
Abstract
Background
The origins and dispersal of Plasmodium vivax to its current worldwide distribution remains controversial. Although progress on P. vivax genetics and genomics has been achieved worldwide, information concerning New World parasites remains fragmented and largely incomplete. More information on the genetic diversity in Latin America (LA) is needed to better explain current patterns of parasite dispersion and evolution.
Methods
Plasmodium vivax circumsporozoite protein gene polymorphism was investigated using polymerase chain reaction amplification and restriction fragment length polymorphism (PCR-RFLP), and Sanger sequencing in isolates from the Pacific Ocean coast of Mexico, Nicaragua, and Peru. In conjunction with worldwide sequences retrieved from the Genbank, mismatch distribution analysis of central repeat region (CRR), frequency estimation of unique repeat types and phylogenetic analysis of the 3′ terminal region, were performed to obtain an integrative view of the genetic relationships between regional and worldwide isolates.
Results
Four RFLP subtypes, vk210a, b, c and d were identified in Southern Mexico and three subtypes vk210a, e and f in Nicaragua. The nucleotide sequences showed that Mexican vk210a and all Nicaraguan isolates were similar to other American parasites. In contrast, vk210b, c and d were less frequent, had a domain ANKKAEDA in their carboxyl end and clustered with Asian isolates. All vk247 isolates from Mexico and Peru had identical RFLP pattern. Their nucleotide sequences showed two copies of GGQAA GGNAANKKAGDAGA at the carboxyl end. Differences in mismatch distribution parameters of the CRR separate vk247 from most vk210 isolates. While vk247 isolates display a homogeneous pattern with no geographical clustering, vk210 isolates display a heterogeneous geographically clustered pattern which clearly separates LA from non-American isolates, except vk210b, c and d from Southern Mexico.
Conclusions
The presence of vk210a in Mexico and vk210e, f and g in Nicaragua are consistent with other previously reported LA isolates and reflect their circulation throughout the continent. The vk210b, c and d are novel genotypes in LA. Their genetic relationships and low variability within these vk210 and/or within the vk247 parasites in Southern Mexico suggest its recent introduction and/or recent expansion to this region. The global analysis of P. vivax csp suggests this parasite introduction to the region and likely LA by different independent events.
Similar content being viewed by others

Background
Plasmodium vivax, is the most widely distributed malaria parasite in Meso- and South America [1], producing 80–90 million cases per year in these regions [2]. Although in Mesoamerica, malaria transmission has significantly decreased during the last decade [3, 4], it persists with fluctuations in several regions. In Mexico, the number of cases peaked during 1998–1999, followed by a gradual decrease until in 2005, when hurricane Stan devastated the southern region resulted in a significant increase in cases in 2006, 2007 and 2008 [5]. Malaria cases in Nicaragua followed a similar gradual pattern, from 23,878 cases in 2000 to 762 in 2008. In Peru, the region of Piura experiences a comparatively high proportion of malaria cases in the coast and a P. vivax outbreak occurred in Sullana province during 2008, while surrounding zones experienced lower rates of transmission [3].
The distribution of P. vivax populations is controversial; some studies indicate a geographical compartmentalization of the New from Old World parasites [6], but this is not supported by others [7]. Consistently, population genetics of parasites in temperate zones from China suggest an ancient population expansion [8]. Although recent relevant progress on genetic features of the parasite has been achieved in various geographical regions worldwide [8–11], information from endemic areas of the Americas remains largely incomplete. Epidemiological and genetic studies covering malaria-endemic countries in the region could provide a better understanding of parasite transmission and dispersion [9].
Plasmodium sporozoites are covered by the circumsporozoite protein (CSP). This protein has multifunctional roles, including sporozoite maturation and salivary gland invasion in mosquitoes and hepatocyte invasion in humans [12]. Motivated by its longstanding as a vaccine candidate, the CSP gene polymorphism has been investigated in parasites from different geographic regions and used to elucidate evolutionary dynamics [13–15]. All CSPs present a central repeat region (CRR) and two conserved domains RI and RII located in the amino and carboxyl ends, respectively. Two main P. vivax CSP CRR phenotypes have been described: vk210 [GDRA(A/D)GQPA] [16] and vk247 [ANGA(G/D)(N/D)QPG] [17]. In southern Mexico, Pvs25/28 ookinete surface protein alleles are partially linked to vk210 and vk247 CSP phenotypes, and associated to the parasite infectivity to the local mosquito vectors Anopheles pseudopuntipennis and Anopheles albimanus[18, 19].
The two P. vivax CSP main CRR genotypes have been reported at variable frequencies in most malaria-endemic regions, including southern Mexico and Peru [20–28]. The vk210 genotype predominates in many affected areas of Brazil and vk247 has been limited to some regions in mixed csp genotype infections [14, 29, 30]. Recently, the predominance of vk247 parasites and a high level of genetic polymorphism of the csp gene were documented on the Pacific Ocean coast of Western Colombia [31]. In the present study, different molecular and bio-informatics approaches were used to analyse P. vivax csp diversity and gene polymorphism of isolates from the Pacific Ocean coast of Mexico, Nicaragua and Peru. Their comparison with other isolates from Latin America (LA) and the rest of the world (OA) identified common genetic features in a group of vk210 LA isolates and the recent introduction and/or dispersal of novel vk210 and vk247 genotypes mainly in the Southern Mexican region.
Methods
Blood sample collection and geographic origin
Following informed consent, infected blood samples were obtained from symptomatic patients living in Tapachula municipality and neighbouring villages in Southern Chiapas, Mexico. Patients were diagnosed by microscopy, using Giemsa-stained thick and thin blood smears. Two groups of samples were collected; one batch of 70 infected whole-blood samples during 2002–2005 and a second batch of 400 infected capillary blood samples dried on filter paper (Whatman No. 2) were collected between 2006 and 2008. Another three samples were collected in other locations in Southern Chiapas [32, 33]. Two other samples were obtained from San Pedro Pochutla, Oaxaca, south-west Mexico in 2007. Other infected dried bloods were collected from affected areas of Nicaragua and Peru. Thirty-seven blood samples were collected in Chinandega, Department of Nicaragua (five from Chinandega town (departmental seat), nine samples from Chichigalpa, 15 from El Viejo, two from Posoltega and six from El Realejo municipalities) from August 2006 to December 2007. The communities are located within 25 km of each other. Nine infected blood samples were obtained from Sullana, Piura, Peru in May, 2008 (Figure 1).

Study sites. (a) Pochutla municipality, Oaxaca, Mexico; (b) Mapastepec; (c) Tapachula municipality and surrounding villages, Chiapas, Mexico; (d) Chinandega department, Nicaragua; (e) Sullana, Piura, Peru.
Polymerase chain reaction and restriction fragment length polymorphism (PCR-RFLP) analysis
PCR-RFLP was used to examine the csp polymorphism of all P. vivax samples. DNA from blood samples was extracted using the QIAamp® DNA mini kit, as per the manufacturer instructions (Qiagen, CA, USA). Briefly, the csp gene amplification was standardized with primers CSPf1 (5′-cgc act gcg ggc aca atg tag atc-3′) and CSPr1 (3′-ggt tac act gca tgg agt cc-5′). The PCR reaction mix was prepared as follows: GoTaqFlexi 1X Buffer, 2 mM magnesium chloride, 0.2 mM dNTPs (Invitrogen, 10297018 Carlsbad, CA, USA), 10 μM each oligonucleotide, 5 U GoTaq® Flexi DNA polymerase (Promega, Madison WI, USA) and ~100 ng of genomic DNA in a 20 μl final volume. PCR conditions were 95°C for 3 min, followed by 35 cycles of 95°C for 30 sec, 58°C for 30 sec, and 72°C for 2 min, and a final extension at 72°C for 10 min. Samples collected as blood spots on filter paper were processed using an alternative nested PCR with primers CSP-f2 (Region I) (5′-aat aag ctg aaa caa cca-3′) and Pv9a (next to region II) (5′-gcc aac ggt agc tct aac ttt- 3′) [24] for the second round amplification. An aliquot (2 μl) of the first amplification sample was added to the second PCR reaction. PCR conditions for the second amplification were the following: first cycle at 95°C for 3 min, followed by 35 cycles each at 95°C for 30 sec, then 57°C (for first PCR) for 30 sec and 72°C for 1 min, followed by a final extension at 72°C for 10 min, using the thermocycler MyCycler (Biorad, Hercules, USA).
The PCR products were analysed using differential restriction enzyme digestion [34]. Enzymes Alu I (New England Biolabs, Beverly, MA, USA) and BstI ( Promega, Madison, WI, USA) cut only the csp repeat Vk247 and Vk210, respectively. DNA fragments were resolved in a 1.5% agarose gel, visualized in a UV-transilluminator (LMS-20E a 254 NM and photographed with a Digital photo-documentation system BioDoc-it™ (UVPInc, Upland, California, USA).
