
Abstract
To improve the properties of low-carbonization of MgO–C refractories, the introduction of composite additives is an effective strategy. Al2O3–SiC composite powder was prepared from clay using electromagnetic induction heating and carbon embedded methods. Further, the Al2O3–SiC composite powder synthesized by electromagnetic induction heating at 600 A was added into low-carbon MgO–C refractories (4 wt.%) to improve their properties. The results showed that when the addition amount of Al2O3–SiC composite powder is within the range of 2.5–5.0 wt.%, the properties of low-carbon MgO–C samples were significantly improved, e.g., the apparent porosity of 7.58%–8.04%, the bulk density of 2.98–2.99 g cm−3, the cold compressive strength of 55.72–57.93 MPa, the residual strength after three air quenching at 1100 °C of 74.86%–78.04%, and the decarburized layer depth after oxidized at 1400 °C for 2 h of 14.03–14.87 mm. Consequently, the idea for the rapid synthesis of Al2O3–SiC composite powder provides an alternative low-carbon MgO–C refractories performance optimization strategy.
Similar content being viewed by others
Avoid common mistakes on your manuscript.
1 Introduction
MgO–C refractories, a kind of the largest market demand carbon-containing refractories, have been widely used in metallurgical kiln linings, such as alternating current/direct current electric arc furnace, converter, Ruhrstal Heraeus refining furnace, and ladle furnace slag line, because of excellent high-temperature performance and outstanding basic slag resistance [1,2,3,4,5]. The rapid development of special steels, including ultra-low-carbon steel, has further promoted the low-carbonization of MgO–C refractories. On the one hand, refractories with high carbon content may cause molten steel to entrap slag inclusions, resulting in carbon content out of standard. On the other hand, although graphite with high thermal conductivity brings better thermal shock resistance to refractories, it reduces strength and increases energy consumption of per ton steel [6,7,8]. Therefore, the low-carbonization of MgO–C refractories, as well as the strengthening and toughening after low-carbonization, is the research focuses currently in the field of refractory materials.
Due to the quantum size effect, surface effect, and high dispersibility of nanocarbon, MgO–C refractories using nanocarbon as a carbon source can form a complete and continuous carbon network inside even with very low-carbon content; thus, they still exhibit high performance in-line with expectation [9]. Liu et al. [10] investigated the effects of different types of carbon black on the properties of low-carbon MgO–C refractories (3 wt.% C) and found that the low-carbon MgO–C refractories added with 0.4 wt.% N220 carbon black showed the almost identical thermal shock resistance better as traditional MgO–C refractories (16 wt.% C). Zhu et al. [11] found that the introduction of graphite oxide nanosheets and carbon nanotubes enhanced the cold compressive strength and thermal shock resistance of low-carbon MgO–C refractories (5 wt.% C), respectively. However, although nanostructured carbon makes up for the shortcomings of thermo-mechanical properties for low-carbon MgO–C refractories, it brings greater challenges in terms of oxidation resistance. Over the past decade, a series of high-quality antioxidants have been reported, including some single-phase powders with higher oxidation resistance like Al–Mg [12], Cr7C3 [6], Ti3AlC2 [8], Al4SiC4 [13], Al4O4C [14], and MgB2 [15], as well as composite powder with both antioxidation and toughening functions like B4C/Al2O3/C [16], Al2O3/SiC [17], C/MgAl2O4 [18], and TiC/Ti3AlC/C [19]. In contrast, composite powder has more development potential because of easier dispersion and lower cost. Among them, Al2O3–SiC composite powder shows the best research potential due to its wide range of sources and efficient application results.
For this purpose, the present work used abundant low-cost clay as the main raw material to prepare Al2O3–SiC composite powder by electromagnetic induction heating method and conventional carbon embedded method. Based on the relevant thermodynamic data, the synthesis mechanism of Al2O3–SiC composite powder was analyzed. Furthermore, the as-prepared Al2O3–SiC composite powder was used to replace the MgO fine powders in low-carbon MgO–C refractories, and the effects of the composite powder addition amounts on their thermal shock resistance and oxidation resistance were investigated in detail.
