
Abstract
Long-term intensive greenhouse production commonly leads to continuous cropping obstacles and therefore declines the productivity of greenhouse system, soil microbiological mechanisms behind which remain poorly understood so far. Here, based on a continuous greenhouse cucumber cropping system, the differences in bacterial community structure of the soils with different cucumber cultivation history were assessed through high-throughput sequencing. The β diversity of bacterial community, but not α diversity, significantly changed as consecutive cucumber cultivation and also positively linked to cucumber yield. As for bacterial community members, at phylum level, prolonged cucumber cultivation increased average relative abundances of Chloroflexi and Gemmatimonadetes while decreasing average relative abundance of Nitrospirae; at genus level, continuous cucumber cultivation decreased average relative abundances of some beneficial microbes (i.e. Bacillus, Solirubrobacter, and Rubrobacter) and N-cycling related microbes (i.e. Nitrospira and Azoarcus) while increasing average relative abundance of some functional microbes (i.e. Agromyces, Thermomicrobium, Desulfotomaculum, Sphaerobacter, and Mycobacterium). Soil available phosphorus and organic carbon contents were the most important contributors to the shifts of bacterial community structure. Co-occurrence network analysis indicated long-term greenhouse cucumber cultivation weakened the interaction of species within bacterial community and led to less complex and connected organization among bacterial taxa. Overall, our results suggested that soil bacterial community structure and the interactions of bacterial taxa may play the important roles in the occurrence of continuous cropping obstacles in a long-term intensive greenhouse production system.
Similar content being viewed by others

Explore related subjects
Discover the latest articles, news and stories from top researchers in related subjects.Avoid common mistakes on your manuscript.
1 Introduction
Greenhouse vegetable cultivation is one of the most important agricultural production patterns worldwide. China is the largest vegetable producer in the world, and greenhouse vegetable cultivation has become a pillar industry in many areas of this country. Due to the growing demand for vegetable consumption and the limited cultivated land, continuous cropping of greenhouse vegetables is becoming increasingly prevalent. However, this type of agricultural practice commonly incurs continuous cropping obstacles and substantially declines the productivity of greenhouse system, which heavily restricts healthy development of vegetable industry (Li and Yang 2016). Increasing the knowledge available regarding the mechanisms behind continuous cropping obstacles can help us to develop effective approaches to overcome this difficult problem present in long-term intensive greenhouse vegetable production.
Soil quality is of crucial importance in the sustainability of agricultural productivity because it controls crop yield and health (Bünemann et al. 2018; Lal 2015). Prolonged greenhouse cultivation commonly contributes to soil quality deterioration (also referred as soil sickness) and then negatively feeds back to crop growth (Mariotte et al. 2018; Huang et al. 2013). Therefore, the alterations in soil properties with increasing greenhouse cultivation history are thought to be the important reasons behind continuous cropping obstacles, such as soil acidification and salinization, soil nutrient depletion and imbalance, release and accumulation of phytotoxic and autotoxic compounds, and build-up of soil-borne pathogen and parasite populations (Chen et al. 2015; Huang et al. 2006). In addition, soil biological properties are also attracting more and more attention, particularly soil microbiome, which occupies vital position in soil quality framework and play a potent role in sustaining agricultural productivity (Mohanram and Kumar 2019; Zhang et al. 2017; Chaparro et al. 2012).
Soil microbial community is an integral part of soil ecosystem and is responsible for maintaining soil quality and mediating plant productivity as it performs heterogeneous functions during soil biological processes (Luo et al. 2018; Chaparro et al. 2012). It has been well recognized that cropping regimes and land use types influence soil microbes forcefully, including their abundances and community structures. However, by comparison with the open field crop production, greenhouse cultivation is commonly featured with high planting intensity, high nutrient inputs, high irrigation frequency, and relatively high soil temperatures, all of which can alter soil physicochemical environment more profoundly and then feed back to soil microbiome (Liu et al. 2019). Therefore, monitoring the shifts in soil microbiome during prolonged greenhouse vegetable production and then exploring their potential links with continuous cropping obstacles are extremely necessary. Our previous study indicated that consecutive greenhouse cucumber cultivation obviously affected soil N turnover through altering the community structures of bacterial nitrifiers (Liu et al. 2019). However, it remains unclear that how soil microbes respond to consecutive greenhouse vegetable cultivation at overall bacterial community level. Although some researchers have reported the responses of soil microbial communities to prolonged greenhouse vegetable cultivation through various traditional methods, such as CLPP, RAPD, Polymerase Chain Reaction-Denaturing Gradient Gel Electrophoresis (PCR-DGGE), and phospholipid fatty acid (PLFA) (Fu et al. 2017; Yao et al. 2016, 2006; Zhou et al. 2014; Zhou and Wu 2012), some important problems, namely, how bacterial community structure respond to the extension of greenhouse cultivation history and even some potentially important shifts in typical or functional community members and species interactions, are still poorly understood.
Cucumber is one of the major vegetables subjected to greenhouse cultivation worldwide, and plant- and soil-related obstacles resulted from prolonged intensive greenhouse production are extremely prevalent in China. Based on a continuous cucumber cropping system located at Henan province of China, this study would assess the differences in bacterial community structures of greenhouse soils with different cucumber cultivation history. The results obtained here could increase the understanding about soil microbiological mechanisms underlying continuous cropping obstacles present in intensive greenhouse production system.
2 Materials and Methods
2.1 Study Region and Experiment Description
The study region was located at Zhuxintun village, Muye district, Xinxiang city, Henan province of central China. Here is always an important greenhouse vegetable production base since 1980s. The region has a warm temperate continental monsoon climate, and the four seasons are distinct; it is very cold in winter and hot in summer. The annual rainfall and air temperature average by 573 mm and 14 °C, respectively. There are about 205 frost-free days and 2400 h of sunshine annually. The soil type is typically classified as the loamy fluvo-aquic soil in China.