Cloning and sequencing
Plasmodium vivax isolates producing different vk210 and vk247 PCR molecular size amplified products and RFLP patterns from different geographic origins were selected for cloning and sequencing to analyse their nucleotide and amino acid sequences. The first PCR amplification yielded sufficient product to carry out the cloning using the TOPO TA cloning® kit with pCR®2.1-TOPO® according to manufacturer instructions (Invitrogen, Carlsbad, CA, USA). Some of the samples collected on filter paper were not amplified using this methodology (csp genes from isolates Mxch10, Mxch11, Mxch13 and Mxch14, MxO and those from Nicaragua). A modified nested PCR amplification was carried out before the ligation reaction using the following modification: 95°C for 3 min, 10 cycles of 95°C for 30 sec, 50°C for 30 sec and 72°C for 2 min followed by 25 cycles of 95°C for 30 sec, 57°C for 30 sec and 72°C for 2 min and, followed by a final extension of 72°C for 10 min. Modification of blunt-ended PCR products was carried out using the Qiagen A-Addition kit as per manufacturer instructions (Qiagen GmbH, Hilden, Germany).
At least two or three different colonies containing the cloned product of each csp allele were sequenced from both directions, using the primers described above. The sequences were obtained using a sequencer ABI PRISM® 3100 Genetic Analyzer at the National Institute for Public Health and the Perkin Elmer/Applied Biosystems 3730 of Biotechnology Institute-UNAM in Cuernavaca, Morelos, Mexico. Some short amplified fragments had few repeat units and were discarded as they could be the result of polymerase failure to amplify the whole repeat fragment.
Data analysis
The RFLP patterns were ordered by the vk genotype accompanied by a lowercase letter (a, b, c, d, etc.). The quality of the electropherogram of each DNA sequence was analysed with both Phred-Phrap-Consed [35] and by manual inspection. The resulting nucleotide sequences were aligned using the six-frame and CLUSTALW (v.3.2) programs, available at the San Diego Supercomputer Center worldwide servers [36]. The allelic consensus sequences were prepared using upstream and downstream nucleotide sequences, both overlapping beyond the central repeat fragment (from RI to RII domains and the other way round). Their nucleotide and amino acid polymorphism were compared to sequences from isolates within and among their geographic origin and to other P. vivax csp nucleotide sequences available in databases. Comparison was carried out using the BLASTN and BLASTX v.2.2.17 program [37] available at the NCBI [38]. The complete gene sequences obtained were submitted to the NCBI [Gen Bank accessions: JQ511263.1-JQ511286.1].
The evolutionary and genetic CSP relationship was investigated by carrying out an analysis of the mismatch distribution of the CRR and a phylogenetic relationship of the 3′ terminal nucleotide sequence. The sequences obtained in the present study along with other vk210 and vk247 alleles from America and other geographic origin obtained from the Gen Bank TM (NCBI) were analysed. The csp-vk210 group sequences were from El Salvador (XM_001613018.1), Honduras (DQ156131.1), Colombia (GU339072.1, GU339085.1), Brazil (M11926.1, DQ156132.1, DQ978648.1, DQ978651.1, DQ978656.1- DQ978658.1, DQ978675.1, EU401924.1, FJ845386.1, FJ845388.1, FJ845389.1, FJ845390.1), Iran (AY367278.1, AY367286.1, AY443706.1, AY443720.1, AY632256.1, AY632287.1, AY632320.1, AY632325.1), India (EU401926.1, FJ491100.1, FJ491117.1, FJ491119.1, FJ491128.1, DQ156140.1), Thailand (M28746.1), South Korea (DQ859754.1, DQ859760.1, DQ859768.1), North Korea (M20670.1, AF316580.1, AF316581.1), Korea (DQ156137.1), Indonesia (DQ156135.1, EU401927.1), Philippines (U08980.1), Solomon (U08982.1), New Guinea (EU401925), Gabon (U09737), Vietnam (EU401929) and Mauritania (AY674050.1).
The csp-vk247 group sequences were from Thailand (M28745.1), Brazil (M69062.1), North Korea (EU401928.1), Colombia (GU339063.1- GU339065.1, GU339067.1- GU339071.1, GU339075.1, GU339076.1, GU339078.1, GU339079.1, GU339082.1, GU339084.1, JN689932.1, JN689933.1), Iran (AY632316.1, AY632330.1, AY632294.1, AY632298.2, AY632299.2, AY443710.2, AY632330.1), Bangladesh (AY843440.1) and Vietnam (DQ156141.1; EU401930.1).
As vk- references, csp sequence of Plasmodium simium I and II isolated from Brazil were incorporated in the analysis: type vk247 (L05069.1) and type vk210 (L05068.1), respectively.
Mismatch distribution analysis of the CRR sequence
A mismatch distribution analysis [13] was carried out in isolates sequenced here, compared to others from LA and OA regions. For an automated identification of vk210 or vk247 repeat allotypes [39] within each csp nucleotide sequence, program HMMER3 were used [40]. First, a HMM profile was created with hmmbuild using as input the aligned unique repeat nucleotide (rat) sequences described by Patil et al.[14] or from vk247 rat sequences identified by eye. The hmm profile was then validated by searching rat s in Patil et al.[14] dataset and the Mexican and Peruvian VK247 alignments. A search for rats in the GenBank sequences was performed with hmmsearch using a per domain inclusion threshold (−−incdomE) of 1.0E-05. The “-A” option was selected as output to obtain the target rat sequence as multiple alignment in Stockholm format, which was transformed to FASTA file. Individual rat sequences were analysed by eye to confirm that were bona fide CRR units [14, 16, 17, 31, 41]. For pairwise comparisons between rats, all sequence comparisons within a CRR sequence were performed with USEARCH version 5.0 [42] with the “--nousort” option. To obtain the mismatch count for the whole CRR sequence, the global alignment option (“--global”) was enabled. Mismatch counts for each pairwise comparisons within each isolate were retrieved from tabular output specified by the “--blast6out” option. The mean proportion of nucleotide difference for all pairwise comparisons among CRR units (p); the proportion of all pairwise comparisons in which p was zero (prop. 0); the proportion of all pairwise comparisons in which p was greater than 0.25 (prop. >0.25) the skewness (Pearson asymmetry coefficient) and the variance of the distribution of p for all pairwise comparisons were calculated [13, 14]. Additionally, the variance of the p distribution values for all pairwise comparisons (σ (p)) was also calculated. Different combinations of MD parameters were plotted to search for clusters. Also, the five mismatch distribution parameters were normalized to its respective highest value and subjected to hierarchical clustering (complete linkage) using uncentred absolute correlation as similarity metric with Cluster 3.0 [43] and visualized in Java TreeView 1.1.6.r2.
The frequency of P. vivax unique repeat types at amino acid (RAT) and nucleotide level (rat) verified by local BLASTn searches were calculated for each group vk210 and vk247 and by geographic region: LA versus OA origins. Two-sample t-test for continuous variables and the Pearson chi-squared test for proportions were used to establish significant differences between the groups vk210 vs vk247 and within vk210 subgroups: C1 vs C2 and C1A vs C1B, and for CSP RATs and nucleotide sequence usage (rats) between LA vs OA isolates, respectively (α = 0.05).
Phylogenetic analysis
To search similarities at the 3′ terminal variable region of the csp gene, an alignment of the nucleotide sequences of the isolates indicated in the previous section, comprising the sequence between last 3′ CRR unit and domain RII, was used to construct a phylogenetic tree using the neighbour-joining method with 1,000 bootstrap replications [44] and conducted in MEGA4 [45].
Ethical clearance
This study was approved by the Ethical Committee of the Mexican National Institute of Public Health. Samples from Nicaragua and Peru were collected as part of projects approved by the Nicaraguan Ministry of Health and PAHO and the Institutional Review Board of NAMRU-6 in Peru, respectively.
Results
DNA amplification and PCR-RFLP analysis of Plasmodium vivax csp gene
A total of 521 samples were analysed. Seven different amplified fragment sizes and/or RFLP patterns were identified in 267 vk210 genes, while only one amplified fragment size and RFLP pattern were identified in 288 vk247 genes.
Samples from Southern Mexico (Chiapas and Oaxaca)
From 475 samples, four different vk210 subtypes were named as a, b, c and d (Tables 1 and 2). Subtype vk210a was present in 208 single and 28 mixed genotype infections; it produced an amplified gene fragment of ~700 bp and a RFLP pattern of ~135/130 and 100 bp. The other subtypes were less frequent, subtype vk210b was present in seven single and six mixed csp genotype infections, and these had a smaller molecular weight to vk210a parasites and had a RFLP pattern of ~190/130 bp. Subtype Vk210c was present in eight mixed genotype infections, had an amplified nucleotide fragment of ~500 bp and digestion products of ~200/130 bp (Figure 2A and 2C). The subtype vk210d was detected in only one isolate (in 2006); it presented an amplified fragment of molecular size similar as vk210a (~700 bp) but a different RFLP pattern of ~170/130 bp (Figure 2C). All vk247 isolates from 215 single and 40 mixed genotype infections from Southern Chiapas and the two samples from Oaxaca produced an amplified product of ~710 bp (slightly larger than the vk210a fragment size) and the Bst I digestion generated products of ~220 bp (Figure 2A and 2D). The csp genotype frequency of vk210 and vk247 in single or mixed genotype infections in samples collected during the 2006–2008 period is presented in Table 2. A high percentage of single genotype infections and similar frequency for both vk210 and vk247 genotypes were observed, but a higher frequency of vk210a compared to vk247 infections was detected throughout 2008.

