
Abstract
Background
Recent studies have demonstrated that autophagy plays a critical role in reducing the drug sensitivity of docetaxel (DTX) therapy. Disulfiram (DSF) has exhibited potent autophagy inducing activity in multiple studies. We hypothesized that DSF co-treatment could sensitize breast cancer cells to DTX therapy via autophagy modulation.
Methods
Breast cancer cells, MCF7, and 4T1, were treated with DTX and DSF, alone and in combination. The effects were analyzed by evaluating cytotoxicity, induction of apoptosis, induction of autophagy, and reactive oxygen species (ROS) generation. In addition, the consequence of autophagy and ROS inhibition on the DTX + DSF mediated cytotoxicity was also evaluated.
Results
Significant synergism in cytotoxicity was observed with DTX + DSF combination in breast cancer cells, MCF7, and 4T1. Hyper induction of ROS and autophagy was also found with the combination treatment. ROS inhibition by N-Acetyl Cysteine (NAC), as well as autophagy inhibition by ATG5 silencing significantly reduced the autophagy level as well as cytotoxicity of the DTX + DSF combination, indicating that the induction of autophagy mediated by high ROS generation played a critical role behind the synergistic cytotoxicity.
Conclusions
This study indicates that DTX + DSF combination therapy can effectively sensitize cancer cells by hyper inducing autophagy through ROS generation and can be developed as a therapeutic strategy for cancer treatment in the future.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Docetaxel (DTX), a microtubule-stabilizing agent, is considered as one of the most potent cancer chemotherapeutic drugs. As a monotherapy, it was found to have high activity against a variety of solid tumors and also against metastatic tumors [1], including breast cancer [2]. However, the development of DTX resistance and treatment failure is common in patients [3, 4]. For improving the anticancer effect and patient survival, different combination therapies of DTX with other chemotherapeutic drugs have been tested with variable success [5,6,7,8]. Previously, most of the study for combination cancer therapy was done using two or more cytotoxic drugs. Recently, the focus has been shifted to non-cytotoxic drug molecules, which can potentiate the activity of chemotherapeutic drugs.
Breast cancer is one of the most prevalent tumors in women. DTX therapy is highly potent and effective at the initial stage of the disease. However, a majority of patients exhibit therapy failure [9]. Many studies have shown that cancer cells induce autophagy as a protective mechanism against DTX treatment that leads to acquired resistance to DTX [10,11,12]. To increase DTX sensitivity, autophagy inhibitors have previously been evaluated [13, 14]; however, their clinical outcome was not very satisfactory [15]. Contrarily, autophagy inducers can also potentiate the cytotoxic efficacy of DTX. In many recent studies, it has been proven that the induction of autophagy can promote cell death [16, 17]. Hence, we aimed to investigate the effect of autophagy induction in the DTX-treated breast cancer cells to establish an alternative treatment strategy.
In the current study, we evaluated the efficacy of disulfiram (DSF) in combination with DTX. DSF, which is used to treat alcoholism, has been shown to have potent autophagy inducing activity through proteasomal inhibition [18, 19]. DSF has also been reported to have synergistic activity and increase the cytotoxicity of many anticancer drugs [20]. In the current study, we have observed that DSF had a high synergistic effect on DTX activity. The mechanism of this synergism and the role of autophagy in the combination effect was also evaluated.
Materials and methods
Cell line and reagents
4T1 cell lines were purchased from American Type Culture Collection (ATCC), and MCF7 Cell lines were purchased from the National Centre for Cell Science (NCCS), Pune. Docetaxel and disulfiram were procured from TCI chemicals (India) Pvt. Ltd. In multiple studies, it has been reported that the anticancer activity of DSF is dependent on the presence of Cu2+ ion [21, 22]. For our study, we have established that 1 µM Cu (Copper (II) chloride dihydrate, Sigma-Aldrich) is non-toxic in our assay system (data available on request), and the same concentration was used with DSF in all the studies.
Cell culture
MCF7 and 4T1 cells were cultured in DMEM media (Gibco) with 10% fetal bovine serum (Himedia), 50 units/ml penicillin, and 50 mg/ml streptomycin (Thermo scientific) at 37 °C, 5% CO2. Trypsin–EDTA solution (0.05%) was used for the detachment of cells.
MTT (3-(4,5-Dimethyl-2-thiazolyl)-2,5-diphenyl-2H-tetrazolium bromide) assay (cell viability assay)
For MTT (3-(4,5-Dimethyl-2-thiazolyl)-2,5-diphenyl-2H-tetrazolium bromide, Sigma) assay, cells were seeded in 96-well plate (4T1 at 1500 cells/well and MCF7 at 2000 cells/well). After 24 h, cells were treated with drug solutions and incubated for 72 h. Then cells were treated with 1 mg/ml MTT in cell culture media for 4 h. After that, the MTT solution was removed, and 200 µl of DMSO was added to dissolve formazan crystals formed in viable cells. Finally, absorbance was measured at 570 nm using ELISA plate reader with a reference wavelength of 630 nm. The percentage of viable cells was calculated using the following formula: Viability (%) = (mean absorbance value of drug-treated cells)/(mean absorbance value of the control) × 100. The results were represented as mean ± SEM values. The analysis of the data was done using two-way ANOVA followed by Bonferroni posttests.
Combination index analysis
Based on the cytotoxicity assay of DTX and DSF individually on 4T1 and MCF7 cell lines, the non-fixed drug ratio of DSF and DTX was used for combination index analysis. The combination index of this drug combination was analyzed using Compusyn software (version 1.0) by the Chou-Talalay method.
Apoptosis assay
The measurement of apoptosis was performed by flow cytometry and PI/Annexin V-FITC staining. MCF7 cells were exposed to DTX (10 nM), DSF (100 nM) and DSF (100 nM) + DTX (10 nM) for 48 h. After completion of treatment, cells were collected by trypsinization into 15 ml centrifuge tubes. Following this, trypsin was removed by centrifugation of cells at 3000 rpm for 10 min. Then the cells were washed twice with 1 ml of cold phosphate-buffered saline and the cells were suspended in 500 μl of 1× binding buffer (Annexin Binding Buffer (5X) for flow cytometry, Thermo fisher scientific). Further, the cells were stained with 4 μl Annexin V-FITC (Annexin V-FITC, Invitrogen™) and 10 μl of PI (propidium iodide solution, FluoroPure™ Grade, Invitrogen™). The samples were incubated for 15 min at room temperature (25 °C) in the dark. The cytometric analysis was performed in a flow cytometer (CytoFLEX, Beckman Coulter), and the data were analyzed by CytExpert software. Apoptosis was calculated over all viable cells and after subtracting the autofluorescence of cells. The results were represented as mean ± SEM values. The analysis was done using two-way ANOVA followed by Bonferroni posttests.