2 Experimental
2.1 Raw materials
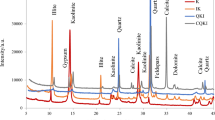
As-received refractory clay (from Jilin, China) and carbon black (N990 type; ≤ 300 nm; from Qingdao, China) were used as the starting materials for synthesizing Al2O3–SiC composite powder. Table 1 and Fig. 1 show the X-ray fluorescence results, X-ray diffraction (XRD) patterns, and particle size distribution of clay, respectively.

XRD pattern (a) and particle size distribution (b) of clay
Fused magnesia (particle sizes of ≤ 0.088 mm, 0–1 mm, 1–3 mm, and 3–5 mm; MgO purity ≥ 97 wt.%; from Yingkou, China), natural flake graphite (≤ 0.088 mm; fixed carbon purity ≥ 97.5 wt.%; from Yingkou, China), aluminum powder (≤ 0.088 mm; Al purity ≥ 98 wt.%; from Yingkou, China), and liquid phenolic resin (carbon yield ≥ 43.0 wt.%; from Yingkou, China) were used as the raw materials for preparing the low-carbon MgO–C refractories.
2.2 Sample preparation
The synthesis process of Al2O3–SiC composite powder is as follows. Clay and carbon black were quantitatively weighed according to the mass ratio of 10:4, and then the raw powders were poured into a high-energy planetary ball mill equipped with cemented carbide jars (QM-3SP4, China) and ball milled at the rate of 200 r min–1 for 4 h. The homogeneous raw powders were pressed into cylindrical green bodies (ϕ20 mm × 20 mm) at 20 MPa. Finally, the green bodies were heated in a high-temperature box resistance furnace (in carbon embedded atmosphere at 1500, 1550, and 1600 °C for 4 h) and an electromagnetic induction heating furnace (in Ar atmosphere of 0.3 L min–1 at 400, 500 and 600 A for 15 min), respectively.
The preparation process of low-carbon MgO–C refractories is as follows. The raw materials were weighed according to the formulas shown in Table 2, and then mixed progressively in a roll-type mixer with addition of fused magnesia aggregate, liquid phenolic resin, flake graphite, magnesia powder, Al powder, and Al2O3–SiC composite powder in sequence. The homogeneous raw materials were pressed into different batches of cylindrical green bodies (ϕ50 mm × 50 mm) at 200 MPa. Finally, the green bodies were cured at 200 °C for 24 h and then coked at 1400 °C for 3 h.
2.3 Characterization
In this work, the formed phases, microstructure, and the elements in the local microstructure of the samples were detected using an X-ray diffractometer (Rigaku-Ultima IV, Japan; 5 (°) min–1, 10°–80°), a scanning electron microscope (SEM, Hitachi-S4800, Japan), and an energy dispersive spectrometer (EDS), respectively. The apparent porosity, bulk density, cold compressive strength, and thermal shock resistance of the samples were measured based on the corresponding Chinese standards (GB/T 2997–2000, 5072–2008, 30873–2014, and 21650.1–2008). Specifically, the thermal shock resistance of the sintered samples was tested by air quenching method and quantitatively characterized by the residual cold compressive strength of the samples after air quenching three times at 1100 °C. In addition, the oxidation test of the samples was carried out at 1400 °C for 2 h [20]. For all the above tests, the average value of the recorded results of three parallel samples was selected as the final test results.
3 Results and discussion
3.1 Synthesis of Al2O3–SiC composite powder from clay by two methods
3.1.1 Phase compositions
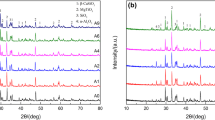
Figure 2 shows XRD patterns of Al2O3–SiC composite powder synthesized by electromagnetic induction heating method at different current densities (Fig. 2a–c) and carbon embedded method at different temperatures (Fig. 2d–f). From Fig. 2a–c, the phases of the samples are mullite and cristobalite at the current intensity of 400 A (Fig. 2a); the phase of the samples are mullite, corundum, and silicon carbide (β-type) at the current intensity of 500 A (Fig. 2b); the phase of the samples are corundum and silicon carbide at the current intensity of 600 A (Fig. 2c). Similarly, with the temperature increasing, the phases of the samples transform from mullite and silicon carbide (1500 °C, Fig. 2d) to mullite, corundum, and silicon carbide (1550 °C, Fig. 2e) and then to silicon carbide and corundum (1600 °C, Fig. 2f). The XRD results suggest that increasing the current intensity or rising the temperature is beneficial to the formation of target phase of Al2O3 and SiC, and when the temperature is 1600 °C or the current intensity is 600 A, the content of the impurity phases in the samples is the lowest.

