
Abstract
Background
Pyridine-2,6-bis(thiocarboxylic acid) (pdtc) is a small secreted metabolite that has a high affinity for transition metals, increases iron uptake efficiency by 20% in Pseudomonas stutzeri, has the ability to reduce both soluble and mineral forms of iron, and has antimicrobial activity towards several species of bacteria. Six GenBank sequences code for proteins similar in structure to MoeZ, a P. stutzeri protein necessary for the synthesis of pdtc.
Results
Analysis of sequences similar to P. stutzeri MoeZ revealed that it is a member of a superfamily consisting of related but structurally distinct proteins that are members of pathways involved in the transfer of sulfur-containing moieties to metabolites. Members of this family of enzymes are referred to here as MoeB, MoeBR, MoeZ, and MoeZdR. MoeB, the molybdopterin synthase activating enzyme in the molybdopterin cofactor biosynthesis pathway, is the most characterized protein from this family. Remarkably, lengths of greater than 73% nucleic acid homology ranging from 35 to 486 bp exist between Pseudomonas stutzeri moeZ and genomic sequences found in some Mycobacterium, Mesorhizobium, Pseudomonas, Streptomyces, and cyanobacteria species.
Conclusions
The phylogenetic relationship among moeZ sequences suggests that P. stutzeri may have acquired moeZ through lateral gene transfer from a donor more closely related to mycobacteria and cyanobacteria than to proteobacteria. The importance of this relationship lies in the fact that pdtc, the product of the P. stutzeri pathway that includes moeZ, has an impressive set of capabilities, some of which could make it a potent pathogenicity factor.
Similar content being viewed by others
Background
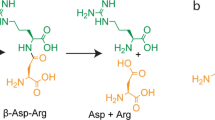
Pyridine-2,6-bis(thiocarboxylic acid) (pdtc) is an iron-regulated metabolite produced by certain pseudomonads that is highly reactive towards metals by virtue of a pair of thiocarboxylate groups and ring nitrogen that combine to form a non-specific, high affinity, tri-dentate ligand (Figure 1)[1, 2]. The formation constants for Co, Cu, and Fe complexes of pdtc were determined to be greater than 1033 [3]. Uptake studies using 59Fe showed that addition of pdtc to the growth medium of two different pdtc-producing pseudomonads increased the efficiency of iron uptake by 20% [1]. The contribution of pdtc to iron uptake may be due to the fact that the copper complex of pdtc (Cu:pdtc) has the ability to reduce both soluble and mineral forms of ferric iron) [1]. Not surprisingly, in light of its high affinity for essential metals, pdtc also exhibits antimicrobial activity [4].

Structure of pdtc.
MoeB, the molybdopterin synthase activating enzyme (MSAE), is part of the well-characterized molybdopterin cofactor (MPT-Mo) synthesis pathway. A dimer of MoeB adenylates the MoaD subunits of molybdopterin synthase that, after the conversion of these AMP moieties to thiocarboxylates by a sulfurtransferase, goes on to convert precursor Z to molybdopterin (MPT) [5, 6]. Molybdopterin synthase is a heterotetrameric protein composed of two units each of MoaD and MoaE. The crystal structure of the E. coli MoeB-MoaD complex clearly shows the interaction between these two proteins and unambiguously confirms the adenylation role of MoeB [7]. After a thiocarboxylate moiety is formed on the terminal glycine of each MoaD subunit, the two sulfur atoms are transferred to precursor Z as sulfhydryl groups in a dithiolene configuration. [8–11]. Molybdenum is added to MPT in subsequent steps to generate MPT-Mo, the active form of the molybdopterin cofactor. Since MPT-Mo is required for nitrate reductase activity [12], the activity of this enzyme can be used as a diagnostic indicator for the presence of the MPT-Mo synthetic pathway [7].
There are a number of genomic sequences deposited in GenBank as MoeB proteins, although only a few have been physiologically or genetically proven to function in MPT-Mo synthesis. Two moeB sequences with evidence for function include a knockout of the open reading frame encoding GenBank accession number BAA35521, which was shown to cause the accumulation of precursor Z in E. coli [13] and accession number Z95150, which was found to be located within a cluster of other MPT-Mo synthesis genes of Mycobacterium tuberculosis [14]. MSAEs from eukaryotic organisms have also been characterized: MOCS3 (GenBank accession number NM_014484) from humans and cnx 5 from Arabidopsis thaliana [15], respectively, have been sequenced, and cnxF from Aspergillus nidulans [16] has been both sequenced and physiologically characterized.
MoeZ is one of several MSAE-like conceptually translated protein sequences identified by the M. tuberculosis genome sequencing group at Sanger Center [14]. Although coding for a putative protein with high sequence similarity to MoeB, moe Z was so named because it had no genetic linkage to other MPT-Mo synthesis genes. Cole et al. [14] state in their GenBank entry (accession number Z95120) that MoeZ is possibly involved in the synthesis of molybdopterin, but there has been no biochemical or physiological characterization of this protein. An open reading frame (ORF-F) identified as a necessary part of the Pseudomonas stutzeri pdt locus coding for the synthesis pathway of pdtc (Figure 1) was found to have 57% identity to M. tuberculosis MoeZ at the amino acid (AA) level. Further analysis revealed that a 521 base pair region of P. stutzeri ORF-F shares 80% homology with M. tuberculosis moe Z [17]. The products of the pdt locus probably function by activating, then sulfurating, a carboxylic acid precursor. The end product of this synthesis is pdtc, a molecule with two thiocarboxylic acid groups that is secreted from the cell.
