
Abstract
Rice kernel smut is an important disease caused by Tilletia horrida that affects the production of rice male sterile lines in most hybrid rice growing regions of the world. In this study, the pathogenicity of seven different strains of T. horrida isolated from different parts of China is described. Whole-genome re-sequencing of six T. horrida strains and transcriptome analysis of the reference genome T. horrida strain JY-521 were conducted at different times post inoculation (8, 12, 24, 48, and 72 h) early in the infection. The highest number of differentially expressed genes (DEGs) occurred at 8 h post inoculation. Based on Kyoto Encyclopedia of Genes and Genomes pathway analysis of the DEGs, autophagy processes and lipid degradation were key pathways for T. horrida pathogenicity. In three weakly pathogenic strains, CN-4, XJ-3, and SN-2, the single nucleotide polymorphisms and expression patterns of pathogenicity genes, including carbohydrate-active enzyme genes, pathogen-host interaction genes, effector genes, secondary metabolism-related genes, cytochrome P450 genes, ATP-binding cassette superfamily transporter genes, and G protein-coupled receptor genes, were also analyzed at different times during the infection. Thus, a new understanding of T. horrida pathogenicity was gained by exploring the potential mechanisms for weakened virulence in different strains of rice kernel smut, and a new foundation was provide for further studies on the infection mechanism and future control of this important rice disease.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Rice kernel smut (RKS) is caused by the soil-borne basidiomycete fungus Tilletia horrida. The disease affects both the yield and quality of hybrid seed by producing masses of dark powdery teliospores. RKS is the most devastating disease of rice male sterile line production in most hybrid rice growing regions of the world (Chen et al. 2016). RKS was once categorized as a minor disease with sporadic occurrence in rice-growing areas. However, as the demand for rice increases worldwide, the extensive planting of high-yielding cultivars and hybrid varieties continues. As a result, the annual prevalence of RKS has reached 40–60% in hybrid rice fields, causing a 5–20% decrease in rice yields (Wang et al. 2015). The incidence of the disease is as high as 87% and 100% in hybrid rice fields in Pakistan and China, respectively (Biswas 2003). RKS is now an increasing threat to hybrid seed production in Asia, Oceania, Europe, America, and Africa (Carris et al. 2006; Brooks et al. 2009). Despite the importance of RKS, the pathogenesis-related genes and pathogenic mechanisms in T. horrida have received little attention (Tsuda et al. 2006).
With the development of high-throughput sequencing technology, the identification and annotation of genes in non-model organisms using genomics and transcriptomics have increased rapidly (Maher et al. 2009; Verbruggen et al. 2015; Zhou et al. 2015). Whole-genome re-sequencing is used to sequence new genomes of different species and can be used to study genetic variation among individuals (Li et al. 2009). For example, whole-genome resequencing can analyze the genetic variation and the pathogenic mechanisms of pathogens. To date, the pathogenic mechanisms of many plant pathogens, including Colletotrichum gloeosporioides, Fusarium graminearum, Valsa mali, Metarhizium anisopliae, V. pyri, Mycosphaerella graminicola, Sclerotinia sclerotiorum, Ustilaginoidea virens, Magnaporthe oryzae, and Neurospora crassa, have been clarified using functional genomics, transcriptomics, and re-sequencing studies (Galagan et al. 2003; Dean et al. 2005; Amselem et al. 2011; Goodwin et al. 2011; Ohm et al. 2012; Xue et al. 2012; Zhao et al. 2013; Yin et al. 2015; Huang et al. 2016). Transcriptomic analyses of pathogenic fungi can increase comprehension of expression patterns of virulence-associated genes at different infection times and can aid in the discovery of many new pathogenic genes (Verbruggen et al. 2015). Thus, functional genomics, transcriptomics, and re-sequencing analysis can provide insights and increase understanding of host-pathogen interactions (Zhang et al. 2014; Yap et al. 2015; Xing et al. 2016; Wang et al. 2019). Smut fungi, such as Ustilago maydis (Kamper et al. 2006; Schuler et al. 2018), Sporisorium scitamineum (Que et al. 2014), and Ustilago hordei (Ali et al. 2014) are parasitic on higher plants, including maize, barley, wheat, rice, sugarcane, and forage grass species (Rogerson 1988). The patterns of gene expression in these fungi, including those of potential pathogenicity genes, have been examined during the course of infection through comparative genomics and transcriptomics.
In this study, transcriptomic analyses and whole genome re-sequencing of T. horrida were conducted using an Illumina high-throughput sequencing system. Gene Ontology (GO) and Kyoto Encyclopedia of Genes and Genomes (KEGG) analyses were conducted on the differentially expressed genes (DEGs) at five times post inoculation. In three weakly pathogenic strains, CN-4, XJ-3, and SN-2, the variation in expression patterns of pathogenicity-related genes, including carbohydrate-active enzyme (CAZy) genes, pathogen-host interaction (PHI) genes, effector genes, secondary metabolism-related genes, cytochrome P450s (CYPs) genes, ATP-binding cassette (ABC) superfamily transporter genes, and G protein-coupled receptor genes (GPCRs), was analyzed at different times during the infection. This study increased the understanding of the genome-level pathogenic variation among different T. horrida strains and identified key genes involved in pathogenic processes.