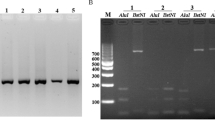
Molecular size variation and RFLP patterns of the csp gene of Plasmodium vivax isolates from the central Pacific Ocean coast region of the Americas A and B, PCR amplified product size. A) Mexican samples; ~700 pb -vk210a (lane 1) and ~700 pb -vk210b (lane 2) had the same molecular size. Lane 3 and 4, show mixed genotype infections: (vk210b and ~500 pb- vk210c) and (>700 pb-vk247 and vk210c), respectively. B) Nicaraguan and Mexican samples; fragments of ~700 bp (lanes 4, 5 and 8; Nir1), ~650 bp (lanes 1, 3 and 9; Nichn), ~600 bp (lanes 2 and 7) and vk210a from Mexico (lane 6) are shown.RFLP patterns: C) Mexican samples; vk210d (lane 1–2), vk210b ( Lane 3–4), vk210c +vk247 (lane 5–6), and vk247+vk210a (lane 7–8). D) Mexican and Peruvian samples; vk210a (lanes 1–4, two samples), vk247 (lanes 5–8, two samples: Mexican and Peruvian); and mixed infection Vk210a-vk247 (lane 9–10). E) Nicaraguan samples; vk210a like - Nir1 isolate (lane 1 and 2), vk210e - Nichn isolate (lane 3–4), vk210f - Nir2 isolate (lane 5 and 6) are shown. C, D and E: odd numbers indicate Alu1 digestion and round numbers indicate BstI digestion. 1% agarose gels and ethidium bromide for staining were used. M, marker 1 kb.
Samples from Nicaragua and Peru
All 37 samples obtained in Nicaragua were single vk210 infections. Three subtypes, a, e and f were identified by the molecular size of the amplified product and RFLP Alu I digestion patterns: (Table 1, Figure 2B). Twenty samples (vk210a) had similar molecular size and RFLP pattern as previously indicated for Mexican isolates, in 12 (vk210e) samples the amplified fragment was of ~650 bp of RFLP pattern ~130 and 105/100 bp and in five samples (vk210f) a shorter fragment of ~600 bp was amplified with RFLP pattern of 135/130 and 100 bp. All of them had slightly different RFLP digestion patterns (Figure 2E). These three RFLP patterns were distributed in all sites at Chinandega department. The nine samples from Peru were vk247, and all had a conserved molecular size and RFLP pattern similar to the Mexican vk247 isolates (Table 1, Figure 2D).
Csp nucleotide sequence analysis
From 20 P. vivax-infected blood samples, 25 csp nucleotide sequences were obtained (Table 1). Sixteen sequences comprised 75 codons at the amino end, the CRR through RII domain. Other nine sequences comprised only from RI to RII domains inclusive.
Pre repeat 5 ′terminal end polymorphism
Gene sequences were compared between 225 nucleotides of the conserved amino region, including region I of three vk210a (isolates Mxch: 1, 2, 3), three vk210b (isolates Mxch: 4, 5, 7), four vk210c (isolates Mxch: 3, 5, 8, 9) and five Mexican vk247 csp (Mxch: 6, 7, 8, 9, 12) and one Peruvian (Peru I). All vk210 and vk247 had identical nucleotide sequence, except for codon 38, which was Asn (AAC) and Gly (GGC) in vk210 and vk247 samples, respectively.
Polymorphism and mismatch distribution (MD) of the CRR
CRR polymorphism
Sixteen csp vk210 consensus sequences were analysed. The PCR-RFLP subtype vk210a produced three different CRR nucleotide sequences named as aI, aII and aIII. Csp subtype vk210aI (Mxch:1, 2, 3) had 20 repeat units; their sequence was identical to that of the Honduras III strain (DQ156131.1), and compared to the Sal I sequence (XM001613018.1) [46] they present a non-synonymous change G→C at the eighth nucleotide codon of the 19th repeat unit (Figure 3). But the nucleotide sequence of isolate vk210aII (Mxch13) was identical to a previously described Brazilian strain (DQ978648.1) [41]. The csp gene of subtypes vk210b, c and d were different to that of Sal I and South American parasites. All one single (Mxch4) and two mixed infections (Mxch5 and Mxch7) vk210b sequences examined, had 18 repeats, and were identical among them (Figure 3A). The vk210c sequences (isolates Mxch: 3, 5, 8, 9) had 12 repeats and match to the first 10 and the last two repeat units of the 18 repeats of vk210b parasites. The vk210d (isolate Mxch14) had 18 repeats and differs from vk210b by only one nucleotide change at the first codon of the ninth repeat (C→A) (Figure 3).

Comparison of amino acid and nucleotide sequences of the Plasmodium vivax circumsporozoite CRR vk210. The sample code and the Gen Bank accession numbers are indicated at top of each repeat block and those from other isolates worldwide are within parenthesis. The amino acid residues are indicated by capital letters, and those corresponding to the nucleotide sequence are underlined; the amino acid substitutions are indicated underneath. On the left side the repeat units are numbered. Dots indicate absence of nucleotide variation. The non-synonymous nucleotide changes are indicated by upper-case letters. As reference for tandem repeat; Honduras III and Belem strains. The dashed lines indicate missing repeats in the vk210c sequences compared to vk210b sequence.
The csp of three different Nicaraguan parasites examined had variable number of repeats and nucleotide substitutions. The csp vk210aIII (isolate Nir1), vk210e (isolate Nichn) and vk210f (isolate Nir2 and Nichg) had 22, 19 and 18, CRR units, respectively (Table 1, Figure 3). The CRR of vk210aIII had a 96% nucleotide identity to the 23 repeats of Brazil I strain (DQ156132.1), and nucleotide variations restricted to the five 3′ terminal repeat units. The csp vk210e and vk210f were also 97% similar to genes previously described in another Brazilian isolates: O10 CSP (DQ978656.1) that had 19 CRR units and O4 CSP (DQ978651.1) that had 18 CRR units, respectively [41]. In these, nucleotide changes were present in most CRR units.
The nine vk247 nucleotide sequences described here had 19 CRR units that were conserved among them. The CRR nucleotide sequences obtained in this study were identical to that of one Colombian (JN689931.1) and one Iranian (AY632330.1) sequences; but the analysis revealed two other different sequences in isolates from Brazil (M69062.1) and Thailand (M28745.1) (Figure 4).

Comparison of amino acid and nucleotide sequences of the Plasmodium vivax circumsporozoite CRR vk247. The sample code and the Gen Bank accession numbers are indicated at top of each repeat block and those from other isolates worldwide are within parenthesis. The amino acid residues are indicated by capital letters, and those corresponding to the nucleotide sequence are underlined; the amino acid substitutions are indicated underneath. On the left side the repeat units are numbered. Dots indicate absence of nucleotide variation. The non-synonymous nucleotide changes are indicated by upper-case letters. Sequences from isolates Mxch6 and PeruI are shown, which are identical to Iranian (AY632330.1) and Colombian (JN689931.1) isolates and different from sequences of Thailand and Brazilian isolates.
MD analysis of the CRR
The MD analysis of worldwide vk210 and vk247 isolates showed low average proportion of mismatches (p) (average 0.086 ± 0.01, range 0.05 - 0.15). According to Hughes [13] negative skew values observed for a distinctive group of vk210 LA isolates, including some from Mexico and Nicaragua (Additional files 1 and 2), could be suggestive of ancient duplication events, but are in contradiction with their low average p, low prop. >0.25 and high prop. 0. It is reasoned that for very recent duplication events, the σ of p distribution (σ (p)) would be low. Accordingly, there were a considerable number of LA isolates having negative skew values but low average p, indicating very few mutations.
At least three main clusters were identified by comparing different MD parameters, which were more clearly resolved by their average p and its σ (p) as an alternative measure of MD (Figure 5). Similar clustering was obtained by the hierarchical clustering of normalized MD parameters (Figure 6). Regardless of their geographical origin, all vk247 CRR sequences formed an unique homogeneous cluster (Figures 5 and 6), displaying higher p, skew, prop. >0.25 and σ (p) values than most vk210 CRR sequences (Table 3A, Additional file 2). The worldwide vk247 isolates grouped with few vk210 OA isolates, mainly recombinant sequences, except one from Thailand, while vk210 CRR sequences present a heterogeneous clustering with highly correlated sub-clusters: most isolates collected in LA showed lower MD values (except prop. 0) (C1) than those from OA (C2) (Figure 5, Table 3B). C1 was further separated in two groups by their σ (p) (Figure 5); LA/C1A-vk210 had the lowest σ (p) included mainly Mesoamerican isolates (Sal I, Honduras III, Mxch3 (vk210aI), Nichn and Nir1 (vk210aIII and e respectively)) but also had four from South America (three from Brazil and one from Colombia) and one from Africa (Gabon isolate). All of the isolates in this group had prop. >0.25 equal to cero; many had negative skewness, the lowest average p and σ (p); LA/sC1B-vk210 includes Mxch13 (vk210aII), Nichg (vk210f) and other isolates from South America (Figure 5, Table 3C). However, Mexican isolates vk210b with and c sequences cluster with OA isolates in C2 (Figure 5) and vk210d was marginal between C1 and C2 groups. The hierarchical clustering of normalized MD parameters not only agreed to a great extent to the groups formed by average p vs σ (p) but also to the carboxyl end genotype (Figure 6). The vk210b and d, two Brazilian isolates and Plasmodium simium I cluster with OA vk210 isolates. The vk210c and vk210aIII sequences clustered separately with other OA group and have the highest prop. 0 values (Figures 5 and 6). In between, two other groups comprised mainly of LA isolates, consistently clustered in C1A and C1B, this sequences of LA parasites do not express the ANKKAEDA domain at carboxyl end, except isolates vk210b, c and d from Southern Mexico as other OA isolates (Figure 6).