XRD patterns of Al2O3–SiC composite powder synthesized by electromagnetic induction heating method (a–c) and carbon embedded method (d–f). a 400 A; b 500 A; c 600 A; d 1500 °C; e 1550 °C; f 1600 °C
3.1.2 Microstructure evolution
Figure 3 presents the SEM images and EDS results of Al2O3–SiC composite powder synthesized by electromagnetic induction heating method at 600 A (Fig. 3a–c) and carbon embedded method at 1600 °C (Fig. 3d–f). The powder samples synthesized by electromagnetic induction heating method are composed of two kinds of particles. Combined with the EDS results, the large particles with a size of 5–10 μm are alumina, and the small particles with a size of 10–20 μm are silicon carbide. Likewise, the powder samples synthesized by carbon embedded method are also composed of large particles of alumina and small particles of silicon carbide. By contrast, the particle size distribution of the powder samples synthesized by electromagnetic induction heating is more uniform than that of the powder samples synthesized by carbon embedded method. The results fully reflect the difference between the two technical methods. The heating rate of electromagnetic induction heating is higher, and the preset temperature can be reached in a very short time [21]. When electromagnetic induction heating was used to treat the clay, the mullite grains inside powder samples were reduced to alumina and silicon carbide by carbon black before they grew up. Naturally, the particle size of reduction products, especially alumina particles, is smaller than that of the samples synthesized by carbon embedded method.

SEM images and EDS results of Al2O3–SiC composite powder synthesized by electromagnetic induction heating method at 600 A (a–c) and carbon embedded method at 1600 °C (d–f)
3.1.3 Analysis of synthesis process of Al2O3–SiC composite powder
From the XRD results in Sect. 3.1.1, when electromagnetic induction heating is used at the current of 400 A (~ 1300 °C), the phases of the powder samples are cristobalite and mullite; as the current increases to 500 A or the temperature rises to 1550 °C (carbon embedded method), the diffraction peaks of cristobalite disappeared while the diffraction peaks of silicon carbide and corundum emerged; when the current increases to 600 A or the temperature rises to 1600 °C, all mullite was transformed into silicon carbide and corundum. Therefore, the reduction process of clay can be more specifically divided into three successive stages: decomposition of kaolinite (reactions (1)–(4)), reduction of cristobalite (reactions (5)–(7)), and reduction of mullite (reactions (8) and (9)) [22].
Among them, reactions (5)–(9) involve gas phases; thus, when calculating their Gibbs free energy values, it is necessary to take into account the influence of the gas phases partial pressure in the real reaction system. Taking reaction (7) for instance, its Gibbs free energy equals \(\Delta G_{7} = \Delta G_{7}^{{\Theta }} + RT\ln J = 16.63T\ln P_{{{\text{CO}}}} /P^{{\Theta }} - 331.98T + 603,150\), where \(\Delta G^{{\Theta }}\) is the standard Gibbs free energy, J mol-1; R is the molar gas constant, 8.314 J mol-1 J-1; T is the initial temperature, K; J is the pressure quotient; PCO is the partial pressure of CO, Pa; and PΘ is the standard atmospheric pressure, 1 × 105 Pa. The calculation results show that in standard atmosphere, the initial temperature of the spontaneous reaction is 1543.68 °C; in Ar atmosphere (PCO = 20 Pa), the initial temperature is reduced to 1000.34 °C; in carbon embedded atmosphere (PCO = 42,000 Pa), the initial temperature is decreased to 1452.22 °C. By comparison, the initial temperature of the reduction reaction in Ar atmosphere is lower, indicating that the electromagnetic induction method is a better choice to synthesize Al2O3–SiC composite powder.