Materials and methods
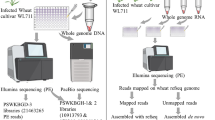
Strains, plant materials, and growth conditions
The RKS samples used for sequencing were collected from several heavily infected rice cultivars grown in different provinces of China. Six T. horrida strains were used for whole genome re-sequencing, and T. horrida strain JY-521 was used as the reference strain (Wang et al. 2018; Supplementary Table 1). For transcriptomic analysis, the moderately pathogenic T. horrida strain JY-521 was used. Rice male sterile line 9311A (which is highly susceptible to RKS) was used for the pathogenicity tests.
Pathogenicity assessment of seven T. horrida strains
Monoconidial cultures of the seven T. horrida strains were grown in potato dextrose agar in an incubator at 28 °C for 4 days. Fungal spores were collected, and a suspension of 106 spores/ml was prepared by suspending the spores in sterilized water. Approximately 1 ml of spore suspension was injected into the immature kernels of field grown rice plants at the booting stage, i.e., 5–7 days before heading. For each strain of T. horrida, 15 immature kernels on different rice plants were inoculated as replicates, and for the control group, the kernels were inoculated with 1 ml of sterilized distilled water. At rice maturity, the disease incidence was examined, and the disease severity was graded on a scale from 0 to 5 on the basis of the number of diseased grains per ear and the loss of output according to the method of Deng et al. (1997). Disease severity grades were Grade 0, with no morbidity or morbidity was extremely low and yield loss was less than 1%; Grade 1, with 1–5 diseased grains per ear and yield loss ~ 3%; Grade 2, with 6–10 diseased grains per ear and yield loss ~ 5%; Grade 3, with 11–15 diseased grains per ear and yield loss ~10%; Grade 4, with 16–20 diseased grains per ear and yield loss ~ 20%; and Grade 5, with more than 21 diseased grains per ear and yield loss ≥30%.
Genomic DNA extraction, library construction and high throughput sequencing
Genomic DNA was extracted from the six T. horrida strains by using the CTAB method (Liu et al. 2005). DNA degradation and contamination were monitored on 1% agarose gel. DNA purity was checked using a NanoPhotometer spectrophotometer (IMPLEN, CA, USA). The DNA concentration was measured using a Qubit DNA Assay Kit in a Qubit® 2.0 Fluorometer (Life Technologies, CA, USA). Each DNA sample was fragmented by sonication to 350 bp fragments, and then, the DNA fragments were end-polished, A-tailed, and ligated with a full-length adapter for Illumina sequencing with further PCR amplification. The PCR products were purified (AMPure XP system), and the libraries were analyzed for size distribution by using an Agilent 2100 Bioanalyzer and quantified using real-time PCR. The whole genome re-sequencing was performed using an Illumina HiSeq 2500 system, and 150 bp paired-end reads were generated with an insert size of approximately 350 bp (Novogene Biological Technology Co., Ltd., China).
Read mapping, single nucleotide polymorphism (SNP) detection and annotation
Fast format raw data (raw reads) were first processed through a series of quality control procedures, and in-house C scripts and clean data (clean reads) were obtained. The clean reads of the six T. horrida strains were mapped to the assembled genome of T. horrida strain JY-521 (Wang et al. 2018) using Burrows-Wheeler Aligner software (version 0.7.12). For this procedure, the command “mem–k 32 -M” was used, and Burrows-Wheeler Aligner alignment files were generated using SAMtools software (version 0.1.19). Single nucleotide polymorphisms (SNPs) and indels were identified using GATK software (version 3.4–46) with a UnifiedGenotyper model and were filtered using a VariantFiltration model and options -Window 4, -filter “QD < 4.0 || FS > 60.0 || MQ < 40.0”; and -G_filter “GQ < 20”. Filtered SNPs and Indels were annotated using ANNOVAR software (version 2014-07-14).
Transcriptome data
Tilletia horrida strain JY-521 was used to infect rice male sterile line 9311A to create the treatment group (Wang et al. 2018). Young panicles from field-grown rice plants (at the booting stage 3–5 days before heading) were collected during late afternoon. The panicles were disinfected with 75% alcohol for 2 min, washed twice with sterile water, and then air-dried for 20 min. A small section of the panicles hull was cut off to allow inoculation of individual colonies of JY-521 on peptone sucrose agar. At six times post-inoculation (0, 8, 12, 24, 48, and 72 h), the mycelium of JY-521 was collected in liquid nitrogen and stored at −80 °C. Total RNA of JY-521 was extracted using the Omega Fungal RNA kit method. Dried RNA samples were dissolved in diethyl pyrocarbonate water. RNA quality was assessed on 1.0% denaturing agarose gels. The total RNA of JY-521 following infection of rice for 0, 8, 12, 24, 48, and 72 h was used for the RNA-seq analysis.
Transcriptome expression analysis of genes
The RNA-seq libraries were constructed using a NEBNext® Ultra™ RNA Library Prep Kit for Illumina® (NE, USA), according to the manufacturer’s instructions. mRNA-seq was used for the expression analysis. The RNA expression analysis was based on the predicted genes in T. horrida. A comparison map of mRNA reads in relation to the genome was generated by Tophat v2.0.14 (Kim et al. 2013; Trapnell et al. 2010), and the number of expected fragments of 1 kb of transcript per million fragments sequenced (FPKM) was calculated using Cufflinks (Trapnell et al. 2014). The resulting P values were adjusted using the Benjamini and Hochberg’s approach for controlling the false discovery rate (FDR) (Benjamini and Hochberg 1995). Genes with adjusted P < 0.05 and |log2fold change (FC)| >1 were designated as DEGs.
