
Abstract
L-Aspartate β-decarboxylase from Acinetobacter radioresistens (ArASD) has been modified to convert 3-methylaspartic acid into 2-aminobutyric acid, which activated a novel process for biosynthesis of 2-aminobutyric acid. However, the process is limited by the low activity of the ArASD. Here, the activity of ArASD was significantly improved by modification based on sequence alignment and structural analysis. The 38th residue of ArASD is speculated to be the key residue for regulating the conformation of the internal aldimine, and site-directed mutagenesis on R38 residue was carried out. A variant, K18A/R38K/V287I, with 2.2 times higher specific activity was isolated. Molecular dynamics simulation indicated that the torsion angle of the imine bond of the variant decreased, which was beneficial to the protonation of the internal aldimine and the increase in the initial energy of the enzyme. Therefore, the energy barrier of the transition state was reduced, resulting in improved catalytic activity toward 3-methylaspartic acid. These results provide a reference and a new point of view for enzyme modification by increasing the energy of the initial state.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
L-Aspartate-β-decarboxylase (ASD) is the only enzyme in nature that catalyzes the reaction of β-decarboxylation and belongs to the family of aspartate aminotransferases. ASD is composed of 12 subunits, each of which combines a coenzyme molecule of pyridoxal 5'-phosphate (PLP) in the active center [1]. The family of PLP-dependent enzymes is divided into five groups (I-IV), and ASD belongs to group I, similar to aspartate aminotransferase, whose structure and catalytic mechanism have been well studied. Furthermore, the structure of the active center of ASD was similar to that of aspartate aminotransferase [2, 3].
The coenzyme PLP covalently formed a Schiff base (the internal aldimine) with lysine (Lys315) in the active site of ASD from Pseudomonas dacunhae (PdASD) [4]. A hydrogen-bond network was formed between a series of key amino acids in the active center and PLP, which was responsible for stabilization and fixation of the phosphate oxygen group of PLP [5]. When the substrate entered the active site, the α and β carboxyl groups of the substrate L-aspartic acid formed ion pairs with Arg497 and Arg375, followed by the internal aldimine undergoing a transaldimine reaction and another Schiff base (the external aldimine) being formed between PLP and the α-amino group of the substrate [4, 5]. The conformation of ASD underwent changes during the transformation of the internal aldimine to the external aldimine, and Lys315 in the active center was involved in controlling the protonation state of the Cα of the substrate L-aspartic acid and the C4' of PLP [6, 7]. In the catalytic process, the β-carboxyl group of L-aspartic acid was perpendicular to the reface of PLP, polarized the β–γ bond and provided electron traps, which was beneficial to the β-decarboxylation of L-aspartic acid [8]. Many PLP-dependent enzymes possess a similar mechanism to break the β–γ bond of the substrate, such as selenocysteine lyase, cysteine desulfurase and kynureninase [9,10,11,12].
Ground state destabilization has been found in many enzymes and generally refers to the unstable interaction (also called force) between the enzyme and the substrate [13]. The force of the internal aldimine of aspartate aminotransferase was maintained by Lys258 and Asn194 in the active center [14, 15]. This force could increase the energy of the initial state (enzyme + substrate), thereby reducing the energy barrier between the initial state and the transition state and increasing the catalytic ability of the enzyme. During the reaction catalyzed by aspartate aminotransferase, the change in the force of the internal aldimine was accompanied by the change in the pKa of the aldimine, which was the proton state of the internal aldimine. The pKa of the internal aldimine of aspartate aminotransferase was 6.8, and the pKa of the internal aldimine increased to 8.8 when the substrate entered the active center. Then the pKa of the external aldimine increased to more than 10 when the internal aldimine was further converted to the external aldimine [14, 15]. Similar forces of the internal aldimines were found in aromatic amino acid transaminase and histidine phosphate transaminase [16, 17].
The Arg37 of PdASD is conserved in the active center of ASD and plays an important role in the protonation regulation of the internal aldimine, which does not exist in aspartate aminotransferase [1, 8]. There was an absorption peak at a wavelength of 425 nm in PdASD, which corresponded to the protonated internal aldimine. However, such an absorption peak at a wavelength of 425 nm was not seen in the mutant R37A, indicating that the internal aldimine was in a nonprotonated state [8]. The proton state of the internal aldimine was determined by the force of the internal aldimine, which has been confirmed in aspartate aminotransferase [15]. Therefore, Arg37 played an important role in controlling the proton state of the internal aldimine and essentially regulated the forces of the internal aldimines. Furthermore, Arg37 could be an important residue for the activation energy between the initial state and the transition state of ASD.