Mismatch distribution of Plasmodium vivax circumsporozoite central repeat sequence: average p versus σ (p). Average p and its variance (σ (p)) clearly cluster central sequences and separate vk247 from vk210 sequences. One worldwide group formed by vk247 parasites with the highest average p and σ (p). Latin American (LA) isolates had a csp central repeat of lower average p than sequences from outside America (OA; other geographic origins). LA isolates were further separated in two groups by their p variance, one with lower σ (p) (C1A; Sal I, Honduras III, Mxch3-vk210aI, Nichn, Nir1-vk210aIII) than the other (C1B; Mxch13-vk210aII, Nichg). The latter was separated by the average p from the OA isolates (C2; Mxch4-vk210b, Mxch3-vk210c). C2 is composed by a tight group and others of higher average p and σ (p) were scattered (see also Additional files 1 and 2). Mxch14-vk210d and P. simium I are marginal between LA and OA isolates. The Mexican vk210b, c and d are indicated by arrows. 1P. simium I and 2P. simium II.

Hierarchical clustering of the CRR mismatch distribution parameters and its relation to vk genotype, geographic origin and carboxyl end. The mismatch distribution parameters (average p, prop. 0, prop. >0.25, skewness and σ (p) were normalized to the highest value and cluster by uncentred absolute correlation. The frequency of sequences by vk genotype and geographic origin are indicated. Samples from this study are indicated in red. Carboxyl type was indicated by the absence or presence of domain ANKKAEDA as type I and II, respectively, and the presence of ANKKAGDA domain as type III. LA, Latin America. OA, outside Latin America; 1 indicate recombinant sequences.
Repeat allotype variation among LA and OA csp sequences
Nine hundred and fifty repeat units of 27- nucleotides each were identified in 53 vk210 isolates, 23 from LA and 30 from OA origin (Additional file 3). Nucleotide sequences were clustered in 47 unique repeat nucleotide sequence allotype (rat) coding for 20 different repeat amino acid allotypes (RAT). Only 14 rats were common in parasites of OA and LA origin, and 29 rats were exclusive for OA versus four rats for LA isolates; from Nicaragua and Brazil. RAT I (GDRADGQPA) and IV (GDRAAGQAA) were more frequent in LA isolates than in the OA, while RATs II (GDRAAGQPA), III (GNGAGGQAA) and V (GDGAAGQPA) were more frequent in OA than LA sequences. RATs I and II represent 87% of the total they were highly variable (Figure 7) and coded by 11 and 13 different nucleotide repeat sequences, respectively. Other RATs were coded by three or fewer nucleotide sequences. In LA isolates, alleles I.1, I.3, I.5 were more frequent and alleles I.2, I.3, II.5 and II.6 were less frequent than in isolates from OA. There were some RATs found in only one isolate: three in LA and 11 in OA sequences. A private RAT XIII (GNRAAGQAA) was expressed in a Nicaraguan isolate. RAT IV (GDRAAGQAA) is coded by an unique rat in most sequences of group C1A (9 of 11) and three other isolates from Korea. RAT IV differs from RAT II (GDRAAGQPA) by a nucleotide change at the eighth codon GCA→ CCA.

Repeat allelic type (RAT) frequency of the circumsporozoite central repeats vk210. A) The 23 Latin American (LA) sequences present seven different RATs; three were exclusive. B) The 30 sequences from outside LA (OA) parasites present 17 RATs and 13 were exclusive. RAT I and IV were more frequent in LA than in OA sequences and RAT II and III were more frequent in OA than LA sequences. I. GDRADGQPA; II. GDRAAGQPA; III. GNGAGGQAA; IV. GDRAAGQAA; V. GDGAAGQPA; VI. GNGAGGQPA (see also Additional file 3A).
Five hundred and twelve repeat units of 27- nucleotide each were obtained from 26 vk247 isolates; 16 from LA and 10 from OA sequences. Fewer amino acid and nucleotide sequences were present than in vk210 origin (Additional file 4). From 30 rats coding for nine RATs only one RAT (I.ANGAGNQPG) was highly variable, coded by 13 different rats. All other RATs were coded by four or fewer nucleotide sequences. Consistently with the MD analysis that showed no geographical clustering for vk247 rats, there were no significant differences in the presence of any RAT between LA and OA. For RAT I, alleles I.2 was more frequent and I.5 was less frequent in LA isolates than in those from OA origin.
Post repeat 3′ terminal end polymorphism and its genetic relationships
The nucleotide sequence variation of the 3′ terminal region was sufficient to generate robust phylogenetic trees (Figures 8 and 9). All Nicaraguan (vk210aIII, e and f) and some Mexican (vk210aIand aII) isolates were similar to the Sal I sequence. The phylogenetic tree confirmed the sequence relationships among Central and South American parasites. These included the Honduras III and Sal I and other from Colombia and Brazil, and all clustered with P. simium in one branch of the tree separated from the origin. In contrast, the Mexican vk210b, c and d types had two synonymous changes in the nucleotide fragment coding for GGNA that flanks the last repeat unit, and it is followed by 57 nucleotides coding the peptide domain ANKKAEDA. The vk210b and c sequences had a triplet of GGNA flanking the carboxyl end, while vk210d had one single copy. These sequences had a Gly residue contiguous to the conserved domain GQGQ. Accordingly, they clustered with vk210 OA isolates having the domain ANKKAEDA on other branches of the phylogenetic tree. The vk210b and c had identical nucleotide sequences to some Korean isolates and vk210d to other OA isolates.

Comparison of amino acid and nucleotide sequences of the circumsporozoite post repeat 3′ terminal region. Key differences linked to vk210 or vk247 CR types are shown. The csp primary structure indicating the interspecies: conserved RI and RII, white boxes. CRR, central repeat region. The variable 3′ terminal region (3′V) between CR region and the conserved domain GQGQ of the csp gene is shown; points indicate absence of nucleotide variation. The dashed lines indicate absence of the fragment. The synonymous and non-synonymous nucleotide changes are indicated by lower and upper case letters respectively. Amino acid substitutions are separated by a slash. Other csp sequences of Sal I strain (X_M001613018), Brazil (DQ978648.1), Belem strain (M11926.1), Korean (DQ156137.1) and Colombian isolates ColS2 (JN689932.1); ColLb (JN689933.1) are indicated as reference. In parenthesis two identical fragments of 57 nucleotide, including last codon of Gly or Ala residue†, are indicated. Nd, no determined. Sm, Southern Mexican isolates. Nicaraguan isolates: Nir1, Nir2, Nichn and Nichg.

Phylogenetic relationship of Plasmodium vivax circumsporozoite 3′ terminal region. The vk210a from Mexico and those from Nicaragua cluster together with other Latin American (LA) isolates in one branch of the tree. However csp sequences vk210 (b and c) cluster with sequences from Korean parasites. As well, the vk210d sequence clustered with parasites from other regions outside LA. All constructions were inferred using the Neighbour-Joining method. The percentages of replicate trees in which the associated taxa clustered together in the bootstrap test (1,000 replicates) are presented next to the branches. Csp sequence analysed in here are in blue.
All csp vk247 nucleotide sequence from samples collected in Mexico and Peru were conserved and clustered together with Iranian and Colombian csp sequences. They present GGNA domains with different synonymous changes than vk210, and display two copies of the domain GGQAAGGNA ANKKAGDAGA plus GA residues flanking the GQGQ conserved domain. These sequences were identical to that of an Iranian isolate (AY632330.1) [47] and closely related to recently reported Colombian parasites (JN689931.1, JN689932.1) [31]). All vk210 and vk247 analysed have a conserved fragment of 38 codons between GQGQ and Region II inclusive, and are identical to that of the Sal I strain. The csp sequences form OA had more variability in their carboxyl end.
Recombinant csp nucleotide sequences
No evidence of recombination events [48] were identified in all sequences vk210 or vk247 from Mexico, Nicaragua and Peru, a feature also found in most sequences analysed worldwide. Residues Asn or Asp (coded by aac or gac respectively) and Gly (coded by ggc) at aminoacid position 38 are consistently distinctive of vk210 and vk247 sequences, respectively. Moreover, the analysis of nucleotide sequences of parasites from around the world indicated that domains GGNA flanking the 3′ terminal CRR of vk210 are coded by alleles ggaggaaac/tgca/g (−ca or −tg), while those of vk247 are coded by a different allele (−tt) (Figure 3). Nucleotide substitutions were used to identify recombination arrangements, e.g., North Korean (EU401928.1) and Indonesian (EU401927.1) present on the amino terminus the residue Gly −38 (vk247 type), followed by 19 vk247 type repeat units and one vk210 type unit (RAT-III) or four vk247 type repeat units and 16 vk210repeat units, respectively. Additionally, the GGNA domain flanking the CRR at the carboxyl terminus resembled vk210 nucleotide sequences coded by allele −tg. This domain is flanked by the ANKKAEDA sequence. A previously described strain from Gabon presents a more complex sequence structure (vk247-vk210-vk247-vk210), had Gly −38, then 19 vk210 repeats followed by one vk247 repeat and the domain GGNA coded by allele −ca.
Discussion
Large human population movements have taken place in ancient and recent times. They have facilitated the spread of infectious diseases, including malaria, throughout the world [49–53]. Within the Americas, current human population movements between countries are very active and comprise migrants from all around the world (passing through South and Mesoamerican regions) into North America [54–56]. This widespread phenomenon could favour the introduction of new malaria strains into new regions with favourable conditions for parasite transmission [11].