Series-clustered analysis
A total of 500 DEGs at different post inoculation time points were classified into 24 clustered profiles based on the basis of the trends observed in gene expression by using the Short Time-series Expression Miner software (STEM) (Ernst and Bar-Joseph 2006). The clustered profiles with P ≤ 0.05 were considered as statistically significant. Then, the DEGs in all or each profile were subjected to KEGG pathway enrichment analysis. Through the hypothesis test of the P value calculation and FDR (Benjamini and Hochberg 1995) correction, the pathways with q-value ≤0.05 were defined as significantly enriched.
GO and KEGG enrichment analysis of DEGs
The GO enrichment analysis of the DEGs was performed in R using the GOseq package, after correction for gene length bias (Wang et al. 2010; Young et al. 2010). GO terms with corrected P < 0.05 were considered as significantly enriched. The KEGG is a database resource used to understand high-level functions and utilities of biological systems, such as the cell, organism, and ecosystem. The KEGG uses molecular information; particularly large-scale molecular datasets generated by genome sequencing and other high-through put experimental technologies (http://www.genome.jp/kegg/). The KOBAS software was used to test the statistical enrichment of DEGs in KEGG pathways (Mao et al. 2005; Kanehisa et al. 2008).
Results and discussion
Pathogenicity assessment of the seven T. horrida strains
The pathogenic effects of seven different T. horrida isolates on rice male sterile line 9311A were identified. As shown in Table 1, three isolates, SN-2, XJ-3, and CN-4 were weakly pathogenic (< 15 diseased grains per ear with loss output <10%); three isolates, JY-521, HN-5, and GZ-6, were moderately strongly pathogenic (~20 diseased grains per ear with loss output ~15%); and strain JS-1 was strongly pathogenic (> 21 diseased grains per ear with loss output >20%). The sprouting ratios of the seven T. horrida strains were all low (<30%); the highest ratio was for strain JS-1 at 23.8%, whereas the lowest was for strain XJ-3 at 15.8% (Fig. 1). These results indicated that pathogenicity was related to the sprouting ratio of the strain.

The sprouting ratios of the seven T. horrida strains. Standard Error of Mean of each sample was noted
Re-sequencing of the six T. horrida strains
From the genome re-sequencing of the six T. horrida strains, 6.54 Gb of data of clean reads were obtained (Supplementary Table 2). The mean GC percentage of the clean reads was 54.98%. The Q20 percentage (proportion of nucleotides with a quality value >20 in the reads) was >97% and the Q30 percentage was >93% (Supplementary Table 2). For all six strains, 8,900,110-8,965,530 clean reads were generated. The portion of the total map varied from 93.78% to 96.09%, with an average depth of 57× and an average genome coverage of 94.84% (Supplementary Table 2). Among the six T. horrida strains, the weakly pathogenic strains CN-4, SN-2, and XJ-3 had the fewest total mapped reads (Supplementary Table 2).
Variation among the six T. horrida strains
In the comparison of the re-sequenced genomes of the six T. horrida strains with the reference genome of the T. horrida strain JY-521, 273,622 high quality SNPs were obtained (Supplementary Table 3; Wang et al. 2018). The weakly pathogenic strains CN-4, XJ-3, and SN-2 had the fewest SNPs, leading to the hypothesis that their weak pathogenicity was because the key genes related to pathogenicity were mutated. Indels in the coding region can alter gene function via frameshift mutations, leading to variation among different individuals. In the whole genomes of the six strains, 32,876 indels were found; thus, fewer indels than SNPs were found. Similarly to SNPs, the three weakly pathogenic strains had the lowest indel variation. Structural variations (SVs) primarily include insertions, deletions, inversions and translocations of large fragments. In this study, the insertions (INS), deletions (DEL), inversions (INV), intra-chromosomal translocations (ITX), and inter-chromosomal translocations (CTX) were identified using Break Dancer software (Chen et al. 2009). Among SVs, INS, DEL, and CTX were abundant in the six re-sequenced T. horrida genomes. The weakly pathogenic strain XJ-3 had the highest number of SVs, suggesting they were related to the pathogenicity of T. horrida (Supplementary Table 4).
Transcriptional sequencing
To investigate the changes in gene expression levels during the early infection stages, RNA-seq analysis of T. horrida strain JY-521 was conducted at 8, 12, 24, 48, and 72 h post inoculation. The RNA-seq analysis of all six samples generated 25.33 Gb of data, and 202,624,224 reads were obtained after quality trimming and adapter removal (Supplementary Table 5). For all 6 samples, the portion of the total map varied from 70.16–73.70% and that of the unique map varied from 69.10% to 72.59% (Supplementary Table 5).
Differentially expressed genes
To identify pathogenicity genes, the changes in gene expression of T. horrida JY-521 were analyzed at different times post inoculation. At 8 h, 337 DEGs were detected (120 up-regulated and 217 down-regulated); at 12 h, 111 DEGs were detected (62 up-regulated and 49 down-regulated); at 24 h, 91 DEGs were detected (29 up-regulated and 62 down-regulated); at 48 h, 89 DEGs were detected (64 up-regulated and 25 down-regulated); and at 72 h, 55 DEGs were detected (32 up-regulated, and 23 down-regulated) (FDR < 0.05 and |log2 FC| > 1; Fig. 2a). Therefore, the highest number of DEGs was detected at 8 h. Based on the overlap analysis of DEGs at different time points, the expression of 110 genes was up-regulated at 8 h (Fig. 2b, c). Thus, more genes played a role in the early infection than at later times. After disease was induced by T. horrida, heat map clustering indicated significant differences in these DEGs (Fig. 2d).

