent and in combination with vemurafenib in BRAF mutant melanoma, has recently been initiated (NCT01673737).
Patient selection
One of the major challenges in the clinical development of PI3K inhibitors is to identify the appropriate patient populations most likely to benefit from the treatment. In the current era where many drug targets are entering clinical evaluation and even more compounds are being developed to interrogate such targets, a rational approach is to intensify biomarker research in the preclinical setting and then incorporate them in early phase clinical trials. Both pharmacodynamic markers to prove biological effect and predictive biomarkers to identify sensitive or resistant populations are of interest, and their exploration in valid preclinical models would inform clinical development.
In preclinical models, cell lines harboring PIK3CA mutation, or amplification of PIK3CA or ERBB2 have shown sensitivity to different PI3K inhibitors, including pan-isoform PI3K inhibitors [89–91] or PI3Kα-specific inhibitors [42, 43]. However, the role of PTEN loss as a predictor of responsiveness to PI3K inhibitors is less clear [40, 90, 92, 93]. In the clinical setting, the retrospective analysis of 217 patients referred to the MD Anderson Cancer Center revealed that those with PIK3CA mutant tumors treated with PI3K-AKT-mTOR axis inhibitors demonstrated a higher objective response rate than patients without such mutations [94, 95]. However, the majority of these patients received combination therapies that included an mTOR inhibitor, and not a PI3K inhibitor. In addition, there are inherent biases to retrospective analyses, and these results should be considered exploratory and interpreted cautiously.
As depicted in Table 3, initially phase I clinical trials with PI3K inhibitors have been developed in unselected patient populations. As preclinical data of sensitivity to pan-PI3K inhibitors in tumors harboring relevant molecular aberrations become available [89–91], different enrichment strategies have been adopted. These strategies range from the selection of patients with any PI3K pathway alterations in the expansion cohort of phase I trials, to the approach utilized in the recent phase I trial of the PI3Kα-specific inhibitor BYL719 in which only patients with PIK3CA mutations or amplifications were enrolled. It is invalid to make a direct comparison between unselected versus selected approaches for patient recruitment, as other factors, such as the anticancer activity of each compound, the number of patients treated at suboptimal doses, pharmacokinetic issues, or the presence of different molecular events that can modify the sensitivity to PI3K inhibitors (such as KRAS mutations), can be confounding. However, preliminary experience from the phase I trial of BYL719 suggests that it is reasonable to select patients based on specific molecular aberrations which are justified by appropriate preclinical models. Importantly, this study has performed large scale screening in local institutions to identify patients with uncommon molecular characteristics without compromising timely enrollment, a finding that supports the feasibility of molecular prescreening already implemented by many large drug development programs [96, 97].
Elucidation of mechanisms of pathway activation and resistance
Results from the first clinical trials (Table 3) of various PI3K inhibitors may shed insight to help identify tumors in which these agents exert sufficient activity to inactivate the PI3K pathway. Unlike BRAF or ALK inhibitors that have demonstrated very early on in their development anticancer activity against patient populations whose tumors are uniquely sensitive to these agents, objective responses seen in the early clinical trials of PI3K inhibitors were less predictable. While some of the responders had PI3K pathway aberrant tumors, there were many who did not respond despite harboring relevant molecular features, as well as others who responded without obvious molecular predisposition. There is clearly a context dependence in which tumor histology may be relevant, as the functionality of the same genomic aberration across different tumor types may vary. However, histology is unlikely the only context as patients with the same tumor type harboring similar molecular aberrations often have different outcomes despite receiving the same matched therapy [98]. A key challenge in the clinical evaluation of PI3K inhibitors is to differentiate patients whose tumors are addicted, dependent, versus resistant, to a PI3K isoform [99].
PI3K isoform-addicted tumors correspond to those in which a dramatic and sustained response is observed with PI3K inhibitors. These tumors may be so vulnerable that even partial pathway inhibition is sufficient to lead to clinical responses. These patients may be extraordinary candidates for treatment with PI3K isoform-selective inhibitors to achieve a high therapeutic index by minimizing off-target adverse effects while obtaining adequate target inhibition.
PI3K-dependent tumors are those which likely require a complete or near complete pathway inhibition to achieve meaningful responses. Tumors which may belong to this categorization include those with upstream RTK hyperactivation, those with simultaneous activation of several points along the PI3K pathway [100, 101], those harboring oncogenic events which can signal through different isoforms [32, 102], or those which were initially addicted to an isoform but have acquired resistance to reactivate the PI3K pathway via alternate mechanisms [99]. Breast cancers with simultaneous human epidermal growth factor receptor 2 (HER2) amplification and PIK3CA mutation represent good examples of simultaneous RTK hyperactivity and activation of the PI3K pathway at several levels [100, 101]. Interesting activity has been reported in the clinical setting with the combination of the PI3K-mTOR inhibitor BEZ235 and trastuzumab, presumably due to the effect of sufficient modulation of both mitogen-activated protein kinase (MAPK) and PI3K pathways [98]. In a preclinical experiment, Liu et al. constructed a PI3K isoform-addicted mouse model of breast cancer conditionally expressing PIK3CA H1047R. Tumor response was observed after suppressing PIK3CA H1047R expression, but spontaneous tumor recurrence was detected in some animals after initial response. Such tumors appeared to have escaped oncogenic addiction and either remained dependent on the PI3K pathway and respond to the pan-isoform PI3K inhibitor GDC-0941 or became totally resistant, with the amplification of c-MET and c-MYC being implicated in these evolutions, respectively [99]. To translate these findings to the clinic, it would be informative to perform tumor biopsies at the time of disease progression in patients who have initially responded to PI3K inhibitors, to determine if the tumor remains dependent on the PI3K pathway and thus may benefit from combinatorial strategies, or whether it has developed resistance through an independent mechanism.
The last group is constituted by those tumors deemed to be resistant to PI3K inhibition, such that interrogation of the PI3K axis alone will be unlikely to yield any clinical benefit. This molecular behavior could either be due to a primary de novo resistance [103] or an acquired resistance after the selective pressure of PI3K inhibition [101]. It is important to recognize resistant subtypes early on in the disease course, as some cases might be appropriate candidates for combination treatment, such as simultaneous inhibition of the PI3K and MAPK pathways [103]. The ability to distinguish various molecular alterations in tumors and their translation to unique biological behaviors would enable a more effective strategy to individualize treatment with PI3K inhibitors.
Therapeutic targeting of the PI3K pathway
The decision of whether PI3K isoform-selective inhibitors are more therapeutically appealing than pan-PI3K inhibitors awaits the maturation of results from ongoing clinical trials. In addition, other challenging questions remain in the clinical development of PI3K inhibitors. For instance, the most optimal drug administration schedule for PI3K inhibition remains elusive. Preclinical models are needed to investigate dosing schedules in tumors which are addicted, dependent, versus resistant to PI3K inhibition to decipher how best to effectively modulate the pathway in each scenario. Dosing schedules may range from the administration of intermittent high doses to completely abrogate the pathway versus continuous low doses to provide sustained but less intense inhibition of the pathway. The availability of both intravenous and oral pan-isoform PI3K inhibitors enables the evaluation of the efficacy and toxicity of this class of agents using different administration schedules. In addition, recent preclinical work has highlighted schedule dependence when combining two different anticancer drugs [104], the relevance of this phenomenon to combinations involving PI3K inhibitors is yet to be assessed. Some early phase trials are evaluating this question in the clinical setting, such as the recently presented study investigating different schedules of the pan-PI3K inhibitor BKM120 in combination with letrozole [105].
Given the lack of significant single agent activity with PI3K inhibitors in many patients tested so far on clinical trials, it is likely that combinatorial approaches incorporating PI3K inhibitors are necessary to achieve meaningful therapeutic effects. Activation of PI3K pathway has been described as a mechanism of resistance to hormone therapy and anti-HER2 therapy in breast cancer [100], clinical trials of combinations of these agents with PI3K inhibitors are currently ongoing. However, KRAS mutation has been described as a resistant factor for PI3K inhibitors, through its activation of the MAPK pathway. Thus, several targeted combination trials of PI3K inhibitors and mitogen-activated protein kinase kinase (MEK) inhibitors are underway in the clinic. However, some KRAS mutations preferentially signal through the PI3K pathway [106], this may explain the partial response observed with BKM120 in a patient with triple negative breast cancer whose tumor harbored a KRAS mutation. A further limitation to finding the most appropriate targeted combination is the inability to readily decipher whether molecular alterations detected represent driver events. Tumor heterogeneity contributes an additional layer of complexity in the selection of targeted combinations [107].
Despite therapeutic advances that have now rendered PI3K a druggable target, many questions remain unanswered. Are alternate pathway activation and tumor heterogeneity the reasons why PI3K inhibitors are not declared as panacea based on the currently available clinical data? Is the pathway so critical in the human organism that compensatory feedback mechanisms emerge very quickly upon inhibition? Are existent PI3K inhibitors in clinical development potent enough with optimal pharmacokinetic and pharmacodynamic properties? Would the early phase clinical results have been superior if all patients had been preselected according to molecular characteristics? As knowledge accumulates in the PI3K pathway and more potent PI3K inhibitors become available, rational application of these agents as monotherapy or in combination is within reach.
Conclusions
Isoform-specific PI3K inhibitors are now entering clinical development; they appear promising by proposing to achieve a greater degree of isoform inhibition with fewer off-target side effects. Tumors differ in their response thresholds to PI3K inhibitors based on their degree of addiction, dependence or resistance to this oncogenic pathway. Characterization of somatic molecular alterations and integration of this information into the treatment algorithm may enable more effective therapeutic targeting using PI3K inhibitors. It is plausible that the best clinical results could only be achieved by deepening the biological knowledge of how each individual tumor would behave upon PI3K pathway interrogation. Only in that context can one most appropriately select the best agent, either as monotherapy or in combination, to administer using the most effective dosing schedule.