In this study, Arg37 of ArASD was modified using semirational modification to regulate the force of the internal aldimine. A previously engineered variant of ArASD, K18A/V287I, was used as a starting enzyme, because the wild type cannot catalyze the conversion of 3-methylaspartate into 2-aminobutyric acid. A variant with higher activity for 3-methylaspartic acid was obtained. The mechanism of the catalytic efficiency improvement, related to the force of the internal aldimine of ArASD variant, was assayed by molecular dynamic simulation.
Materials and methods
Bacterial strains and plasmids
Escherichia coli JM109 and BL21 (DE3) were used for plasmid construction and enzyme expression, respectively. Luria–Bertani (LB) medium (10 g/L tryptone, 5 g/L yeast extract and 10 g/L NaCl) was used for cell growth, and 2YT medium (16 g/L tryptone, 10 g/L yeast extract and 5 g/L NaCl) was used for enzyme expression. All chemicals used in this study were of reagent grade.
Genetic manipulation
The plasmids containing the ArASD K18A/V287I gene were used as the template for variant construction, which was constructed in our previous study [18]. Site-directed mutagenesis was carried out using whole plasmid PCR, and the primers are listed in Table S1. The PCR product was transformed into E. coli JM109 competent cells. The variants were further verified by DNA sequencing.
Expression and purification of ArASD variants
A single colony of E. coli BL21(DE3) harboring the plasmid was inoculated into 2YT medium with 50 μg/mL kanamycin and cultivated at 37 °C and 200 rpm. The inducer of 0.2 mM IPTG was added to induce expression of the enzyme when OD600 reached 0.8, and cells were cultivated at 25 °C for 16 h. Cells were harvested and lysed by sonication in potassium phosphate buffer (10 mM, pH 7.0), followed by centrifugation at 10 krpm for 10 min at 4 °C.
Cell lysates were passed through a HisTrap HP column (GE Heathcare), and enzymes were purified using a linear gradient of imidazole (0–500 mM). Purified enzymes were dialyzed against potassium phosphate buffer (10 mM, pH 7.0) at 4 °C overnight to remove imidazole. The concentration of purified enzyme was measured by the Bradford method.
Characterization of ArASD variants
The activity of the ArASD variant was detected in a mixture of the corresponding buffer (10 mM) with 1 mM α-ketoglutaric acid and 0.5 mM PLP. After preincubation at the corresponding temperature for 5 min, substrate was added to start the reaction for 10 min, which was subsequently inactivated at 100 °C for 10 min. The specific activities and kinetic parameters of ArASD variants toward L-aspartic acid were determined at 50 °C and pH 5.4, and those toward 3-methylasparic acid were determined at 50 °C and pH 3.8. Statistical analyses were performed using GraphPad Prism 7 software.
One unit of decarboxylation activity of the ArASD variant toward L-aspartic acid (or 3-methylasparic acid) was defined as the amount of enzyme that catalyzed the formation of 1 μmol of L-alanine (or 2-aminobutyric acid) per min. The concentrations of L-alanine and 2-aminobutyric acid were measured by high performance liquid chromatography using a Diamonsil C18 column (5 μm, 250 × 4.6 mm, Dikma, China) after derivatization by phenyl isosulfate (PITC). The derivative conditions were as follows: the volume of the reaction was made up to 500 μL by adding 250 μl PITC–acetonitrile solution (0.1 mM) and 250 μl triethylamine–acetonitrile solution (1 mM). The detection conditions were as follows: mobile phase A was 80% acetonitrile, mobile phase B was a mixture of 97% 0.1 M sodium acetate and 3% acetonitrile, the detection wavelength was 254 nm, the flow rate was 0.6 mL/min, the injection volume was 10 μL, and the column temperature was 40 °C.