MDC (Monodansylcadaverine) assay for autophagy
MDC, an autophagosomal marker, was used to analyze autophagy induction. Cells were cultured on glass coverslips and treated with DTX (10 nM), DSF (100 nM) and DSF (100 nM) + DTX (10 nM) for 24 h, then cells were incubated with 0.05 mM MDC (D4008, Sigma) at 37 °C for 10 min in dark. After that, the coverslips with cells were washed with PBS and mounted with antifade DAPI (S36938, ThermoFisher Scientific). The resultant MDC punctate dots were captured at 40× or 63× magnification under a fluorescence microscope (ZEISS, India).
For flow cytometric analysis of MDC fluorescence, cells were grown in a 6-well plate. After treatment, cells were incubated with MDC for 10 min in the dark, followed by PBS wash, and then collected by trypsinization. Samples were then acquired using a flow cytometer (CytoFLEX, Beckman Coulter).
Immunoblotting
After treatment, the MCF7 cells were lysed in a modified RIPA buffer (Sigma-Aldrich), and the protein content was measured using the Bradford reagent. Then 5× loading buffer was added to the lysates followed by heat denaturation (100 °C for 10 min) and cooling on ice. Equal concentrations of protein lysates were loaded in denaturing polyacrylamide gels (5% stacking and 12% resolving). Subsequently, it was transferred to the PVDF membrane (Millipore), then blocked with 5% skimmed milk (HiMedia). The blots were probed with primary antibody of protein of interest at their specified dilution including LC3B-II (3868S-100uL, Cell Signaling Technology) or Bax (BB-AB-0250, Biobharthi) or Bcl-2 (BB-AB0230, Biobharthi) or p62 (BB-AB0130, Biobharthi) or ATG5 (BB-AB0225, Biobharthi) and loading control GAPDH (sc-365062, Santa Cruz Biotechnology) at 1:2000 dilution. The secondary antibodies used were horseradish peroxide-conjugated goat anti-rabbit IgG or goat anti-mouse IgG. The protein intensity was detected using the enhanced chemiluminescence by the Bio-Rad ChemiDoc™ XRS + imaging system. The protein expression was densitometrically quantified using Image-J software and normalized to control. Results were represented as mean ± SEM values and the analysis was done using one-way ANOVA followed by post hoc Tukey’s test.
Reactive Oxygen Species (ROS) assay
ROS production was detected by DCFH-DA (2′,7′-dichlorofluorescein diacetate) fluorogenic probe. Once inside the cell, the de-esterified product becomes the fluorescent compound 2′,7′-dichloro- fluorescin (DCF) on oxidation by ROS and the fluorescent signal (Ex. 490 nm/Em. 535 nm) is proportional to ROS production [23]. N-acetyl cysteine (NAC), a ROS scavenger (5 mM), was added 1 h before treatments. Cells were treated with DTX (10 nM), DSF (100 nM) and DSF (100 nM) + DTX (10 nM) in triplicate in a 96 well plate for 24 h. Later, cells were washed with PBS, and then they were treated with 10 µM DCFH-DA solution in PBS for 60 min. Readings were taken at the fluorescent signal (Ex. 490 nm/Em. 535 nm) using BioTek fluorescent plate reader. Fluorescent microscopic images of the cells were also taken at 40× magnification using Inverted microscope Axio Vert.A1 FL-LED (ZIESS, India). Results were represented as mean ± SEM values. The analysis was done using one-way ANOVA followed by post hoc Tukey’s test.
ATG5 silencing
To inhibit macroautophagy, cells were transiently transfected with ATG5 shRNA (procured from Indian Institute of Science, shRNA repository) by using Lipofectamine 3000 Reagent (Thermo Scientific) according to reagents protocol. Studies were performed in MCF7 cells pre-incubated with 2 µg shRNA of ATG5 for 48 h, followed by exposure to drugs.
Autophagy flux inhibition
Chloroquine diphosphate (CQ) was used to inhibit autophagy flux as it inhibits the fusion of the autophagosome with the lysosome. CQ was used at the concentration of 10 µM in all the experiments. In the case of co-treatment with other drugs, cells were pretreated with CQ, 1 h before the addition of other drugs.
Statistical analysis
The obtained data were analyzed using GraphPad Prism software version 8. One-way ANOVA was used when ≥ 2 groups were compared to the same control; posthoc Tukey’s test was performed to compare the means of each group with every other group. Two-way ANOVA was used to compare groups that have been split into two independent variables; posthoc Bonferroni multiple comparison tests were performed to compare between means of each group. Data were represented in mean ± SEM. In the figures, the level of significance was indicated using different symbols as * (comparison with control), # (comparison with DTX), $ (comparison with DSF), @ (comparison with DTX + DSF).
Results
Evaluation of DSF for potentiating the anticancer activity of DTX
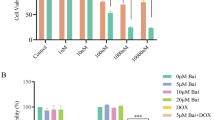
To determine the effect of DSF on the cytotoxicity of DTX, we first compared the cytotoxicity of DTX alone, DSF alone, and DTX + DSF combination against two different breast cancer cell lines, 4T1 (murine breast cancer) and MCF7 (human breast cancer). We have used DSF at the constant dose of 100 nM (non-cytotoxic dose, based on previous reports [24,25,26]) and three different doses of DTX (1, 10, and 50 nM). The percentage of cell death was analyzed using the MTT assay. DSF at 100 nM dose exhibited 70–80% cell viability in both the cell lines. DTX + DSF combination exhibited significantly enhanced cytotoxicity compared to DTX alone in both the cell lines indicating synergistic activity (MCF7: F(2,12) = 746.8; 4T1: F(2,12) = 241.07) (Fig. 1a, b).

DSF synergistically potentiates the cytotoxic activity of DTX both in 4T1 and MCF7 cells. a, b MTT assay on 4T1 and MCF7 cells with constant dose of DSF (100 nM) and variable concentration of DTX (1, 10 and 50 nM). In both the cell lines, DSF (100 nM) + DTX (1 nM) (MCF7: p = 0.000003; 4T1: p = 0.000001) and DSF (100 nM) + DTX (10 nM) (MCF7: p = 0.000002; 4T1: p = 0.000001) showed significant increase in cytotoxicity compared to DTX alone at same dose. Results were mean ± SEM values. Analysis was done using two-way ANOVA followed by Bonferroni posttests. c, d MTT assay on 4T1 and MCF7 cell lines with DTX, DSF and DTX (10 nM) + DSF (variable dose), results were obtained by experiments conducted twice independently in triplicates
Combination index analysis of DTX and DSF co-treatment to find the effective dose ratio
After observing synergistic cytotoxicity of DTX + DSF, to find the most effective dose ratio of DTX and DSF, MTT assay on 4T1 and MCF7 cells was performed using a varied concentration of DSF (1 nM, 10 nM, 100 nM, 1000 nM, and 10,000 nM) with a fixed concentration of DTX (10 nM) (Fig. 1c, d).The resulted cytotoxicity of the combination therapy was used to analyze the combination index (CI) by the Chou-Talalay method [27] using CompuSyn software (version 1.0). In both 4T1 and MCF7 cell lines strong synergy was observed at 1:10 ratio of DTX and DSF (DTX [10 nM] + DSF [100 nM]) (Table 1). Based on this observation, in all further experiments, DTX and DSF were taken in a 1:10 molar ratio (DTX 10 nM and DSF 100 nM).