A total of 15 greenhouse plots in Zhuxintun village were randomly selected for this study. These plots had different histories of continuous cucumber cultivation, namely, over 1, 5, 10, 15, and 20 years, respectively, and there were three plots for each cultivation history as three replicates. Hence, five cultivation year treatments (denoted by CC1, CC5, CC10, CC15, and CC20, respectively) in triplicate were constructed. To maximumly remove potential impacts of planting and management regimes across different plots and years on soil sampling and data analysis, greenhouse plots tested were from the same farmers’ planting cooperatives and were relatively close to each other. Moreover, these tested plots were applied to identical local practices of cucumber cultivation in past two decades. The details on management and cultivation regimes have been described in our previous report (Liu et al. 2019).
Soil samples were collected on 18 October 2017 (the harvest time of cucumber in autumn). Using auger ten soil cores (depth in 0–20 cm) were randomly taken in each plot and then were thoroughly mixed after removing the debris as a composite sample. Fresh composite sample was divided into three subsamples. One subsample was stored at − 80 °C for molecular ecological assay, one subsample was allowed to air dry at room temperature for physicochemical analysis, and one subsample was immediately used for biochemical analysis. Soil physicochemical and biochemical properties under different treatments have been reported by Liu et al. (2019). Besides, cucumber yield in each plot within this growing season was obtained based on local farmers’ records.
2.2 Soil DNA Extraction, High-Throughput Sequencing, and Bioinformatic Analysis
Total microbial DNA in each soil sample was extracted via a PowerSoil® DNA Isolation Kit (MOBIO, Carlsbad, CA, USA) according to manufacturer’s protocol. DNA quality and purity were checked using 1% agarose gel electrophoresis and spectrophotometry. The genomic DNA was dissolved in 50 μl of elution buffer and stored at − 20 °C until further analysis.
High-throughput amplicon sequencing was performed to assess the changes in bacterial community structures with increasing cucumber cultivation history. The primer pair 338f/806r (Xu et al. 2016) was used to amplify the fragments of V3-V4 region of bacterial 16S rRNA gene for Illumina MiSeq sequencing (PE300 platform). The preliminary experiment, containing some randomly selected DNA templates plus four negative controls without DNA templates, was carried out to explore optimal PCR condition and ensure sufficient amount of amplification product. Finally, PCR was performed in a 20-μl reaction mixture containing 4 μl FastPfu buffer (5×), 2 μl dNTPs (2.5 mM), 0.4 μl FastPfu polymerase, 0.2 μl bovine serum albumin, 0.8 μl each primer (5 μM), 10 ng template DNA, and 11.2 μl ddH2O. PCR condition was as follows: pre-denaturation at 95 °C 3 min, 95 °C 30 s, 55 °C 30 s, 72 °C 40 s, 27 cycles, a final extension at 72 °C 10 min. Triplicate reaction mixtures for each sample were pooled together, purified through Agarose Gel DNA Purification Kit, and quantified using NanoDrop 2000. PCR products were finally sent to Shanghai Majorbio Bio-pharm Technology Co., Ltd. for sequencing. The samples were individually barcoded to enable multiplex sequencing. The barcoded PCR products from all samples were normalized in equimolar amounts before MiSeq sequencing. Sequencing reads recovered in this study have been deposited in NCBI Sequence Read Archive (SRA) database under accession number SRP174960.
Unprocessed FASTQ files were obtained for the analysis. The overlapping paired-end reads were assembled using FLASH software. Quality filtration and the analysis of sequences generated by Miseq sequencing were performed using Mothur software. Low-quality sequences were removed, including front primers containing two-base mismatch, sequences with at least eight successive identical bases, sequences with length shorter than 200 bp, sequences containing unidentifiable bases, and sequences with an average quality score less than 20. In total 805,765 high-quality sequences with average length 438 bp were obtained, which were further assigned to different samples and demultiplexed. Based on sequence similarity of 97%, the sequences were divided into different OTUs, and the most abundant sequence of each OTU was extracted as representative sequence. All representative sequences were classified with Silva 128/16S_bacteria database with 70% confidence threshold. A few representative sequences failed to classification were manually matched in NCBI database. The methodological information on bioinformatics analysis and calculation procedures applied here were fully described in I-Sanger cloud platform constructed by Shanghai Majorbio (http://www.isanger.com/).
2.3 Statistical Analysis
Statistical analysis was carried out by one-way ANOVA (Duncan’s test at 95% level of probability) to test significant differences between treatments using SPSS 21.0 software for windows. The mean values and standard deviations (n = 3) were expressed in this study. The occurrence of relationships between different data was assessed using one-variable regression models. The alpha and beta diversities for bacterial communities were assessed based on identical sequence number per soil sample. The weighted Unifrac algorithm-based hierarchical clustering tree was conducted to compare evolutionary distance of bacterial communities among tested soil samples. ANOSIM (analysis of similarities) test was applied to determine whether statistical differences in bacterial community structures were significant among treatments. By means of an algorithm for Bray-Curtis dissimilarity, principal co-ordinates analysis (PCoA) was applied to analyse the shifts in bacterial community structures as increasing greenhouse cultivation years. In addition, redundancy analysis (RDA) was performed to determine the relationships between bacterial community structures and environmental variables. Meanwhile, co-occurrence network analysis was conducted to compare the complexity of the interactions among bacterial taxa through R programming language and Gephi software.
3 Results
3.1 The α Diversity of Soil Bacterial Community
For each soil sample, 23,975 reads were randomly sampled from the original high-quality read pool to assess bacterial community. Rarefaction curve indicated that the numbers of reads sampled were enough to reflect the actual states of soil bacterial communities (Fig. 1). The α diversity of soil bacterial community was insensitive to consecutive greenhouse cucumber cultivation (Fig. 2). The number of observed OTU and Shannon index in CC5 was significantly lower than those in CC1, whereas no significant difference was observed for other treatments. Compared to CC1, increasing cucumber cultivation history did not alter Simpson index and Chao1 index.