The optimal pH of the ArASD variant was detected at 50 °C in various buffers (pH 2.0–8.0), and the optimal temperature was detected at various temperatures (30–60 °C) in acetate buffer (pH 3.8). The same amount of the ArASD variant was incubated in a water bath at 40, 45, 50 °C for 120 min, and the residual activity was detected to determine the thermal stability of the ArASD variant. The same amount of the ArASD variant in different pH buffers (pH 3.0–8.0) was kept on ice for more than 12 h, and the residual activity was detected to determine the pH stability of the ArASD variant.
Molecular dynamic simulations assay
The structures of ArASD and its variant were built by SWISS-MODEL software using Alcaligenes faecali ASD (PDB ID: 2yz4) as the template. Molecular dynamics (MD) simulations were performed using NAMD 2.13 with the force field Charmm27. The protonation state of residues was determined with the PDB2PQR server (http://apbs-rest-test.westus2.cloudapp.azure.com/). The topology and parameters of PLP for simulation were generated by the swissparam server (http://www.swissparam.ch/). The MD simulations included 1 ns of water equilibration, 60 ps of heating protein from 0 K up to 300 K, and 50 ns classical MD simulations.
Results and discussion
The key residue of ArASD for regulating the internal aldimine
As described above, the residue Arg37 essentially regulated the internal aldimine conformation of PdASD, which could enhance the catalytic efficiency of PdASD. Based on the amino acid sequence alignment of PdASD and ArASD, Arg38 of ArASD corresponded to Arg37 of PdASD (Fig. S1). Therefore, Arg38 was selected as a modification candidate that was expected to improve the catalytic ability of ArASD toward 3-methylasparic acid. The starting enzyme was K18A/V287I that catalyzed the conversion of 3-methylaspartate into 2-aminobutyric acid [18].
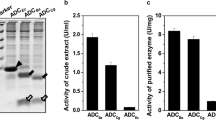
Arg38 of K18A/V287I was modified as seven representative amino acids: alanine (Ala), leucine (Leu), glutamic acid (Glu), glutamine (Gln), lysine (Lys), histidine (His) and phenylalanine (Phe). The activities of crude enzymes showed that the variant K18A/R38K/V287I significantly increased the activity toward 3-methylaspartic acid, whereas the other variants exhibited decreased activities toward 3-methylaspartic acid (Fig. 1). No activity toward 3-methylaspartic acid was detected when the positively charged Arg38 was mutated into the negatively charged Glu, which might be caused by the hydrogen bonds between the phosphate group of the coenzyme PLP and ArASD being broken and the force of the internal aldimine being destroyed. Similarly, the activities of variants decreased sharply as Arg38 was mutated into uncharged Ala, Leu, Gln and Phe. Compared with Arg, both Lys and His are positively charged residues and have one amino group on the side chain, which might weaken the interactions between the enzyme and PLP and affect the conformation of the internal aldimine. The amino acid at position 38 has an important influence on the activity of ArASD.

The β-decarboxylation activity of variants toward 3-methylaspartic acid. The activity was determined at 45 °C for 10 min using crude enzymes. Reaction solution: 10 mM potassium phosphate buffer (pH 7.0), 0.5 mM PLP, 0.5 mM α-ketoglutarate, and 50 mM DL-threo-3-methylasparic acid. The activity of K18A/V287I was defined as 100%
Enzymatic properties of K18A/R38K/V287I
As the variant K18A/R38K/V287I exhibited the highest activity toward 3-methylaspartic acid, it was purified for further study. The optimal catalytic temperature and pH for K18A/R38K/V287I toward 3-methylaspartic acid were 50 °C (Fig. 2a) and pH 3.4–3.8 (Fig. 2b), which was similar to that of K18A/V287I [18]. K18A/R38K/V287I retained 63% of its activity after treatment at 50 °C for 120 min (Fig. 2c). Compared with K18A/V287I, which only retained less than 20% of its activity after treatment at 50 °C for 120 min, the thermostability of K18A/R38K/V287I was much better than that of K18A/V287I. K18A/R38K/V287I was stable at pH 3.4–7.8 (Fig. 2d), which was also similar to that of K18A/V287I.