Authors' information
IB is a clinical research fellow at the Drug Development Program at the Princess Margaret Cancer Center. The focus of her research is the evaluation of new investigational drugs in early phases of clinical development, as well as preclinical evaluation of combination of targeted agents, with a special focus on PI3K inhibitors. LLS is a Professor of Medicine and a Senior Medical Oncologist at Princess Margaret Cancer Centre. She holds a Cancer Care Ontario Chair in Experimental Therapeutics. Her research focus is in the area of new drug development and cancer genomics.
Abbreviations
- ALK:
-
anaplastic lymphoma receptor tyrosine kinase
- BRAF:
-
v-raf murine sarcoma viral oncogene homolog B1
- CLL:
-
chronic lymphocytic leukemia
- EML4:
-
echinoderm microtubule-associated protein-like 4
- GPCR:
-
G protein-coupled receptor
- KRAS:
-
v-Ki-ras2 Kirsten rat sarcoma viral oncogene homolog
- MAPK:
-
mitogen-activated protein kinase
- MEK:
-
mitogen-activated protein kinase (MAPK) kinase
- PI3K:
-
phosphatidylinositol 3-kinase
- PIK3CA:
-
phosphatidylinositol 3-kinase p110α isoform gene
- PIP2:
-
phosphatidylinositol 4,5-bisphosphate
- PIP3:
-
phosphatidylinositol 3,4,5-trisphosphate
- PTEN:
-
phosphatase and tensin homologue deleted on chromosome 10
- TKR:
-
tyrosine kinase receptor.
References
Engelman JA: Targeting PI3K signalling in cancer: opportunities, challenges and limitations. Nat Rev Cancer. 2009, 9: 550-562. 10.1038/nrc2664.
Vanhaesebroeck B, Guillermet-Guibert J, Graupera M, Bilanges B: The emerging mechanisms of isoform-specific PI3K signalling. Nat Rev Mol Cell Biol. 2010, 11: 329-341. 10.1038/nrm2882.
Flaherty KT, Puzanov I, Kim KB, Ribas A, McArthur GA, Sosman JA, O'Dwyer PJ, Lee RJ, Grippo JF, Nolop K, Chapman PB: Inhibition of mutated, activated BRAF in metastatic melanoma. N Engl J Med. 2010, 363: 809-819. 10.1056/NEJMoa1002011.
Falchook GS, Long GV, Kurzrock R, Kim KB, Arkenau TH, Brown MP, Hamid O, Infante JR, Millward M, Pavlick AC, O'Day SJ, Blackman SC, Curtis CM, Lebowitz P, Ma B, Ouellet D, Kefford RF: Dabrafenib in patients with melanoma, untreated brain metastases, and other solid tumours: a phase 1 dose-escalation trial. Lancet. 2012, 379: 1893-1901. 10.1016/S0140-6736(12)60398-5.
Kwak EL, Bang YJ, Camidge DR, Shaw AT, Solomon B, Maki RG, Ou SH, Dezube BJ, Janne PA, Costa DB, Varella-Garcia M, Kim WH, Lynch TJ, Fidias P, Stubbs H, Engelman JA, Sequist LV, Tan W, Gandhi L, Mino-Kenudson M, Wei GC, Shreeve SM, Ratain MJ, Settleman J, Christensen JG, Haber DA, Wilner K, Salgia R, Shapiro GI, Clark JW, et al: Anaplastic lymphoma kinase inhibition in non-small-cell lung cancer. N Engl J Med. 2010, 363: 1693-1703. 10.1056/NEJMoa1006448.
Edelman G, Bedell C, Shapiro G, Pandya SS, Kwak EL, Scheffold C, Nguyen LT, Laird A, Baselga J, Rodon J: A phase I dose-escalation study of XL147 (SAR245408), a PI3K inhibitor administered orally to patients (pts) with advanced malignancies [abstract]. ASCO Annual Meeting Proceedings: June 4-8 2010; Chicago, IL. 2010, Alexandria, VA: American Society of Clinical Oncology, Abstract 3004
Bendell JC, Rodon J, Burris HA, De Jonge M, Verweij J, Birle D, Demanse D, De Buck SS, Ru QC, Peters M, Goldbrunner M, Baselga J: Phase I, dose-escalation study of BKM120, an oral pan-class I PI3K inhibitor, in patients with advanced solid tumors. J Clin Oncol. 2012, 30: 282-290. 10.1200/JCO.2011.36.1360.
Moreno Garcia V, Baird RD, Shah KJ, Basu B, Tunariu N, Blanco M, Cassier PA, Pedersen JV, Puglisi M, Sarker D, Papadatos-Pastos D, Omlin AG, Biondo A, Ware JA, Koeppen H, Levy GG, Mazina KE, De Bono JS: A phase I study evaluating GDC-0941, an oral phosphoinositide-3 kinase (PI3K) inhibitor, in patients with advanced solid tumors or multiple myeloma [abstract]. ASCO Annual Meeting Proceedings June 4-8 2011; Chicago, IL. 2011, Alexandria, VA: American Society of Clinical Oncology, Abstract 30211
Von Hoff DD, LoRusso P, Demetri GD, Weiss GJ, Shapiro G, Ramanathan RK, Ware JA, Raja R, Jin J, Levy GG, Mazina KE, Wagner AJ: A phase I dose-escalation study to evaluate GDC-0941, a pan-PI3K inhibitor, administered QD or BID in patients with advanced or metastatic solid tumors [abstract]. ASCO Annual Meeting Proceedings: June 4-8 2011; Chicago, IL. 2011, Alexandria, VA: American Society of Clinical Oncology, Abstract 3052
Patnaik A, Appleman LJ, Mountz JM, Ramanathan RK, Beeram M, Tolcher AW, Papadopoulos KP, Lotze MT, Petro DP, Laymon C, Paige L, Rajagopalan P, Jeffers M, Roth D, Dubowy RL: A first-in-human phase I study of intravenous PI3K inhibitor BAY 80-6946 in patients with advanced solid tumors: results of dose-escalation phase [abstract]. ASCO Annual Meeting Proceedings: June 4-8 2011; Chicago, IL. 2011, Alexandria, VA: American Society of Clinical Oncology, Abstract 3035
Lotze MT, Appleman LJ, Ramanathan RK, Tolcher AW, Beeram M, Papadopoulos KP, Rasco DW, Weiss GJ, Mountz JM, Toledo FGS, Alvarez RJ, Oborski MJ, Rajagopalan P, Jeffers M, Roth D, Dubowy RL, Patnaik A: Phase I study of intravenous PI3K inhibitor BAY 80-6946: activity in patients (pts) with advanced solid tumors and non-Hodgkin lymphoma treated in MTD expansion cohorts [abstract]. ASCO Annual Meeting Proceedings: June 1-5 2012; Chicago, IL. 2012, Alexandria, VA: American Society of Clinical Oncology, Abstract 3019
Omlin AG, Spicer JF, Sarker D, Pinato DJ, Agarwal R, Cassier PA, Stavraka C, Blanco M, Suder A, Allan S, Heaton S, Decordova S, Pope L, Prince J, Noguchi K, Jones K, Inatani M, Shiokawa R, Banerji U, Blagden SP: A pharmacokinetic (PK) pharmacodynamic (PD) driven first-in-human study of the oral class I PI3K inhibitor CH5132799, in patients with advanced solid tumors [abstract]. ASCO Annual Meeting Proceedings: June 1-5 2012; Chicago, IL. 2012, Alexandria, VA: American Society of Clinical Oncology, Abstract 3022
Hong DS, Bowles DW, Falchook GS, Messersmith WA, George GC, O'Bryant CL, Vo AC, Klucher K, Herbst RS, Eckhardt SG, Peterson S, Hausman DF, Kurzrock R, Jimeno A: A multicenter phase I trial of PX-866, an oral irreversible phosphatidylinositol 3-kinase inhibitor, in patients with advanced solid tumors. Clin Cancer Res. 2012, 18: 4173-4182. 10.1158/1078-0432.CCR-12-0714.