Apoptotic cell death analysis with DTX + DSF treatment
Next, we wanted to find out the contribution of apoptosis in the cell death observed with the DTX + DSF combination. Apoptotic cell death was analyzed by flow cytometry using Annexin V-FITC/PI staining. MCF7 cells were treated with either DTX alone (10 nM), or DSF (100 nM) alone, or DTX (10 nM) + DSF (100 nM) combination. Treatment with DSF alone showed a lower level of apoptosis. In contrast, a moderate increase in % of total apoptotic cells (early + late apoptosis, total Annexin V positive cells) was found with DTX + DSF combination (60.5 ± 0.5) over DTX treatment (57.3 ± 0.3%). However, a significant increase in the percentage of the late apoptotic cells (Annexin V + PI-positive cells) was observed with the combination treatment compared to DTX alone (DTX: 7.1 ± 0.1%; DTX + DSF: 13.7 ± 0.2%; p = 0.000021) (F(9,24) = 4365.06) (Fig. 2a, b). These data indicated that DSF co-treatment augmented the DTX mediated apoptosis. Such synergistic activity of DTX and DSF combination on apoptosis supports and complements the MTT assay results, as recorded earlier (Fig. 1).

Effect of DTX + DSF on apoptosis. a Dot-plot of the FACS analysis for the apoptosis assay on MCF7 cells with control, DTX (10 nM), DSF (100 nM), and DTX (10 nM) + DSF (100 nM). b Analysis of the % of cells at various stages of apoptotic cell death. % of late apoptotic cells were significantly high in DTX + DSF compared to DTX and DSF alone (p = 0.000021). Results were mean ± SEM values. The analysis was done using two-way ANOVA followed by Bonferroni posttests. c Immunoblotting of Bax protein with DTX (10 nM), DSF (100 nM), and DTX + DSF treated MCF7 cells after 24 h. Compared to control there was no significant difference in Bax protein expression in DTX (p = 0.316266), in DSF there was significant decrease (p = 0.004183) and in DTX + DSF there was significant increase (p = 0.000009). d Immunoblotting of Bcl-2 protein with DTX, DSF, and DTX + DSF treated cells. Bcl-2 protein expression was not significantly different in DTX and DSF (DTX: p = 0.981617, DSF: p = 0.974966) whereas it was significantly decreased in DTX + DSF (p = 0.000005) compared to control. e Bax/Bcl-2 ratio in DTX, DSF and DTX + DSF. Bax/Bcl-2 ratio was not changed significantly in DTX (p = 0.999797) and DSF (p = 0.998654) compared to control whereas with DTX + DSF treatment it was significantly increased compared to control (p = 0.000002). Results were mean ± SEM values. The analysis was done using one-way ANOVA followed by post hoc Tukey’s test
Analysis of apoptosis induction by Bax/Bcl-2 ratio
After observing an increase in the apoptotic cell population, we further analyzed the Bax/Bcl-2 ratio to study the effect of DTX + DSF on the induction of apoptosis using MCF7 cells. With docetaxel (10 nM), we did not observe any significant change in Bax and Bcl-2 expression. With DSF (100 nM) treatment, we detected a moderate reduction in Bax while Bcl-2 expression remained unaltered in comparison to control. In contrast, with DTX + DSF treatment there was significant increase in Bax (p = 0.000009) along with significant reduction in Bcl-2 (p = 0.000005) (Bax: F(3,8) = 108.8; Bcl-2: F(3,8) = 84.6), thereby increasing the Bax/Bcl-2 ratio 72 ± 4-fold compared to control (p = 0.000002) (F(3,8) = 254.4). These data indicated that there was a significant apoptosis induction in DTX + DSF treatment by upregulating pro-apoptotic Bax and downregulating anti-apoptotic Bcl-2 (Fig. 2c–e).
Determination of the effect of DTX + DSF combination on autophagy
As both DTX and DSF can induce autophagy, we performed MDC assay using both MCF-7 and 4T1 cells treated with DTX, or DSF, or their combination (10 nM, 100 nM, 10 nM + 100 nM, respectively). Fluorescent microscopic imaging of the MDC stained cells demonstrated a similar response in both MCF-7 and 4T1 cell lines. The majority of the cells in the DTX + DSF group were found to show MDC fluorescence, whereas, with DSF treatment, a limited number of cells were found to have MDC fluorescence. With DTX treatment, the number of cells with MDC fluorescence was negligible (Fig. 3a). To quantify the number of cells having MDC fluorescence, we further analyzed MDC stained MCF7 cells by flow cytometry. A significant shift in the MDC positive cells was observed in DTX + DSFtreated cells (10.6 ± 0.1%) compared to DTX (0.3 ± 0.02%, p = 0.000002) and DSF (1.3 ± 0.13%, p = 0.000001) treatment, indicating hyper-induction of autophagy with the DTX + DSF combination treatment (F(3,8) = 3468) (Fig. 3b).

MDC assay and LC3B-II expression together indicate the induction of autophagy in DTX + DSF treatment. a MDC assay using fluorescent microscopy with DTX, DSF, and DTX + DSF treated cells showing increased green fluorescence in DTX + DSF treatment compared to control, DTX, and DSF in both MCF7 and 4T1 cell lines. The scale bar in the image indicates 50 µm. b MDC assay using flow cytometry with DTX, DSF, and DTX + DSF treated MCF7 cells, indicating increased MDC fluorescent cells in DTX + DSF treatment compared to DTX and DSF alone. c Immunoblotting of LC3-II protein with DTX, DSF, and DTX + DSF treated MCF7 cells. LC3B-II expression in DTX group was not significantly different from control (p = 0.930440) whereas there was significant increase in DSF (p = 0.000550) and DTX + DSF (p = 0.000217). Results were mean ± SEM values. The analysis was done using one-way ANOVA followed by post hoc Tukey’s test. d Immunoblotting of p62 protein with DTX, DSF, and DTX + DSF treated MCF7 cells. p62 expression in DTX was significantly decreased (p = 0.000720), in DSF it was increased significantly (p = 0.000002) and in DTX + DSF it was significantly decreased (p = 0.000884) compared to control. Results were mean ± SEM values. The analysis was done using one-way ANOVA followed by post hoc Tukey’s test
An increase in the MDC fluorescence is an indicator of the accumulation of autophagic vacuoles in the cell [28]. To further confirm the effect of DTX + DSF treatment on autophagy, we quantified the level of the autophagy marker LC3B-II in drug-treated MCF7 cells through western blotting. With DTX treatment, there was no significant change in LC3B-II expression compared to control (p = 0.930440), whereas with DSF (p = 0.000550) or DTX + DSF (p = 0.000217), a significant increase in LC3B-II expression was observed compared to control (F(3,8) = 34.28) (Fig. 3c).