Enzymatic properties of K18A/R38K/V287I using 3-methylaspartic acid as the substrate. The optimal temperature (a) and pH (b) of K18A/R38K/V287I toward 3-methylaspartic acid were measured, and the highest activity was plotted as 100%. c The thermostability of K18A/R38K/V287I. Purified K18A/R38K/V287I was treated at different temperatures, and the activity of the untreated enzyme was plotted as 100%. d The pH stability of K18A/R38/V287I. The purified K18A/R38K/V287I was treated in the corresponding buffers at 4 °C overnight, and the highest enzyme activity was plotted as 100%. Error bars indicate the standard deviation of three independent experiments
The β-decarboxylation activities of K18A/V287I and K18A/R38K/V287I toward 3-methylaspartic acid and L-aspartic acid were compared (Table 1). The specific activity of K18A/R38K/V287I toward 3-methylaspartic acid was 41.5 ± 0.51 U/mg, which was 2.24-fold higher than that of K18A/V287I. The results of enzyme kinetic parameters showed that the turnover number (kcat) of K18A/R38K/V287I for both substrates was significantly increased, but the affinities for both substrates were decreased. The variant K18A/V287I had a similar affinity toward 3-methylaspartic acid and L-aspartic acid; however, K18A/R38K/V287I had a higher affinity (Km) toward 3-methylaspartic acid than L-aspartic acid. We considered that K18A/R38K/V287I was switched to 3-methylaspartate β-decarboxylase from L-aspartate β-decarboxylase, on the basis of the increased discrimination between 3-methylaspartic acid and the natural substrate L-aspartic acid. The catalytic efficiency of K18A/R38K/V287I (kcat/Km) toward 3-methylaspartic acid was higher than that of K18A/V287I. However, kcat/Km of K18A/R38K/V287I toward L-aspartic acid was much lower than that of K18A/V287I, which might be due to the lower affinity.
Structural basis of K18A/R38K/V287I for improved catalytic efficiency
Hayashi et al. speculated that the torsion angle of the aldimine bond (C3–C4–C4'–Nε, θ') in aspartate aminotransferase played an important role in force formation by analyzing the structures of the protonated and deprotonated internal aldimine [14]. The active center of ArASD is similar to that of aspartate aminotransferase, while C4' of PLP (C4'PLP) is linked by the imine bond with ε-N of Lys314 (Nε314). Meanwhile, hydrogen bonds were formed between the oxygen atom of the hydroxyl group on C3' of PLP (O3'PLP) and Nδ2 of Asn256 (Nδ2256) (Fig. 3).

Structural analysis of K18A/V287I and K18A/R38K/V287I. a The active sites of K18A/V287I. b The active sites of K18A/R38K/V287I. The yellow dotted lines represent hydrogen bonds. The distances of Nδ2256–O3'PLP and Nε314–O3'PLP are displayed and designated by red dotted lines
Two hydrogen bonds were formed between the side chain of Arg38 and O3'PLP in K18A/V287I (Fig. 3a). However, only one hydrogen bond was formed between Lys38 and O3'PLP in K18A/R38K/V287I (Fig. 3b), and the interaction between Lys38 and PLP was weakened. The conformation of the internal aldimine in K18A/R38K/V287I was changed, leading to a change in the catalytic efficiency.
MD simulation analysis of K18A/V287I and K18A/R38K/V287I showed that the distance between the Cα of Lys314 (Cα314) and C4'PLP in K18A/R38K/V287I was shorter (Fig. 4a). As shown in Fig. 4b, the distance of Cα314–C4'PLP in K18A/V287I was mainly distributed at approximately 7.0 Å, while that in K18A/R38K/V287I was mainly distributed at approximately 6.0 Å. The shorter distance would cause the imine bond of the internal aldimine in K18A/R38K/V287I to be close to the pyridine ring of PLP, and it resulted in an easily protonated internal aldimine and increased the free energy of the enzyme and substrate. The distance between Nδ2256 and O3'PLP in K18A/R38K/V287I was longer (Fig. 4c) and was mainly distributed at approximately 5–6 Å, which was 1.5–2 times that of K18A/V287I (Fig. 4d). The longer distance between Nδ2256 and O3'PLP meant that the interaction between PLP and Asn256 in K18A/R38K/V287I was weakened, thereby promoting protonation of the internal aldimine.