Rarefaction curve

Effect of consecutive greenhouse cucumber cultivation on OTU-based α diversity of bacterial community structure
3.2 The β Diversity of Soil Bacterial Community
A total of 3078 OTUs were obtained in this study. The shared OTUs by tested soil samples were 1770 (Fig. 3), accounting for about 57.50% of total observed OTUs. Besides, the numbers of unique OTUs in CC1, CC5, CC10, CC15, and CC20 were 79, 12, 15, 15, and 46, respectively. These results revealed soil bacterial community composition was altered by consecutive greenhouse cucumber cultivation. Hierarchical clustering and PCoA analysis further displayed that continuous cucumber cultivation obviously altered the β diversity of bacterial community (Fig. 4), which could also be demonstrated by ANOSIM test. In PCoA plot, soil samples tested could be divided into three individual groups (CC1 samples as a single group, CC5 and CC10 and CC15 samples as the second group, and CC20 samples as the third group), and these three groups clearly separated each other on both PC1 and PC2 axes.

The shared and unique OTUs by tested soil samples (Venn diagram)

OTU-based hierarchical clustering tree (a) and PCoA analysis (b)
3.3 Soil Bacterial Community Members
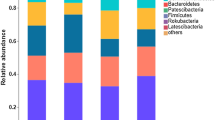
There were nine bacterial phyla in average relative abundances exceeding 1%, and these nine phyla accounted for more than 95% of total sequences recovered by MiSeq sequencing (Fig. 5a). Across tested soil samples, the most abundant phyla in bacterial community were Proteobacteria, followed by Actinobacteria, Chloroflexi, and Acidobacteria, average relative abundances of which were about 27.14%, 16.73%, 12.81%, and 11.88%, respectively. There was no significant change in average relative abundance between treatments for Acidobacteria, Actinobacteria, Bacteroidetes, and Planctomycetes. Average relative abundances of Firmicutes and Proteobacteria in CC1 were no statistical difference with those in other treatments. Average relative abundance of Chloroflexi in CC20 obviously rose by 53.69% compared to that in CC1. Average relative abundance of Gemmatimonadetes in CC15 obviously increased by 29.57% compared to that in CC1. As for Nitrospirae, its relative abundance in CC5, CC15, and CC20 significantly decreased by 31.76%, 25.18%, and 31.42% relative to that in CC1, respectively. Besides, greenhouse cultivation history was positively correlated with average relative abundances of both Chloroflexi and Gemmatimonadetes (Fig. 5b, c), but negatively with that of Nitrospirae (Fig. 5d). These results confirmed phylum level-based bacterial community members were regulated by prolonged greenhouse cucumber cultivation.

Changes in average relative abundances of bacterial phyla with the extension of cucumber cultivation history. Phylum-based bacterialcommunity structure (a), linear correlations between cucumber cultivation history and average relative abundances of Chloroflexi (b) and Gemmatimonadetes (c) and Nitrospirae (d)
The top 10 abundant bacterial genera (i.e. Gp6, Bacillus, Nitrospira, Gemmatimonas, Anaerolinea, Thermomicrobium, Clostridium sensu stricto 1, Arboricoccus, Paenisporosarcina, and Gaiella) altogether accounted for about 30.94% of total recovered sequences (Table 1). The top 50 abundant bacterial genera occupied more than 65% of total recovered sequences. We assessed the alterations in average relative abundances of these 50 genera with consecutive cucumber cultivation. There were 38 genera existing significant differences in average relative abundance between treatments, also indicating that genus level-based bacterial community members were substantially affected by continuous cucumber production. Besides, as for these significantly changed bacterial genera, some of which existed significant correlations between cucumber cultivation history and average relative abundances (Fig. 6). In brief, Bacillus, Nitrospira, Azoarcus, Solirubrobacter, and Rubrobacter negatively correlated to cucumber cultivation history, but the opposites for Thermomicrobium, Desulfotomaculum, Agromyces, Sphaerobacter, and Mycobacterium.

Changes in average relative abundances of some selected bacterial genera with the extension of cucumber cultivation history. Bacillus (a), Nitrospira (b), Thermomicrobium (c), Azoarcus (d), Solirubrobacter (e), Rubrobacter (f), Desulfotomaculum (g), Agromyces (h), Sphaerobacter (i) and Mycobacterium (j)
3.4 Linking Bacterial Community to Soil Physicochemical Variables
We analysed the correlations between average relative abundances of the top 50 abundant genera and soil physicochemical properties through Pearson linear model (Table 2). There were 27 bacterial genera significantly correlated to one or more soil physicochemical properties. Also, 15 out of these 27 bacterial genera positively linked to soil physicochemical properties (Thermomicrobium, Clostridium sensu stricto 1, Paenisporosarcina, Desulfotomaculum, Romboutsia, Turicibacter, Agromyces, Nocardioides, Sphaerobacter, Mycobacterium, Ohtaekwangia, Tumebacillus, Alkaliphilus, Chryseolinea and Haliangium), and the others displayed negative correlations with soil physicochemical properties (Bacillus, Nitrospira, Arboricoccus, H16, Xanthomonas, Azoarcus, Longimicrobium, Solirubrobacter, Rubrobacter, Methyloligella, Methyloversatilis, and Ilumatobacter). These results revealed that bacterial community members were powerfully manipulated by significantly changed soil physicochemical environment originated from long-term intensive greenhouse production.
At overall community level, OTU-based RDA analysis was applied to test which of soil physicochemical variables significantly affected bacterial community structure and their relative contributions to the alterations in community structures (Fig. 7). Soil physicochemical variables explained 50.5% of the variation in community structure by the first two constrained axes of RDA, with the first axis 35.1% and the second 15.4%, respectively. Five soil physicochemical variables affected community structure significantly. In brief, soil available phosphorus and organic carbon contents were the most important contributors, with followed by total nitrogen, available potassium, and C/N.