Recently, data obtained from genome sequencing projects have made it clear that bacteria exchange genetic information by means of transducing bacteriophages, conjugative plasmids, and natural transformation [18] at a much higher rate than previously thought [19]. There seem to be few insurmountable barriers to interspecies gene transfer, with transfers being documented between such diverse organisms as archea and eubacteria [20]. Synechococcus sp. [21] and some P. stutzeri strains [22] are two examples of the many bacterial species known to undergo natural transformation. Of the completely sequenced genomes, all show some level of lateral gene transfer (LGT). For example, 3.3% and 16.6% of the M. tuberculosis and Synechocystis sp. PCC6803 genomes, respectively, are estimated to be composed of sequences acquired by LGT [19].
We report here on the structural, functional, and evolutionary relationships of moeZ and discuss the possibility that the discontinuous distribution of moeZ is the result of a LGT event between unrelated bacteria.
Results and Discussion
Structure
We assembled a set of sequences similar to the product of Pseudomonas stutzeri pdt locus ORF-F (Ps-moeZ) (Table 1). Protein BLAST searches using Ps-MoeZ as the query yielded more than 100 sequences with significant similarity (BLAST E value < 0.01). Interestingly, Ps-MoeZ had higher similarity to seven sequences of mycobacterial, streptomycete, and cyanobacterial origin than to the most similar pseudomonad sequence (Table 2). NA BLAST searches showed that Ps-moeZ had higher homology, as measured by length of homologous overlap, to four of these same sequences than to pseudomonad sequences (Table 2, Figure 2a). We found that Mlo-moeZdR, Mle-moeZ, Mt-moeZ, Pa-moeB, Pa-moeBR, Sc-moeBR, and Syc-moeZ had significant homology (> 35 bp of > 75% identity) to Ps-moeZ (Table 2). To characterize the structure, function, and evolution of Ps-MoeZ, we chose to study these seven genes along with the P. stutzeri sequence. Ec-moeB was included in the study because its function has been extensively characterized. Mt-moeBR, Nos-moeZ, and Syy-moeZ were included for reasons discussed later. BLAST searches of finished and unfinished microbial genomes http://www.ncbi.nlm.nih.gov/Microb_blast/ yielded additional sequences in the genomes of Corynebacterium diphtheriae, Mycobacterium avium, M. bovis, and M. smegmatis with significant NA homology to Ps-moeZ, but these were not included in this study because of their potentially provisional nature, pending completion of the genome sequencing projects. Inclusion of these sequences in the analyses presented here did not change any of our conclusions.

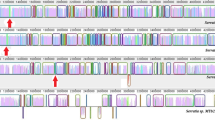
Domain and homology map of MoeB/BR/Z sequences. A. Regions of NA homology between Ps-moeZ and other sequences in this study. Homologous regions between the members of the high-homology group and Ps-moeZ are depicted as solid lines. Regions of Ps-moeZ homology to other strains are shown with dotted lines. The horizontal scale is based on the NA alignments with 0 being the first base of Mle- moeZ. B. Moving average of the ratio of the synonymous to non-synonymous nucleotide substitutions (dS/dN) per codon found among the high homology group using an 18 bp window. Scale is based on the AA alignment with 0 being the first residue of Mle- moeZ. C. Structure of MoeB/BR/Z conceptual proteins. The bars labeled with protein designations indicate the length of each structural class and the inclusion or omission of domains and motifs. ThiF = ThiF family domain; 2X CXXC = MoeB C-terminal domain containing tandem cysteine pairs, MoeBR central domain containing tandem cysteine pairs, or MoeZ central domain with modified regions in place of cysteine pairs; RHOD = Rhodanese-like domain. Locations of CXXC motifs are indicated by vertical arrows. **** = dinucleotide binding motif. (2X CXXC) = modified 2X CXXC domain. Location of E. coli MoeB residue 155 is labeled with substitutions found in each structural class within each bar. PP = Polyproline motif of MoeBR and MoeZ proteins.
Using each protein sequence listed in Table 1 as a query to search the Conserved Domain Databases, we found that each contained an N-terminal domain with high similarity (BLAST E value < 6-30) to the ThiF family domain (Pfam00899) (Table 2, Figure 2c), suggesting that this portion of all the proteins in this study performed a similar function. The ThiF protein adenylates ThiS prior to sulfuration of that protein by a separate sulfurtransferase [23, 24]. Pfam00899 encompasses the nucleotide binding site of ThiF. All 12 sequences in this region have high similarity to the derived consensus sequence GXGXXG(7X)A(SX)GXGXXG(4X)D(9X)R, which defines a nucleotide binding motif [16, 25]. In addition, all sequences have five residues, or conservative substitutions in the case of Pa-MoeB, shown to participate in the adenylation reaction catalyzed by Ec-MoeB (Figure 3) [7]. The fact that such high similarities are retained in the ThiF coding region across three bacterial divisions suggests that this domain functions more or less autonomously. If the ThiF domain interacts only with dinucleotides and had evolved to a high degree prior to the speciation of the strains in our study, then there would be little selective pressure to fix additional modifications. The high degree of AA similarity that all 12 sequences have to the ThiF family domain probably indicates that these proteins, at least in part, perform a similar function, most likely activating a substrate by adenylation.