Brana I, LoRusso P, Baselga J, Heath EI, Patnaik A, Gendreau S, Laird A, Papadopoulos K: A phase I dose-escalation study of the safety, pharmacokinetics (PK), and pharmacodynamics of XL765 (SAR245409), a PI3K/TORC1/TORC2 inhibitor administered orally to patients (pts) with advanced malignancies [abstract]. ASCO Annual Meeting Proceedings: June 4-8 2010; Chicago, IL. 2010, Alexandria, VA: American Society of Clinical Oncology, Abstract 3030
Papadopoulos KP, Abrisqueta P, Chambers G, Rasco D, Patnaik A, Tabernero J, Rajangam K, Rockich K, Egile C, Kelly A, Xu Y, Lager J, Vose JM: A phase I dose expansion cohort study of the safety, pharmacokinetics and pharmacodynamics of SAR245409 (S09), an orally administered PI3K/mTOR inhibitor, in patients with lymphoma [abstract]. ASH Annual Meeting Abstracts: November 18 2011; San Diego, CA. 2011, Washington, DC: The American Society of Hematology, Abstract 1608
Burris H, Rodon J, Sharma S, Herbst RS, Tabernero J, Infante JR, Silva A, Demanse D, Hackl W, Baselga J: First-in-human phase I study of the oral PI3K inhibitor BEZ235 in patients (pts) with advanced solid tumors [abstract]. ASCO Annual Meeting Proceedings: June 4-8 2010; Chicago, IL. 2010, Alexandria, VA: American Society of Clinical Oncology, Abstract 3005
Arkenau H-T, Jones SF, Kurkjian C, Infante JR, Pant S, Burris HA, Moore KN, McMeekin DS, Greco FA, Ramsey S, Bendell JC: The PI3K/mTOR inhibitor BEZ235 given twice daily for the treatment of patients (pts) with advanced solid tumors [abstract]. ASCO Annual Meeting Proceedings: June 1-5 2012; Chicago, IL. 2012, A Alexandria, VA: American Society of Clinical Oncology, Abstract 3097
Munster PN, van der Noll R, Voest EE, Dees EC, Tan AR, Specht JM, Falchook GS, Daud A, Lolkema MP, Grilley-Olson JE, Yu EY, Fu S, Bergsland EK, Kleha J, Peng S, Smith DA, Lampkin TA, Schellens JHM, Morris SR, Kurzrock R: Phase I first-in-human study of the PI3 kinase inhibitor GSK2126458 (GSK458) in patients with advanced solid tumors (study P3K112826) [abstract]. ASCO Annual Meeting Proceedings: June 3-7 2011; Chicago, IL. 2011, Alexandria, VA: American Society of Clinical Oncology, Abstract 3018
Wagner AJ, Bendell JC, Dolly S, Morgan JA, Ware JA, Fredrickson J, Mazina KE, Lauchle JO, Burris HA, De Bono JS: A first-in-human phase I study to evaluate GDC-0980, an oral PI3K/mTOR inhibitor, administered QD in patients with advanced solid tumors [abstract]. ASCO Annual Meeting Proceedings: June 3-7 2011; Chicago, IL. 2011, Alexandria, VA: American Society of Clinical Oncology, Abstract 3020
Hollebecque A, Clamp A, Horsley L, Morgan JA, Bahleda R, George S, Shaw D, Lauchle JO, Ware J, Desai R, Wu J, Fu L, Jayson GC, Soria JC, Wagner AJ: A phase I study evaluating the pharmacokinetics (PK) and pharmacodynamic (PD) activity of the dual PI3K/mTOR inhibitor GDC-0980 administered once weekly (QW) [abstract]. AACR-NCI-EORTC International Conference: Molecular Targets and Cancer Therapeutics: November 12-16 2011; San Francisco, CA. 2011, Philadelphia, PA: American Association for Cancer Research, Abstract B153
Mahadevan D, Chiorean EG, Harris W, Von Hoff DD, Younger A, Rensvold DM, Cordova F, Qi W, Shelton CF, Becker MD, Garlich JR, Ramanathan RK: Phase I study of the multikinase prodrug SF1126 in solid tumors and B-cell malignancies [abstract]. ASCO Annual Meeting Proceedings: June 3-7 2011; Chicago, IL. 2011, Alexandria, VA: American Society of Clinical Oncology, Abstract 3015
LoRusso P, Britten C, Millham R, Borzillo G, Houk B, Wainberg Z, Marburg L, Dy G, Adjei A: First-in-patient study of PF-04691502, a small molecule intravenous dual inhibitor of PI3K and mTOR in patients with advanced cancer: update on safety, efficacy, and pharmacology [abstract]. AACR-NCI-EORTC International Conference: Molecular Targets and Cancer Therapeutics: November 12 2011; San Francisco, CA. 2011, Philadelphia, PA: American Association for Cancer Research, Abstract B163
Tabernero J, Bell-McGuinn K, Bendell J, Molina J, Kwak E, Millham R, Houk B, Borzillo G, Shapiro G: First-in-patient study of PF-05212384, a small molecule intravenous dual inhibitor of PI3K and mTOR in patients with advanced cancer: update on safety, efficacy, and pharmacology [abstract]. AACR-NCI-EORTC International Conference: Molecular Cancer Therapy: November 12-16 2011; San Francisco, CA. 2011, Philadelphia, PA: American Association for Cancer Research, Abstract A167
Markman B, Tabernero J, Krop I, Shapiro GI, Siu L, Chen LC, Mita M, Melendez Cuero M, Stutvoet S, Birle D, Anak O, Hackl W, Baselga J: Phase I safety, pharmacokinetic, and pharmacodynamic study of the oral phosphatidylinositol-3-kinase and mTOR inhibitor BGT226 in patients with advanced solid tumors. Ann Oncol. 2012, 23: 2399-2408. 10.1093/annonc/mds011.
Juric D, Rodon J, Gonzalez-Angulo AM, Burris HA, Bendell J, Berlin JD, Middleton MR, Bootle D, Boehm M, Schmitt A, Rouyrre N, Quadt C, Baselga J: BYL719, a next generation PI3K alpha specific inhibitor: Preliminary safety, PK, and efficacy results from the first-in-human study [abstract]. Proceedings of the 103rd Annual Meeting of the American Association for Cancer Research: March 31-April 4 2012; Chicago, IL. 2012, Philadelphia, PA: American Association for Cancer Research, Abstract CT-01
Coutre SE, Byrd JC, Furman RR, Brown JR, Benson DM, Wagner-Johnston ND, Flinn IW, Kahl BS, Spurgeon SEF, Lannutti BJ, Hsu HKW, Ulrich R, Peterman S, Holes L, Miller LL, Yu AS: Phase I study of CAL-101, an isoform-selective inhibitor of phosphatidylinositol 3-kinase P110d, in patients with previously treated chronic lymphocytic leukemia [abstract]. ASCO Annual Meeting Proceedings: June 3-7 2011; Chicago, IL. 2011, Alexandria, VA: American Society of Clinical Oncology, Abstract 6631
Kahl B, Byrd JC, Flinn IW, Wagner-Johnston N, Spurgeon S, Benson DM, Furman RR, Brown JR, Coutre S, Lannutti B, Giese NA, Ulrich RG, Webb HK, Peterman S, Holes L, Yu AS: Clinical safety and activity in a phase 1 study of CAL-101, an isoform-selective inhibitor of phosphatidylinositol 3-kinase P110{delta}, in patients with relapsed or refractory non-Hodgkin lymphoma [abstract]. ASH Annual Meeting Abstracts: December 4-7 2010; Orlando, FL. 2010, Washington, DC: The American Society of Hematology, Abstract 1777
Chantry D, Vojtek A, Kashishian A, Holtzman DA, Wood C, Gray PW, Cooper JA, Hoekstra MF: p110δ, a novel phosphatidylinositol 3-kinase catalytic subunit that associates with p85 and is expressed predominantly in leukocytes. J Biol Chem. 1997, 272: 19236-19241. 10.1074/jbc.272.31.19236.
Vanhaesebroeck B, Welham MJ, Kotani K, Stein R, Warne PH, Zvelebil MJ, Higashi K, Volinia S, Downward J, Waterfield MD: p110δ, A novel phosphoinositide 3-kinase in leukocytes. Proc Natl Acad Sci USA. 1997, 94: 4330-4335. 10.1073/pnas.94.9.4330.
Falasca M, Hughes WE, Dominguez V, Sala G, Fostira F, Fang MQ, Cazzolli R, Shepherd PR, James DE, Maffucci T: The role of phosphoinositide 3-kinase C2α in insulin signaling. J Biol Chem. 2007, 282: 28226-28236. 10.1074/jbc.M704357200.
Sopasakis VR, Liu P, Suzuki R, Kondo T, Winnay J, Tran TT, Asano T, Smyth G, Sajan MP, Farese RV, Kahn CR, Zhao JJ: Specific roles of the p110alpha isoform of phosphatidylinsositol 3-kinase in hepatic insulin signaling and metabolic regulation. Cell Metabol. 2010, 11: 220-230. 10.1016/j.cmet.2010.02.002.
Zhao JJ, Cheng H, Jia S, Wang L, Gjoerup OV, Mikaini A, Roberts TM: The p110α isoform of PI3K is essential for proper growth factor signaling and oncogenic transformation. Proc Natl Acad Sci USA. 2006, 103: 16296-16300. 10.1073/pnas.0607899103.
Knight ZA, Gonzalez B, Feldman ME, Zunder ER, Goldenberg DD, Williams O, Loewith R, Stokoe D, Balla A, Toth B, Balla T, Weiss WA, Williams RL, Shokat KM: A pharmacological map of the PI3-K family defines a role for p110alpha in insulin signaling. Cell. 2006, 125: 733-747. 10.1016/j.cell.2006.03.035.
Zhao L, Vogt PK: Class I PI3K in oncogenic cellular transformation. Oncogene. 2008, 27: 5486-5496. 10.1038/onc.2008.244.
Ikenoue T, Kanai F, Hikiba Y, Obata T, Tanaka Y, Imamura J, Ohta M, Jazag A, Guleng B, Tateishi K, Asaoka Y, Matsumura M, Kawabe T, Omata M: Functional analysis of PIK3CA gene mutations in human colorectal cancer. Cancer Res. 2005, 65: 4562-4567. 10.1158/0008-5472.CAN-04-4114.
Isakoff SJ, Engelman JA, Irie HY, Luo J, Brachmann SM, Pearline RV, Cantley LC, Brugge JS: Breast cancer-associated PIK3CA mutations are oncogenic in mammary epithelial cells. Cancer Res. 2005, 65: 10992-11000. 10.1158/0008-5472.CAN-05-2612.