OTU-based RDA analysis between bacterial community structure and soil physicochemical variables. SOC soil organic carbon, TN total nitrogen, CN the ratio of soil organic carbon to total nitrogen, AN NH4+, NN NO3−, AP available phosphorus, AK available potassium, EC electrical conductance, pH pH value. The data of soil physicochemical properties are from our previous report using the same soil samples (Liu et al. 2019)
3.5 Network Structure of Soil Bacterial Community
We used co-occurrence network analysis to explore the complexity of the interactions of soil bacterial taxa under various cucumber cultivation histories. Here, the tested soil samples were divided into two groups. The first group represented short cucumber cultivation history (SCH) and six soil samples in this group were from CC1 and CC5; the second group meant long cucumber cultivation history (LCH); and six soil samples in this group came from CC15 and CC20. The soil samples from CC10 were abandoned in order to maintain identical sample number within the two groups during network construction and analysis. OTU-based visualized network plots representing SCH and LCH are shown in Fig. 8a, b, respectively, and the topological properties and correlations of obtained networks were calculated to determine differences between SCH and LCH (Table 3). Our results indicated that SCH showed the high level of complexity while LCH displayed a less complex (Fig. 8). The number of nodes in SCH was almost comparable to that in LCH (210 versus 208). However, notable difference in number of edges between SCH and LCH was observed, which was demonstrated by that number of edges in LCH declined about 13.03% relative to that in SCH. Further, numbers of positive and negative edges in LCH reduced about 12.61% and 13.54% compared to those in SCH, respectively. Besides, the network of SCH presented higher number of connections per node (average degree = 21.562) and a lower average path length (2.68) in comparison with those of LCH, effectively indicating a highly connected community (Table 3). Although numbers of modules in SCH and LCH networks were identical (number of communities), SCH displayed a more modular network structure relative to LCH due to relatively high of average clustering coefficient.

OTU-based visualized co-occurrence network plots representing short-term (a) and long-term (b) greenhouse cucumber cultivation. A connection stands for Spearman correlation with 0.6 < r < 0.93 (positive correlation–blue edges) or − 0.93 < r < − 0.6 (negative correlation–red edges) and statistically significant (p < 0.05). Each node represents taxa affiliated at OTU level (based on 16S rRNA), and the size of node is proportional to the number of connections (that is, degree)
We analysed phylum level-based composition of the nodes detected in SCH and in LCH co-occurrence networks, respectively (Fig. 9). These nodes mainly belonged to Acidobacteria, Actinobacteria, Chloroflexi, Firmicutes, and Proteobacteria. On the whole, there was no remarkable difference in phylum level-based composition of the nodes between SCH and LCH except Nitrospirae and SBR1093. In SCH network (210 nodes totally), only 4 nodes were unique, and in LCH network (208 nodes totally), just 2 nodes were unique, also meaning that almost all the nodes were shared by SCH and LCH networks. Still, obvious changes in keystone taxa between SCH and LCH networks were observed (Fig. 10). Based on the network properties, the keystone taxa possess more betweenness centrality, which is defined to the number of times a node acts as a bridge along the shortest path between two other nodes and may be interpreted as key taxa inside a connected community. We identified five bacterial OTUs with the highest of betweenness centrality in SCH and LCH networks, respectively. As listed in Fig. 10, only one key OTU was shared by SCH and LCH networks, and this OTU was affiliated within Chloroflexi. These results suggested that the composition of keystone taxa in soil bacterial co-occurrence network was substantially altered by prolonged greenhouse cucumber production.

Phylum level-based composition of the nodes detected in SCH and LCH networks

Keystone OTU in SCH and LCH networks. BC value means betweenness centrality of each keystone OTU
3.6 Linking Soil Bacterial Community to Cucumber Yield
Long-term greenhouse cultivation significantly decreased cucumber productivity (Fig. 11A). Compared to CC1, cucumber yield in CC15 and CC20 significantly decreased about 26.17% and 23.49%, respectively. We also observed that cucumber yield was negatively correlated with greenhouse cultivation history (Fig. 11B). Besides, cucumber yield positively linked to the β diversity rather than α diversity of soil bacterial community (Fig. 11C, D).

Cucumber yield (A), linear correlation between cucumber yield and greenhouse cultivation history (B), and linear correlations between cucumber yield and both the α diversity (C) and β diversity (D) of soil bacterial community
4 Discussion
In our study, the α diversity of bacterial community was relatively stable as the extension of cucumber cultivation history (Fig. 2), which agreed to previous findings through PLFA and PCR-DGGE methods (Yao et al. 2016; Li et al. 2010). However, the β diversity of bacterial community obviously responded to consecutive greenhouse cucumber production (Fig. 4). These results indicated that bacterial community structure was more sensitive to continuous cropping practice in the tested condition. Indeed, OTU-based hierarchical clustering and PCoA analysis clearly demonstrated that bacterial community structure was altered by continuous cucumber cultivation powerfully. Further, the β diversity of bacterial community positively correlated to greenhouse cucumber productivity (Fig. 11D), also highlighting the important role of bacterial community structure in sustaining crop production. It is well known that soil microorganisms are of fundamental importance in energy flow, organic matter transformation, nutrient cycling, pollutant degradation, stress resistance of crops, and suppression of soil-borne diseases (Xiong et al. 2017; Zhang et al. 2017; Lugtenberg and Kamilova 2009; Raaijmakers et al. 2009). Soil microbial community structure directly maps to soil biological functions and therefore links to crop productivity closely (Lemanceau et al. 2015; Philippot et al. 2013). Hence, improving soil microbial community structure to unblocked soil inherent biological functions might be crucial to overcome continuous cropping obstacles in long-term intensive greenhouse production systems.
Compared to some traditional methods applied previously, high-throughput sequencing supplied finer profile regarding bacterial community members in response to consecutive cucumber cultivation. Our results confirmed that a few bacterial phyla were significantly affected by cucumber cultivation history (Fig. 5). Chloroflexi, Gemmatimonadetes, and Nitrospirae sensitively responded to prolonged greenhouse cucumber cultivation. It has been reported that prolonged potato monoculture increased average relative abundances of Chloroflexi and Gemmatimonadetes while reducing that of Nitrospirae (Liu et al. 2014), which were in good line with the results obtained here. In addition, similar results were also observed by Xiong et al. (2015a, b), Wu et al. (2018), and Zhao et al. (2018) in continuous cropping systems of other crops. By comparison with bacterial phyla, more clear alterations in average relative abundances of most of bacterial genera were revealed. Here, we concentrated on some typical bacterial genera, because these microbial taxa are commonly correlated to soil functioning and crop productivity via driving soil important biological processes directly.