Alignment of sequences included in this study. The entire lengths of the MoeB and MoeZdR proteins and all except the rhodanese domains of MoeBR and MoeZ proteins are included. The existence rhondanese-like domains (RHOD) are indicated by lines for the sequences that include them. Positions of CXXC motifs are marked with vertical arrows. Positions of Ec-MoeB active sites residues that participate in adenylation reactions are marked with filled circles. Position 155 of Ec-MoeB is highlighted with a star. The polyprotein motif found in MoeBRs and MoeZs is boxed.
All of the predicted amino acid sequences except Ec-MoeB, Mlo-MoeZdR, and Pa-MoeB also have C-terminal similarity (BLAST E value < 1-12) to a rhodanese-like domain (Pfam00581) (Figure 2c, Table 2) [26]. Rhodanese, a thiosulfate:cyanide sulfurtransferase (EC 2.8.1.1), is composed of two domains that are similar in three-dimensional structure but divergent in sequence [27]. All residues contributing to the active site of rhodanese are contained within its C-terminal domain. The cysteine that covalently bonds to the transferred S atom is conserved in all nine of the sequences that contain the rhodanese-like domain included in this study (data not shown). The similarity scores to the rhodanese-like domain, for those sequences that contain it, were lower than the ThiF similarity scores. However, since the rhodanese domain is characterized by conservation in terms of three dimensional structure rather than primary sequence, these lower similarity scores are not surprising.
Examination of the Clustal X-alignment of all the protein sequences revealed significant structural differences (Figure 3). In addition to the ThiF family domain, all sequences except Mlo-MoeZdR also possessed a region similar to the C-terminal half of ThiF. Ec-MoeB, Mt-MoeBR, Pa-MoeB, Pa-MoeBR, and Sc-MoeBR all contain two pairs of cysteine residues, each arranged in a CXXC motif and spaced 68–70 residues apart, like those in ThiF proteins (arrows in Figures 2c and 3). But the Ps-MoeZ sequence, along with Mlo-MoeZdR, Mle-MoeZ, Mt-MoeZ, Nos-MoeZ, Syc-MoeZ, and Syy-MoeZ, lacks these four cysteine residues. In all these sequences except Mlo-MoeZdR, the first CXXC motif is replaced with NYRD. In Mlo-MoeZdR the first CXXC is substituted with GYRD. All these sequences except Mlo-MoeZdR also have a gap in place of the second CXXC. Mlo-MoeZdR ends prior to the position where the second CXXC would occur. This dual CXXC motif, found in MoeB, ThiF, and HesA proteins, forms a metal center with a zinc atom [7, 9]. The presence of all four of these cysteines has been shown to be required for activity in MoeB). [5]. Therefore, the proteins lacking them are unlikely to function as MSAEs. We designated the region containing these two cysteine pairs as 2X CXXC for the purposes of our discussion (Figure 2C).
MoeBR, MoeZdR, and MoeZ proteins can be further differentiated from MoeBs by the inclusion of a polyproline motif immediately following the first CXXC location (boxed area in Figure 3). This polyproline tract is immediately adjacent to a disordered surface loop found in MoeB proteins thought to be involved in protein-protein interactions [7]. The consensus for this motif (RXLYPEPPPP) closely corresponds to a sequence (RSLYPPPPPP) found in rat and human metabotropic 1 glutamate receptors [28, 29]. This motif is reminiscent of protein-protein interaction domains found in some proteins [30]; however, interactions of this type have not been shown to be facilitated by the polyproline motif in rat and human metabotropic 1 glutamate receptors.
An AA substitution found in MoeBRs and MoeZs may alter the substrate specificity of these enzymes. In Ec-MoeB, β sheet 5 (residues 149–155 of Ec-MoeB, Figure 3) forms part of the pocket which accepts the C-terminal portion of MoaD, of which the terminal glycine is subsequently adenylated [7]. Ec-MoeB I155, located at the C-terminal end of this beta loop, is substituted in MoeBR proteins by Y and in MoeZ proteins by F or Y. These bulky substitutions may restrict access of the C-terminus of MoeD-like proteins and may indicate that a different type of substrate is adenylated by MoeBR and MoeZ.
The presence or absence of two features, the two pairs of cysteines in the 2X CXXC domain and the rhodanese-like domain, form the primary basis for differentiation between the four different protein structures among the sequences found in Table 1 (Figure 2c). MoeB, as represented by the functionally characterized Ec-MoeB, consists of an N-terminal ThiF domain followed by a 2X CXXC domain with both CXXC pairs intact and no rhodanese-like domain. MoeBs also lack the polyproline motif and have I at position 155. MoeBR has the ThiF and 2X CXXC domains configured like MoeB but includes an N-terminal rhodanese-like domain, the polyproline motif, and has a Y substitution at position 155 (Ec-MoeB numbering). MoeZ has all the components of MoeBR, but all the cysteines in the CXXC motifs are absent and position 155 contains either F or Y. Finally, MoeZdR (MoeZ, deleted Rhodanese) contains the ThiF domain followed by an N-terminal portion of the 2X CXXC domain that has GYRD substituted for the first CXXC and terminates prior to the second CXXC pair. (MoeZdR probably does not function as a MoeB, BR, or Z protein because of its substantial truncation. It is included in this study only because of its homology to Ps-MoeZ.) Once these structural definitions were defined, we decided to add Nos-MoeZ and Syy-MoeZ to our data set since they were MoeZs that we had not included in our dataset by reason of homology. We also included Mt-MoeBR because we were interested in the relationships between MoeBR and MoeZ sequences that were coresident in the same genome.