Zhao JJ, Liu Z, Wang L, Shin E, Loda MF, Roberts TM: The oncogenic properties of mutant p110alpha and p110beta phosphatidylinositol 3-kinases in human mammary epithelial cells. Proc Natl Acad Sci USA. 2005, 102: 18443-18448. 10.1073/pnas.0508988102.
Niedermeier M, Hennessy BT, Knight ZA, Henneberg M, Hu J, Kurtova AV, Wierda WG, Keating MJ, Shokat KM, Burger JA: Isoform-selective phosphoinositide 3'-kinase inhibitors inhibit CXCR4 signaling and overcome stromal cell-mediated drug resistance in chronic lymphocytic leukemia: a novel therapeutic approach. Blood. 2009, 113: 5549-5557. 10.1182/blood-2008-06-165068.
Jamieson S, Flanagan JU, Kolekar S, Buchanan C, Kendall JD, Lee WJ, Rewcastle GW, Denny WA, Singh R, Dickson J, Baguley BC, Shepherd PR: A drug targeting only p110alpha can block phosphoinositide 3-kinase signalling and tumour growth in certain cell types. Biochem J. 2011, 438: 53-62. 10.1042/BJ20110502.
Dan S, Okamura M, Seki M, Yamazaki K, Sugita H, Okui M, Mukai Y, Nishimura H, Asaka R, Nomura K, Ishikawa Y, Yamori T: Correlating phosphatidylinositol 3-kinase inhibitor efficacy with signaling pathway status: in silico and biological evaluations. Cancer Res. 2010, 70: 4982-4994. 10.1158/0008-5472.CAN-09-4172.
Torbett NE, Luna-Moran A, Knight ZA, Houk A, Moasser M, Weiss W, Shokat KM, Stokoe D: A chemical screen in diverse breast cancer cell lines reveals genetic enhancers and suppressors of sensitivity to PI3K isoform-selective inhibition. Biochem J. 2008, 415: 97-110. 10.1042/BJ20080639.
Huang A, Fritsch C, Wilson C, Reddy A, Liu M, Lehar J, Quadt C, Hofmann F, Schlegel R: Single agent activity of PIK3CA inhibitor BYL719 in a broad cancer cell line panel [abstract]. Proceedings of the 103rd Annual Meeting of the American Association for Cancer Research: March 31-April 4 2012; Chicago, IL. 2012, Philadelphia, PA: American Association for Cancer Research, Abstract 3749
Jessen K, Kessler L, Kucharski J, Guo X, Staunton J, Janes M, Elia M, Banerjee U, Lan L, Wang S, Stewart J, Luzader A, Darjania L, Li L, Chan K, Martin M, Ren P, Rommel C, Liu Y: A potent and selective PI3K inhibitor, INK1117, targets human cancers harboring oncogenic PIK3CA mutations [abstract]. AACR-NCI-EORTC International Conference: Molecular Targets and Cancer Therapeutics: November 12-16 2011; San Francisco, CA. 2011, Philadelphia, PA: American Association for Cancer Research, Abstract A171
Herman SEM, Gordon AL, Wagner AJ, Heerema NA, Zhao W, Flynn JM, Jones J, Andritsos L, Puri KD, Lannutti BJ, Giese NA, Zhang X, Wei L, Byrd JC, Johnson AJ: Phosphatidylinositol 3-kinase- inhibitor CAL-101 shows promising preclinical activity in chronic lymphocytic leukemia by antagonizing intrinsic and extrinsic cellular survival signals. Blood. 2010, 116: 2078-2088. 10.1182/blood-2010-02-271171.
Ikeda H, Hideshima T, Fulciniti M, Perrone G, Miura N, Yasui H, Okawa Y, Kiziltepe T, Santo L, Vallet S, Cristea D, Calabrese E, Gorgun G, Raje NS, Richardson P, Munshi NC, Lannutti BJ, Puri KD, Giese NA, Anderson KC: PI3K/p110 is a novel therapeutic target in multiple myeloma. Blood. 2010, 116: 1460-1468. 10.1182/blood-2009-06-222943.
Lannutti BJ, Meadows SA, Herman SEM, Kashishian A, Steiner B, Johnson AJ, Byrd JC, Tyner JW, Loriaux MM, Deininger M, Druker BJ, Puri KD, Ulrich RG, Giese NA: CAL-101, a p110δ selective phosphatidylinositol-3-kinase inhibitor for the treatment of B-cell malignancies, inhibits PI3K signaling and cellular viability. Blood. 2011, 117: 591-594. 10.1182/blood-2010-03-275305.
Meadows SA, Kashishian A, Johnson D, Ulrich RG, Miller LL, Lannutti BJ: CAL-101 (GS-1101), a specific inhibitor of phosphatidylinositol-3-kinase-delta (PI3K{delta}), disrupts signals from the microenvironment, induces apoptosis, and enhances the antitumor activity of everolimus (RAD001), an inhibitor of mammalian target of rapamycin (mTOR), in mantle cell lymphoma (MCL) [abstract]. ASH Annual Meeting Abstracts: December 10-13 2011; San Diego, CA. 2011, Washington, DC: The American Society of Hematology, Abstract 3730
Iyengar S, Clear AJ, Owen A, Maharaj L, Matthews J, Calaminici M, Auer R, Ghazaly E, Iqbal S, Gribben JG, Joel S: PI3K inhibition with GDC-0941 has greater efficacy compared to p110{delta}-selective inhibition with CAL-101 in mantle cell lymphoma and may be particularly advantageous in multiply relapsed patients [abstract]. ASH Annual Meeting Abstracts: December 10-13 2011; San Diego, CA. 2011, Washington, DC: The American Society of Hematology, Abstract 1654
Meadows SA, Kashishian A, Johnson D: CAL-101 a potent selective inhibitor of the p110{delta} Isoform of phosphatidylinositol 3-kinase attenuates pathway signaling induces apoptosis and overcomes signals from the microenvironment in cellular models of Hodgkin lymphoma [abstract]. ASH Annual Meeting Abstracts: December 4-7 2010; Orlando, FL. 2010, Washington, DC: The American Society of Hematology, Abstract 3926
Hoellenriegel J, Meadows SA, Sivina M, Wierda WG, Kantarjian H, Keating MJ, Giese N, O'Brien S, Yu A, Miller LL, Lannutti BJ, Burger JA: The phosphoinositide 3'-kinase delta inhibitor, CAL-101, inhibits B-cell receptor signaling and chemokine networks in chronic lymphocytic leukemia. Blood. 2011, 118: 3603-3612. 10.1182/blood-2011-05-352492.
Davids MS, Lannutti BJ, Brown JR, Letai AG: BH3 profiling demonstrates that CAL-101 restores apoptotic priming in stroma-exposed chronic lymphocytic leukemia cells [abstract]. XIV International Workshop on Chronic Lymphocytic Leukemia; October 28-30 2011; Houston, TX. 2011, Clinical Lymphoma, Myeloma and Leukemia, S174-S175. Abstract 172.125
Sinclair A, Metz D, Cushing T, Liu L, Brake R, Starnes C, Means G, Henne K, Archibeque I, Mattson B, Drew A, Busse L, Wang L, Al-Assaad A-S, Molineux G: Phosphatidylinositol-3 kinase delta (PI3K{delta}) inhibitor AMG 319 is a potent, selective and orally bioavailable small molecule inhibitor that suppresses PI3K-mediated signaling and viability in neoplastic B cells [abstract]. ASH Annual Meeting Abstracts: December 10-13 2011; San Diego, CA. 2011, Washington, DC: The American Society of Hematology, Abstract 4964
Liu L, Sun B-C, Pistillo J, Payton M, Wang L, Archibeque I, Molineux G, Sinclair A: Phosphatidylinsoitol-3 kinase delta (PI3K{delta}) inhibitor AMG 319 combined with vincristine enhances G2/M arrest and apoptotic death in neoplastic B cells [abstract]. ASH Annual Meeting Abstracts: December 10-13 2011; San Diego, CA. 2011, Washington, DC: The American Society of Hematology, Abstract 4963
Flinn IW, Byrd JC, Furman RR, Brown JR, Benson DM, Coutre SE, Kahl BS, Smith BD, Wagner-Johnston ND, Spurgeon SE, Giese NA, Yu AS: Evidence of clinical activity in a phase 1 study of CAL-101, an oral P110{Delta} isoform-selective inhibitor of phosphatidylinositol 3-kinase, in patients with relapsed or refractory B-cell malignancies [abstract]. ASH Annual Meeting Abstracts: December 5-8 2009; New Orleans, LA. 2009, Washington, DC: The American Society of Hematology, Abstract 922
Flinn IW, Byrd JC, Furman RR, Brown JR, Lin TS, Bello C, Giese NA, Yu AS: Preliminary evidence of clinical activity in a phase I study of CAL-101, a selective inhibitor of the p1108 isoform of phosphatidylinositol 3-kinase (P13K), in patients with select hematologic malignancies [abstract]. ASCO Annual Meeting Proceedings: May 29-June 2 2009; Orlando, FL. 2009, Alexandria, VA: American Society of Clinical Oncology, Abstract 3543
Hallek M, Cheson BD, Catovsky D, Caligaris-Cappio F, Dighiero G, Döhner H, Hillmen P, Keating MJ, Montserrat E, Rai KR, Kipps TJ: Guidelines for the diagnosis and treatment of chronic lymphocytic leukemia: a report from the International Workshop on Chronic Lymphocytic Leukemia updating the National Cancer Institute-Working Group 1996 guidelines. Blood. 2008, 111: 5446-5456. 10.1182/blood-2007-06-093906.