To explore whether the DTX + DSF combination activates autophagic flux, we analyzed p62 expression in DTX, or DSF, or DTX + DSF treated cells. With DTX treatment, no change in LC3B-II expression was found; nonetheless, a significant reduction in p62 expression (p = 0.000720) was observed, indicating low autophagic induction and increased autophagic clearance. With DSF treatment, there was a significant increase in both LC3B-II, and p62 (p = 0.000002), which signifies a high level of autophagosome formation, yet decreased activity in the downstream autophagy. In DTX + DSF treatment, a significant increase in LC3B-II expression and significant reduction in p62 (p = 0.000884) expression was observed, implying induction of high level of functional autophagy (F(3,8) = 199.5) (Fig. 3d).
Both MDC data and LC3B-II/p62 expression analysis collectively indicate that there is a significant increase in autophagy with DTX + DSF treatment compared to DTX and DSF alone.
Estimation of ROS with DTX + DSF treatment
Intracellular ROS is a major inducer of autophagy [29, 30]. ROS induction has been reported previously with both DSF as well as DTX treatment [31,32,33]. To study whether ROS has any role in the synergistic cytotoxic activity of the DTX + DSF combination, we have analyzed ROS levels in MCF7 and 4T1 cells after treatment with DTX or DSF or DTX + DSF combination (10 nM, 100 nM, 10 nM + 100 nM, respectively) for 24 h. ROS generated was quantified using DCFH-DA fluorescent probe by fluorescent microscopy as well as by fluorescent plate reader (Fig. 4a–c). Total fluorescence was normalized with respect to control cells. A significant increase in ROS levels in the cells treated with the combination of DTX + DSF (MCF7: 2.8 ± 0.06; 4T1: 3.6 ± 0.03) was observed compared to DTX (MCF7: 1.8 ± 0.09, p = 0.000001; 4T1: 1.4 ± 0.02, p = 0.000002) or DSF (MCF7: 2.3 ± 0.1, p = 0.001664; 4T1: 2.5 ± 0.17, p = 0.000005) alone in both the cell lines. When the cells were pretreated with ROS inhibitor NAC (N-Acetyl-l-Cysteine), ROS was found to decrease to the basal level (MCF7: F(5,12) = 142.9; 4T1: F(5,12) = 216). These data indicate that enhanced ROS generation with the DTX + DSF combination may play a critical role in their synergistic activity.

Hyper induction of ROS in DTX + DSF treatment. a Fluorescent microscopic images of DCFDA stained MCF7 and 4T1 cells after treatment with DTX, DSF, DTX + DSF, NAC and NAC pretreated DTX + DSF showing increased DCF fluorescence in DTX + DSF compared to control, DTX, and DSF. Scale bar in the image indicates 100 µm. b, c ROS analysis by DCFDA assay using fluorescent plate reader with DTX, DSF, DTX + DSF, NAC and NAC pretreated DTX + DSF of MCF7 and 4T1 respectively. In case of MCF7, DCF fluorescence increased significantly in DTX (p = 0.000012), DSF (p = 0.000001) and DTX + DSF (p = 0.000001) compared to control. DCF fluorescence in DTX + DSF also increased significantly compared to DTX (p =0.000001) and DSF (p =0.001664) alone. In case of 4T1, DCF fluorescence increased significantly in DTX (p = 0.046288), DSF (p = 0.000001) and DTX + DSF (p = 0.000001) compared to control. DCF fluorescence in DTX + DSF also increased significantly compared to DTX (p = 0.000001) and DSF (p = 0.000005) alone. With NAC + DTX + DSF the DCF fluorescence decreased almost equal to control in both 4T1 and MCF7 indicating NAC inhibition of DCF fluorescence in DTX + DSF. Results were mean ± SEM values. Analysis was done using one-way ANOVA followed by post hoc Tukey’s test
Effect of ROS and autophagy on cell death observed with DTX and DSF
After observing a significant increase in ROS, autophagy, and cell death with DTX + DSF combination therapy, we wanted to find out whether this ROS had any potential role in cell death. For this, we inhibited ROS by using NAC (5 mM), 1 h before treating cells with DTX or DSF or DTX + DSF combination (10 nM, 100 nM, 10 nM + 100 nM, respectively) and analyzed the cell death using MTT assay after 72 h treatment, on 4T1 and MCF7 cell lines (Fig. 5a, b). NAC treatment produced a differential effect on the cytotoxicity of the DTX or DSF or DTX + DSF treated cells. No significant change in the % survival was observed with NAC co-treatment in the DTX treated cells (MCF7: p = 0.536127; 4T1: p = 0.094975), indicating ROS generation was not the major pathway for DTX cytotoxicity. With DSF, a moderate increase in cell survival was noted with NAC co-treatment (MCF7: p = 0.000004; 4T1: p = 0.000003). However, NAC co-treatment was found to have a significant impact on the DTX + DSF treated cells, where it almost completely reversed the cell death observed in both MCF-7 and 4T1 cell lines (MCF7: p = 0.000001; 4T1: p = 0.000001) (MCF7: F(6,14) = 223; 4T1: F(6,14) = 251.6). This data indicate that cell death with DTX + DSF treatment was mainly via ROS generation.