Different expression genes (DEGs) analysis of T. horrida strain JY-521 at different inoculation time points. a: Number of DEGs at 8, 12, 24, 48, and 72 h in T. horrida strain JY-521; b: Venn diagrams showing the overlapping of the down-regulated DEGs modulated at 8, 12, 24, 48, and 72 h in T. horrida strain JY-521; c: Venn diagrams showing the overlapping of the up-regulated DEGs modulated at 8, 12, 24, 48, and 72 h in T. horrida strain JY-521; d: Heat map cluster analysis of 500 DEGs. The expression level was indicated by different colors, red indicates a high expression and blue indicates a low expression
Enrichment analysis of DEGs
GO classification of the DEGs at different infection times during the infection was conducted to determine whether these DEGs were significantly associated with a specific biological process, molecular function, or cellular component. In the biological process category, GO term annotations showed that most enriched genes were associated with biological regulation, cellular component organization or biogenesis, cellular process, localization, metabolic process classified, response to stimulus, and single-organism process. In the molecular function category, most DEGs were associated with binding, catalytic activity, and transporter activity. In the cellular component category, most DEGs were associated with cell, cell part, and membrane and organelle (Fig. 3). Based on the KEGG pathways analysis, the following pathways were most enriched in DEGs: degradation of aromatic compounds, amino sugar and nucleotide sugar metabolism, fatty acid metabolism, linoleic acid metabolism, starch and sucrose metabolism, fatty acid biosynthesis, galactose metabolism, regulation of autophagy, nitrogen metabolism, fatty acid degradation, steroid biosynthesis, phenylalanine metabolism, and protein processing in endoplasmic reticulum (Fig. 4). Among these pathways, autophagy processes are highly conserved pathways that play important roles in the pathogenicity of filamentous fungi (Zhu et al. 2018)

GO enrichment analysis of DEGs in T. horrida strain JY-521. Gene Ontology terms are classified into three main categories: biological process, cellular component, and molecular function

KEGG enrichment analysis of DEGs in T. horrida strain JY-521
Differences in gene expression patterns
The resistance to T. horrida results from the common effect of multiple genes. A total of 500 DEGs at different time points were detected in T. horrida and categorized into 24 subclustered profiles (Fig. 5). The trend analysis showed that genes in T. horrida primarily sustained up-regulated expression (profile 23), or down-regulated expression (profile 0), or down-regulated expression at 8 h after infection (profiles 6 and 2). The metabolic pathways were also analyzed using the KEGG to elucidate the biological functions of DEGs in each profile. Among all profiles, the expression of profile 23 showed an upward trend at all time points, and those sustained up-regulated genes appeared to be biologically important in the resistance to T. horrida. According to KEGG enrichment analysis, most enriched genes in profile 23 were associated with transport and catabolism; lipid metabolism; global and overview; folding, sorting, and degradation; and carbohydrate metabolism (Fig. 6). The transport and catabolism pathway suggested a general response or a basic role in the process of T. horrida inoculation. In a previous study the carbohydrate metabolism and lipid degradation pathways produced secondary metabolites associated with pathogenesis (Cao et al. 2018). The DEGs clustered in profiles 17, 19, 14, and 18 in T. horrida were up-regulated at 8 h and down-regulated at subsequent time points. The KEGG enrichment analysis revealed that the enriched genes in those profiles were involved in folding, sorting, and degradation; global and overview; lipid degradation; metabolism of cofactors and vitamins; translation; amino acid metabolism; transport and catabolism; and replication and repair. These results suggest that these pathways play important role in T. horrida pathogenicity; however, further research is needed to understand the mechanisms of pathogenicity.

Clusters of genes with unique expression patterns in T. horrida strain JY-521. Clusters were ordered according the number of genes assigned to them

KEGG annotation of genes in each profile
Carbohydrate-active enzyme genes
Pathogenic fungi secrete carbohydrate-active enzymes (CAZymes) that can degrade plant cell walls (Cantarel et al. 2009). Among 1424 total putative CAZyme genes, 874 genes produced 6,820 nonsynonymous SNPs in the re-sequenced genomes of the six T. horrida strains. Among these nonsynonymous SNPs, 396 were common to the six T. horrida strains, whereas 312 (GZ-6), 642 (HN-5), 591 (JS-1), 613 (SN-2), 701 (XJ-3), and 607 (CN-4) nonsynonymous SNPs were unique to the re-sequenced genomes (Fig. 7). Fifty-one nonsynonymous SNPs were common to the re-sequenced genomes of the three weakly pathogenic strains CN4, XJ3, and SN-2, but none were found in the other strains (Supplementary Table 6). Those nonsynonymous SNPs were found in 21 genes, and their expression patterns at different times during the infection are shown in Fig. 8. The smut_2980 that encoded chitin deacetylase was up-regulated at 12 h after infection, and chitin deacetylase can prevent pathogenic fungi from being recognized by the plant immune system during infection (Cord-Landwehr et al. 2016). The nonsynonymous SNP in smut_2980 of the three weakly pathogenic strains could change the function of the protein encoded and as a result, the host could identify the pathogen during early infection. Similarly, smut_1230, which encoded an uncharacterized protein, and smut_1222, which encoded a retrotransposon ty3-gypsy subclass were up-regulated at 8 h after infection and may play an important role during early infection. These nonsynonymous SNPs may be responsible for the poor infection rate of weak pathogens.