Sharman J, de Vos S, Leonard JP, Furman RR, Coutre SE, Flinn IW, Schreeder MT, Barrientos JC, Wagner-Johnston ND, Boyd T, Fowler NH, Holes L, Lannutti B, Johnson D, Jahn TM, Miller LL: A phase 1 study of the selective phosphatidylinositol 3-kinase-delta (PI3K{delta}) inhibitor, CAL-101 (GS-1101), in combination with rituximab and/or bendamustine in patients with relapsed or refractory chronic lymphocytic leukemia (CLL) [abstract]. ASH Annual Meeting Abstracts: December 10-13 2011; San Diego, CA. 2011, Washington, DC: The American Society of Hematology, Abstract 1787
de Vos S, Schreeder MT, Flinn IW, Coutre SE, Leonard JP, Wagner-Johnston ND, Fowler NH, Boccia RV, Barrientos JC, Boyd T, Sharman J, Holes L, Lannutti B, Johnson DM, Jahn TM, Miller LL: A phase 1 study of the selective phosphatidylinositol 3-kinase-delta (PI3K{delta}) inhibitor, Cal-101 (GS-1101), in combination with rituximab and/or bendamustine in patients with previously treated, indolent non-Hodgkin lymphoma (iNHL) [abstract]. ASH Annual Meeting Abstracts: December 10-13 2011; San Diego, CA. 2011, Washington, DC: The American Society of Hematology, Abstract 2699
Furman RR, Barrientos JC, Sharman JP, De Vos S, Leonard J, Coutre SE, Schreeder MT, Wagner-Johnston ND, Boyd TE, Fowler NH, Flinn IW, Boccia RV, Holes L, Lannutti BJ, Johnson D, Jahn TM, Miller LL: A phase I/II study of the selective phosphatidylinositol 3-kinase-delta (PI3K{delta}) inhibitor, GS-1101 (CAL-101), with ofatumumab in patients with previously treated chronic lymphocytic leukemia (CLL) [abstract]. ASCO Annual Meeting Proceedings: June 1-5 2012; Chicago, IL. 2012, Alexandria, VA: American Society of Clinical Oncology, Abstract 6518
García-Martínez JM, Wullschleger S, Preston G, Guichard S, Fleming S, Alessi DR, Duce SL: Effect of PI3K- and mTOR-specific inhibitors on spontaneous B-cell follicular lymphomas in PTEN/LKB1-deficient mice. Br J Cancer. 2011, 104: 1116-1125. 10.1038/bjc.2011.83.
Amrein L, Shawi M, Panasci L: Phosphatidylinositol-3 kinase I inhibitor BKM120 induces cell death in B-chronic lymphocytic leukemia cells in-vitro [abstract]. European Multidisciplinary Cancer Congress: September 23-27 2011; Brussels. 2011, Eur J Cancer, S649-S650. Abstract 9236
Garlich J, Qi W, Becker M, Stejskal A, Mahadevan D: Translational studies of the novel prodrug PI3K/mTOR/PIM1 inhibitor SF1126 in DLBCL [abstract]. Proceedings of the 102nd Annual Meeting of the American Association for Cancer Research: April 2-6 2011; Orlando, FL. 2011, Philadelphia, PA: American Association for Cancer Research, Abstract nr LB-383
Mahadevan D, Qi W, Stejskal A, Cooke L, Garlich JR: SF1126, a pan-PI3K inhibitor has superior preclinical activity to CAL-101 a PI3K delta-specific inhibitor in aggressive B-cell non-Hodgkin's lymphoma [abstract]. ASH Annual Meeting Abstracts: December 10-13 2011; San Diego, CA. 2011, Washington, DC: The American Society of Hematology, Abstract 2720
Brown JR, Davids MS, Rodon J, Abrisqueta P, DeCillis AP, Rockich K, Egile C, Kelly A, Xu Y, Lager J, Awan FT: Phase I trial of SAR245408 (S08), a pan-phosphatidylinositol 3 kinase (PI3K) inhibitor, in patients with chronic lymphocytic leukemia (CLL) and lymphoma [abstract]. ASH Annual Meeting Abstracts: December 10-13 2011; San Diego, CA. 2011, Washington, DC: The American Society of Hematology, Abstract 2683
Jia S, Liu Z, Zhang S, Liu P, Zhang L, Lee SH, Zhang J, Signoretti S, Loda M, Roberts TM, Zhao JJ: Essential roles of PI(3)K-p110beta in cell growth, metabolism and tumorigenesis. Nature. 2008, 454: 776-779.
Guillermet-Guibert J, Bjorklof K, Salpekar A, Gonella C, Ramadani F, Bilancio A, Meek S, Smith AJH, Okkenhaug K, Vanhaesebroeck B: The p110β isoform of phosphoinositide 3-kinase signals downstream of G protein-coupled receptors and is functionally redundant with p110γ. Proc Natl Acad Sci USA. 2008, 105: 8292-8297. 10.1073/pnas.0707761105.
Hazeki O, Okada T, Kurosu H, Takasuga S, Suzuki T, Katada T: Activation of PI 3-kinase by G protein betagamma subunits. Life Sci. 1998, 62: 1555-1559. 10.1016/S0024-3205(98)00106-4.
Kubo H, Hazeki K, Takasuga S, Hazeki O: Specific role for p85/p110β in GTP-binding-protein-mediated activation of Akt. Biochem J. 2005, 392: 607-614. 10.1042/BJ20050671.
Roche S, Downward J, Raynal P, Courtneidge SA: A function for phosphatidylinositol 3-kinase beta (p85alpha-p110beta) in fibroblasts during mitogenesis: requirement for insulin- and lysophosphatidic acid-mediated signal transduction. Mol Cell Biol. 1998, 18: 7119-7129.
Yart A, Roche S, Wetzker R, Laffargue M, Tonks N, Mayeux P, Chap H, Raynal P: A function for phosphoinositide 3-kinase beta lipid products in coupling beta gamma to Ras activation in response to lysophosphatidic acid. J Biol Chem. 2002, 277: 21167-21178. 10.1074/jbc.M110411200.
Edgar KA, Wallin JJ, Berry M, Lee LB, Prior WW, Sampath D, Friedman LS, Belvin M: Isoform-specific phosphoinositide 3-kinase inhibitors exert distinct effects in solid tumors. Cancer Res. 2010, 70: 1164-1172. 10.1158/0008-5472.CAN-09-2525.
Crowder RJ, Phommaly C, Tao Y, Hoog J, Luo J, Perou CM, Parker JS, Miller MA, Huntsman DG, Lin L, Snider J, Davies SR, Olson JA, Watson MA, Saporita A, Weber JD, Ellis MJ: PIK3CA and PIK3CB inhibition produce synthetic lethality when combined with estrogen deprivation in estrogen receptor-positive breast cancer. Cancer Res. 2009, 69: 3955-3962. 10.1158/0008-5472.CAN-08-4450.
Wu X, Senechal K, Neshat MS, Whang YE, Sawyers CL: The PTEN/MMAC1 tumor suppressor phosphatase functions as a negative regulator of the phosphoinositide 3-kinase/Akt pathway. Proc Natl Acad Sci USA. 1998, 95: 15587-15591. 10.1073/pnas.95.26.15587.
Hollander MC, Blumenthal GM, Dennis PA: PTEN loss in the continuum of common cancers, rare syndromes and mouse models. Nat Rev Cancer. 2011, 11: 289-301. 10.1038/nrc3037.
Alvarez-Nuñez F, Bussaglia E, Mauricio D, Ybarra J, Vilar M, Lerma E, De Leiva A, Matias-Guiu X: PTEN promoter methylation in sporadic thyroid carcinomas. Thyroid. 2006, 16: 17-23. 10.1089/thy.2006.16.17.
García JM, Silva J, Peña C, Garcia V, Rodriguez R, Cruz MA, Cantos B, Provencio M, España P, Bonilla F: Promoter methylation of the PTEN gene is a common molecular change in breast cancer. Genes Chromosomes Cancer. 2004, 41: 117-124. 10.1002/gcc.20062.
Ho CM, Lin MC, Huang SH, Huang CJ, Lai HC, Chien TY, Chang SF: PTEN promoter methylation and LOH of 10q22-23 locus in PTEN expression of ovarian clear cell adenocarcinomas. Gynecol Oncol. 2009, 112: 307-313. 10.1016/j.ygyno.2008.09.040.
Kang YH, Lee HS, Kim WH: Promoter methylation and silencing of PTEN in gastric carcinoma. Lab Invest. 2002, 82: 285-291. 10.1038/labinvest.3780422.
Soria JC, Lee HY, Lee JI, Wang L, Issa JP, Kemp BL, Liu DD, Kurie JM, Mao L, Khuri FR: Lack of PTEN expression in non-small cell lung cancer could be related to promoter methylation. Clin Cancer Res. 2002, 8: 1178-1184.
Huse JT, Brennan C, Hambardzumyan D, Wee B, Pena J, Rouhanifard SH, Sohn-Lee C, Le Sage C, Agami R, Tuschl T, Holland EC: The PTEN-regulating microRNA miR-26a is amplified in high-grade glioma and facilitates gliomagenesis in vivo. Genes Dev. 2009, 23: 1327-1337. 10.1101/gad.1777409.
Zhang JG, Wang JJ, Zhao F, Liu Q, Jiang K, Yang GH: MicroRNA-21 (miR-21) represses tumor suppressor PTEN and promotes growth and invasion in non-small cell lung cancer (NSCLC). Clin Chim Acta. 2010, 411: 846-852. 10.1016/j.cca.2010.02.074.