Therefore, the overall reaction of the clay reduction process is as follows [23]:
3.2 Effects of as-synthesized Al2O3–SiC composite powder on properties of low-carbon MgO–C refractories
3.2.1 Phase compositions and microstructures
Figure 4a shows the XRD patterns of samples MC and MC-5AS after coked at 1400 °C for 3 h. The diffraction peaks of graphite and periclase phases were found in the XRD pattern of sample MC. In comparison, in addition to graphite and periclase phases, the diffraction peaks of magnesia-alumina spinel and forsterite phases were also detected in sample MC-5AS. In order to further investigate the influence of Al2O3–SiC composite powder on the microstructure of the low-carbon MgO–C samples, the SEM images of the samples are displayed in Fig. 4b–e. MgO aggregates (light grey) and flake graphite (dark grey) together form the matrix structure of the samples, and some pores (black) are dispersed between the particles. With the increase in the addition amount of Al2O3–SiC composite powder, the number of pores on the surface of the samples (from sample MC to sample MC-5AS) decreases, and the pore size becomes smaller. However, bigger and more pores reappear on the surface of sample MC-7.5AS, especially where the spinel and forsterite phases are enriched (confirmed by EDS).

XRD patterns (a) and SEM images (b–e) of MgO–C samples. b Sample MC; c sample MC-2.5AS; d sample MC-5AS; e sample MC-7.5AS. FM Fused magnesia; FG flake graphite; MA spinel; M2S forsterite; P pore
3.2.2 Physical properties
Figure 5 shows the apparent porosity and bulk density of the MgO–C samples after coked at 1400 °C for 3 h. The apparent porosity of the MgO–C samples first decreases and then increases with the increase in the Al2O3–SiC composite powder addition, and the sample MC-2.5AS achieve the minimum apparent porosity of 7.58%. In contrast, the bulk density of the MgO–C sample rises first and then decreases, and the corresponding maximum value of the bulk density is obtained at the 2.5 wt.% point. This change can be mainly attributed to two reasons. One is that the micron-sized Al2O3–SiC composite powder has good dispersibility and can be better filled between the aggregates, thereby bringing the microstructure with high packing to the MgO–C samples [24]. The other is that the Al2O3–SiC composite powder can react with MgO in the MgO–C sample to form spinel. According to theoretical density calculations, ~ 8% volume expansion will generate when Al2O3 and MgO react to form stoichiometric spinel, which is beneficial to offsetting some pore defects inside the material [25]. However, when the volume expansion caused by the solid–solid reaction is too large, the original structure of the material will inevitably be destroyed, resulting in the formation of some new cracks and pores. This is also the reason why the apparent porosity of samples MC-5AS and MC-7.5AS is slightly higher than that of sample MC-2.5AS (but still better than sample MC).

Al2O3–SiC composite powder addition amounts versus apparent porosity and bulk density of samples
3.2.3 Cold compressive strength and thermal shock resistance
Figure 6 shows the cold compressive strength of the MgO–C samples before and after quenched at 1100 °C for 3 times and the corresponding residual strength ratio. The strength of the MgO–C samples, as expected, displays a trend of first increasing and then decreasing, same as the apparent porosity of the samples. The change in the cold compressive strength of the MgO–C samples can be explained by their microstructures. With the introduction of Al2O3–SiC composite powder, the aggregate and matrix (fine powders) of the MgO–C samples achieve a more ideal close packed structure, and meanwhile, the in-situ formed ceramic phases (spinel and forsterite) further enhance the bond between the aggregates (Fig. 4b–e). As shown in Fig. 6, the residual strength ratio of the MgO–C samples increases from 67.86% (MC) to 74.86% (MC-2.5AS), 78.04% (MC-5AS), and 71.47% (MC-7.5AS), respectively. This can be attributed to the effect of microcracks and micropores formed due to volume expansion. For ceramics and refractories, generally, although the generation of microcracks and micropores will inevitably increase the apparent porosity of the material to a certain extent, they can even paly a toughening effect to improve the high-temperature properties of the material if controlled properly. Therefore, for sample MC-5AS, although the microcracks and micropores caused the increase in its apparent porosity, these microcracks and pores brought it better high-temperature properties including thermal shock resistance. In addition, for refractory materials, thermal shock damage and fracture are dominated by crack propagation rather than generation. Therefore, the pre-forming microcracks and micropores inside samples MC-2.5AS and MC-5AS effectively delay the propagation of cracks by increasing the surface energy. Conversely, the size of the cracks inside sample MC-7.5AS exceeds the critical stable crack length, which causes the crack propagation process to be accelerated [26, 27].