Firstly, average relative abundances of some beneficial and putative biocontrol microbes were significantly inhibited by prolonged cucumber cultivation, such as Bacillus, Solirubrobacter, and Rubrobacter (Table 1; Fig. 6). Bacillus is well known as the most common biocontrol and plant growth-promoting rhizobacteria (Santoyo et al. 2012; Handelsman and Stabb 1996). Bacillus not only positively influences plant growth through synthesis and excretion of auxin or cytokinin type phytostimulating substances but also forms stable and extensive biofilm and secretes many antifungal compounds protecting plants against attack by soil-borne pathogens (Shen et al. 2018; Santoyo et al. 2012). Previous research in different replanted apple orchards revealed that Solirubrobacter positively affected plant growth (Franke-Whittle et al. 2015), suggesting its potential being a beneficial agent. Rubrobacter is from Actinobacteria, and previous researchers reported that, compared to Fusarium wilt conducive soil, suppressive soil recruited more Rubrobacter for disease suppressiveness (Siegel-Hertz et al. 2018). The reductions of relative shares of these beneficial microbes could incur the invasions of soil-borne pathogens and thus affect crop health, particularly for Bacillus, which was the second most abundant genus next to Gp6 within bacterial community (Table 1). Although we did not investigate the data involved in soil-borne diseases in the tested greenhouse plots, field observations from local farmers indeed confirmed more serious crop diseases with prolonged cucumber cultivation.
Secondly, average relative abundances of some functional genera also significantly decreased after long-term cucumber cultivation, such as Nitrospira and Azoarcus (Table 1; Fig. 6). Nitrospira performs soil nitrite oxidation because it carries specific nxr gene encoding nitrite oxidoreductase and therefore affects crop N uptake and soil N availability (Daims et al. 2016). Azoarcus harbours nitrogenase reductase (nifH) gene encoding the iron protein subunit of nitrogenase and is capable of reducing N2 into NH4+ (i.e. biological nitrogen fixation) (Perez et al. 2014; Soares et al. 2006; Wartiainen et al. 2008; Reinhold-Hurek and Hurek 1997). These results suggested that soil N turnover was inhibited by consecutive cucumber cultivation.
Thirdly, average relative abundances of some functional genera significantly increased after prolonged greenhouse cucumber cultivation, such as Thermomicrobium, Desulfotomaculum, Agromyces, Sphaerobacter, and Mycobacterium. Thermomicrobium potentially contributes to soil CO2 emissions through oxidizing CO aerobically as energy source (de Miera et al. 2014; Wu et al. 2009). Desulfotomaculum is well recognized as sulfate-reducing bacteria, which also plays an important role in geochemical transformations of heavy metals (Fan et al. 2017; Lin et al. 2010; Liu et al. 2009). Agromyces plays an important role in the degradation of xylan and cellulose (Wang et al. 2017; Pepe-Ranney et al. 2016) and therefore affects soil C cycling through decomposing crop residues and straw (Chávez-Romero et al. 2016; Fan et al. 2014; Xu et al. 2018; Brennan and Acosta-Martine 2017). Sphaerobacter is recognized as methylating microbes and involved in soil arsenic turnover (Jia et al. 2013; Zhai et al. 2017). Mycobacterium is capable of degrading and removing environmental pollutants, such as PAHs (Kumari et al. 2018; Ma et al. 2018; Chen et al. 2018), pesticides (Fang et al. 2018), and antibiotics (Wang et al. 2018; Fang et al. 2016). These results suggested that some particular soil biological functions/ecological services potentially changed after consecutive greenhouse production due to the alterations in their agents. Due to high inputs of agricultural chemicals and manures year after year, prolonged greenhouse production may lead to the accumulations of soil pollutants (i.e. heavy metals, PAHs, pesticide residues, antibiotics), which probably correlated to the increases in average relative abundances of these functional genera above mentioned.
As the most important habitat, the properties of soils regulate soil microorganisms fundamentally. Our results indicated that most of bacterial genera highly linked to soil physicochemical variables (Table 2). The alterations in bacterial community members could be attributable to significantly changed soil physicochemical environment in long-term intensive greenhouse production (Liu et al. 2019). In addition, soil overall bacterial community structure was also induced by some physicochemical variables obviously, such as available phosphorus and soil organic carbon (Fig. 7). Different groups of soil bacterial taxa prefer different niches, and differentially adapt to soil physicochemical environment corresponding (Mendes et al. 2014; Uroz et al. 2010; Dumbrell et al. 2010). Meanwhile, environmental pressures/changes also drove the assembly of the microbial community effectively (Hargreaves et al. 2015; Wang et al. 2013).
Previous studies involved in soil microbial communities more focused on microbial abundance, community diversity, and structure. However, the interactions among community members were commonly overlooked. Here, bacterial OTU-based co-occurrence networks representing short and long cucumber cultivation history were constructed respectively, and their topological properties were also analysed (Fig. 8; Table 3). Although both networks were highly modular and had comparable the numbers of modules and nodes as well as the similar composition of nodes, our results clearly confirmed that prolonged cucumber cultivation contributed to less complex and connected co-occurrence network pattern in bacterial community compared with that of short-term cultivation. Previous study in a continuous potato monoculture system also pointed out that the interactions and associations of soil fungal species were powerfully weakened by continuous cropping practice (Lu et al. 2013). In this study, higher number of edges in SCH network relative to that in LCH network suggested that there existed stronger strength in functional interrelation among soil bacterial members in the condition of short-term greenhouse cucumber cultivation. Besides, a relatively higher topological clustering coefficient and network connectivity (i.e. number of edges and average degree) existed in the soil with short-term cucumber cultivation shown that OTUs had a higher tendency to share neighbours and benefit to more effective resource utilization (Zhao et al. 2019, Morrieën et al. 2017). Further researches proposed that a highly connected network provided more functional redundancy (Mougi and Kondoh, 2012) and led to greater community stability, which therefore benefited to stronger resistance to abiotic and biotic disturbances (Yang et al. 2019, Mendes et al. 2018; Wei et al. 2015, 2018). These findings plus our results meant that prolonged greenhouse cucumber cultivation drove a relatively advantage-lacked organization among soil bacterial members when suffering soil environmental changes and pathogen invasion, and contrast bacterial taxa were used as crucial nodes (i.e. keystone taxa) to re-construct the associations among bacterial community members as continuous greenhouse cucumber cultivation. Co-occurrence networks obtained here and their topological properties provide a new insight into bacterial community assembly in long-term intensive cultivated greenhouse soils.