Poliseno L, Salmena L, Riccardi L, Fornari A, Song MS, Hobbs RM, Sportoletti P, Varmeh S, Egia A, Fedele G, Rameh L, Loda M, Pandolfi PP: Identification of the miR-106b~25 microRNA cluster as a proto-oncogenic PTEN-targeting intron that cooperates with its host gene MCM7 in transformation. Sci Signal. 2010, 3: ra29-10.1126/scisignal.2000594.
Wang X, Jiang X: Post-translational regulation of PTEN. Oncogene. 2008, 27: 5454-5463. 10.1038/onc.2008.242.
Trotman LC, Wang X, Alimonti A, Chen Z, Teruya-Feldstein J, Yang H, Pavletich NP, Carver BS, Cordon-Cardo C, Erdjument-Bromage H, Tempst P, Chi SG, Kim HJ, Misteli T, Jiang X, Pandolfi PP: Ubiquitination regulates PTEN nuclear import and tumor suppression. Cell. 2007, 128: 141-156. 10.1016/j.cell.2006.11.040.
Wee S, Wiederschain D, Maira S-M, Loo A, Miller C, deBeaumont R, Stegmeier F, Yao Y-M, Lengauer C: PTEN-deficient cancers depend on PIK3CB. Proc Natl Acad Sci USA. 2008, 105: 13057-13062. 10.1073/pnas.0802655105.
Rivero RA, Hardwicke MA: Identification of GSK2636771, a potent and selective, orally bioavailable inhibitor of phosphatidylinositol 3-kinase-beta (PI3K{alpha}) for the treatment of PTEN deficient tumors [abstract]. Proceedings of the 103rd Annual Meeting of the American Association for Cancer Research: March 31-April 4 2012; Chicago, IL. 2012, Philadelphia, PA: American Association for Cancer Research, Abstract 2913
Ni J, Liu Q, Xie S, Carlson C, Von T, Vogel K, Riddle S, Benes C, Eck M, Roberts T, Gray N, Zhao J: Functional characterization of an isoform-selective inhibitor of PI3K-p110β as a potential anticancer agent. Cancer Discov. 2012, 2: 425-433. 10.1158/2159-8290.CD-12-0003.
Certal V, Halley F, Virone-Oddos A, Delorme C, Karlsson A, Rak A, Thompson F, Filoche-Romme B, El-Ahmad Y, Carry JC, Abecassis PY, Lejeune P, Vincent L, Bonnevaux H, Nicolas JP, Bertrand T, Marquette JP, Michot N, Benard T, Below P, Vade I, Chatreaux F, Lebourg G, Pilorge F, Angouillant-Boniface O, Louboutin A, Lengauer C, Schio L: Discovery and optimization of new benzimidazole- and benzoxazole-pyrimidone selective PI3Kbeta inhibitors for the treatment of phosphatase and TENsin homologue (PTEN)-deficient cancers. J Med Chem. 2012, 55: 4788-4805. 10.1021/jm300241b.
Tanaka H, Yoshida M, Tanimura H, Fujii T, Sakata K, Tachibana Y, Ohwada J, Ebiike H, Kuramoto S, Morita K, Yoshimura Y, Yamazaki T, Ishii N, Kondoh O, Aoki Y: The selective class I PI3K inhibitor CH5132799 targets human cancers harboring oncogenic PIK3CA mutations. Clin Cancer Res. 2011, 17: 3272-3281. 10.1158/1078-0432.CCR-10-2882.
Ihle NT, Lemos R, Wipf P, Yacoub A, Mitchell C, Siwak D, Mills GB, Dent P, Kirkpatrick DL, Powis G: Mutations in the phosphatidylinositol-3-kinase pathway predict for antitumor activity of the inhibitor PX-866 whereas oncogenic ras is a dominant predictor for resistance. Cancer Res. 2009, 69: 143-150. 10.1158/0008-5472.CAN-07-6656.
Serra V, Markman B, Scaltriti M, Eichhorn PJ, Valero V, Guzman M, Botero ML, Llonch E, Atzori F, Di Cosimo S, Maira M, Garcia-Echeverria C, Parra JL, Arribas J, Baselga J: NVP-BEZ235, a dual PI3K/mTOR inhibitor, prevents PI3K signaling and inhibits the growth of cancer cells with activating PI3K mutations. Cancer Res. 2008, 68: 8022-8030. 10.1158/0008-5472.CAN-08-1385.
Shoji K, Oda K, Kashiyama T, Ikeda Y, Nakagawa S, Sone K, Miyamoto Y, Hiraike H, Tanikawa M, Miyasaka A, Koso T, Matsumoto Y, Wada-Hiraike O, Kawana K, Kuramoto H, McCormick F, Aburatani H, Yano T, Kozuma S, Taketani Y: Genotype-dependent efficacy of a dual PI3K/mTOR inhibitor, NVP-BEZ235, and an mTOR inhibitor, RAD001, in endometrial carcinomas. PLoS ONE. 2012, 7: e37431-10.1371/journal.pone.0037431.
Koul D, Fu J, Shen R, LaFortune TA, Wang S, Tiao N, Kim Y-W, Liu J-L, Ramnarian D, Yuan Y, Garcia-Echevrria C, Maira S-M, Yung WKA: Antitumor activity of NVP-BKM120 - a selective pan class I PI3 kinase inhibitor showed differential forms of cell death based on p53 status of glioma cells. Clin Cancer Res. 2012, 18: 184-195. 10.1158/1078-0432.CCR-11-1558.
Janku F, Tsimberidou AM, Garrido-Laguna I, Wang X, Luthra R, Hong DS, Naing A, Falchook GS, Moroney JW, Piha-Paul SA, Wheler JJ, Moulder SL, Fu S, Kurzrock R: PIK3CA mutations in patients with advanced cancers treated with PI3K/AKT/mTOR axis inhibitors. Mol Cancer Ther. 2011, 10: 558-565. 10.1158/1535-7163.MCT-10-0994.
Janku F, Wheler JJ, Westin SN, Moulder SL, Naing A, Tsimberidou AM, Fu S, Falchook GS, Hong DS, Garrido-Laguna I, Luthra R, Lee JJ, Lu KH, Kurzrock R: PI3K/AKT/mTOR inhibitors in patients with breast and gynecologic malignancies harboring PIK3CA mutations. J Clin Oncol. 2012, 30: 777-782. 10.1200/JCO.2011.36.1196.
Rodon J, Saura C, Dienstmann R, Vivancos A, Cajal SR, Baselga J, Tabernero J: Molecular prescreening to select patient population in early clinical trials. Nat Rev Clin Oncol. 2012, 9: 359-366. 10.1038/nrclinonc.2012.48.
Tsimberidou AM, Iskander NG, Hong DS, Wheler JJ, Fu S, Piha-Paul SA, Naing A, Falchook GS, Janku F, Luthra R, Wen S, Kurzrock R: Personalized medicine in a phase I clinical trials program: The M. D. Anderson Cancer Center Initiative [abstract]. ASCO Annual Meeting Proceedings: June 3-7 2011; Chicago, IL. 2011, Alexandria, VA: American Society of Clinical Oncology, Abstract CRA2500
Krop IE, Saura C, Rodon Ahnert J, Becerra C, Britten CD, Isakoff SJ, Demanse D, Hackl W, Quadt C, Silva AP, Burris HA, Abu-Khalaf MM, Baselga J: A phase I/IB dose-escalation study of BEZ235 in combination with trastuzumab in patients with PI3-kinase or PTEN altered HER2+ metastatic breast cancer [abstract]. ASCO Annual Meeting Proceedings: June 1-5, 2012; Chicago, IL. 2012, Alexandria, VA: American Society of Clinical Oncology, Abstract 508
Liu P, Cheng H, Santiago S, Raeder M, Zhang F, Isabella A, Yang J, Semaan DJ, Chen C, Fox EA, Gray NS, Monahan J, Schlegel R, Beroukhim R, Mills GB, Zhao JJ: Oncogenic PIK3CA-driven mammary tumors frequently recur via PI3K pathway-dependent and PI3K pathway-independent mechanisms. Nat Med. 2011, 17: 1116-1121. 10.1038/nm.2402.
Eichhorn PJ, Gili M, Scaltriti M, Serra V, Guzman M, Nijkamp W, Beijersbergen RL, Valero V, Seoane J, Bernards R, Baselga J: Phosphatidylinositol 3-kinase hyperactivation results in lapatinib resistance that is reversed by the mTOR/phosphatidylinositol 3-kinase inhibitor NVP-BEZ235. Cancer Res. 2008, 68: 9221-9230. 10.1158/0008-5472.CAN-08-1740.
Serra V, Scaltriti M, Prudkin L, Eichhorn PJ, Ibrahim YH, Chandarlapaty S, Markman B, Rodriguez O, Guzman M, Rodriguez S, Gili M, Russillo M, Parra JL, Singh S, Arribas J, Rosen N, Baselga J: PI3K inhibition results in enhanced HER signaling and acquired ERK dependency in HER2-overexpressing breast cancer. Oncogene. 2011, 30: 2547-2557. 10.1038/onc.2010.626.
Inukai K, Funaki M, Anai M, Ogihara T, Katagiri H, Fukushima Y, Sakoda H, Onishi Y, Ono H, Fujishiro M, Abe M, Oka Y, Kikuchi M, Asano T: Five isoforms of the phosphatidylinositol 3-kinase regulatory subunit exhibit different associations with receptor tyrosine kinases and their tyrosine phosphorylations. FEBS Lett. 2001, 490: 32-38. 10.1016/S0014-5793(01)02132-9.