The Conserved Domain Database was screened for additional sequences related to the Ps-MoeZ sequence. This search yielded 19 proteins structurally similar to MoeBR and MoeZ proteins. Five of these were eukaryotic sequences of unknown function; five others were eukaryotic MSAE proteins; and one was a ThiF ORF within an operon containing two other thiamin biosynthesis genes in Erwinia amylovora [31]. The remaining eight sequences were of bacterial origin and were already included in our dataset. A similar search using Pa-MoeB as the query yielded 146 sequences with a domain structure similar to MoeB, most of which were entered into the database as MoeB, ThiF, HesA, or ubiquitin-activating enzymes from archaea, bacteria, and eukaryota. In terms of distribution, the MoeB structure (146 sequences) was much more common than the MoeBR structure (12 sequences), while MoeZ was represented by only six sequences in the GenBank non-redundant (nr) database.
Based on our sequence analysis, some moeZ sequences have been misidentified. Syc-MoeZ, Syy-MoeZ, and Nos-MoeZ are identified as a MoeBs in GenBank accession numbers CAA76284, BAA18438, and NP_486946, respectively. The lack of all essential cysteines in both copies of the CXXC motifs suggest that these MoeZs probably do not function like MSAE proteins.
Conserved NA sequences
When the regions of greater than 75% NA homology between each individual NA sequence and Ps-moeZ were plotted, it became apparent that these homologies clustered in defined regions (Figure 2a). Five sequences had a homologous region within the N-terminal half of the ThiF domain; six sequences had a homologous area in the C-terminal half of the same domain; and four sequences showed homology to areas of the 2X CXXC domain. Five sequences with greater than 200 bp of more than 75% NA acid identity to Ps-moeZ (Mle-moeZ, Mt-moeZ, Pa-moeBR, Sc-moeBR, and Syc-moeZ) along with Ps-moeZ were designated the "high homology group" and used in subsequent analyses of the homologous regions.
Using a codon-based alignment of the NA sequences in the high homology group, we analyzed the occurrence of synonymous and nonsynonymous substitutions over the entire coding region. The plot of the ratio of synonymous/nonsynonymous substitutions (dS/dN) identified five regions where the synonymous substitution rate exceeds the nonsynonymous substitution rate (dS / dN > 1), suggesting that homology in these areas was driven by the need for AA conservation (Figure 2b). Analysis of a plot of the degree of identical amino acid residues among the high homology group showed that a high degree of AA similarity existed in these same regions (data not shown). Visual inspection of the NA alignment of the high homology group revealed that substitutions in the areas where dS exceeded dN tended to involve the second and third bases of the codons, confirming the previous suggestion (data not shown). There was also a correlation between the regions of homology to Ps-moeZ and the areas where synonymous substitutions exceeded nonsynonymous substitutions (Figure 2a and 2b). These correlations support the supposition that the NA homologies among the sequences are driven by functional constraints of the proteins and not by the need to maintain secondary nucleic acid structure. Analysis of predicted secondary mRNA structure did not reveal any conserved stems or loops in the regions of NA conservation among the high homology group (data not shown).
Function
The functionality of MoeB, MoeBR, and MoeZ are known. MoeB, exemplified by Ec-MoeB, and MoeBR, exemplified by cnxF of Aspergillis nidulans [16], participate in the synthesis of MPT-Mo as MSAEs. This is a housekeeping function for most organisms. MoeZ, on the other hand, has been shown to be necessary for the synthesis of pdtc, an unusual metabolite. However, since CTN1, a mutant of P. stutzeri, missing a 171 kb genomic fragment that includes moeZ [17], reduces nitrate as does the wild-type (data not shown), it is evident that Ps-moeZ is not necessary for MPT-Mo synthesis.
Both moe BR and moe Z coexist in the M. tuberculosis genome, which exhibits nitrate reductase activity, but moe BR is missing from M. leprae, which has no nitrate reductase activity. The fact that these two separate genes are maintained in M. tuberculosis supports the supposition that they perform different functions. On the other hand, the sequencing of the nitrate-reducing cyanobacteria Synechocystis sp. PCC6803 and Nostoc sp. PCC7120 is complete, but no MSAE sequences have been found. Perhaps MoeZ fulfills MSAE function as part of a modified MPT-Mo pathway in these species.