Analysis of the effect of ROS and autophagy inhibition on the cytotoxic potency of DTX + DSF combination. a, b MTT assay on 4T1 and MCF7 cells with DTX, DSF, DTX + DSF, and NAC pretreated DTX + DSF. In both 4T1 and MCF7 there was no significant difference in cytotoxicity of DTX and NAC + DTX (MCF7: p = 0.536127; 4T1: p = 0.094975) indicating there was no effect of ROS on cytotoxicity of DTX (10 nM). In case of DSF (MCF7: p = 0.000004; 4T1: p = 0.000003) and DTX + DSF (MCF7: p = 0.000001; 4T1: p = 0.000001), NAC treatment significantly decreased the cytotoxicity, indicating ROS was playing a significant role in their cytotoxicity. Results were analyzed using one-way ANOVA followed by post hoc Tukey’s test. c MTT assay on MCF7 cells with DTX, DSF, DTX + DSF, and along with CQ (10 µM) co-treatment. CQ co-treatment significantly increased the cytotoxicity of DTX (p = 0.000038), DSF (p = 0.009377) and DTX + DSF (p = 0.001817). Results were analyzed using one-way ANOVA followed by post hoc Tukey’s test. d ATG5 silencing by shRNA in MCF7 cell lines. When ATG5 was silenced by transient transfection of shRNA, expression of ATG5 decreased to almost 30% compared to control as well as scrambled. Results were analyzed using one-way ANOVA followed by post hoc Tukey’s test; error bars indicate SEM. e MTT assay on MCF7 cells with DTX, DSF, DTX + DSF, and after ATG5 silencing with ATG5 shRNA. With DTX, there was a significant increase in cytotoxicity after ATG5 silencing (p = 0.001185). With DSF, there was no significant change after ATG5si (p =0.343044), whereas with DTX + DSF, there was a significant decrease in cytotoxicity with ATG5si (p = 0.000543). There was no significant change in cytotoxicity between DTX + DSF and DTX + DSF + Scambled (p = 0.863452), indicating there was no effect of scrambled on cytotoxicity. Results were mean ± SEM values. It was analyzed using one-way ANOVA followed by post hoc Tukey’s test
To study the contribution of autophagy in the enhanced cell death seen with the DTX + DSF combined treatment, we have inhibited autophagy using CQ (10 µM) 1 h before treating MCF7 cells with DTX or DSF or DTX + DSF combination (10 nM, 100 nM, 10 nM + 100 nM, respectively) and analyzed the cell death using MTT assay. CQ pre-treatment was found to enhance cell death in all the treatments, DTX (p = 0.000038), DSF (p = 0.009377) and DTX + DSF combination (p = 0.001817) (F(5,12) = 390.8) (Fig. 5c).
CQ treatment results in autophagy inhibition at a late stage, preventing auto-lysosomal fusion. To understand the effect of autophagy inhibition at an early stage, we have used ATG5 silencing to prevent the formation of autophagic vacuoles, which interrupt the whole autophagy process [34]. We have transiently inhibited autophagy in MCF7 cells using ATG5 shRNA which resulted in significant reduction in ATG5 expression to 0.29 ± 0.011 in comparison to control (p = 0.000003, F(2,6) = 290.9) (Fig. 5d). We treated those cells with DTX (10 nM) or DSF (100 nM) or DTX + DSF (10 nM + 100 nM), and % cell viability was analyzed using MTT assay after 72 h of treatment. Interestingly, it was observed that with ATG5 silencing, a significant increase in survival of the DTX + DSF treated cells (p = 0.000543), which indicates that autophagy is playing a pivotal role for the increased cell death found with DTX + DSF co-treatment (F(9,20) = 544.5) (Fig. 5e). With both DTX alone and DSF alone treatment, there was a significant increase in cell death with ATG5 silencing (p = 0.001185), indicating that the autophagy induction in the DTX or DSF treatment was a pro-survival autophagy.
Effect of different autophagy inhibitors in the DTX and DSF treated cells
To confirm the differential effect of CQ and ATG5 shRNA treatment on cell survival, we have analyzed the level of autophagy using MDC fluorescence after pretreating MCF7 cells with CQ or ATG5 shRNA followed by DTX + DSF treatment for 24 h. A significant increase in MDC fluorescence was observed with CQ + DTX + DSF as compared to DTX + DSF, indicating a higher level of autophagosome accumulation. On the contrary, with ATG5 shRNA treatment, there was a significant reduction in MDC fluorescence, implying a reduction in autophagy (Fig. 6a). To further confirm this observation, we have analyzed the LC3B-II expression with the same treatments. A significant increase in LC3B-II expression in CQ + DTX + DSF (27.08 ± 0.528, p = 0.000001) compared to DTX + DSF (5.42 ± 0.218) was detected, whereas ATG5-shRNA + DTX + DSF (2.44 ± 0.052, p = 0.000016) has shown decrease in LC3B-II expression compared to DTX + DSF; (F(5,12) = 1757) (Fig. 6b). Both MDC fluorescence study and LC3B-II expression analysis indicated that DTX + DSF treatment resulted in hyper induction of autophagy and accumulation of autophagic vacuoles, which triggered apoptotic cell death. The autophagic flux inhibition with CQ led to even higher accumulation of autophagic vacuoles, whereas autophagy inhibition with ATG5 shRNA reduced autophagosome formation and cytotoxicity.

Differential effects of CQ and ATG5 silencing on the autophagosome formation in the DTX + DSF combination-treated MCF7 cells. a MDC fluorescence microscopic images showing an increase in MDC fluorescence in DTX + DSF + CQ treatment and a decrease in DTX + DSF + ATG5Si compared to DTX + DSF. The scale bar in the image indicates 50 µm. b Analysis of LC3B-II expression in DTX + DSF, CQ + DTX + DSF, and ATG5-ShRNA + DTX + DSF. With DTX + DSF + CQ, there was a significant increase in LC3B-II compared to DTX + DSF (p = 0.000001). With DTX + DSF + ATG5Si, there was a significant decrease in LC3B-II expression compared to DTX + DSF (p = 0.000016). Results were mean ± SEM values. The analysis was done using one-way ANOVA followed by post hoc Tukey’s test
Analysis of ROS and autophagy in synergistic cytotoxicity of DTX + DSF
ROS generation is a known trigger for the induction of autophagy [35]. Hence, we wanted to find out whether autophagy induction by the DTX + DSF combination is ROS dependent or not. MCF7 cells were given NAC pre-treatment along with DTX + DSF treatment, and MDC fluorescence was analyzed using fluorescent microscopy. NAC pre-treatment was found to reduce MDC fluorescence in the DTX + DSF treated cells (Fig. 7a). Concurrently, LC3B-II expression analysis by western blotting had also shown ROS inhibition by NAC pre-treatment, which restored the LC3B-II to the basal level in the DTX + DSF treated cells, suggesting ROS was responsible for the overall autophagy state in the DTX + DSF treated cells. LC3B-II expression in DTX + DSF was 6.21 ± 0.865 and NAC + DTX + DSF was 1.14 ± 0.08 (p = 0.000174, F(3,8) = 33.42) (Fig. 7b). Both these data together indicated ROS was the major pathway for autophagy induction with this treatment.

Inhibition of ROS with NAC resulted in inhibition of autophagy in DTX + DSF treatment in MCF7 cells. a Fluorescent microscopic images showing a decrease in MDC fluorescence with DTX + DSF + NAC compared to DTX + DSF. The scale bar in the image indicates 50 µm. b Analysis of the level of LC3B-II expression. LC3B-II expression decreased significantly in DTX + DSF + NAC compared to DTX + DSF (p = 0.000174). Results were mean ± SEM values. The analysis was done using one-way ANOVA followed by post hoc Tukey’s test
Discussion
The most common problem of cancer chemotherapy is the desensitization of the tumor cell towards the drug, which leads to therapy failure and relapse of the disease. DTX is a potent anticancer agent, against which tumor cells can develop resistance by inducing autophagy [10,11,12]. The importance of autophagy for the generation of drug resistance has been reviewed extensively [36]. It has been proposed that autophagy modulation can sensitize the cancer cells towards chemotherapeutic drugs [36]. Mostly autophagy inhibitors have been tested for this purpose as autophagy helps in the survival of the cancer cells. However, it has been demonstrated that autophagy acts as a double-edged sword in tumor cells. Low autophagy induction has been linked with the development of resistance, whereas hyper-induction of autophagy has shown to promote cell death [37]. Thus, multiple autophagy inducers are under clinical trial or clinically used for cancer treatment [38].