Molecular biology, phylogenetic and bioinformatics approaches were integrated to analyse P. vivax csp diversity and the genetic relationships of parasites from the Pacific Ocean coast of Latin America with other isolates of worldwide origin. As a screening approach, the PCR-RFLP results suggested that csp vk210 and vk247 genotypes had a non-homogeneous geographic distribution among the study areas. In Southern Mexico, both genotypes were present as previously reported [28], while in Nicaragua and Peru only vk210 and vk247 parasites were detected, respectively. Parasite sampling in Nicaragua was carried out in five different villages, suggesting a low prevalence or non-existence of csp vk247 parasites, at least during the year-round sampling effort. In agreement with these observations, 17 P. vivax samples collected during 2004 and 2005 in the North, North-West and South of Guatemala [57] and 84 isolates collected between 2010 and 2011 in 20 municipalities of Honduras [58] were all of the vk210 genotype. Similarly, the predominance of a single vk247 genotype in infections reported in a Colombian region close to the Peruvian border [31] concurs with the occurrence of this genotype in Piura, Peru. While no variation was detected among vk247 sequences, genetic variation within csp vk210 genotypes was detected in Southern Mexico and Nicaragua.
The PCR-RFLP analysis provided enough resolution to allow an initial classification of vk210 parasites. Six different vk210 RFLP subtypes were identified; four in Mexico and three in Nicaragua. Subtype vk210a was observed in both countries and was the most frequent, the e and f from Nicaragua were less frequent, and could be local variations from subtype vk210a. The better resolution was given by the nucleotide sequence to vk210aI and aII (detected only in Mexico) and subtype aIII (detected only in Nicaragua). As Alu I restriction enzyme digest the whole CRR, it is suggested the presence of other genotypes likely at the CRR of vk210a[59]. No PCR-RFLP polymorphism was detected in the vk247 isolates.
The integration of the DNA polymorphism examination, MD analysis of the CRR and the phylogenetic trees of the 3′ terminal nucleotide sequences provided a better understanding into the P. vivax csp genetic relationships of Mexican, Nicaraguan and Peruvian parasites with those from other geographic origins. The genetic diversity of the CSP gene was mostly restricted to the CRR and the 3′ terminal variable region, and was due to nucleotide changes and variability in the repeat numbers as reported in other studies. This gene has been useful in molecular epidemiological studies, understanding transmission dynamics and evolutionary relationships [13, 14, 60–63]. However, being under selective pressure, phylogenetic studies using this marker must be cautiously interpreted [15]. Moreover, the repetitive nature of the CRR is not amenable to traditional phylogenetic analysis. To overcome this limitation, we implemented the MD analysis as suggested by Hughes [13]. Although this analysis is not time calibrated, parameters such as average p, prop. 0, prop. >0.25 and σ (p) suggest temporal relations in the occurrence of rat duplication/deletion events. The MD of CRR is in accordance with a relatively recent rat duplication and deletion as a possibly consequence of slipped strand mispairing during DNA replication, as previously suggested for Plasmodium repeat regions [14]. However, differences of the MD parameters enabled the identification of distinctive level of concerted evolution and geographical segregation, strongly supported by hierarchical clustering: 1) based on higher values of average p, σ (p) and prop. >0.25 as well as the carboxyl end, vk247 isolates form a unique cluster with no geographical correlation; 2) vk210 parasites could be subdivided into three distinct clusters (C1 (A and B) and C2) that partially correlated with the RFLP genotype, and where C1 cluster was composed mainly from LA isolates, whereas C2 was composed mainly by OA isolates; 3) phylogenetic similarities of the 3′ terminal region, particularly the allele coding for GGNA domain and the presence or absence of the ANKKAEDA in C2 and C1, respectively, supported MD analysis based sub-clustering; and 4) the difference of the MD parameters between vk247 and vk210 may reflect the different levels of concerted evolution. Considerations that address each of the four points listed above are as follows:
All vk247 sequences cluster into a single group by MD parameters and all of them express at least one copy of the ANKKAGDA domain at the carboxyl end. The vk247 sequences from different distant locations (Oaxaca and Chiapas) of Mexico and Peru are highly conserved and they are identical, both at CRR and the 3′ terminal end, to that of Iranian parasites [47] and highly similar to Colombian parasites (JN689931.1, JN689932.1, [31]). It is noteworthy that a divergence occurs between rats of the CRR of Mexican, Colombian and Iranian parasites compared to those of Brazilian and Thai isolates (Figure 4). While Brazilian and Thai isolates have the same genotype at the 3′ terminal end, they seem to have undergone different differentiation processes at the CRR. Zakeri et al.[47] suggested that an extra copy of the domain GGQAAGGNAANKKAGDAGA at the variable carboxyl end was originated by a nucleotide fragment duplication by replication slippage, which includes five nucleotides of the last repeat up to the A residue that flanks at the GQGQ conserved domain. Such duplication is unique and increased the inter CRR-3′ terminal region variability. Further investigation would be necessary to determine the origin and dispersal of this genotype. The large csp polymorphism in vk247 parasites from the Pacific Ocean coast of Colombia [31] and Iran [47] is probably associated with prolonged and/or more complex transmission patterns of this genotype in these regions. The csp genotype vk247, first reported in Mexico and Peru in 1992 [24] might have been restricted to some locations and probably spread through the foothills of Southern Mexico after the environmental disturbances caused by hurricane Paulina in 1997 [19, 28]. This hypothesis is consistent with microsatellite analysis of parasites collected in this region between 1997–2005, which revealed three populations with significant genetic differentiation and suggested that at least one population (f1 and/or f2) was recently introduced into the foothills [64]. The f1 seem to comprise parasites vk210 detected from 2001 and onwards and f2 comprised the vk247 [19, 64].
Although MD analysis does not directly provide information on the type and position of nucleotide changes, it is outstanding that the main vk210 hierarchical clustering was strongly supported by the 3′ terminal polymorphism analysis. Two LA clusters of similar p distinguished by their (σ (p)) (C1A and C1B respectively) and one mainly OA of higher p (C2). Accordingly, higher numbers of exclusive repeat units were detected in OA than in LA isolates. In fact, LA isolates of cluster C1A (Mxch: 1–3, Nichn, Nir1, Sal I and Honduras III and South American isolates) display very limited rat variation (lower σ (p), prop. >0.25 and skew values) and probable a more recent repeat duplication than C1B that includes Nichg and Mxch13 and other South American isolates, that probably result from an independent introduction to the Americas and a consequent population bottleneck. These results further expand those from a hypo-endemic region of Brazil [14] as result of clonal mode of reproduction especially when a low rate of mixed genotype infection prevails [14, 15]. In Southern Mexico, the systematic blood collection during 2006–2008 revealed more than 90% single csp genotype infections similar to all parasite samples from Nicaraguan and Peru.
The similarity of the 3′ terminal nucleotide sequence among csp vk210 aI and aII of Mexican and all Nicaraguan isolates of C1 are consistent with previously reported LA isolates [14, 31, 41]. In contrast, the MD parameters of the CRR from Mexican sequences vk210d, c and d grouped them closely to OA isolates of higher variation (p and σ) than those from LA. Additionally, these sequences formed a closely related group, alleles vk210b and d, had almost identical CRR, and vk210c allele could likely be the result of a single or various deletion events from vk210b. In agreement with the CRR, the domain ANKKAEDA at the carboxyl end of these isolates indicated a close relationship to Asian parasites. In fact, the phylogenetic tree confirmed that the 3′ terminal region of the csp of these parasites was highly similar to those from various OA locations, including China, North Korea (NK), Philippines [65], Thailand [66], South Korea [67], and Iran [47]. Mitochondrial genome sequencing analysis suggests that these parasites are likely the result of an ancient population expansion in those regions [8].
Successful transmission of the parasite depends basically on the competence of the mosquito vectors present in each geographic region [6, 19, 68]. The recent predominance of both csp genotypes (vk247 and vk210) at similar frequencies in the foothills of Southern Mexico could be explained by the susceptibility of Anopheles pseudopuntipennis, the main vector in this region [19, 28, 64]. This vector is also present in Piura, Peru. While An. Albimanus is likely the main vector on the coastal regions of Mexico and Central America [19, 28, 58, 69]. Interestingly, two P. vivax lineages (Old and New World) were previously proposed, based on mosquito susceptibility and ribosomal gene isoforms [6]. Extensive work only on the ribosomal variation with Colombian and Indian isolates partially contradicted the existence of such dichotomy [7]. It is possible to speculate that at least the isolates classified by Li et al. [6] as “New World” might be related to the C1A cluster, a subgroup of the 18S ribosomal RNA type Sal-I likely adapted to local An. albimanus. But further studies on vector susceptibility and parasite genomics are necessary to clarify of these conflicting results.
According to Hughes [13] the higher mismatch distribution values (average p, σ (p) and prop. >0.25) of all vk247 repeats (including the three divergent CRR types) suggest that they are likely more ancestral than vk210. Furthermore, the vk247 genotype displays high identity at the carboxyl end to the csp sequence reported for Plasmodium cynomolgi (Gen Bank AB524342.1). This contrasts with the higher rat diversity of vk210 than vk247 CRRs, possibly related to the higher worldwide prevalence of vk210 than vk247 [14, 15, 20, 30, 47, 58, 70–74]. In Southern Mexico vk247 became more prevalent from 1997 onwards [19, 28]. However, the possibility that the vk247 rats are genetically more stable (i.e., less prone to replication slippage) than vk210 rats cannot be excluded.