MD simulation analysis of K18A/V287I and K18A/R38K/V287I. a The distance between Cα314 and C4'PLP. b The distribution of the distance between Cα314 and C4'PLP. c The distance between Nδ2256 and O3'PLP. d The distribution of the distance between Nδ2256 and O3'PLP
The torsion angle of the imine bond (θ) was calculated and it was mainly distributed at approximately 40° in K18A/V287I; however, it was distributed at approximately 30° in K18A/R38K/V287I (Fig. 5). The bond of the internal aldimine in K18A/R38K/V287I more easily formed a coplanar structure, which was beneficial to the protonation of the internal aldimine. All three factors (the longer distance between Nδ2256 and O3'PLP, Cα314 and C4'PLP, θ) resulted in hydrogen bonds between Nε314 and O3'PLP in K18A/R38K/V287I being more easily formed to stabilize the coplanar structure and the protonation of the internal aldimine state [14]. The weakened hydrogen bonds between Lys38 and O3'PLP in K18A/R38K/V287I led to a greater force generated by the internal aldimine and an increasing catalytic efficiency.

Distribution of the torsion angle of the imine bond in ArASD variants calculated by MD simulations
In addition, the hydrogen-bond network between the α- and β-carboxyl groups of the substrate and Arg386, Arg292 and Asn194 of the active center of aspartate aminotransferase played an important role in maintaining the affinity of the substrate [15, 19, 20]. Based on the similarity of the active centers of ASD and aspartate aminotransferase, it was speculated that a similar hydrogen-bond network existed between ASD and the substrate. Based on sequence alignment, hydrogen bonds between the substrate and Arg496 and Arg374 of the active center and the α-carboxyl group of the substrate and Nδ2256 are formed [4, 5]. Compared with K18A/V287I, the longer distance of Nδ2256-O3'PLP in K18A/R38K/V287I (Fig. 4D) may destroy the hydrogen bonds between Nδ2256 and the α-carboxyl oxygen atom of the substrate. The hydrogen-bond network between the substrate and the active center of ArASD could be further destroyed, resulting in a decrease in the affinity for the substrate, especially for the natural substrate L-aspartic acid.
Conclusion
In this study, a key residue of ArASD for maintaining the conformation of the internal aldimine was modified, and a variant K18A/R38K/V287I that had higher catalytic efficiency for 3-methylaspartic acid was obtained. Compared with K18A/V287I, which was constructed in our previous study, the hydrogen bonds between the 38th residue and PLP were weakened and the conformation of the internal aldimine was changed in K18A/R38K/V287I, leading to the increased energy of the initial state and the reduced energy barrier for the transition state, further significantly improving the catalytic ability of ArASD toward 3-methylaspartic acid.
Availability of data and material
The data generated during and/or analyzed during the current study are available from the corresponding author on reasonable request.
Code availability
Not applicable.
References
Wang NC, Lee CY. Molecular cloning of the aspartate 4-decarboxylase gene from Pseudomonas sp. ATCC 19121 and characterization of the bifunctional recombinant enzyme. Appl Microbiol Biotechnol. 2006;73:339–48.
Jansonius JN. Structure, evolution and action of vitamin B6-dependent enzymes. Curr Opin Struct Biol. 1998;8:759–69.
Dajnowicz S, Parks MJ, Hu X, Gesler K, Andrey Y, Kovalevsky YA, Mueser C. Direct evidence that an extended hydrogen-bonding network influences activation of pyridoxal 5′-phosphate in aspartate aminotransferase. J Biol Chem. 2017;292:5970–80.
Chen HJ, Ko TP, Lee CY, Wang NC, Wang AH. Structure, assembly, and mechanism of a PLP-dependent dodecameric L-aspartate beta-decarboxylase. Structure. 2009;17:517–29.
Lima S, Sundararaju B, Huang C, Khristoforov R, Momany C, Phillips SR. The crystal structure of the Pseudomonas dacunhae aspartate-b-decarboxylase dodecamer reveals an unknown oligomeric assembly for a pyridoxal-5′-phosphate-dependent enzyme. J Mol Biol. 2009;388:98–108.
Chang CC, Laghai A, O’Leary MH, Floss HG. Some stereochemical features of aspartate beta-decarboxylase. J Biol Chem. 1982;257:3564–9.