Number of nonsynonymous SNP in six T. horrida strains. Common: number of nonsynonymous SNP common in six T. horrida; GZ: number of nonsynonymous SNP unique in strain GZ-6; HN: number of nonsynonymous SNP unique in strain HN-5; JS: number of nonsynonymous SNP unique in strain JS-1; SN: number of nonsynonymous SNP unique in strain SN-2; XJ: number of nonsynonymous SNP unique in strain XJ-3; CN: number of nonsynonymous SNP unique in strain CN-4; Weak common: number of nonsynonymous SNP common in three weak pathogenicity T. horrida, but not found in other strains. ABC transporter denotes ABC transporter genes; CAZy denotes carbohydrate-active enzymes genes; Effectors denote Effector protein genes; GPCRs denote G protein-coupled receptors genes; p450 denotes cytochrome P450 genes; PHI denotes pathogen-host interaction genes; Secondary metabolites denote secondary metabolism-related genes

Expression pattern of genes related to pathogenicity. The expression level was indicated by different colors, red indicates a high expression and blue indicates a low expression
Pathogen-host interaction genes
Of the 1697 putative pathogen-host interaction (PHI) genes, 1,429 genes produced 11,340 nonsynonymous SNPs in the re-sequenced genomes of the six T. horrida strains. A total of 777 nonsynonymous SNPs were common to the six T. horrida strains, whereas 535 (GZ-6), 1026 (HN-5), 906 (JS-1), 1018 (SN-2), 1221 (XJ-3), and 930 (CN-4) were unique to the re-sequenced genomes (Fig. 7). Fifty-two nonsynonymous SNPs were common to the re-sequenced genomes of the three weakly pathogenic strains CN4, XJ3, and SN-2, but none were found in the other strains (Supplementary Table 7). Those nonsynonymous SNPs were found in 36 genes, and their expression patterns at different times during the infection are shown in Fig. 8. Four genes (smut_1409, smut_3510, smut_6708, and smut_2974) were up-regulated at 8 h, and five genes (smut_0201, smut_6433, smut_4096, smut_7557, and smut_3993) were up-regulated at 24 h. Those genes may have an important role during the process of infection and the nonsynonymous SNPs found in SN-2, XJ-3, and CN-4 were likely the cause of the weak pathogenicity.
Effector protein genes
Effectors are the virulence factors encoded by fungal avirulence genes that play an important role in the process of host colonization (Faris et al. 2010; Ma et al. 2010). Most fungal secreted effectors are cysteine-rich proteins that play a role during early infection; for example, Avr4 of Cladosporium fulvum protects chitin in fungal cell walls against plant chitinases (Stergiopoulos and de Wit 2009). From 366 candidate effector proteins, 289 genes produced 1,129 nonsynonymous SNPs in the re-sequenced genomes of the six T. horrida strains. Among the nonsynonymous SNPs, 76 were in common to the six T. horrida strains, wherea 48 (GZ-6), 95 (HN-5), 79 (JS-1), 95 (SN-2), 121 (XJ-3), and 74 (CN-4) were unique to the re-sequenced genomes (Fig. 7). Six nonsynonymous SNPs were common to the re-sequenced genomes of the three weakly pathogenic strains CN4, XJ3, and SN-2, but none were found in the other strains (Supplementary Table 8). Those SNPs were found in five genes, and their expression patterns at different times during the infection are shown in Fig. 8. Of those five genes, smut_1985, smut_1637, and smut_4897 were up-regulated during early infection; thus, the three candidate effector genes may be important in the colonization of rice by T. horrida, encoding eIF-2alpha kinase GCN2, putative isp4-oligopeptide transporter, and an uncharacterized protein, respectively, according to annotation of the NR database.
Secondary metabolism-related genes
Plant pathogenic fungi produce some secondary metabolites that are associated with pathogenicity, such as host-selective toxins (Keller et al. 2005). Of 77 putative secondary metabolism-related genes, 72 genes produced 1128 nonsynonymous SNPs in the re-sequenced genomes of the six T. horrida strains. Forty-four of the nonsynonymous SNPs were common to the six T. horrida strains, and 39 (GZ-6), 25 (HN-5), 62 (JS-1), 66 (SN-2), 85 (XJ-3), and 68 (CN-4) were unique to the re-sequenced genomes (Fig. 7). Six nonsynonymous SNPs were common to the re-sequenced genomes of the three weakly pathogenic strains CN4, XJ3, and SN-2, but none were found in the other strains (Supplementary Table 9). Those nonsynonymous SNPs were found in four genes, and their expression patterns at different times during the infection are shown in Fig. 8. Smut_2974 was up-regulated at 8 h and was associated with sterigmatocystin biosynthesis, according to NR annotation. The toxic effects of sterigmatocystin, with a xanthone nucleus attached to a bifuran structure, are similar to those of aflatoxins (Struna 2016). Therefore, the nonsynonymous SNPs found in SN-2, XJ-3, and CN-4 likely affected the biosynthesis of sterigmatocystin, resulting in weak pathogenicity.
Cytochrome P450 genes
Cytochrome P450s (CYPs) play an important role in pathogenesis and in the production of toxins (Nelson 1999; Que et al. 2014). Of 19 CYP genes, 18 genes produced 107 nonsynonymous SNPs in the re-sequenced genomes of the six T. horrida strains. Fifteen of the nonsynonymous SNPs were common to the six T. horrida strains, and 5 (GZ-6), 7 (HN-5), 5 (JS-1), 16 (SN-2), 7 (XJ-3), and 5 (CN-4) were unique to the re-sequenced genomes (Fig. 7). In the three weakly pathogenic strains CN4, XJ3, and SN-2, no common nonsynonymous SNPs were found.