Engelman JA, Chen L, Tan X, Crosby K, Guimaraes AR, Upadhyay R, Maira M, McNamara K, Perera SA, Song Y, Chirieac LR, Kaur R, Lightbown A, Simendinger J, Li T, Padera RF, García-Echeverría C, Weissleder R, Mahmood U, Cantley LC, Wong KK: Effective use of PI3K and MEK inhibitors to treat mutant Kras G12D and PIK3CA H1047R murine lung cancers. Nat Med. 2008, 14: 1351-1356. 10.1038/nm.1890.
Lee MJ, Ye AS, Gardino AK, Heijink AM, Sorger PK, Macbeath G, Yaffe MB: Sequential application of anticancer drugs enhances cell death by rewiring apoptotic signaling networks. Cell. 2012, 149: 780-794. 10.1016/j.cell.2012.03.031.
Mayer IA, Abramson VG, Balko JM, Isakoff SJ, Kuba MG, Sanders M, Forero-Torres A, Yap JT, Van Den Abbeele AD, Li Y, Arteaga CL, Winer EP, Stand Up to Cancer - PI3K in Women's Cancers: SU2C phase Ib study of pan-PI3K inhibitor BKM120 with letrozole in ER+/HER2- metastatic breast cancer (MBC) [abstract]. ASCO Annual Meeting Proceedings: June 1-5 2012; Chicago, IL. 2012, Alexandria, VA: American Society of Clinical Oncology, Abstract 510
Ihle NT, Byers LA, Kim ES, Saintigny P, Lee JJ, Blumenschein GR, Tsao A, Liu S, Larsen JE, Wang J, Diao L, Coombes KR, Chen L, Zhang S, Abdelmelek MF, Tang X, Papadimitrakopoulou V, Minna JD, Lippman SM, Hong WK, Herbst RS, Wistuba II, Heymach JV, Powis G: Effect of KRAS oncogene substitutions on protein behavior: implications for signaling and clinical outcome. J Natl Cancer Inst. 2012, 104: 228-239. 10.1093/jnci/djr523.
Gerlinger M, Rowan AJ, Horswell S, Larkin J, Endesfelder D, Gronroos E, Martinez P, Matthews N, Stewart A, Tarpey P, Varela I, Phillimore B, Begum S, McDonald NQ, Butler A, Jones D, Raine K, Latimer C, Santos CR, Nohadani M, Eklund AC, Spencer-Dene B, Clark G, Pickering L, Stamp G, Gore M, Szallasi Z, Downward J, Futreal PA, Swanton C: Intratumor heterogeneity and branched evolution revealed by multiregion sequencing. N Engl J Med. 2012, 366: 883-892. 10.1056/NEJMoa1113205.
COSMIC database: Distribution of somatic mutations in PIK3CA. [http://www.sanger.ac.uk/perl/genetics/CGP/cosmic?action=bygene&ln=PIK3CA&start=1&end=1069&coords=AA:AA]
Andersson P, Kolaric A, Windahl T, Kirrander P, Soderkvist P, Karlsson MG: PIK3CA, HRAS and KRAS gene mutations in human penile cancer. The Journal of urology. 2008, 179: 2030-2034. 10.1016/j.juro.2007.12.040.
Shi J, Yao D, Liu W, Wang N, Lv H, Zhang G, Ji M, Xu L, He N, Shi B, Hou P: Highly frequent PIK3CA amplification is associated with poor prognosis in gastric cancer. BMC Cancer. 2012, 12: 50-10.1186/1471-2407-12-50.
Byun DS, Cho K, Ryu BK, Lee MG, Park JI, Chae KS, Kim HJ, Chi SG: Frequent monoallelic deletion of PTEN and its reciprocal associatioin with PIK3CA amplification in gastric carcinoma. Int J Cancer. 2003, 104: 318-327. 10.1002/ijc.10962.
Abubaker J, Jehan Z, Bavi P, Sultana M, Al-Harbi S, Ibrahim M, Al-Nuaim A, Ahmed M, Amin T, Al-Fehaily M, Al-Sanea O, Al-Dayel F, Uddin S, Al-Kuraya KS: Clinicopathological analysis of papillary thyroid cancer with PIK3CA alterations in a Middle Eastern population. J Clin Endocrinol Metab. 2008, 93: 611-618. 10.1210/jc.2007-1717.
Pedrero JM, Carracedo DG, Pinto CM, Zapatero AH, Rodrigo JP, Nieto CS, Gonzalez MV: Frequent genetic and biochemical alterations of the PI 3-K/AKT/PTEN pathway in head and neck squamous cell carcinoma. Int J Cancer. 2005, 114: 242-248. 10.1002/ijc.20711.
Woenckhaus J, Steger K, Werner E, Fenic I, Gamerdinger U, Dreyer T, Stahl U: Genomic gain of PIK3CA and increased expression of p110alpha are associated with progression of dysplasia into invasive squamous cell carcinoma. J Pathol. 2002, 198: 335-342. 10.1002/path.1207.
Ji M, Guan H, Gao C, Shi B, Hou P: Highly frequent promoter methylation and PIK3CA amplification in non-small cell lung cancer (NSCLC). BMC Cancer. 2011, 11: 147-10.1186/1471-2407-11-147.
Massion PP, Kuo WL, Stokoe D, Olshen AB, Treseler PA, Chin K, Chen C, Polikoff D, Jain AN, Pinkel D, Albertson DG, Jablons DM, Grays JW: Genomic copy number analysis of non-small cell lung cancer using array comparative genomic hybridization: Implications of the phosphatidylinositol 3-kinase pathway. Cancer Res. 2002, 62: 3636-3640.
Ma YY, Wei SJ, Lin YC, Lung JC, Chang TC, Whang-Peng J, Liu JM, Yang DM, Yang WK, Shen CY: PIK3CA as an oncogene in cervical cancer. Oncogene. 2000, 19: 2739-2744. 10.1038/sj.onc.1203597.
Bertelsen BI, Steine SJ, Sandvei R, Molven A, Laerum OD: Molecular analysis of the PI3K-AKT pathway in uterine cervical neoplasia: frequent PIK3CA amplification and AKT phosphorylation. Int J Cancer. 2006, 118: 1877-1883. 10.1002/ijc.21461.
Abubaker J, Bavi P, Al-Haqawi W, Jehan Z, Munkarah A, Uddin S, Al-Kuraya KS: PIK3CA alterations in Middle Eastern ovarian cancers. Mol Cancer. 2009, 8: 51-10.1186/1476-4598-8-51.
Agell L, Hernandez S, Salido M, de Muga S, Juanpere N, Arumi-Uria M, Menendez S, Lorenzo M, Lorente JA, Serrano S, Lloreta J: PI3K signaling pathway is activated by PIK3CA mRNA overexpression and copy gain in prostate tumors, but PIK3CA, BRAF, KRAS and AKT1 mutations are infrequent events. Mod Pathol. 2011, 24: 443-452. 10.1038/modpathol.2010.208.
Konopka B, Janiec-Jankowska A, Kwiatkowska E, Najmola U, Bidzinski M, Olszewski W, Goluda C: PIK3CA mutations and amplification in endometrioid endometrial carcinomas: Relation to other genetic defects and clinicopathologic status of the tumors. Hum Pathol. 2011, 42: 1710-1719. 10.1016/j.humpath.2010.01.030.
Salvesen HB, Carter SL, Mannelqvist M, Dutt A, Getz G, Stefansson IM, Raeder MB, Sos ML, Engelsen IB, Trovik J, Wik E, Greulich H, Bo TH, Jonassen I, Thomas RK, Zander T, Garraway LA, Oyan AM, Sellers WR, Kalland KH, Meyerson M, Akslen LA, Beroukhim R: Integrated genomic profiling of endometrial carcinoma associates aggressive tumors with indicators of PI3 kinase activation. Proc Natl Acad Sci USA. 2009, 106: 4834-4839. 10.1073/pnas.0806514106.
Wu G, Xing M, Mambo E, Huang X, Liu J, Guo Z, Chatterjee A, Goldenberg D, Gollin S, Sukumar S, Trink B, Sidransky D: Somatic mutation and gain of copy number of PIK3CA in human breast cancer. Breast Cancer Research. 2005, 7: R609-R616. 10.1186/bcr1262.
Oliveira MC, Pereira SR, Ribeiro EMF, Lima RS, Urban CA, Sebastiao APM, Marian C, Haddad BR, Cavalli IJ, Cavalli LR: DNA copy number changes in the PI3K signaling pathway genes in triple negative breast cancer [Abstract]. Proceedings of the 102nd Annual Meeting of the American Association for Cancer Research: April 2-6 2011; Orlando, FL. 2011, Philadelphia, PA: American Association for Cancer Research, Abstract 3822
Brown JR, Hanna M, Tesar B, Werner L, Pochet N, Asara JM, Wang YE, Dal Cin P, Fernandes SM, Thompson C, MacConaill L, Wu CJ, Van De Peer Y, Correll M, Regev A, Neuberg D, Freedman AS: Integrative genomic analysis implicates PIK3CA and myc in chronic lymphocytic leukemia. Clin Lymphoma Myeloma Leuk. 2011, 11: S147-S148.
Chiariello E, Roz L, Albarosa R, Magnani I, Finocchiaro G: PTEN/MMAC1 mutations in primary glioblastomas and short-term cultures of malignant gliomas. Oncogene. 1998, 16: 541-545. 10.1038/sj.onc.1201689.
Feilotter HE, Nagai MA, Boag AH, Eng C, Mulligan LM: Analysis of PTEN and the 10q23 region in primary prostate carcinomas. Oncogene. 1998, 16: 1743-1748. 10.1038/sj.onc.1200205.