5 Conclusions
This study evaluated the shifts in soil bacterial community structure with increasing greenhouse cucumber cultivation years via high-throughput amplicon sequencing. Prolonged greenhouse cucumber production disturbed bacterial community structure, which was powerfully revealed by the following: (1) significant alterations in average relative abundances of soil beneficial microbes and some functional genera and (2) less complex and connected co-occurrence network among bacterial taxa. Our findings suggested that the alteration in soil bacterial community structure and the advantage-lacked organization between bacterial taxa may play an important role in the occurrence of continuous cropping obstacles in long-term intensive greenhouse production system. Exploring the mechanisms behind soil bacterial community assembly in long-term intensive cultivated greenhouse soils is required in the next work.
References
Brennan EB, Acosta-Martine V (2017) Cover cropping frequency is the main driver of soil microbial changes during six years of organic vegetable production. Soil Biol Biochem 109:188–204
Bünemann EK, Bongiorno G, Bai ZG, Creamer RE, De Deyn G, de Goede R, Fleskens L, Geissen V, Kuyper TW, Mäder P, Pulleman M, Sukkel W, van Groenigen JW, Brussaard L (2018) Soil quality – a critical review. Soil Biol Biochem 120:105–125
Chaparro JM, Sheflin AM, Manter DK, Vivanco JM (2012) Manipulating the soil microbiome to increase soil health and plant fertility. Plant Soil 48:489–499
Chávez-Romero Y, Navarro-Noya YE, Reynoso-Martínez SC, Sarria-Guzmán Y, Govaerts B, Verhulst N, Dendooven L, Luna-Guido M (2016) 16S metagenomics reveals changes in the soil bacterial community driven by soil organic C, N-fertilizer and tillage-crop residue management. Soil Till Res 159:1–8
Chen T, Lin S, Wu LK, Lin WX, Sampietro DA (2015) Soil sickness: current status and future perspectives. Allelopathy J 36:167–196
Chen SC, Duan GL, Ding K, Huang FY, Zhu YG (2018) DNA stable-isotope probing identifies uncultivated members of Pseudonocardia associated with biodegradation of pyrene in agricultural soil. FEMS Microbiol Ecol 94(3):fiy026
Daims H, Lücker S, Wagner M (2016) A new perspective on microbes formerly known as nitrite-oxidizing bacteria. Trends Microbiol 24:699–712
de Miera LES, Arroyo P, de Luis CE, Falagán J, Ansola G (2014) High-throughput sequencing of 16S RNA genes of soil bacterial communities from a naturally occurring CO2 gas vent. Int J Greenh Gas Con 29:176–184
Dumbrell AX, Nelson M, Helgason T, Dytham C, Fitter AH (2010) Relative roles of niche and neutral processes in structuring a soil microbial community. ISME J 4:337–345
Fan FL, Yin C, Tang YJ, Li ZJ, Song AL, Wakelin SA, Zou J, Liang YC (2014) Probing potential microbial coupling of carbon and nitrogen cycling during decomposition of maize residue by 13C-DNA-SIP. Soil Biol Biochem 70:12–21
Fan FQ, Zhang BY, Morrill PL, Husain T (2017) Profiling of sulfate-reducing bacteria in an offshore oil reservoir using phospholipid fatty acid (PLFA) biomarkers. Water Air Soil Pollut 228:410
Fang H, Han LX, Cui YL, Xue YF, Cai L, Yu YL (2016) Changes in soil microbial community structure and function associated with degradation and resistance of carbendazim and chlortetracycline during repeated treatments. Sci Total Environ 572:1203–1212
Fang H, Zhang HP, Han LX, Mei JJ, Ge QQ, Long ZN, Yu YL (2018) Exploring bacterial communities and biodegradation genes in activated sludge from pesticide wastewater treatment plants via metagenomic analysis. Environ Pollut 243:1206–1216
Franke-Whittle IH, Manici LM, Blaz Stres HI (2015) Rhizosphere bacteria and fungi associated with plant growth in soils of three replanted apple orchards. Plant Soil 395:317–333
Fu HD, Zhang GX, Zhang F, Sun ZP, Geng GM, Li TL (2017) Effects of continuous tomato monoculture on soil microbial properties and enzyme activities in a solar greenhouse. Sustainability 9:317
Handelsman J, Stabb EV (1996) Biocontrol of soilborne plant pathogens. Plant Cell 8:1855–1869
Hargreaves SK, Williams RJ, Hofmockel KS (2015) Environmental filtering of microbial communities in agricultural soil shifts with crop growth. PLoS One 10(7):e0134345
Huang H, Chou C, Erickson R (2006) Soil sickness and its control. Allelopathy J 18:1–21
Huang LF, Song LX, Xia XJ, Mao WH, Shi K, Zhou YH, Yu JQ (2013) Plant-soil feedbacks and soil sickness: from mechanisms to application in agriculture. J Chem Ecol 39:232–242
Jia Y, Huang H, Zhong M, Wang FH, Zhang LM, Zhu YG (2013) Microbial arsenic methylation in soil and rice rhizosphere. Environ Sci Technol 47:3141–3148
Kumari S, Regar RK, Manickam N (2018) Improved polycyclic aromatic hydrocarbon degradation in a crude oil by individual and a consortium of bacteria. Bioresour Technol 254:174–179
Lal R (2015) Restoring soil quality to mitigate soil degradation. Sustainability 7:5875–5895
Lemanceau P, Maron PA, Mazurier S, Mougel C, Pivato B, Plassart P, Ranjard L, Revellin C, Tardy V, Wipf D (2015) Understanding and managing soil biodiversity: a major challenge in agroecology. Agron Sustain Dev 35:67–81