As the use of autophagy inhibitors with DTX co-treatment did not result in favorable clinical outcomes [15], we wanted to evaluate the other extreme: hyper-induction of autophagy to potentiate DTX activity. The potential of an autophagy inducer to enhance DTX activity has been demonstrated previously. Buoncervello et al. have shown that combination treatment of DTX with apicidin, which is an autophagy inducer, improves cytotoxic potency of DTX [39]. However, apicidin is not very potent: it is effective at a very high dose of 1000 nM. Also, it is not a clinically approved drug. To find an alternative to such drawbacks, we wanted to evaluate a clinically available, potent autophagy inducer to enhance DTX activity.
DSF is a clinically approved drug. Though it is primarily used for the treatment of alcoholism, it is reported to have autophagy inducing activity, targeting different pathways such as proteasomal inhibition, NFĸB inhibition, ALDH inhibition, etc. [20]. DSF has shown to synergistically improve the cytotoxic efficacy of multiple anticancer drugs, including 5-fluorouracil [40], doxorubicin [41], and gemcitabine [42]. In a Phase IIb clinical trial, DSF co-treatment exhibited enhanced efficacy with cisplatin and vinorelbine against non-small cell lung cancer [43].
In the current study, we have observed highly synergistic cytotoxic activity with DTX and DSF co-treatment (Fig. 1, Table 1). DSF was found out to be highly potent (with 100 nM dose) in augmenting the cytotoxic activity of DTX, compared to apicidin. Analysis of the % of apoptotic cells pointed out the increased conversion of early apoptotic cells to the late apoptotic cells with this combination, pointing towards higher apoptosis induction (Fig. 2a, b). While analyzing the involvement of Bax/Bcl-2, it was observed that DTX treatment did not cause any significant change in the Bax/Bcl-2 ratio (Fig. 2c, d), indicating Bcl-2 independent cell death. Bcl-2 independent cytotoxicity of DTX has been reported earlier [44, 45]. With DSF treatment, no significant change in the Bax/Bcl-2 ratio was observed, as the tested dose of DSF did not cause any significant apoptosis (Fig. 2a, b). However, with DTX + DSF, there was a significant increase in the Bax/Bcl-2 ratio indicating the induction of Bax and Bcl-2 dependent apoptosis. Bcl-2 plays an important role in both apoptosis and autophagy: it can also inhibit autophagy by binding to Beclin-1, an autophagy inducer [46]. The decreased Bcl-2 level has been associated with the induction of autophagy [47].
In our study, an increased accumulation of autophagic vacuoles was found with this combination compared to both DTX and DSF alone (Fig. 3a, b). DTX treatment was found to induce autophagic flux (no change in LC3B-II and decreased p62), whereas, with DSF treatment, there was higher autophagosome formation, and reduced autophagic flux (increase in both LC3B-II and p62). An increase in both LC3B-II and p62 with DSF treatment has been reported earlier as well [48]. In the DTX + DSF treated cells, both autophagosome formation and increased flux were observed (increase in LC3B-II and reduction in p62) (Fig. 3c, d). This combined treatment was also found to induce intracellular ROS generation, and inhibition of ROS by NAC was found to abolish almost all the cytotoxicity of the combination (Figs. 4 and 5a, b), pointing a major role of ROS. ROS is known to be a major inducer of autophagy [29], and this combination may work through that pathway.
Increased autophagy is observed in many cell death situations, which may be a failed attempt to attenuate cell damage [49]. On the other hand, autophagy itself can also induce cell death [50]. It is critical to understand whether any autophagy observed is a survival attempt or it would lead to autophagy-dependent cell death (ADCD) [51]. In the case of the ADCD, inhibition of autophagy would protect the cell from death. To evaluate whether the autophagy induced by the DTX + DSF combination was responsible for ADCD, we tested two autophagy inhibitors, one autophagic flux inhibitor (CQ) and another autophagosome formation inhibitor (ATG5 silencing [ATG5Si]). Surprisingly, inhibition of autophagy at these two different stages resulted in two opposite outcomes: inhibition with CQ resulted in higher cell death, whereas ATG5Si produced lower cell death (Fig. 5c, e). To decipher this contradiction, we analyzed the number of autophagic vacuoles formed with these two inhibitors. Autophagic flux inhibition with CQ resulted in a significantly higher vacuole accumulation, whereas inhibition of autophagosome formation with ATG5Si led to lower vacuole accumulation (Fig. 6a). The same observation was found with LC3B-II level as well: CQ produced increased LC3B-II level, and with ATG5Si, LC3B-II level decreased (Fig. 6b).
CQ inhibits autophagosome fusion with the lysosome, thereby preventing clearance of the autophagic vacuoles [52]. Hence, CQ pre-treatment would result in hyper-accumulation of autophagic vacuoles, which can cause oxidative stress, DNA damage, and cytotoxicity [53]. Whereas, ATG5 silencing would prevent the formation of autophagosome, which interrupts the whole autophagy process [34]. Higher cell death with the CQ treatment may be attributed to increased accumulation of autophagic vacuole and enhanced autophagic load. Similar observation with CQ treatment has been reported previously as well [54]. On the other hand, autophagy inhibition at an early stage with ATG5 silencing produced decreased autophagic vacuole and reduced cell death. This data indicate that autophagy is playing a pivotal role in the cell death found with DTX + DSF co-treatment. ATG5 silencing in the DTX treated cells resulted in a significant increase in cell death, indicating DTX produced pro-survival autophagy, which is also supported by previous studies [55].
Lastly, we found out that the autophagy induction by DTX + DSF treatment was dependent on ROS and ROS inhibition resulted in decreased autophagosome formation (Fig. 7a). Also, ROS inhibition restored the LC3B-II to the basal level in the DTX + DSF treated cells (Fig. 7b), suggesting ROS was the major pathway for autophagy induction with this treatment. It has been demonstrated that ROS can also downregulate Bcl-2 expression [56]. Altogether, these data indicate that the DTX + DSF treatment-induced ROS production, which resulted in both autophagy and apoptosis mediated cell death.
This study demonstrated that DSF could effectively sensitize breast cancer cells for DTX therapy by modulating ROS and autophagy. Hence, this strategy can be developed as a therapeutic approach for cancer treatment in the future.
References
Hortobagyi G. Docetaxel in breast cancer and a rationale for combination therapy. Oncology (Williston Park). 1997;11:11–5.