MoeBR incorporates a rhodanese-like domain, but MoeB does not. Examples of both MoeB (E. coli) and MoeBR (CNXF of A. nidulans) have been characterized as MSAEs, indicating that the C-terminal rhodanese-like domain addition does not inhibit MSAE activity. The presence of a rhodanese domain in MoeBR and MoeZ suggests that these members of the family may perform an additional function compared to MoeB. P. aeruginosa has both a moe B and a moe BR, also suggesting that the products of these two genes have different functions or perform the same function in a slightly different manner. However, there is no physiological evidence for the functional significance of the rhodanese domain in MoeBR. Rhodanese extracts a sulfur from sulphane sulphur containing anions (e.g., thiosulfate), forms a temporary covalent linkage between the S atom and a conserved cysteine residue, then transfers the sulfur to a thiophilic acceptor (e.g., cyanide). Whether sulfur transferase functionality is retained in the rhodanese-like domains of MoeBR and MoeZ and whether these proteins can function to activate a substrate as well as transfer a sulfur-containing moiety is unknown.
Evolution
Because of the definitive structural features shared among MoeZs and the homologies among their coding regions, we decided to investigate the phylogenetic relationships of moeZ sequences. To infer baseline interspecies evolutionary relationships we performed a phylogenetic analysis of the 16S rDNA (SSU) sequences from the seven MoeZ-containing strains included in our study. We obtained sequences for all SSU alleles for each strain from GenBank. Analysis of all the SSU alleles for strains with multiple copies revealed that there was no substantial variation among them. We chose one representative full-length SSU sequence for each strain for our analyses (Table 1). The tree of SSU sequences shows the two proteobacteria (P. stutzeri and M. loti) at one end of the tree with cyanobacteria (Synechococcus sp. and Synechocystis sp.) and mycobacteria (M. leprae and M. tuberculosis) clades branching off from a common node (Figure 4). This tree topology agrees with those that have been previously published [32].

Maximum likelihood, neighbor joining trees of moeZ ThiF domain and associated SSU sequences used in this study. The incongruent placement of P. stutzeri between the trees is highlighted with a dotted line. Confidence estimates are placed near the branches that they apply to.
The moeB/BR/Z sequences under study are a mixture of orthologs and paralogs, and a combined study of their phylogeny is not possible, so we limited our study to moeZ sequences. We constructed the tree from a gapless region of the NA alignment spanning the entire ThiF domain except for the last two bases. The topology of the ThiF domain tree matches the SSU tree except that Ps-moeZ, which was adjacent to M. loti in the SSU tree, appeared between the cyanobacteria and mycobacteria clades (Figure 4). Parsimony, likelihood, and distance methods all resulted in trees with the same topology. Moreover, the placement of Ps-moeZ in the ThiF tree was not changed when full-length MoeZ AA sequences were analyzed using the same methods.
Additional support for the topology of the ThiF domain tree was sought by conducting a manual analysis of the insertions and deletions (indels) in the alignment of full-length moeZ sequences. Ps-moeZ contains five indels in the codon-based NA alignment (Table 3). Of these, one is unique to Ps-moeZ; four are shared with members of the mycobacteria clade; and none are shared with members of the cyanobacteria clade or Mlo-moeZdR. Thus the results of the indel analysis supported the position of Ps-moeZ in the ThiF domain tree.
The incongruence between the SSU and the ThiF domain trees suggests that Ps-moeZ may have been acquired by P. stutzeri strain KC by lateral gene transfer (LGT) from an ancestor more closely related to mycobacteria and cyanobacteria than to proteobacteria. The results of the indel analysis also suggest that Ps-moeZ is more closely related to the mycobacteria moeZ sequences than to the cyanobacteria and proteobacteria moeZ sequences. BLAST homology results also support this close relationship to the mycobacteria.
Although they were not included in the analysis presented here, the homologies of Ps-moeZ to sequences in the unfinished microbial genomes of M. avium, M. bovis, and M. smegmatis were also very high and thus provide additional support for the placement of Ps-moeZ in the ThiF tree and suggest that the direction of LGT was from a mycobacterial species to P. stutzeri.
Comparison of the GC/AT percentages and codon bias profiles of Ps-moeZ to data from other known P. stutzeri strain KC coding regions indicates that acquisition of Ps-moeZ was ancient. The GC/AT percentages for the 1st, 2nd, 3rd, and all three codon bases combined compiled from the seven published coding regions of P. stutzeri strain KC were 66.0%, 46.8%, 72.2%, and 61.7%, respectively. For Ps-moeZ, these values were similar (64.8%, 42.6%, 75.3%, and 60.9%, respectively). A comparison of the Ps-moeZ codon bias table with the one compiled from the seven published strain-KC coding regions did not reveal any divergent trends. Many of these substitutions could be due to GC/AT percentage adjustment to the P. stutzeri genome, but since the donor is unknown and only seven ORFs have been sequenced from strain KC, a definitive analysis is not possible.
It is not unreasonable that a pseudomonad coexisting in the soil with cyanobacteria, mesorhizobium, streptomycete, and free-living mycobacteria could acquire moeZ from one of these species. P. stutzeri has been characterized as having an exceptionally plastic genome with high genotypic diversity within the species [33]. LGT events are thought to contribute to this plasticity and, consistent with this, many P. stutzeri strains also exhibit a natural transformation ability [34]. The fact that incorporation of DNA into the genome through homologous recombination requires a region of homology usually limits LGT mediated by natural transformation to closely related species [34, 35]. The ThiF domains of the high homology group do provide a region of homology suitable for homologous recombination. This homology is shared across a wide range of species and thus could have contributed to a natural transformation-mediated LGT event in P. stutzeri. This mechanism of acquisition is supported by the fact that no phage genes, transposase coding sequences, IS elements, or repeat sequences were found in the 25.7 kb cosmid clone that contains Ps-moeZ [17].