ABC transporter genes
Transporters not only obtain carbon and nitrogen sources from their host plants, but also contribute to toxin and effector secretion (Mueller et al. 2008). Of 40 ABC transporter genes, 36 genes produced 461 nonsynonymous SNPs in the re-sequenced genomes of the six T. horrida strains. Nineteen of the nonsynonymous SNPs were common to the six T. horrida strains, and 20 (GZ-6), 45 (HN-5), 32 (JS-1), 31 (SN-2), 69 (XJ-3), and 51 (CN-4) were unique re-sequenced genomes (Fig. 7). Two nonsynonymous SNPs were common to the re-sequenced genomes of the three weak pathogenic strains CN4, XJ3, and SN-2, but none were found in the other strains (Supplementary Table 10). Those nonsynonymous SNPs were found in two genes, and their expression patterns at different times during the infection are shown in Fig. 8. Smut_4787 was up-regulated at 72 h, and annotation with NR data showed that this gene was a pleiotropic drug resistance ABC transporter. The nonsynonymous SNPs found in SN-2, XJ-3, and CN-4 may weaken the resistance of those three strains to plant immune systems and the external environment.
G protein-coupled receptor genes
To infect a host plant, plant pathogenic fungi must make appropriate responses to a variety of host plant surface environmental receptors. These receptors function via the mitogen-activated protein kinase (MAPK) pathway, which sends a signal from a receptor on the surface of a cell to the nucleus. G protein-coupled receptors (GPCRs) and G-proteins are important components of the MAPK pathway, which has seven transmembrane domain receptors, constitutes a large family of cell surface receptors and is responsible for transducing extracellular signals into intracellular responses that involve complex intracellular-signalling networks (Dean et al. 2005; Soanes et al. 2007). Of 67 GPCRs, 57 genes produced 423 nonsynonymous SNPs in the re-sequenced genomes of the six T. horrida strain. Seventeen of the nonsynonymous SNPs were common to the six T. horrida strains, and 21 (GZ-6), 29 (HN-5), 29 (JS-1), 38 (SN-2), 39 (XJ-3), and 47 (CN-4) were unique to re-sequenced genomes (Fig. 7). Smut_1863 and smut_4953 produced two nonsynonymous SNPs that were common to the re-sequenced genomes of the three weakly pathogenic strains CN4, XJ3, and SN-2, but none were found in the other strains (Supplementary Table 11). The expression patterns of the two genes at different times during the infection are shown in Fig. 8. Smut_4953 was up-regulated at 24 h after infection, which was associated with the ATM1-mitochondrial inner membrane ABC transporter involved in the maturation of cytosolic iron-sulfur cluster-containing proteins, according to annotation of NR data.
Conclusions
Next generation sequencing technologies are low in cost and high in accuracy and have been used to study the genomics and transcriptomics of a wide variety of organisms (Bentley 2006; Maher et al. 2009). However, RKS has been previously overlooked, and few studies have been performed to differentiate and investigate the pathogenicity of T. horrida. In this study, the pathogenicity of seven RKS T. horrida strains from different districts in China was evaluated, and then, whole genome re-sequencing of high and low pathogenic T. horrida strains was performed using the Illumina Hiseq 2500 high-throughput sequencing technology. The genome assembly size (23.2 Mb) of T. horrida strains was smaller than that of other fungi in the genus Tilletia, such as T. caries (29.5 Mb), T. controversa (28.8 Mb), T. indica (30.4 Mb), and T. walkeri (24.3 Mb) as well as for other rice pathogenic fungi such as Magnaporthe grisea (38.8 Mb) (Dean et al. 2005). These studies provided an opportunity to analyze the mechanisms underlying the diverse range of rice- T. horrida interactions.
SNPs, small indels, and SVs were identified between the re-sequenced genomes of the six T. horrida strains and the genome of the reference strain JY-521 through comparative genomic and phylogenetic analyses. These results showed that polymorphisms exist in the RKS pathogen. These mutant sites can provide important resources for the development of relevant markers and the discovery of pathogenicity related genes and are therefore important in studies of T. horrida pathogenic mechanisms. Furthermore, this variation might be caused by significant variations in pathogenicity related genes. The nonsynonymous SNPs of virulence-associated genes (CAZyme genes, PHI genes, effector protein genes, secondary metabolism-related genes, CYP genes, ABC transporter genes, and GPCR genes) in the three weakly pathogenic strains and the other strains were compared, and some important virulence-associated genes contained the nonsynonymous SNPs in the genomes of the weakly pathogenic strains. However, in the genomes of the other more pathogenic strains, none were found. The genomic and transcriptomic data of the six T. horrida strains generated in this study provide a foundation for future studies on pathogenic mechanisms. In addition, the transcriptome data recorded at different times during infection can be used to further study the pathogenicity related genes in T. horrida.
Data availability
Sequencing data of Tilletia horrida were deposited at the National Center for Biotechnology (NCBI) Sequence Read Archive (SRA) under bioproject PRJNA494098 (for RNA-Seq) and bioproject PRJNA494149 (for genome sequencing).