Palmeri L, Vaglica M, Palmeri S. Weekly docetaxel in the treatment of metastatic breast cancer. Ther Clin Risk Manag. 2008;4:1047–59.
Rivera E, Gomez H. Chemotherapy resistance in metastatic breast cancer: the evolving role of ixabepilone. Breast Cancer Res. 2010;12(Suppl 2):S2.
Hansen SN, Westergaard D, Thomsen MB, Vistesen M, Do KN, Fogh L, et al. Acquisition of docetaxel resistance in breast cancer cells reveals upregulation of ABCB1 expression as a key mediator of resistance accompanied by discrete upregulation of other specific genes and pathways. Tumour Biol. 2015;36:4327–38.
Burris HA 3rd, Fields S, Peacock N. Docetaxel (Taxotere) in combination: a step forward. Semin Oncol. 1995;22:35–40.
O’Shaughnessy J, Miles D, Vukelja S, Moiseyenko V, Ayoub JP, Cervantes G, et al. Superior survival with capecitabine plus docetaxel combination therapy in anthracycline-pretreated patients with advanced breast cancer: phase III trial results. J Clin Oncol. 2002;20:2812–23.
von Minckwitz G, Rezai M, Loibl S, Fasching PA, Huober J, Tesch H, et al. Capecitabine in addition to anthracycline- and taxane-based neoadjuvant treatment in patients with primary breast cancer: phase III GeparQuattro study. J Clin Oncol. 2010;28:2015–23.
Perez EA, Suman VJ, Fitch TR, Mailliard JA, Ingle JN, Cole JT, et al. A phase II trial of docetaxel and carboplatin as first-line chemotherapy for metastatic breast cancer: NCCTG study N9932. Oncology. 2005;69:117–21.
Gonzalez-Angulo AM, Morales-Vasquez F, Hortobagyi GN. Overview of resistance to systemic therapy in patients with breast cancer. Adv Exp Med Biol. 2007;608:1–22.
Pan B, Chen D, Huang J, Wang R, Feng B, Song H, et al. HMGB1-mediated autophagy promotes docetaxel resistance in human lung adenocarcinoma. Mol Cancer. 2014;13:165.
Vera-Ramirez L, Vodnala SK, Nini R, Hunter KW, Green JE. Autophagy promotes the survival of dormant breast cancer cells and metastatic tumour recurrence. Nat Commun. 2018;9:1944.
Jain K, Paranandi KS, Sridharan S, Basu A. Autophagy in breast cancer and its implications for therapy. Am J Cancer Res. 2013;3:251–65.
Gong C, Hu C, Gu F, Xia Q, Yao C, Zhang L, et al. Co-delivery of autophagy inhibitor ATG7 siRNA and docetaxel for breast cancer treatment. J Control Release. 2017;266:272–86.
Wang Q, He WY, Zeng YZ, Hossain A, Gou X. Inhibiting autophagy overcomes docetaxel resistance in castration-resistant prostate cancer cells. Int Urol Nephrol. 2018;50:675–86.
Hansen AR, Tannock IF, Templeton A, Chen E, Evans A, Knox J, et al. Pantoprazole affecting docetaxel resistance pathways via autophagy (PANDORA): phase II Trial of High Dose Pantoprazole (Autophagy Inhibitor) with docetaxel in metastatic castration-resistant prostate cancer (mCRPC). Oncologist. 2019;24:1188–94.
Russo M, Russo GL. Autophagy inducers in cancer. Biochem Pharmacol. 2018;153:51–61.
Fulda S. Autophagy in cancer therapy. Front Oncol. 2017;7:128.
Park YM, Go YY, Shin SH, Cho JG, Woo JS, Song JJ. Anti-cancer effects of disulfiram in head and neck squamous cell carcinoma via autophagic cell death. PLoS One. 2018;13:e0203069.
Wu X, Xue X, Wang L, Wang W, Han J, Sun X, et al. Suppressing autophagy enhances disulfiram/copper-induced apoptosis in non-small cell lung cancer. Eur J Pharmacol. 2018;827:1–12.
Jiao Y, Hannafon BN, Ding WQ. Disulfiram’s Anticancer Activity: evidence and Mechanisms. Anticancer Agents Med Chem. 2016;16:1378–84.
Rae C, Tesson M, Babich JW, Boyd M, Sorensen A, Mairs RJ. The role of copper in disulfiram-induced toxicity and radiosensitization of cancer cells. J Nucl Med. 2013;54:953–60.
Liu P, Brown S, Channathodiyil P, Kannappan V, Armesilla AL, Darling JL, et al. Reply: cytotoxic effect of disulfiram/copper on human glioblastoma cell lines and ALDH-positive cancer-stem-like cells. Br J Cancer. 2013;108:994.
Osseni RA, Debbasch C, Christen MO, Rat P, Warnet JM. Tacrine-induced reactive oxygen species in a human liver cell line: the role of anethole dithiolethione as a scavenger. Toxicol Vitro. 1999;13:683–8.
Fasehee H, Ghavamzadeh A, Alimoghaddam K, Ghaffari SH, Faghihi S. A comparative cytotoxic evaluation of disulfiram encapsulated PLGA nanoparticles on MCF-7 cells. Int J Hematol Oncol Stem Cell Res. 2017;11:102–7.
Wiggins HL, Wymant JM, Solfa F, Hiscox SE, Taylor KM, Westwell AD, et al. Disulfiram-induced cytotoxicity and endo-lysosomal sequestration of zinc in breast cancer cells. Biochem Pharmacol. 2015;93:332–42.
Yip NC, Fombon IS, Liu P, Brown S, Kannappan V, Armesilla AL, et al. Disulfiram modulated ROS-MAPK and NFkappaB pathways and targeted breast cancer cells with cancer stem cell-like properties. Br J Cancer. 2011;104:1564–74.
Chou TC. Drug combination studies and their synergy quantification using the Chou-Talalay method. Cancer Res. 2010;70:440–6.
Biederbick A, Kern HF, Elsasser HP. Monodansylcadaverine (MDC) is a specific in vivo marker for autophagic vacuoles. Eur J Cell Biol. 1995;66:3–14.
Cordani M, Donadelli M, Strippoli R, Bazhin AV, Sanchez-Alvarez M. Interplay between ROS and autophagy in cancer and aging: from molecular mechanisms to novel therapeutic approaches. Oxid Med Cell Longev. 2019;2019:8794612.
Chen YF, Liu H, Luo XJ, Zhao Z, Zou ZY, Li J, et al. The roles of reactive oxygen species (ROS) and autophagy in the survival and death of leukemia cells. Crit Rev Oncol Hematol. 2017;112:21–30.
Hassani S, Ghaffari P, Chahardouli B, Alimoghaddam K, Ghavamzadeh A, Alizadeh S, et al. Disulfiram/copper causes ROS levels alteration, cell cycle inhibition, and apoptosis in acute myeloid leukaemia cell lines with modulation in the expression of related genes. Biomed Pharmacother. 2018;99:561–9.