Three other P. stutzeri isolates, obtained from the American Type Culture Collection, have been shown by Southern analysis to lack pdtc synthesis genes. These same strains plus P. aeruginosa PAO1, P. putida mt2, and P. fluorescens F113 were also shown not to produce pdtc [17]. Pdtc production in bacteria other than P. stutzeri strain KC is limited to P. putida strain To8 and one other unclassified pseudomonad strain [36]. P. stutzeri and P. putida are closely related, both belonging to rRNA homology group I [37], so they may be descendants of the original pseudomonad that acquired moeZ by LGT. The relative rarity of pdtc production among pseudomonads, especially the P. stutzeri strains, is consistent with the hypothesis that Ps-moeZ was acquired through LGT by strain KC. Alternatively, a pseudomonad ancestor could have contained the genes for and produced pdtc, but this function could have been lost from most strains. Loss of this function is plausible if pdtc production is a highly specialized function or is only required for survival in extreme or competitive environments.
Conclusions
It is not known what function MoeZ performs in the mycobacteria and cyanbacteria species that contain it. Indications are that in mycobacteria, MSAE function is performed by MoeBR. But in the fully-sequenced cyanobacteria there are only MoeZs and no MSAE, so in these organisms MSAE function may be fulfilled by MoeZ. In either case, MoeZ is essential to pdtc synthesis in P. stutzeri strain KC, and pdtc is a potent and versatile molecule. Its capabilities of enhancing iron uptake and iron reduction make it a good candidate for a pathogenicity factor. The role of MoeZ in the pathogenic mycobacteria and its impact on virulence of these strains should be investigated.
Materials and Methods
Sequences were obtained from the GenBank non-redundant (nr) sequence database at the National Center for Biotechnology Information. BLAST searches were used to obtain the initial set of sequences similar to P. stutzeri strain KC ORF-F [38]. The program, BLAST 2 Sequences, was used to obtain the degrees of similarity and homology among amino acid (AA) and nucleic acid (NA) sequences [39]. Protein domains, as codified by Pfam [26], were identified by searching the Conserved Domain Database using the Domain Architecture Retrieval Tool provided by GenBank.
Protein alignments were generated using the Clustal X program and checked manually [40]. SSU NA sequences were aligned using Clustal W as implemented by the OMIGA package and checked manually [41, 42]. NA alignments of full-length moeZ sequences for indel analysis were aligned manually based on the Clustal X protein sequence alignment.
Codon bias tables and GC/AT percentages were calculated using the Countcodon program available from the Codon Usage Database [43].
Synonymous and non-synonymous substitution rates were calculated by the Synonymous/non-synonymous Analysis Program (SNAP) provided by Los Alamos National Laboratory http://hiv-web.lanl.gov/ using the codon-based NA alignment of the high homology group. Substitution rates were averaged over an 18 codon window to smooth the data for graphing.
Phylogenic trees were inferred using PAUP 4.0 [44]. Three data sets were analyzed: those portions of the NA alignment coding for the ThiF domains of the MoeZ conceptual proteins, the alignment of the complete MoeZ sequences, and the alignment of the full length 16S rDNA (SSU) sequences. Maximum likelihood, parsimony, and distance methods were used for analysis. Confidence estimates were determined by bootstrap analysis based on 1000 replicates.
RNA secondary structure of the ThiF domain coding regions of the sequences in the high homology group was predicted using the GeneBee web server http://www.genebee.msu.su/ using both individual sequences as well as alignments [45].
References
Cortese MS, Paszczynski A, Lewis TA, Borek V, Crawford RL: Metal chelating properties of pyridine-2,6-bis(thiocarboxylic acid) produced by Pseudomonas spp. and the biological activities of the formed complexes. Biometals. 2001, 15: 103-120. 10.1023/A:1015241925322.
Lewis TA, Paszczynski A, Gordon-Wylie SW, Jeedigunta S, Lee C-H, Crawford RL: Carbon tetrachloride dechlorination by the bacterial transition metal chelator pyridine-2,6-bis(thiocarboxylic acid). Environ Sci Technol. 2001, 35: 552-559. 10.1021/es001419s.
Stolworthy JC, Paszczynski A, Korus R, Crawford RL: Metal binding by pyridine-2,6-bis(monothiocarboxylic acid), a biochelator produced by Pseudomonas stutzeri and Pseudomonas putida. Biodegradation. 2001,
Sebat JL, Paszczynski AJ, Cortese MS, Crawford RL: Antimicrobial properties of pyridine-2,6-dithiocarboxylic acid, a metal chelator produced by Pseudomonas spp. Appl Environ Microbiol. 2001, 67: 3934-3942. 10.1128/AEM.67.9.3934-3942.2001.
Leimkuhler S, Wuebbens MM, Rajagopalan KV: Characterization of Escherichia coli MoeB and its involvement in the activation of molybdopterin synthase for the biosynthesis of the molybdenum cofactor. J Biol Chem. 2001, 276: 34695-34701. 10.1074/jbc.M102787200.
Leimkuhler S, Rajagopalan KV: A sulfurtransferase is required in the transfer of cysteine sulfur in the in vitro synthesis of molybdopterin from precursor Z in Escherichia coli. J Biol Chem. 2001, 276: 22024-22031. 10.1074/jbc.M102072200.