Cold compressive strength and thermal shock resistance of samples versus Al2O3–SiC composite powder addition amounts versus
3.2.4 Oxidation resistance
Figure 7a and b presents the optical images, the corresponding oxidation ratio and decarburized layer depth of the MgO–C samples after oxidized at 1400 °C for 2 h. With the increase in the addition amount of Al2O3–SiC composite powder, the decarburized layer area (light color) of the MgO–C samples decreases first and then increases. The results calculated by image analysis software indicate that sample MC-5A has the best oxidation resistance, which is represented by the lowest oxidation ratio and thickness of the decarburized layer. It is generally believed that the oxidation resistance of refractories is directly related to the apparent porosity, because the open pores provide the main channel for the infiltration of oxygen [28]. The apparent porosity of samples MC-2.5AS and MC-5AS is lower than that of samples MC and MC-7.5AS; thus, there are fewer channels for oxygen to enter them. In addition, the chemical reaction that occurs in the decarburized layer will also affect the subsequent oxidation. The XRD pattern of sample MC-5AS after oxidation is shown in Fig. 7c. Except for MgO phase, both MgAl2O4 and Mg2SiO4 phases are newly formed oxidation products, and their reaction formula is as follows:

Optical images of cross-section (a), oxidation ratio and decarburized layer depth (b) of samples after oxidized at 1400 °C for 2 h, and XRD pattern of decarburized layer of sample MC-5AS (c)
On the one hand, the volume expansion of reactions (11) and (12) filled the pores of the oxide layer and formed a dense layer in the oxide area of sample MC-5AS, thereby reducing the possibility of continued contact between oxygen and graphite. On the other hand, reaction (12) made up for the loss of carbon inside sample MC-5AS, thereby further delaying the oxidation process.
4 Conclusions
-
1.
As the current intensity increased, the phases of clay transformed from mullite and cristobalite (400 A) to mullite, corundum, β-type silicon carbide (500 A) and then to corundum and silicon carbide (600 A); similarly, when the carbon embedded was used, silicon carbide was detected at 1500 °C, and the phases of the samples were transformed into the target phases of Al2O3 and SiC at 1600 °C. In comparison, the electromagnetic induction heating method can save more time and energy, and meanwhile, the particle size distribution and morphology of the Al2O3–SiC composite powder synthesized by electromagnetic induction heating method are superior to those of the Al2O3–SiC composite powder synthesized by carbon embedded method.
-
2.
When 2.5 wt.% Al2O3–SiC composite powder is added, the sintering properties of low-carbon MgO–C samples are the best, which can be attributed to the volume expansion effect generated by the reaction between Al2O3 and MgO matrix. When 5 wt.% Al2O3–SiC composite powder is introduced, the microcrack toughening effect makes sample MC-5AS exhibit the best thermal shock resistance. In addition, Al2O3–SiC composite powder improves the oxidation resistance of low-carbon MgO–C samples by forming a spinel- and forsterite-containing dense protective layer in the oxide layer.
References
Y. Chen, C.J. Deng, X. Wang, J. Ding, C. Yu, H.X. Zhu, Constr. Build. Mater. 240 (2020) 117964.
C. Wöhrmeyer, S. Gao, Z.F. Ping, C. Parr, C.G. Aneziris, P. Gehre, Steel Res. Int. 91 (2020) 1900436.
S.E. Gass, P.G. Galliano, A.G. Tomba Martinez, J. Eur. Ceram. Soc. 41 (2021) 3769–3781.
M. Chen, S. Gao, L. Xu, N. Wang, Ceram. Int. 45 (2019) 21023–21028.
X.M. Ren, B.Y. Ma, S.M. Li, H.X. Li, G.Q. Liu, W.G. Yang, F. Qian, S.X. Zhao, J.K. Yu, J. Iron Steel Res. Int. 28 (2021) 38–45.
Y. Yang, J. Yu, H.Z. Zhao, H. Zhang, P.D. Zhao, Y.C. Li, X.Q. Wang, G.P. Li, Ceram. Int. 46 (2020) 19743–19751.
T.B. Zhu, Y.W. Li, S.B. Sang, J. Alloy. Compd. 783 (2019) 990–1000.
J.F. Chen, N. Li, J. Hubálková, C.G. Aneziris, J. Eur. Ceram. Soc. 38 (2018) 3387–3394.