The genome sequence analysis of P. vivax strains, including Sal I, Peru, Brazil I, India VII, North Korea and Mauritania [11, 46], partially agrees with the MD of CRR analysis and the separation of clusters LA and OA isolates, and supported by the presence of higher number of private repeat units in OA isolates than in LA. However, more extensive genome sequencing analysis is required for a better insight into parasite evolution and its dispersal into Meso and South America. The marked genetic structure determined in LA regions, but the microheterogeneity [11, 64, 67, 74, 75] revealed the presence of scenarios epidemiologically diverse. P. vivax genetics and biological characteristics, host immunity and local vectors may contribute to their different patterns of demographic expansion favoured or limited by the eco-epidemiological conditions.
In Southern Mexico the low frequent genotypes vk210b, c and d, first time reported in America, and the lack of vk247 polymorphism suggest, a relatively recent introduction and/or expansion events of these genotypes at least into this region. Between 1970 and 1985, a constant decline in malaria cases, transmission focalization, and likely a limited gene flow between microregions occurred as a result of intensified control measures [76]. Genotype vk210d was detected by an intensify sampling between 2006 and 2008, and although vk210b and c were more prevalent between 2002 and 2005, their prevalence declined in 2006–2008, possible due to a significant reduction of malaria incidence in 2005.
This study contributes to the molecular epidemiology of P. vivax in the Americas, suggesting that the introduction of parasite’s genotypes vk247, vk210b, c and d (OA like) and LA clusters C1A and/or C1B might have occurred at different time points [77, 78]. Whether they were introduced by ancestral migration events or more recent ones (pre- or during colonization of the American continent) [8, 11, 77, 78] is beyond the scope of the present work. A recent study suggest that some Central American P. vivax were the most recently introduced, with TMRCA of 23 Kya, as expected were related to Mid-Eastern, Asian and South American parasites, their TMRCA of 158, ≥199 and 309 Kya were calculated, respectively, and several events of P. vivax introductions into South America were suggested [78]. Similarly, Yalcindag et al.[79] reported that P. falciparum were introduced into South America by various independent events from Africa by the slave trade. It emphasizes the need to extend and intensify genetic studies in the affected regions; a larger sample in more LA locations is required for a better understanding of their genetic epidemiology and evolutionary relationships, their transmission patterns and dispersion. The availability of the genome sequences of many P. vivax isolates [10, 11, 46] and the analysis of the haplotype structure will help to reveal the complex distribution of this parasite in Latin America, as well as the relationships with the worldwide genetic diversity.
Abbreviations
- CSP:
-
Circumsporozoite protein
- csp:
-
Circumsporozoite gene
- CRR:
-
Central repeat region
- MD:
-
Mismatch distribution
- RAT:
-
Repeat allelic amino acid type
- rat:
-
Repeat allelic nucleotide type
- domains RI and RII:
-
Conserved regions I and II at the 5′ and 3′ terminal of the csp gene, respectively
- LA:
-
Latin America
- OA:
-
Outside Latin America.
References
Guerra CA, Howes RE, Patil AP, Gething PW, Van Boeckel TP, Temperley WH, Kabaria CW, Tatem AJ, Manh BH, Elyazar IR, Baird JK, Snow RW, Hay SI: The international limits and population at risk of Plasmodium vivax transmission in 2009. PLoS Negl Trop Dis. 2010, 4: e774-10.1371/journal.pntd.0000774.
De Souza-Neiras WC, De Melo LM, Machado RL: The genetic diversity of Plasmodium vivax–a review. Mem Inst Oswaldo Cruz. 2007, 102: 245-254.
PAHO: Report on the situation of Malaria in the Americas, 2008. 2008, Washington: Pan American Health Organization
WHO: World Malaria report 2010. 2010, Geneva: World Health Organization
CENAVECE: Boletín Epidemiológico 2006–2008. 2006–2008, Mexico: Dirección de Información Epidemiológica
Li J, Collins WE, Wirtz RA, Rathore D, Lal A, McCutchan TF: Geographic subdivision of the range of the malaria parasite Plasmodium vivax. Emerg Infect Dis. 2001, 7: 35-42. 10.3201/eid0701.010105.
Prajapati SK, Joshi H, Shalini S, Patarroyo MA, Suwanarusk R, Kumar A, Sharma SK, Eapen A, Dev V, Bhatt RM, Velecha N, Nosten F, Rizvi MA, Dash AP: Plasmodium vivax lineages: geographical distribution, tandem repeat polymorphism, and phylogenetic relationship. Malar J. 2011, 10: 374-10.1186/1475-2875-10-374.
Miao M, Yang Z, Patch H, Huang Y, Escalante AA, Cui L: Plasmodium vivax populations revisited: mitochondrial genomes of temperate strains in Asia suggest ancient population expansion. BMC Evol Biol. 2012, 12: 22-10.1186/1471-2148-12-22.
Arnott A, Barry AE, Reeder JC: Understanding the population genetics of Plasmodium vivax is essential for malaria control and elimination. Malar J. 2012, 11: 14-10.1186/1475-2875-11-14.
Chan ER, Menard D, David PH, Ratsimbasoa A, Kim S, Chim P, Do C, Witkowski B, Mercereau-Puijalon O, Zimmerman PA, Serre D: Whole Genome Sequencing of Field Isolates Provides Robust Characterization of Genetic Diversity in Plasmodium vivax. PLoS Negl Trop Dis. 2012, 6: e1811-10.1371/journal.pntd.0001811.
Neafsey DE, Galinsky K, Jiang RH, Young L, Sykes SM, Saif S, Gujja S, Goldberg JM, Young S, Zeng Q, Chapman SB, Dash AP, Anvikar AR, Sutton PL, Birren BW, Escalante AA, Barnwell JW, Carlton JM: The malaria parasite Plasmodium vivax exhibits greater genetic diversity than Plasmodium falciparum. Nat Genet. 2012, 44: 1046-1050. 10.1038/ng.2373.
Coppi A, Natarajan R, Pradel G, Bennett BL, James ER, Roggero MA, Corradin G, Persson C, Tewari R, Sinnis P: The malaria circumsporozoite protein has two functional domains, each with distinct roles as sporozoites journey from mosquito to mammalian host. J Exp Med. 2011, 208: 341-356. 10.1084/jem.20101488.
Hughes AL: The evolution of amino acid repeat arrays in Plasmodium and other organisms. J Mol Evol. 2004, 59: 528-535. 10.1007/s00239-004-2645-4.
Patil A, Orjuela-Sanchez P, Da Silva-Nunes M, Ferreira MU: Evolutionary dynamics of the immunodominant repeats of the Plasmodium vivax malaria-vaccine candidate circumsporozoite protein (CSP). Infect Genet Evol. 2010, 10: 298-303. 10.1016/j.meegid.2010.01.006.
Dias S, Wickramarachchi T, Sahabandu I, Escalante AA, Udagama PV: Population genetic structure of the Plasmodium vivax circumsporozoite protein (Pvcsp) in Sri Lanka. Gene. 2013, 518: 381-387. 10.1016/j.gene.2013.01.003.
Arnot DE, Barnwell JW, Tam JP, Nussenzweig V, Nussenzweig RS, Enea V: Circumsporozoite protein of Plasmodium vivax: gene cloning and characterization of the immunodominant epitope. Science. 1985, 230: 815-818. 10.1126/science.2414847.
Rosenberg R, Wirtz RA, Lanar DE, Sattabongkot J, Hall T, Waters AP, Prasittisuk C: Circumsporozoite protein heterogeneity in the human malaria parasite Plasmodium vivax. Science. 1989, 245: 973-976. 10.1126/science.2672336.
Gonzalez-Ceron L, Rodriguez MH, Nettel JC, Villarreal C, Kain KC, Hernandez JE: Differential susceptibilities of Anopheles albimanus and Anopheles pseudopunctipennis to infections with coindigenous Plasmodium vivax variants VK210 and VK247 in Southern Mexico. Infect Immun. 1999, 67: 410-412.
Gonzalez-Ceron L, Alvarado-Delgado A, Martinez-Barnetche J, Rodriguez MH, Ovilla-Munoz M, Perez F, Hernandez-Avila JE, Sandoval MA, Rodriguez Mdel C, Villarreal-Trevino C: Sequence variation of ookinete surface proteins Pvs25 and Pvs28 of Plasmodium vivax isolates from Southern Mexico and their association to local anophelines infectivity. Infect Genet Evol. 2010, 10: 645-654. 10.1016/j.meegid.2010.03.014.
Bonilla JA, Validum L, Cummings R, Palmer CJ: Genetic diversity of Plasmodium vivax Pvcsp and Pvmsp1 in Guyana, South America. Am J Trop Med Hyg. 2006, 75: 830-835.
Cui L, Mascorro CN, Fan Q, Rzomp KA, Khuntirat B, Zhou G, Chen H, Yan G, Sattabongkot J: Genetic diversity and multiple infections of Plasmodium vivax malaria in Western Thailand. Am J Trop Med Hyg. 2003, 68: 613-619.