Lake MW, Wuebbens MM, Rajagopalan KV, Schindelin H: Mechanism of ubiquitin activation revealed by the structure of a bacterial MoeB-MoaD complex. Nature. 2001, 414: 325-329. 10.1016/S0168-9002(98)00634-2.
Rudolph MJ, Wuebbens MM, Rajagopalan KV, Schindelin H: Crystal structure of molybdopterin synthase and its evolutionary relationship to ubiquitin activation. Nat Struct Biol. 2001, 8: 42-46. 10.1038/83034.
Rajagopalan KV: Biosynthesis and processing of the molybdenum cofactors. Biochem Soc Trans. 1997, 25: 757-761.
Mendel RR: Molybdenum cofactor of higher plants: Biosynthesis and molecular biology. Planta. 1997, 203: 399-405. 10.1007/s004250050206.
Gutzke G, Fischer B, Mendel RR, Schwarz G: Thiocarboxylation of molybdopterin synthase provides evidence for the mechanism of dithiolene formation in metal-binding pterins. J Biol Chem. 2001, 276: 36268-36274. 10.1074/jbc.M105321200.
Richardson DJ, Berks BC, Russell DA, Spiro S, Taylor CJ: Functional, biochemical and genetic diversity of prokaryotic nitrate reductases. Cell Mol Life Sci. 2001, 58: 165-178.
Johnson ME, Rajagopalan KV: Involvement of c hlA, E, M, and N loci in Escherichia coli molybdopterin biosynthesis. J Bacteriol. 1987, 169: 117-125.
Cole ST, Brosch R, Parkhill J, Garnier T, Churcher C, Harris D: Deciphering the biology of Mycobacterium tuberculosis from the complete genome sequence. Nature. 1998, 393: 537-544. 10.1038/31159.
Nieder J, Stallmeyer B, Brinkmann H, Mendel RR: Molybdenum cofactor biosynthesis: Identification of A. thaliana cDNAs homologous to the E. coli sulphotransferase MoeB. In Sulphur Metabolism in Higher Plants. Edited by: Cram WJ, De Kok LJ, Stulen I, Brunold C, Rennenberg H. 1997, Leiden, The Netherlands: Backhuys, 275-277.
Appleyard MV, Sloan J, Kana'n GJ, Heck IS, Kinghorn JR, Unkles SE: The Aspergillus nidulans cnxF gene and its involvement in molybdopterin biosynthesis. Molecular characterization and analysis of in vivo generated mutants. J Biol Chem. 1998, 273: 14869-14876. 10.1074/jbc.273.24.14869.
Lewis TA, Cortese MS, Sebat JL, Green TL, Lee C-H, Crawford RL: A Pseudomonas stutzeri gene cluster encoding biosynthesis of the CCl4-dechlorination agent pyridine-2,6-bis(thiocarboxylic acid). Environ Microbiol. 2000, 2: 407-416. 10.1046/j.1462-2920.2000.00122.x.
Paul JH: Microbial gene transfer: an ecological perspective. J Mol Microbiol Biotechnol. 1999, 1: 45-50.
Ochman H, Lawrence JG, Groisman EA: Lateral gene transfer and the nature of bacterial innovation. Nature. 2000, 405: 299-304. 10.1002/(SICI)1096-9861(19990315)405:3<299::AID-CNE2>3.0.CO;2-6.
Faguy DM, Doolittle WF: Horizontal transfer of catalase-peroxidase genes between archaea and pathogenic bacteria. Trends Genet. 2000, 16: 196-197. 10.1016/S0168-9525(00)02007-2.
Stevens SE, Porter RD: Heterospecific transformation among cyanobacteria. J Bacteriol. 1986, 167: 1074-1076.
Stewart GJ, Sinigalliano CD: Exchange of chromosomal markers by natural transformation between the soil isolate, Pseudomonas stutzeri JM300, and the marine isolate, Pseudomonas stutzeri strain ZoBell. Antonie Van Leeuwenhoek. 1991, 59: 19-25.
Lauhon CT, Kambampati R: The iscS gene in Escherichia coli is required for the biosynthesis of 4-thiouridine, thiamin, and NAD. J Biol Chem. 2000, 275: 20096-20103. 10.1074/jbc.M002680200.
Taylor SV, Kelleher NL, Kinsland C, Chiu HJ, Costello CA, Backstrom AD: Thiamin biosynthesis in Escherichia coli. Identification of ThiS thiocarboxylate as the immediate sulfur donor in the thiazole formation. J Biol Chem. 1998, 273: 16555-16560. 10.1074/jbc.273.26.16555.
Bellamacina CR: The nicotinamide dinucleotide binding motif: a comparison of nucleotide binding proteins. FASEB J. 1996, 10: 1257-1269.
Bateman A, Birney E, Durbin R, Eddy SR, Howe KL, Sonnhammer EL: The Pfam protein families database. Nucleic Acids Res. 2000, 28: 263-266. 10.1093/nar/28.1.263.
Ray WK, Zeng G, Potters MB, Mansuri AM, Larson TJ: Characterization of a 12-kilodalton rhodanese encoded by glpE of Escherichia coli and its interaction with thioredoxin. J Bacteriol. 2000, 182: 2277-2284. 10.1128/JB.182.8.2277-2284.2000.