References
Ali S, Laurie JD, Linning R, Cervantes-Chávez JA, Gaudet D, Bakkeren G (2014) An immunity-triggering effector from the barley smut fungus Ustilago hordei resides in an Ustilaginaceae-specific cluster bearing signs of transposable element-assisted evolution. PLoS Pathog 10(7):e1004223
Amselem J, Cuomo CA, van Kan JA, Viaud M, Benito EP, Couloux A, Coutinho PM, de Vries RP (2011) Genomic analysis of the necrotrophic fungal pathogens Sclerotinia sclerotiorum and Botrytis cinerea. PLoS Genet 7(8):e1002230
Benjamini Y, Hochberg Y (1995) Controlling the false discovery rate: a practical and powerful approach to multiple hypothesis testing. Journal of Royal Statistical Society. Series B 57:289–300
Bentley DR (2006) Whole genome re-sequencing. Curr Opin Genet Dev 16(6):545–552
Biswas A (2003) Kernel smut disease of rice: current status and future challenges. Environ Ecol 21:336–351
Brooks SA, Anders MM, Yeater KM (2009) Effect of cultural management practices on the severity of false smut and kernel smut of Rice. Plant Dis 93:1202–1208
Cantarel BL, Coutinho PM, Rancurel C, Bernard T, Lombard V, Henrissat B (2009) The carbohydrate-active EnZymes database (CAZy): an expert resource for glycogenomics. Nucleic Acids Res 37:D233–D238
Cao HJ, Huang PY, Yan YX, Shi YK, Dong B, Liu XH, Ye LD, Lin FC, Lu JP (2018) The basic helix-loop-helix transcription factor Crf1 is required for development and pathogenicity of the rice blast fungus by regulating carbohydrate and lipid metabolism. Environ Microbiol 20(9):3427–3441
Carris LM, Castlebury LA, Goates BJ (2006) Nonsystemic bunt Fungi-Tilletia indica and T. horrida: a review of history, systematics, and biology. Annu Rev Phytopathol 44:113–133
Chen K, Wallis JW, McLellan MD, Larson DE, Kalicki JM, Pohl CS, McGrath SD, Wendl MC, Zhang Q, Locke DP, Shi X, Fulton RS, Ley TJ, Wilson RK, Ding L, Mardis ER (2009) Break dancer: an algorithm for high-resolution mapping of genomic structural variation. Nat Methods 6:677–681
Chen Y, Yang X, Yao J, Kyaw EP, Zhang AF, Li YF, Gu CY, Zang HY, Gao TC (2016) Simple and rapid detection of Tilletia horrida causing rice kernel smut in rice seeds. Sci Rep 6:33258
Cord-Landwehr S, Melcher RLJ, Kolkenbrock S, Moerschbacher BM (2016) A chitin deacetylase from the endophytic fungus Pestalotiopsis sp. efficiently inactivates the elicitor activity of chitin oligomers in rice cells. Sci Rep 6:38018
Dean RA, Talbot NJ, Ebbole DJ, Farman ML, Mitchell TK, Orbach MJ, Thon M, Kulkarni R (2005) The genome sequence of the rice blast fungus Magnaporthe grisea. Nature 434:980–986
Deng GH, Yang ZH, Cheng JF (1997) Study on standardized index for grain smut of rice. China Journal of Plant Protection 24(2):189–190
Ernst J, Bar-Joseph Z (2006) STEM: a tool for the analysis of short time series gene expression data. BMC bioinformatics 7:191
Faris JD, Zhang ZC, Lu HJ, Lu SW, Reddy L, Cloutier S, Fellers JP, Meinhardt SW, Rasmussen JB, Xu SS, Oliver RP, Simons KJ, Friesen TL (2010) A unique wheat disease resistance-like gene governs effector-triggered susceptibility to necrotrophic pathogens. Proc Natl Acad Sci U S A 107:13544–13549
Galagan JE, Calvo SE, Borkovich KA, Selker EU, Read ND, Jaffe D, FitzHugh W (2003) The genome sequence of the filamentous fungus Neurospora crassa. Nature 422(6934):859–868
Goodwin SB, M'barek SB, Dhillon B, Wittenberg AH, Crane CF, Hane JK, Foster AJ, der Lee V (2011) Finished genome of the fungal wheat pathogen Mycosphaerella graminicola reveals dispensome structure, chromosome plasticity, and stealth pathogenesis. PLoS Genet 7(6):e1002070
Huang HP, Ma S, Huang JH, Zheng JL, Yi KX (2016) Whole genome re-sequencing and transcriptome analysis of the Stylosanthes Anthracnose pathogen Colletotrichum gloeosporioides reveal its characteristics. Mycosphere 7(8):1124–1146
Kamper J, Kahmann R, Bolker M, Ma LJ, Brefort T, Saville BJ, Banuett F, Kronstad JW, Gold SE, Mulle O (2006) Insights from the genome of the biotrophic fungal plant pathogen Ustilago maydis. Nature 444:97–101
Kanehisa M, Araki M, Gota S, Hattori M, Hirakawa M, Itoh M, Katayama T, Kawashima S, Okuda S, Tokimatsu T, Yamanishi Y (2008) KEGG for linking genomes to life and the environment. Nucleic Acids Res 36(Database issue):D480–D484
Keller NP, Turner G, Bennett JW (2005) Fungal secondary metabolism-from biochemistry to genomics. Nat Rev Microbiol 3:937–947
Kim D, Pertea G, Trapnell C, Pimentel H, Kelley R, Salzberg SL (2013) TopHat 2: accurate alignment of transcriptomes in the presence of insertions, deletions and gene fusions. Genome Biol 14:R36