Gray IC, Stewart LMD, Phillips SMA, Hamilton JA, Gray NE, Watson GJ, Spurr NK, Snary D: Mutation and expression analysis of the putative prostate tumour-suppressor gene PTEN. Br J Cancer. 1998, 78: 1296-1300. 10.1038/bjc.1998.674.
Pesche S, Latil A, Muzeau F, Cussenot O, Fournier G, Longy M, Eng C, Lidereau R: PTEN/MMAC1/TEP1 involvement in primary prostate cancers. Oncogene. 1998, 16: 2879-2883. 10.1038/sj.onc.1202081.
Wang SI, Parsons R, Ittmann M: Homozygous deletion of the PTEN tumor suppressor gene in a subset of prostate adenocarcinomas. Clin Cancer Res. 1998, 4: 811-815.
Freihoff D, Kempe A, Beste B, Wappenschmidt B, Kreyer E, Hayashi Y, Meindl A, Krebs D, Wiestler OD, von Deimling A, Schmutzler RK: Exclusion of a major role for the PTEN tumour-suppressor gene in breast carcinomas. Br J Cancer. 1999, 79: 754-758. 10.1038/sj.bjc.6690121.
Feilotter HE, Coulon V, McVeigh JL, Boag AH, Dorion-Bonnet F, Duboue B, Latham WC, Eng C, Mulligan LM, Longy M: Analysis of the 10q23 chromosomal region and the PTEN gene in human sporadic breast carcinoma. Br J Cancer. 1999, 79: 718-723. 10.1038/sj.bjc.6690115.
Celebi JT, Shendrik I, Silvers DN, Peacocke M: Identification of PTEN mutations in metastatic melanoma specimens. J Med Genet. 2000, 37: 653-657. 10.1136/jmg.37.9.653.
Abdel-Rahman MH, Yang Y, Zhou XP, Craig EL, Davidorf FH, Eng C: High frequency of submicroscopic hemizygous deletion is a major mechanism of loss of expression of PTEN in uveal melanoma. J Clin Oncol. 2006, 24: 288-295.
COSMIC database: Distribution of somatic mutations in PTEN. [http://www.sanger.ac.uk/perl/genetics/CGP/cosmic?action=bygene&ln=PTEN&start=1&end=404&coords=AA:AA]
Holway AH, Rieger-Christ KM, Miner WR, Cain JW, Dugan JM, Pezza JA, Silverman ML, Shapter A, McLellan R, Summerhayes IC: Somatic mutation of PTEN in vulvar cancer. Clin Cancer Res. 2000, 6: 3228-3235.
COSMIC database: Distribution of somatic mutations in PTEN in grade IV astrocytoma and oligoastrocytoma. [http://www.sanger.ac.uk/perl/genetics/CGP/cosmic?action=bycancer&ln=PTEN&sn=central_nervous_system&ss=NS;&ss=basal_ganglia;&ss=brain;&ss=brainstem;&ss=cerebellum;&ss=cerebral_cortex;&ss=cerebral_hemisphere;&ss=cerebrum;&ss=choroid_plexus;&ss=corpus_callosum;&ss=diencephalon;&ss=filum;&ss=fourth_ventricle;&ss=frontal_lobe;&ss=frontobasal;&ss=frontoparietal;&ss=frontotemporal;&ss=hypothalamus;&ss=infratentorial;&ss=intraventricular;&ss=lateral_ventricle;&ss=medulla;&ss=meninges;&ss=midbrain;&ss=occipital_lobe;&ss=optic_nerve;&ss=paracentral;&ss=parietal_lobe;&ss=parietooccipital;&ss=pineal_gland;&ss=posterior_fossa;&ss=sella_turcica;&ss=septum_pellucidum;&ss=spinal_cord;&ss=supratentorial;&ss=tectum;&ss=temporal_lobe;&ss=temporobasal;&ss=temporooccipital;&ss=temporoparietal;&ss=thalamus;&ss=third_ventricle;&ss=trigone&sh=astrocytoma_Grade_IV;&sh=oligoastrocytoma_Grade_IV&hn=glioma]
Maira SM, Pecchi S, Huang A, Burger M, Knapp M, Sterker D, Schnell C, Guthy D, Nagel T, Wiesmann M, Brachmann S, Fritsch C, Dorsch M, Chène P, Shoemaker K, De Pover A, Menezes D, Martiny-Baron G, Fabbro D, Wilson CJ, Schlegel R, Hofmann F, García-Echeverría C, Sellers WR, Voliva CF: Identification and characterization of NVP-BKM120, an orally available pan-class I PI3-kinase inhibitor. Mol Cancer Ther. 2012, 11: 317-328. 10.1158/1535-7163.MCT-11-0474.
Folkes AJ, Ahmadi K, Alderton WK, Alix S, Baker SJ, Box G, Chuckowree IS, Clarke PA, Depledge P, Eccles SA, Friedman LS, Hayes A, Hancox TC, Kugendradas A, Lensun L, Moore P, Olivero AG, Pang J, Patel S, Pergl-Wilson GH, Raynaud FI, Robson A, Saghir N, Salphati L, Sohal S, Ultsch MH, Valenti M, Wallweber HJA, Nan CW, Wiesmann C, et al: The identification of 2-(1H-indazol-4-yl)-6-(4-methanesulfonyl-piperazin-1- ylmethyl)-4-morpholin-4-yl-thieno[3,2-d]pyrimidine (GDC-0941) as a potent, selective, orally bioavailable inhibitor of class I PI3 kinase for the treatment of cancer. J Med Chem. 2008, 51: 5522-5532. 10.1021/jm800295d.
Maira SM, Stauffer F, Brueggen J, Furet P, Schnell C, Fritsch C, Brachmann S, Chene P, De Pover A, Schoemaker K, Fabbro D, Gabriel D, Simonen M, Murphy L, Finan P, Sellers W, Garcia-Echeverria C: Identification and characterization of NVP-BEZ235, a new orally available dual phosphatidylinositol 3-kinase/mammalian target of rapamycin inhibitor with potent in vivo antitumor activity. Mol Cancer Ther. 2008, 7: 1851-1863. 10.1158/1535-7163.MCT-08-0017.
Yuan J, Mehta PP, Yin MJ, Sun S, Zou A, Chen J, Rafidi K, Feng Z, Nickel J, Engebretsen J, Hallin J, Blasina A, Zhang E, Nguyen L, Sun M, Vogt PK, McHarg A, Cheng H, Christensen JG, Kan JL, Bagrodia S: PF-04691502, a potent and selective oral inhibitor of PI3K and mTOR kinases with antitumor activity. Mol Cancer Ther. 2011, 10: 2189-2199. 10.1158/1535-7163.MCT-11-0185.
Mallon R, Feldberg LR, Lucas J, Chaudhary I, Dehnhardt C, Delos Santos E, Chen Z, Dos Santos O, Ayral-Kaloustian S, Venkatesan A, Hollander I: Antitumor efficacy of PKI-587, a highly potent dual PI3K/mTOR kinase inhibitor. Clin Cancer Res. 2011, 17: 3193-3203. 10.1158/1078-0432.CCR-10-1694.
Sutherlin DP, Bao L, Berry M, Castanedo G, Chuckowree I, Dotson J, Folks A, Friedman L, Goldsmith R, Gunzner J, Heffron T, Lesnick J, Lewis C, Mathieu S, Murray J, Nonomiya J, Pang J, Pegg N, Prior WW, Rouge L, Salphati L, Sampath D, Tian Q, Tsui V, Wan NC, Wang S, Wei B, Wiesmann C, Wu P, Zhu BY, et al: Discovery of a potent, selective, and orally available class i phosphatidylinositol 3-kinase (PI3K)/Mammalian target of rapamycin (mTOR) kinase inhibitor (GDC-0980) for the treatment of cancer. J Med Chem. 2011, 54: 7579-7587. 10.1021/jm2009327.
Fritsch CM, Schnell C, Chatenay-Rivauday C, Guthy DA, Pover AD, Wartmann M, Brachmann S, Maira S-M, Huang A, Quadt C, Hofmann F, Caravatti G: NVP-BYL719, a novel PI3Kalpha selective inhibitor with all the characteristics required for clinical development as an anti-cancer agent [abstract]. Proceedings of the 103rd Annual Meeting of the American Association for Cancer Research: March 31-April 4 2012; Chicago, IL. 2012, Philadelphia, PA: American Association for Cancer Research, Abstract 3748
Pre-publication history
The pre-publication history for this paper can be accessed here:http://www.biomedcentral.com/1741-7015/10/161/prepub
Acknowledgements
IB is a recipient of a '2012 Conquer Cancer Foundation Young Investigator Award' and a 'Spanish Society of Medical Oncology (SEOM) Translational Research Grant'.
Author information
Authors and Affiliations
Corresponding author
Additional information
Competing interests
LLS: research support from Novartis, GSK, Bristol-Myers Squibb, Pfizer, Genentech/Roche.
Authors' contributions
IB and LLS: conception and design; manuscript writing; final approval of manuscript.
Authors’ original submitted files for images
Below are the links to the authors’ original submitted files for images.
Rights and permissions
This article is published under license to BioMed Central Ltd. This is an Open Access article distributed under the terms of the Creative Commons Attribution License (http://creativecommons.org/licenses/by/2.0), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
About this article
Cite this article
Brana, I., Siu, L.L. Clinical development of phosphatidylinositol 3-kinase inhibitors for cancer treatment. BMC Med 10, 161 (2012). https://doi.org/10.1186/1741-7015-10-161
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/1741-7015-10-161