Li TL, Yang LJ (2016) Overcoming continuous cropping obstacles-the difficult problem. Sci Agr Sin 49(5):916–918
Li CG, Li XM, Kong WD, Wu Y, Wang JG (2010) Effect of monoculture soybean on soil microbial community in the Northeast China. Plant Soil 330:423–433
Lin HR, Shi JY, Chen XL, Yang JJ, Chen YX, Zhao YD, Hu TD (2010) Effects of lead upon the actions of sulfate-reducing bacteria in the rice rhizosphere. Soil Biol Biochem 42:1038–1044
Liu XZ, Zhang LM, Prosser JI, He JZ (2009) Abundance and community structure of sulfate reducing prokaryotes in a paddy soil of southern China under different fertilization regimes. Soil Biol Biochem 41:687–694
Liu X, Zhang JL, Gu TY, Zhang WM, Shen QR, Yin SX, Qiu HZ (2014) Microbial community diversities and taxa abundances in soils along a seven-year gradient of potato monoculture using high throughput pyrosequencing approach. PLoS One 9(1):e86610
Liu X, Zhang Y, Ren XJ, Chen BH, Shen CW, Wang F (2019) Long-term greenhouse vegetable cultivation alters the community structures of soil ammonia oxidizers. J Soils Sediments 19:883–902
Lu LH, Yin SX, Liu X, Zhang WM, Gu TY, Shen QR, Qiu HZ (2013) Fungal networks in yield-invigorating and -debilitating soils induced by prolonged potato monoculture. Soil Biol Biochem 65:186–194
Lugtenberg B, Kamilova F (2009) Plant-growth-promoting rhizobacteria. Annu Rev Microbiol 63:541–556
Luo GW, Rensing C, Chen H, Liu MQ, Wang M, Guo SW, Ling N, Shen QR (2018) Deciphering the associations between soil microbial diversity and ecosystem multifunctionality driven by long-term fertilization management. Funct Ecol 32:1103–1116
Ma L, Deng FC, Yang C, Guo CL, Dang Z (2018) Bioremediation of PAH-contaminated farmland: field experiment. Environ Sci Pollut Res 25:64–72
Mariotte P, Mehrabi Z, Bezemer TM, De Deyn GB, Kulmatiski A, Drigo B, Veen GF, van der Heijden MGA, Kardol P (2018) Plant–soil feedback: bridging natural and agricultural sciences. Trends Ecol Evol 33:129–142
Mendes LW, Kuramae EE, Navarrete AA, van Veen JA, Tsai SM (2014) Taxonomical and functional microbial community selection in soybean rhizosphere. ISME J 8:1577–1587
Mendes LW, Raaijmakers JM, de Hollander M, Mendes R, Tsai SM (2018) Influence of resistance breeding in common bean on rhizosphere microbiome composition and function. ISME J 12:212–224
Mohanram S, Kumar P (2019) Rhizosphere microbiome: revisiting the synergy of plant-microbe interactions. Ann Microbiol 69:307–320
Morrieën E, Hannula SE, Snoek LB, Helmsing NR, Zweers H, de Hollander M, Soto RL, Bouffaud ML, Buée M, Dimmers W, Duyts H, Geisen S, Girlanda M, Griffiths RI, Jørgensen HB, Jensen J, Plassart P, Redecker D, Schmelz RM, Schmidt O, Thomson BC, Tisserant E, Uroz S, Winding A, Bailey MJ, Bonkowski M, Faber JH, Martin F, Lemanceau P, de Boer W, van Veen JA, van der Putten WH (2017) Soil networks become more connected and take up more carbon as nature restoration progresses. Nat Commun 8:14349
Mougi A, Kondoh M (2012) Diversity of interaction types and ecological community stability. Science 337:349–351
Pepe-Ranney C, Campbell AN, Koechli CN, Berthrong S, Buckley DH (2016) Unearthing the ecology of soil microorganisms using a high resolution DNA-SIP approach to explore cellulose and xylose metabolism in soil. Front Microbiol 7:703
Perez PG, Ye J, Wang S, Wang XL, Huang DF (2014) Analysis of the occurrence and activity of diazotrophic communities in organic and conventional horticultural soils. Appl Soil Ecol 79:37–48
Philippot L, Raaijmakers JM, Lemanceau P, van der Putten WH (2013) Going back to the roots: the microbial ecology of the rhizosphere. Nat Rev Microbiol 11:789–799
Raaijmakers JM, Paulitz TC, Steinberg C, Alabouvette C, Moënne-Loccoz Y (2009) The rhizosphere: a playground and battlefield for soilborne pathogens and beneficial microorganisms. Plant Soil 321:341–361
Reinhold-Hurek B, Hurek T (1997) Azoarcus spp. and their interactions with grass roots. Plant Soil 194:57–64
Santoyo G, del Carmen O-MM, Govindappa M (2012) Mechanisms of biocontrol and plant growth-promoting activity in soil bacterial species of Bacillus and Pseudomonas: a review. Biocontrol Sci Tenhn 8:855–872
Shen ZZ, Xue C, Taylor PWJ, Ou YN, Wang BB, Zhao Y, Ruan YZ, Li R, Shen QR (2018) Soil pre-fumigation could effectively improve the disease suppressiveness of biofertilizer to banana Fusarium wilt disease by reshaping the soil microbiome. Biol Fert Soils 54:793–806
Siegel-Hertz K, Edel-Hermann V, Chapelle E, Terrat S, Raaijmakers JM, Steinberg C (2018) Comparative microbiome analysis of a Fusarium Wilt suppressive soil and a Fusarium Wilt conducive soil from the Châteaurenard Region. Front Microbiol 9:568
Soares RA, Roesch LFW, Zanatta G, de Oliveira Camargo FA, Passaglia LMP (2006) Occurrence and distribution of nitrogen fixing bacterial community associated with oat (Avena sativa) assessed by molecular and microbiological techniques. Appl Soil Ecol 33:221–234
Uroz S, Buée M, Murat C, Frey-Klett P, Martin F (2010) Pyrosequencing reveals a contrasted bacterial diversity between oak rhizosphere and surrounding soil. Env Microbiol Rep 2:281–288