Ngo HPT, Cerqueira MFSAN, Kim JK, Hong MK, Fernandes PA, Ramos JM, et al. PLP undergoes conformational changes during the course of an enzymatic reaction. Acta Crystallogr A. 2014;70:596–606.
Phillips SR, Lima S, Khristoforov R, Sudararaju B. Insights into the mechanism of Pseudomonas dacunhae aspartate beta-decarboxylase from rapid-scanning stopped-flow kinetics. Biochemistry. 2010;49:5066–73.
Esaki N, Karai N, Nakamura T, Tanaka H, Soda K. Mechanism of reactions catalyzed by selenocysteine beta-lyase. Arch Biochem Biophys. 1985;238:418–23.
Zheng L, White HR, Cash LV, Dean RD. Mechanism for the desulfurization of L-cysteine catalyzed by the nifS gene product. Biochemistry. 1994;33:4714–20.
Toney MD. Reaction specificity in pyridoxal phosphate enzymes. Arch Biochem Biophys. 2005;433:279–87.
Christen P, Mehta PK. From cofactor to enzymes. The molecular evolution of pyridoxal-5’-phosphate-dependent enzymes. Chem Record. 2010;1:436–47.
Hayashia H, Mizuguchia H, Miyaharab I, MainulIslama M, Hiroko I, Nakajimab Y, et al. Strain and catalysis in aspartate aminotransferase. Biochem Biophys Acta. 2003;1647:103–9.
Hayashi H, Mizuguchi H, Kagamiyama H. The imine-pyridine torsion of the pyridoxal 5’-phosphate Schiff base of aspartate aminotransferase lowers its pKa in the unliganded enzyme and is crucial for the successive increase in the pKa during catalysis. Biochemistry. 1998;37:15076–85.
Mizuguchi H, Hayashi H, Okada K, Miyahara I, Hirotsu K, Kagamiyama H. Strain is more important than electrostatic interaction in controlling the pKa of the catalytic group in aspartate aminotransferase. Biochemistry. 2001;40:353–60.
Islam MM, Hayashi H, Mizuguchi H, Kagamiyama H. The substrate activation process in the catalytic reaction of Escherichia coli aromatic amino acid aminotransferase. Biochemistry. 2000;39:15418–28.
Mizuguchi H, Hayashi H, Miyahara I, Hirotsu K, Kagamiyama H. Characterization of histidinol phosphate aminotransferase from Escherichia coli. Biochem Biophys Acta. 2003;1647:321–4.
Liu Y, Han L, Cheng Z, Liu Z, Zhemin Z. Enzymatic biosynthesis of L-2-aminobutyric acid by glutamate mutase coupled with L-aspartate-β-decarboxylase using L-glutamate as the sole substrate. ACS Catal. 2020;10:13913–7.
Danishefsky TA, Onnufer JJ, Petsko GA, Ringe D. Activity and structure of the active-site mutants R386Y and R386F of Escherichia coli aspartate aminotransferase. Biochemistry. 1991;30:1980–5.
Jäger J, Moser M, Sauder U, Jansonius JN. Crystal structures of Escherichia coli aspartate aminotransferase in two conformations: comparison of an unliganded open and two liganded closed forms. J Mol Biol. 1994;239:285–305.
Funding
This work was supported by National Key R&D Program of China (2017YFE0129600), the National Natural Science Foundation of China (21878125), and the Priority Academic Program Development of Jiangsu Higher Education Institutions, the 111 Project (No. 111-2-06).
Author information
Authors and Affiliations
Contributions
Yufeng Liu has a major contribution to the study and performed experiments of construction, characterization, MD analysis of variants. Mingzhu Hao contributed to enzymatic properties analysis of variants. Zhongmei Liu conceived the project and designed the experiments. The manuscript was drafted by Yufeng Liu and Zhongmei Liu, revised by Zhemin Zhou, and approved by all co-authors.
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that there is no conflict of interest regarding the publication of this article.
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
About this article

Cite this article
Liu, Y., Hao, M., Zhou, Z. et al. Isolation and mechanism analysis of a catalytic efficiency improved L-aspartate β-decarboxylase toward 3-methylaspartic acid. Syst Microbiol and Biomanuf 2, 157–164 (2022). https://doi.org/10.1007/s43393-021-00049-5
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s43393-021-00049-5