S. Behera, R. Sarkar, Prot. Met. Phys. Chem. Surf. 52 (2016) 467–474.
B. Liu, J.L. Sun, G.S. Tang, K.Q. Liu, L. Li, Y.F. Liu, J. Iron Steel Res. Int. 17 (2010) No. 10, 75–78.
T.B. Zhu, Y.W. Li, S.B. Sang, S.L. Jin, Y.B. Li, L. Zhao, X. Liang, Ceram. Int. 40 (2014) 4333–4340.
A.P. Luz, T.M. Souza, C. Pagliosa, M.A.M. Brito, V.C. Pandolfelli, Ceram. Int. 42 (2016) 9836–9843.
H.B. Yao, X.M. Xing, E.H. Wang, B. Li, J.H. Chen, J.L. Sun, X.M. Hou, Coatings 7 (2017) 85.
C. Yu, H.X. Zhu, W.J. Yuan, C.J. Deng, P. Cui, S.M. Zhou, J. Alloy. Compd. 579 (2013) 348–354.
K.S. Campos, G.F.B. Lenz e Silva, E.H.M. Nunes, W.L. Vasconcelos, Ceram. Int. 38 (2012) 5661–5667.
D.H. Ding, X.C. Chong, G.Q. Xiao, L.H. Lv, C.K. Lei, J.Y. Luo, Y.F. Zang, Ceram. Int. 45 (2019) 16433–16441.
B.Y. Ma, Q. Zhu, Y. Sun, J.K. Yu, Y. Li, J. Mater. Sci. Technol. 26 (2010) 715–720.
L.H. Lv, G.Q. Xiao, D.H. Ding, Y. Ren, S.L. Yang, P. Yang, X. Hou, Int. J. Appl. Ceram. Technol. 16 (2019) 1253–1263.
X.G. Liu, S.W. Zhang, Y. Li, X.Y. Yang, Y. Zhang, Ceram. Int. 44 (2018) 22567–22573.
X.M. Ren, B.Y. Ma, G.L. Zhang, G.F. Fu, J.K. Yu, G.Q. Liu, Mater. Chem. Phys. 252 (2020) 123309.
J. Chen, D. Chen, H.Z. Gu, A. Huang, H.W. Ni, J. Alloy. Compd. 870 (2021) 159463.
B.Y. Ma, X.M. Ren, Y. Yin, L. Yuan, Z. Zhang, Z.Q. Li, G.Q. Li, Q. Zhu, J.K. Yu, Ceram. Int. 43 (2017) 11830–11837.
A.C.D. Chaklader, S.D. Gupta, E.C.Y. Lin, B. Gutowski, J. Am. Ceram. Soc. 75 (1992) 2283–2285.
D.H. Ding, L.H. Lv, G.Q. Xiao, J.Y. Luo, C.K. Lei, Y. Ren, S.L. Yang, P. Yang, X. Hou, Int. J. Appl. Ceram. Technol. 17 (2020) 645–656.
M.T. Li, N.N. Zhou, X.D. Luo, G.D. Zhang, Z.P. Xie, L.C. Xu, P.C. Liu, Mater. Chem. Phys. 175 (2016) 6–12.
I. Khlifi, O. Pop, J.C. Dupré, P. Doumalin, M. Huger, J. Eur. Ceram. Soc. 39 (2019) 3893–3902.
Y.J. Dai, Y.W. Li, X.F. Xu, Q.Y. Zhu, W. Yan, S.L. Jin, H. Harmuth, J. Eur. Ceram. Soc. 39 (2019) 5433–5441.
Y.M. Qin, X.G. Liu, Q. Zhang, F. Zhao, X.H. Liu, Q.L. Jia, Corros. Sci. 166 (2020) 108446.
Acknowledgements
This work was financially supported by the National Natural Science Foundation of China (Grant Nos. U20A20239 and U1908227), the Fundamental Research Funds for the Central Universities (Grant No. N2125002), and the open research fund for State Key Laboratory of Advance Refractories (Grant No. SKLAR202001).
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article

Cite this article
Ma, By., Ren, Xm., Gao, Z. et al. Influence of pre-synthesized Al2O3–SiC composite powder from clay on properties of low-carbon MgO–C refractories. J. Iron Steel Res. Int. 29, 1080–1088 (2022). https://doi.org/10.1007/s42243-021-00653-8
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s42243-021-00653-8