Wang JJ, Shen J, Wu YC, Tu C, Soininen J, Stegen JC, He JZ, Liu XQ, Zhang L, Zhang EL (2013) Phylogenetic beta diversity in bacterial assemblages across ecosystems: deterministic versus stochastic processes. ISME J 7:1310–1321
Wang R, Zhang HC, Sun LG, Qi GF, Chen S, Zhao XY (2017) Microbial community composition is related to soil biological and chemical properties and bacterial wilt outbreak. Sci Rep 7:343
Wang L, You LX, Zhang JM, Yang T, Zhang W, Zhang ZX, Liu PX, Wu S, Zhao F, Ma J (2018) Biodegradation of sulfadiazine in microbial fuel cells: reaction mechanism, biotoxicity removal and the correlation with reactor microbes. J Hazard Mater 360:402–411
Wartiainen I, Eriksson T, Zheng WW, Rasmussen U (2008) Variation in the active diazotrophic community in rice paddy—nifH PCR-DGGE analysis of rhizosphere and bulk soil. Appl Soil Ecol 39:65–75
Wei Z, Yang T, Friman VP, Xu Y, Shen Q, Jousset A (2015) Trophic network architecture of root-associated bacterial communities determines pathogen invasion and plant health. Nat Commun 6:8413
Wei Z, Hu J, Gu YA, Yin SX, Xu YC, Jousset A, Shen QR, Friman VP (2018) Ralstonia solanacearum pathogen disrupts bacterial rhizosphere microbiome during an invasion. Soil Biol Biochem 118:8–17
Wu DY, Raymond J, Wu M, Chatterji S, Ren QH, Graham JE, Bryant DA, Robb F, Colman A, Tallon LJ, Badger JH, Madupu R, Ward NL, Eisen JA (2009) Complete genome sequence of the aerobic CO-oxidizing Thermophile Thermomicrobium roseum. PLoS One 4(1):e4207
Wu LK, Chen J, Xiao ZG, Zhu XG, Wang JY, Wu HM, Wu YH, Zhang ZY, Lin WX (2018) Barcoded pyrosequencing reveals a shift in the bacterial community in the rhizosphere and rhizoplane of Rehmannia glutinosa under consecutive monoculture. Int J Mol Sci 19:50
Xiong W, Li ZG, Liu HJ, Xue C, Zhang RF, Wu HS, Li R, Shen QR (2015a) The effect of long-term continuous cropping of black pepper on soil bacterial communities as determined by 454 pyrosequencing. PLoS One 10(8):e0136946
Xiong W, Zhao QY, Zhao J, Xun WB, Li R, Zhang RF, Wu HS, Shen QR (2015b) Different continuous cropping spans significantly affect microbial community membership and structure in a vanilla-grown soil as revealed by deep pyrosequencing. Microb Ecol 70:209–218
Xiong W, Guo S, Jousset A, Zhao QY, Wu HS, Li R, Kowalchuk GA, Shen QR (2017) Bio-fertilizer application induces soil suppressiveness against Fusarium wilt disease by reshaping the soil microbiome. Soil Biol Biochem 114:238–247
Xu N, Tan GC, Wang HY, Gai XP (2016) Effect of biochar additions to soil on nitrogen leaching, microbial biomass and bacterial community structure. Eur J Soil Biol 74:1–8
Xu QC, Dai RB, Ruan Y, Rensing C, Liu MQ, Guo SW, Ling N, Shen QR (2018) Probing active microbes involved in Bt-containing rice straw decomposition. Appl Microbiol Biot 102:10273–10284
Yang W, Jing XY, Guan YP, Zhai C, Wang T, Shi DY, Sun WP, Gu SY (2019) Response of fungal communities and co-occurrence network patterns to compost amendment in black soil of Northeast China. Front Microbiol 10:1562
Yao HY, Jiao XD, Wu FZ (2006) Effects of continuous cucumber cropping and alternative rotations under protected cultivation on soil microbial community diversity. Plant Soil 284:195–203
Yao ZY, Xing JJ, Gu HP, Wang HZ, Wu JJ, Xu JM, Brookes PC (2016) Development of microbial community structure in vegetable-growing soils from open-field to plastic-greenhouse cultivation based on the PLFA analysis. J Soils Sediments 16:2041–2049
Zhai WW, Wong MT, Luo F, Hashmi MZ, Liu XM, Edwards EA, Tang XJ, Xu JM (2017) Arsenic methylation and its relationship to abundance and diversity of arsM genes in composting manure. Sci Rep 7:42198
Zhang RF, Vivanco JM, Shen QR (2017) The unseen rhizosphere root-soil-microbe interactions for crop production. Curr Opin Microbiol 37:8–14
Zhao QY, Xiong W, Xing YZ, Sun Y, Lin XJ, Dong YP (2018) Long-term coffee monoculture alters soil chemical properties and microbial communities. Sci Rep 8:6116
Zhao XR, Wu HY, Song XD, Yang SH, Dong Y, Yang JL, Zhang GL (2019) Intra-horizon differentiation of the bacterial community and its co-occurrence network in a typical Plinthic horizon. Sci Total Envriron 678:692–701
Zhou XG, Wu FZ (2012) Dynamics of the diversity of fungal and Fusarium communities during continuous cropping of cucumber in the greenhouse. FEMS Microbiol Ecol 80:469–478
Zhou XG, Gao DM, Liu J, Qiao PL, Zhou XL, Lu HT, Wu X, Liu D, Jin X, Wu FZ (2014) Changes in rhizosphere soil microbial communities in a continuously monocropped cucumber (Cucumis sativus L.) system. Eur J Soil Biol 60:1–8
Funding
We received support for this research from the National Natural Science Foundation of China (grant number: 41601250).
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of Interest
The authors declare that they have no competing interests.
Additional information
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
About this article

Cite this article
Liu, X., Li, Y., Ren, X. et al. Long-Term Greenhouse Cucumber Production Alters Soil Bacterial Community Structure. J Soil Sci Plant Nutr 20, 306–321 (2020). https://doi.org/10.1007/s42729-019-00109-9
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s42729-019-00109-9