Desai MA, Burnett JP, Mayne NG, Schoepp DD: Cloning and expression of a human metabotropic glutamate receptor 1 alpha: enhanced coupling on co-transfection with a glutamate transporter. Mol Pharmacol. 1995, 48: 648-657.
Houamed KM, Kuijper JL, Gilbert TL, Haldeman BA, O'Hara PJ, Mulvihill ER: Cloning, expression, and gene structure of a G protein-coupled glutamate receptor from rat brain. Science. 1991, 252: 1318-1321.
Kay BK, Williamson MP, Sudol M: The importance of being proline: The interaction of proline-rich motifs in signaling proteins with their cognate domains. FASEB J. 2000, 14: 231-241.
McGhee GC, Jones AL: Complete nucleotide sequence of ubiquitous plasmid pEA29 from Erwinia amylovora strain Ea88: Gene organization and intraspecies variation. Appl Environ Microbiol. 2000, 66: 4897-4907. 10.1128/AEM.66.11.4897-4907.2000.
Brown JR, Douady CJ, Italia MJ, Marshall WE, Stanhope MJ: Universal trees based on large combined protein sequence data sets. Nat Genet. 2001, 28: 281-285. 10.1038/90129.
Ginard M, Lalucat J, Tummler B, Romling U: Genome organization of Pseudomonas stutzeri and resulting taxonomic and evolutionary considerations. Int J Syst Bacteriol. 1997, 47: 132-143.
Lorenz MG, Sikorski J: The potential for intraspecific horizontal gene exchange by natural genetic transformation: Sexual isolation among genomovars of Pseudomonas stutzeri. Microbiology. 2000, 146: 3081-3090.
Dubnau D: DNA uptake in bacteria. Annu Rev Microbiol. 1999, 53: 217-244. 10.1146/annurev.micro.53.1.217.
Ockels W, Römer A, Budzikiewicz H: An Fe(III) complex of pyridine-2,6-di-(monothiocarboxylic acid) – A novel bacterial metabolic product. Tetrahedron Lett. 1978, 36: 3341-3342. 10.1016/S0040-4039(01)85634-3.
Byng GS, Johnson JL, Whitaker RJ, Gherna RL, Jensen RA: The evolutionary pattern of aromatic amino acid biosynthesis and the emerging phylogeny of pseudomonad bacteria. J Mol Evol. 1983, 19: 272-282.
Altschul SF, Gish W, Miller W, Myers EW, Lipman DJ: Basic local alignment search tool. J Mol Biol. 1990, 215: 403-410. 10.1006/jmbi.1990.9999.
Tatusova TA, Madden TL: BLAST 2 Sequences, a new tool for comparing protein and nucleotide sequences. FEMS Microbiol Lett. 1999, 174: 247-250. 10.1016/S0378-1097(99)00149-4.
Jeanmougin F, Thompson JD, Gouy M, Higgins DG, Gibson TJ: Multiple sequence alignment with Clustal X. Trends Biochem Sci. 1998, 23: 403-405. 10.1016/S0968-0004(98)01285-7.
Thompson JD, Higgins DG, Gibson TJ: Improving the sensitivity of progressive multiple sequence alignment through sequence weighting, position-specific gap penalties and weight matrix choice. Nucleic Acids Res. 1994, 22: 4673-4680.
Kramer JA: Omiga. A PC-based sequence analysis tool. Methods Mol Biol. 2000, 132: 31-45.
Nakamura Y, Gojobori T, Ikemura T: Codon usage tabulated from international DNA sequence databases: status for the year 2000. Nucleic Acids Res. 2000, 28: 292-10.1093/nar/28.1.292.
Swofford DL: PAUP: Phylogenetic analysis using parsimony, version 3.1. Champaign, Ill., Computer program distributed by the Illinois Natural History Survey. 1993
Brodskii LI, Ivanov VV, Kalaidzidis IL, Leontovich AM, Nikolaev VK, Feranchuk SI: GeneBee-NET: An Internet based server for biopolymer structure analysis. Biokhimiia. 1995, 60: 1221-1230.
Acknowledgements
The authors would like to thank Sarah Koerber for her editorial assistance. This material is based on work supported by, or in part by, the U.S. Army Research Laboratory and the U.S. Army Research Office under the contract/grant number DAAD19-00-1-0078.
Author information
Authors and Affiliations
Corresponding author
Additional information
Authors' Contributions
MSC conceived and designed the study, performed the molecular genetic analyses, produced the sequence alignments, and drafted the manuscript. All authors read and approved the final manuscript.
Authors’ original submitted files for images
Below are the links to the authors’ original submitted files for images.
Rights and permissions
This article is published under an open access license. Please check the 'Copyright Information' section either on this page or in the PDF for details of this license and what re-use is permitted. If your intended use exceeds what is permitted by the license or if you are unable to locate the licence and re-use information, please contact the Rights and Permissions team.
About this article
Cite this article
Cortese, M.S., Caplan, A.B. & Crawford, R.L. Structural, functional, and evolutionary analysis of moeZ, a gene encoding an enzyme required for the synthesis of the Pseudomonas metabolite, pyridine-2,6-bis(thiocarboxylic acid). BMC Evol Biol 2, 8 (2002). https://doi.org/10.1186/1471-2148-2-8
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/1471-2148-2-8