Zhang J, Wang J, Wong YK, Sun X, Chen Y, Wang L, et al. Docetaxel enhances lysosomal function through TFEB activation. Cell Death Dis. 2018;9:614.
Hung CH, Chan SH, Chu PM, Tsai KL. Docetaxel facilitates endothelial dysfunction through oxidative stress via modulation of protein kinase C Beta: the protective effects of sotrastaurin. Toxicol Sci. 2015;145:59–67.
Ye X, Zhou XJ, Zhang H. Exploring the role of autophagy-related gene 5 (ATG5) yields important insights into autophagy in autoimmune/autoinflammatory diseases. Front Immunol. 2018;9:2334.
Azad MB, Chen Y, Gibson SB. Regulation of autophagy by reactive oxygen species (ROS): implications for cancer progression and treatment. Antioxid Redox Signal. 2009;11:777–90.
Li YJ, Lei YH, Yao N, Wang CR, Hu N, Ye WC, et al. Autophagy and multidrug resistance in cancer. Chin J Cancer. 2017;36:52.
Morikawa Y, Koike H, Sekine Y, Matsui H, Shibata Y, Ito K, et al. Rapamycin enhances docetaxel-induced cytotoxicity in a androgen-independent prostate cancer xenograft model by survivin downregulation. Biochem Biophys Res Commun. 2012;419:584–9.
Bishop E, Bradshaw TD. Autophagy modulation: a prudent approach in cancer treatment? Cancer Chemother Pharmacol. 2018;82:913–22.
Buoncervello M, Borghi P, Romagnoli G, Spadaro F, Belardelli F, Toschi E, et al. Apicidin and docetaxel combination treatment drives CTCFL expression and HMGB1 release acting as potential antitumor immune response inducers in metastatic breast cancer cells. Neoplasia. 2012;14:855–67.
Wang W, McLeod HL, Cassidy J. Disulfiram-mediated inhibition of NF-kappaB activity enhances cytotoxicity of 5-fluorouracil in human colorectal cancer cell lines. Int J Cancer. 2003;104:504–11.
Duan X, Xiao J, Yin Q, Zhang Z, Yu H, Mao S, et al. Smart pH-sensitive and temporal-controlled polymeric micelles for effective combination therapy of doxorubicin and disulfiram. ACS Nano. 2013;7:5858–69.
Guo X, Xu B, Pandey S, Goessl E, Brown J, Armesilla AL, et al. Disulfiram/copper complex inhibiting NFkappaB activity and potentiating cytotoxic effect of gemcitabine on colon and breast cancer cell lines. Cancer Lett. 2010;290:104–13.
Nechushtan H, Hamamreh Y, Nidal S, Gotfried M, Baron A, Shalev YI, et al. A phase IIb trial assessing the addition of disulfiram to chemotherapy for the treatment of metastatic non-small cell lung cancer. Oncologist. 2015;20:366–7.
Ogura T, Tanaka Y, Tamaki H, Harada M. Docetaxel induces Bcl-2- and pro-apoptotic caspase-independent death of human prostate cancer DU145 cells. Int J Oncol. 2016;48:2330–8.
Hernandez-Vargas H, Palacios J, Moreno-Bueno G. Molecular profiling of docetaxel cytotoxicity in breast cancer cells: uncoupling of aberrant mitosis and apoptosis. Oncogene. 2007;26:2902–13.
Marquez RT, Xu L. Bcl-2: beclin 1 complex: multiple, mechanisms regulating autophagy/apoptosis toggle switch. Am J Cancer Res. 2012;2:214–21.
Pattingre S, Levine B. Bcl-2 inhibition of autophagy: a new route to cancer? Cancer Res. 2006;66:2885–8.
Jivan R, Peres J, Damelin LH, Wadee R, Veale RB, Prince S, et al. Disulfiram with or without metformin inhibits oesophageal squamous cell carcinoma in vivo. Cancer Lett. 2018;417:1–10.
Denton D, Nicolson S, Kumar S. Cell death by autophagy: facts and apparent artefacts. Cell Death Differ. 2012;19:87–95.
Gump JM, Thorburn A. Autophagy and apoptosis: what is the connection? Trends Cell Biol. 2011;21:387–92.
Bialik S, Dasari SK, Kimchi A. Autophagy-dependent cell death—where, how and why a cell eats itself to death. J Cell Sci. 2018;131. https://doi.org/10.1242/jcs.215152
Mauthe M, Orhon I, Rocchi C, Zhou X, Luhr M, Hijlkema KJ, et al. Chloroquine inhibits autophagic flux by decreasing autophagosome-lysosome fusion. Autophagy. 2018;14:1435–55.
Singh SS, Vats S, Chia AY, Tan TZ, Deng S, Ong MS, et al. Dual role of autophagy in hallmarks of cancer. Oncogene. 2018;37:1142–58.
Lohitesh K, Saini H, Srivastava A, Mukherjee S, Roy A, Chowdhury R. Autophagy inhibition potentiates SAHAmediated apoptosis in glioblastoma cells by accumulation of damaged mitochondria. Oncol Rep. 2018;39:2787–96.
Cristofani R, Montagnani Marelli M, Cicardi ME, Fontana F, Marzagalli M, Limonta P, et al. Dual role of autophagy on docetaxel-sensitivity in prostate cancer cells. Cell Death Dis. 2018;9:889.
Hildeman DA, Mitchell T, Aronow B, Wojciechowski S, Kappler J, Marrack P. Control of Bcl-2 expression by reactive oxygen species. Proc Natl Acad Sci USA. 2003;100:15035–40.
Acknowledgements
We express our sincere gratitude to Dr. Kumar Sankar Bhattacharya and Dr. Syamantak Majumder for helping us with copy-editing the manuscript.
Funding
The authors like to acknowledge financial support from ‘Department of Science and Technology (DST), Govt. of India’ under the project (ECR/2016/000566/LS), as well as to BITS-Pilani for core research grant support. KLS acknowledges fellowship support from the Council of Scientific & Industrial Research (CSIR), India.
Author information
Authors and Affiliations
Contributions
All the data presented in the study were generated in-house. No data was used from any other source. KLS and SS performed the experiments and analyzed the data. KLS, RC, and AR designed the study plan and wrote the manuscript.
Corresponding author
Ethics declarations
Conflict of interest
The authors declare no conflicts of interest.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
About this article

Cite this article
Swetha, K.L., Sharma, S., Chowdhury, R. et al. Disulfiram potentiates docetaxel cytotoxicity in breast cancer cells through enhanced ROS and autophagy. Pharmacol. Rep 72, 1749–1765 (2020). https://doi.org/10.1007/s43440-020-00122-1
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s43440-020-00122-1