Gonzalez JM, Hurtado S, Arevalo-Herrera M, Herrera S: Variants of the Plasmodium vivax circumsporozoite protein (VK210 and VK247) in Colombian isolates. Mem Inst Oswaldo Cruz. 2001, 96: 709-712. 10.1590/S0074-02762001000500023.
Kain KC, Keystone J, Franke ED, Lanar DE: Global distribution of a variant of the circumsporozoite gene of Plasmodium vivax. J Infect Dis. 1991, 164: 208-210. 10.1093/infdis/164.1.208.
Kain KC, Wirtz RA, Fernandez I, Franke ED, Rodriguez MH, Lanar DE: Serologic and genetic characterization of Plasmodium vivax from whole blood-impregnated filter paper discs. Am J Trop Med Hyg. 1992, 46: 473-479.
Kim TS, Kim HH, Lee SS, Na BK, Lin K, Cho SH, Kang YJ, Kim DK, Sohn Y, Kim H, Lee HW: Prevalence of Plasmodium vivax VK210 and VK247 subtype in Myanmar. Malar J. 2010, 9: 195-10.1186/1475-2875-9-195.
Moon SU, Lee HW, Kim JY, Na BK, Cho SH, Lin K, Sohn WM, Kim TS: High frequency of genetic diversity of Plasmodium vivax field isolates in Myanmar. Acta Trop. 2009, 109: 30-36. 10.1016/j.actatropica.2008.09.006.
Qari SH, Goldman IF, Povoa MM, Oliveira S, Alpers MP, Lal AA: Wide distribution of the variant form of the human malaria parasite Plasmodium vivax. J Biol Chem. 1991, 266: 16297-16300.
Rodriguez MH, Gonzalez-Ceron L, Hernandez JE, Nettel JA, Villarreal C, Kain KC, Wirtz RA: Different prevalences of Plasmodium vivax phenotypes VK210 and VK247 associated with the distribution of Anopheles albimanus and Anopheles pseudopunctipennis in Mexico. Am J Trop Med Hyg. 2000, 62: 122-127.
Machado RL, De Figuereido Filho AF, Calvosa VS, Figueredo MC, Nascimento JM, Povoa MM: Correlation between Plasmodium vivax variants in Belem, Para State, Brazil and symptoms and clearance of parasitaemia. Braz J Infect Dis. 2003, 7: 175-177. 10.1590/S1413-86702003000300002.
Machado RL, Povoa MM: Distribution of Plasmodium vivax variants (VK210, VK247 and P. vivax-like) in three endemic areas of the Amazon region of Brazil and their correlation with chloroquine treatment. Trans R Soc Trop Med Hyg. 2000, 94: 377-381. 10.1016/S0035-9203(00)90110-X.
Hernandez-Martinez MA, Escalante AA, Arevalo-Herrera M, Herrera S: Antigenic diversity of the Plasmodium vivax circumsporozoite protein in parasite isolates of Western Colombia. Am J Trop Med Hyg. 2011, 84: 51-57. 10.4269/ajtmh.2011.09-0785.
Gonzalez-Ceron L, Rodriguez MH, Chavez-Munguia B, Santillan F, Nettel JA, Hernandez-Avila JE: Plasmodium vivax: impaired escape of Vk210 phenotype ookinetes from the midgut blood bolus of Anopheles pseudopunctipennis. Exp Parasitol. 2007, 115: 59-67. 10.1016/j.exppara.2006.06.001.
Gonzalez-Ceron L, Rodriguez MH, Santillan F, Chavez B, Nettel JA, Hernandez-Avila JE, Kain KC: Plasmodium vivax: ookinete destruction and oocyst development arrest are responsible for Anopheles albimanus resistance to circumsporozoite phenotype VK247 parasites. Exp Parasitol. 2001, 98: 152-161. 10.1006/expr.2001.4626.
Imwong M, Pukrittayakamee S, Gruner AC, Renia L, Letourneur F, Looareesuwan S, White NJ, Snounou G: Practical PCR genotyping protocols for Plasmodium vivax using Pvcsp and Pvmsp1. Malar J. 2005, 4: 20-10.1186/1475-2875-4-20.
Ewing B, Hillier L, Wendl MC, Green P: Base-calling of automated sequencer traces using phred I. Accuracy assessment. Genome Res. 1998, 8: 175-185. 10.1101/gr.8.3.175.
Biology Workbench.http://workbench.sdsc.edu,
Altschul SF, Madden TL, Schaffer AA, Zhang J, Zhang Z, Miller W, Lipman DJ: Gapped BLAST and PSI-BLAST: a new generation of protein database search programs. Nucleic Acids Res. 1997, 25: 3389-3402. 10.1093/nar/25.17.3389.
Basic Local Alignment Search Tool.http://blast.ncbi.nlm.nih.gov/Blast.cgi,
Rich SM, Hudson RR, Ayala FJ: Plasmodium falciparum antigenic diversity: evidence of clonal population structure. Proc Natl Acad Sci USA. 1997, 94: 13040-13045. 10.1073/pnas.94.24.13040.
Finn RD, Clements J, Eddy SR: HMMER web server: interactive sequence similarity searching. Nucleic Acids Res. 2011, 39: W29-W37. 10.1093/nar/gkr367.
Santos-Ciminera PD, Alecrim MG, Roberts DR, Quinnan GV: Molecular epidemiology of Plasmodium vivax in the State of Amazonas, Brazil. Acta Trop. 2007, 102: 38-46. 10.1016/j.actatropica.2007.02.013.
Edgar RC: Search and clustering orders of magnitude faster than BLAST. Bioinformatics. 2010, 26: 2460-2461. 10.1093/bioinformatics/btq461.
Eisen MB, Spellman PT, Brown PO, Botstein D: Cluster analysis and display of genome-wide expression patterns. Proc Natl Acad Sci USA. 1998, 95: 14863-14868. 10.1073/pnas.95.25.14863.
Saitou N, Nei M: The neighbor-joining method: a new method for reconstructing phylogenetic trees. Mol Biol Evol. 1987, 4: 406-425.
Tamura K, Dudley J, Nei M, Kumar S: MEGA4: Molecular Evolutionary Genetics Analysis (MEGA) software version 4.0. Mol Biol Evol. 2007, 24: 1596-1599. 10.1093/molbev/msm092.
Carlton J: The Plasmodium vivax genome sequencing project. Trends Parasitol. 2003, 19: 227-231. 10.1016/S1471-4922(03)00066-7.
Zakeri S, Abouie Mehrizi A, Djadid ND, Snounou G: Circumsporozoite protein gene diversity among temperate and tropical Plasmodium vivax isolates from Iran. Trop Med Int Health. 2006, 11: 729-737. 10.1111/j.1365-3156.2006.01613.x.
Lim CS, Tazi L, Ayala FJ: Plasmodium vivax: recent world expansion and genetic identity to Plasmodium simium. Proc Natl Acad Sci USA. 2005, 102: 15523-15528. 10.1073/pnas.0507413102.
Choi KM, Choi YK, Kang YA, Seo SY, Lee HW, Cho SH, Lee WJ, Rhie HG, Lee HS, Kim JY: Study of the genetic discrimination between imported and autochthonous cases of malaria in South Korea. J Travel Med. 2011, 18: 63-66. 10.1111/j.1708-8305.2010.00473.x.
Choi YK, Choi KM, Park MH, Lee EG, Kim YJ, Lee BC, Cho SH, Rhie HG, Lee HS, Yu JR, Lee JS, Kim TS, Kim JY: Rapid dissemination of newly introduced Plasmodium vivax genotypes in South Korea. Am J Trop Med Hyg. 2010, 82: 426-432. 10.4269/ajtmh.2010.09-0245.
Snowden FM: Emerging and reemerging diseases: a historical perspective. Immunol Rev. 2008, 225: 9-26. 10.1111/j.1600-065X.2008.00677.x.
Vatandoost H, Ashraf H, Lak SH, Mahdi RE, Abai MR, Nazari M: Factors involved in the re-emergence of malaria in borderline of Iran, Armenia, Azerbaijan and Turkey. Southeast Asian J Trop Med Public Health. 2003, 34 (Suppl 2): 6-14.
WHO: World Malaria Report 2008. 2008, Geneva: World Health Organization
First Report of the Continuous Reporting System on International Migration in the Americas.http://www.oecd.org/dataoecd/50/36/48423814.pdf,
Bérubé M: In the Crossfire. 2005, Migration Information Source: Colombia
Jokisch B: Diversity in Migration. 2007, Migration Information Source: Ecuador
Mendizábal-Cabrera R, Padilla N: Diversidad genética de Plasmodium vivax en regiones de alto riesgo de malaria en Guatemala [Genetic Diversity of Plasmodium vivax in areas of high risk of malaria in Guatemala]. 2006, Guatemala: Revista de la Universidad del Valle de Guatemala, 62-79.
Lopez AC, Ortiz A, Coello J, Sosa-Ochoa W, Torres RE, Banegas EI, Jovel I, Fontecha GA: Genetic diversity of Plasmodium vivax and Plasmodium falciparum in Honduras. Malar J. 2012, 11: 391-10.1186/1475-2875-11-391.
Souza-Neiras WC, Storti-Melo LM, Cassiano GC, Couto VS, Couto AA, Soares IS, Carvalho LH, Cunha MG, Povoa MM, Herrera S, Herrera MA, Rossit AR, Carareto CM, Machado RL: Plasmodium vivax circumsporozoite genotypes: a limited variation or new subspecies with major biological consequences?. Malar J. 2010, 9: 178-10.1186/1475-2875-9-178.
Schodel F, Wirtz R, Peterson D, Hughes J, Warren R, Sadoff J, Milich D: Immunity to malaria elicited by hybrid hepatitis B virus core particles carrying circumsporozoite protein epitopes. J Exp Med. 1994, 180: 1037-1046. 10.1084/jem.180.3.1037.
McCutchan TF, Kissinger JC, Touray MG, Rogers MJ, Li J, Sullivan M, Braga EM, Krettli AU, Miller LH: Comparison of circumsporozoite proteins from avian and mammalian malarias: biological and phylogenetic implications. Proc Natl Acad Sci USA. 1996, 93: 11889-11894. 10.1073/pnas.93.21.11889.
Brito CF, Ferreira MU: Molecular markers and genetic diversity of Plasmodium vivax. Mem Inst Oswaldo Cruz. 2011, 106 (Suppl 1): 12-26.
Tripathi V, Gupta D: Evolutionary analysis of circumsporozoite surface protein and merozoite surface protein-1 (CSP and MSP-1) sequences of malaria parasites. Bioinformation. 2011, 6: 320-323. 10.6026/97320630006320.
Joy DA, Gonzalez-Ceron L, Carlton JM, Gueye A, Fay M, McCutchan TF, Su XZ: Local adaptation and vector-mediated population structure in Plasmodium vivax malaria. Mol Biol Evol. 2008, 25: 1245-1252. 10.1093/molbev/msn073.
Mann VH, Huang T, Cheng Q, Saul A: Sequence variation in the circumsporozoite protein gene of Plasmodium vivax appears to be regionally biased. Mol Biochem Parasitol. 1994, 68: 45-52. 10.1016/0166-6851(94)00148-0.
Rongnoparut P, Supsamran N, Sattabongkot J, Suwanabun N, Rosenberg R: Phenotype and genotype diversity in the circumsporozoite proteins of Plasmodium vivax in Thailand. Mol Biochem Parasitol. 1995, 74: 201-210. 10.1016/0166-6851(95)02504-9.
Lim CS, Kim YK, Lee KN, Kim SH, Hoffman KJ, Song KJ, Song JW: The analysis of circumsporozoite-protein gene sequences from South Korean isolates of Plasmodium vivax. Ann Trop Med Parasitol. 2001, 95: 229-235. 10.1080/00034980120053997.
Cohuet A, Harris C, Robert V, Fontenille D: Evolutionary forces on Anopheles: what makes a malaria vector?. Trends Parasitol. 2010, 26: 130-136. 10.1016/j.pt.2009.12.001.
Rodriguez MH: Malaria and dengue vector biology and control in Latin America. En: Bridging Laboratory and Field Research for Genetic Control of Disease Vectors. Wageningen U. R. Frontis vol. 11. Edited by: Knols BGJ, Louis C, Bogers RJ. 2006, Dordrecht, The Netherlands: Springer, 129-141.
Cerutti C, Boulos M, Coutinho AF, Hatab Mdo C, Falqueto A, Rezende HR, Duarte AM, Collins W, Malafronte RS: Epidemiologic aspects of the malaria transmission cycle in an area of very low incidence in Brazil. Malar J. 2007, 6: 33-10.1186/1475-2875-6-33.
Chenet SM, Schneider KA, Villegas L, Escalante AA: Local population structure of Plasmodium: impact on malaria control and elimination. Malar J. 2012, 11: 412-10.1186/1475-2875-11-412.
Raza A, Ghanchi NK, Thaver AM, Jafri S, Beg MA: Genetic diversity of Plasmodium vivax clinical isolates from Southern Pakistan using pvcsp and pvmsp1 genetic markers. Malar J. 2013, 12: 16-10.1186/1475-2875-12-16.
Kho WG, Park YH, Chung JY, Kim JP, Hong ST, Lee WJ, Kim TS, Lee JS: Two new genotypes of Plasmodium vivax circumsporozoite protein found in the Republic of Korea. Korean J Parasitol. 1999, 37: 265-270. 10.3347/kjp.1999.37.4.265.
Kim JR, Imwong M, Nandy A, Chotivanich K, Nontprasert A, Tonomsing N, Maji A, Addy M, Day NP, White NJ, Pukrittayakamee S: Genetic diversity of Plasmodium vivax in Kolkata. India. Malar J. 2006, 5: 71-10.1186/1475-2875-5-71.
Van den Eede P, Van der Auwera G, Delgado C, Huyse T, Soto-Calle VE, Gamboa D, Grande T, Rodriguez H, Llanos A, Anne J, Erhart A, D'Alessandro U: Multilocus genotyping reveals high heterogeneity and strong local population structure of the Plasmodium vivax population in the Peruvian Amazon. Malar J. 2010, 9: 151-10.1186/1475-2875-9-151.
Betanzos AF: La malaria en México. Progresos y desafíos hacia su eliminación [Challenges and progress in the elimination of malaria in Mexico]. Bol Med Hosp Infant Mex. 2011, 68: 159-168.
Carter R: Speculations on the origins of Plasmodium vivax malaria. Trends Parasitol. 2003, 19: 214-219. 10.1016/S1471-4922(03)00070-9.
Taylor JE, Pacheco MA, Bacon DJ, Beg MA, Dantas Machado RL, Fairhurst RM, Herrera S, Kim JY, Menard D, Povoa MM, Villegas L, Mulyanto , Snounou G, Cui L, Zeyrek FY, Escalante AA: The evolutionary history of Plasmodium vivax as inferred from mitochondrial genomes: parasite genetic diversity in the Americas. Mol Biol Evol. 2013, [Epub ahead of print] PMID: 23733143
Yalcindag E, Elguero E, Arnathau C, Durand P, Akiana J, Anderson TJ, Aubouy A, Balloux F, Besnard P, Bogreau H, Carnevale P, D'Alessandro U, Fontenille D, Gamboa D, Jombart T, Le Mire J, Leroy E, Maestre A, Mayxay M, Menard D, Musset L, Newton PN, Nkoghe D, Noya O, Ollomo B, Rogier C, Veron V, Wide A, Zakeri S, Carme B, Legrand E, Chevillon C, Ayala FJ, Renaud F, Prugnolle F: Multiple independent introductions of Plasmodium falciparum in South America. Proc Natl Acad Sci USA. 2012, 76: 511-516.
Acknowledgements
This work was supported by funds of CONACYT salud-2004-119 and CB- 2009-01-131247, and given to L Gonzalez-Ceron. Blood samples from Nicaragua were collected by the project PMSGF/4030-03-22- Grant 195002¸ PNUMA/GEF, OPS/OMS – Ministry of Health of Nicaragua. Blood samples from Peru were collected by the project NMRCD.2008.0004. Samples from Oaxaca were collected by the project PAHO-162 / OPS/OMS and were kindly provided by Dr Jorge Armenta (Sanitary Jurisdiction IV of Oaxaca). Thanks to José A Nettel and Olga L Palomeque for their technical assistance. Special thanks to R Cerritos for his comments that improved the manuscript.
Author information
Authors and Affiliations
Corresponding author
Additional information
Competing interests
The authors declare that they have no competing interests.
Authors’ contributions
LGC conceived, designed, deployed the study, and participated in data analysis, preparation of figures and tables and interpretation, and drafted the manuscript. JMB carried out the mismatch distribution analysis and hierarchical clustering, organized information in figures and tables, interpreted data and drafted the manuscript. CMS and FS participated in RFLP analysis and PCR amplification and cloning, nucleotide sequence alignment and phylogenetic analysis, and drafted the manuscript. AS and OCH participated in study design and sample collection of P. vivax samples from Chinandega, Nicaragua, its storage and drafted the manuscript. MHR participated in data organization and interpretation and drafted the manuscript. BE participated in study design and collection of P. vivax samples from Peru, and drafted the manuscript. All authors read and approved the final manuscript.
Electronic supplementary material
12936_2013_2870_MOESM1_ESM.docx
Additional file 1: A. Mismatch distribution analysis of the P. vivax circumsporozoite: CRR-vk210 from different geographic origins.(DOCX 34 KB)
12936_2013_2870_MOESM2_ESM.docx
Additional file 2: B. Mismatch distribution analysis of the P. vivax circumsporozoite CRR-vk247 from different geographic origins.(DOCX 30 KB)
12936_2013_2870_MOESM3_ESM.docx
Additional file 3: A. Frequency of amino acid and nucleotide repeat types in the circumsporozoite CR sequence, of the P. vivax vk210 from Latin America and outside America.(DOCX 39 KB)
12936_2013_2870_MOESM4_ESM.docx
Additional file 4: B. Frequency of amino acid and nucleotide repeat types in the circumsporozoite CR sequence, of the P. vivax vk247 from Latin America and outside America.(DOCX 37 KB)
Authors’ original submitted files for images
Below are the links to the authors’ original submitted files for images.
Rights and permissions
This article is published under license to BioMed Central Ltd. This is an Open Access article distributed under the terms of the Creative Commons Attribution License (http://creativecommons.org/licenses/by/2.0), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
About this article
Cite this article
González-Cerón, L., Martinez-Barnetche, J., Montero-Solís, C. et al. Molecular epidemiology of Plasmodium vivax in Latin America: polymorphism and evolutionary relationships of the circumsporozoite gene. Malar J 12, 243 (2013). https://doi.org/10.1186/1475-2875-12-243
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/1475-2875-12-243