Li R, Li Y, Fang X, Yang H, Wang J, Kristiansen K, Wang J (2009) SNP detection for massively parallel whole-genome re-sequencing. Genome Res 19(6):545–552
Liu SH, Lu JP, Zhu RL, Dai FM (2005) A rapid and simple extraction method for plant pathogenic fungi. Acta Phytopathologica Sinica 35(4):362–365
Ma LJ, van der DC, Borkovich KA, Coleman JJ, Daboussi MJ, Di Pietro A, Dufresne M, Freitag M, Grabherr M (2010) Comparative genomics reveals mobile pathogenicity chromosomes in Fusarium. Nature 464:367–373
Maher CA, Palanisamy N, Brenner JC, Cao X, Kalyana-Sundaram S, Luo S, Khrebtukova I, Barrette TR, Grasso C, Yu J, Lonigro RJ, Schroth G, Kumar-Sinha C, Chinnaiyan AM (2009) Chimeric transcript discovery by paired-end transcriptome sequencing. Proc Natl Acad Sci U S A 106(30):12353–12358
Mao X, Cai T, Olyarchuk JG, Wei L (2005) Automated genome annotation and pathway identification using the KEGG Orthology (KO) as a controlled vocabulary. Bioinformatics. 21:3787–3793
Mueller O, Kahmann R, Aguilar G, Wu A (2008) The secretome of the maize pathogen Ustilago maydis. Fungal Genet Biol 45:S63–S70
Nelson DR (1999) Cytochrome P450 and the individuality of species. Arch Biochem Biophys 369:1–10
Ohm RA, Feau N, Henrissat B, Schoch CL, Horwitz BA, Barry KW, Condon BJ, Copeland AC (2012) Diverse lifestyles and strategies of plant pathogenesis encoded in the genomes of eighteen Dothideomycetes fungi. PLoS Pathog 8(12):e1003037
Que YX, Xu LP, Wu QB, Ling H, Liu YH, Zhang YY, Guo JL, Su YC, Chen JB, Wang SS, Zhang CG (2014) Genome sequencing of Sporisorium scitamineum provides insights into the pathogenic mechanisms of sugarcane smut. BMC Genomics 15:996
Rogerson CT (1988) Illustrated genera of smut fungi. Brittonia 40:107
Schuler D, Höll C, Grün N, Ulrich J, Dillner B, Klebl F, Ammon A, Voll LM, Kämper J (2018) Galactose metabolism and toxicity in Ustilago maydis. Fungal Genet Biol 114:42–52
Soanes DM, Richards TA, Talbot NJ (2007) Insights from sequencing fungal and oomycete genomes: what can we learn about plant disease and the evolution of pathogenicity? Plant Cell 19:3318–3326
Stergiopoulos L, de Wit PJG (2009) Fungal effector proteins. Annu Rev Phytopathol 47:233–263
Struna A (2016) Toxic effects of aflatoxin B1, sterigmatocystin and ochratoxina a on a HaCaT human keratinocyte cell line. Univerza v Ljubljani
Trapnell C, Williams BA, Pertea G, Mortazavi A, Kwan G, van Baren MJ, Salzberg SL, Wold BJ, Pachter L (2010) Transcript assembly and quantification by RNA-Seq reveals unannotated transcripts and isoform switching during cell differentiation. Nat Biotechnol 28:511–515
Trapnell C, Roberts A, Goff L, Pertea G, Kim D, Kelley DR, Pimentel H, Salzberg SL, Rinn JL, Pachter L (2014) Differential gene and transcript expression analysis of RNA-seq experiments with TopHat and cufflinks. Nat Protoc 9(10):2513
Tsuda M, Sasahara M, Ohara T, Kato S (2006) Optimal application timing of simeconazole granules for control of rice kernel smut and false smut. J Gen Plant Pathol 72:301–304
Verbruggen B, Bickley LK, Santos EM, Tyler CR, Stentiford GD, Bateman KS, van Aerle R (2015) De novo assembly of the Carcinus maenas transcriptome and characterization of innate immune system pathways. BMC Genomics 16(1):1–17
Wang L, Feng Z, Wang X, Zhang X (2010) DEGseq: an R package for identifying differentially expressed genes from RNA-seq data. Bioinformatics 26:136–138
Wang N, Ai P, Tang Y, Zhang J, Dai XJ, Li P, Zheng AP (2015) Draft genome sequence of the rice kernel smut Tilletia horrida strain QB-1. Genome Announc 3:e00621–e00615
Wang AJ, Pan LX, Wang N, Ai P, Yin DS, Li SC, Deng QM, Zhu J, Liang YY, Zhu JQ, Li P, Zheng AP (2018) The pathogenic mechanisms of Tilletia horrida as revealed by comparative and functional genomics. Sci Rep 8:15413
Wang Z, Li Y, Li C, Song X, Lei J, Gao Y, Liang Q (2019) Comparative transcriptome profiling of resistant and susceptible sugarcane genotypes in response to the airborne pathogen Fusarium verticillioides. Mol Biol Rep:1–13
Xing M, Lv H, Ma J, Xu D, Li H, Yang L, Kang J, Wang X, Fang Z (2016) Transcriptome profiling of resistance to Fusarium oxysporum f. sp. conglutinans in cabbage (Brassica oleracea) roots. PLoS One 11(2):e0148048
Xue M, Yang J, Li Z, Hu S, Yao N, Dean RA, Zhao W, Shen M, Zhang H, Li C, Liu L, Cao L (2012) Comparative analysis of the genomes of two field isolates of the rice blast fungus Magnaporthe oryzae. PLoS Genet 8(8):e1002869
Yap HY, Chooi YH, Fung SY, Ng ST, Tan CS, Tan NH (2015) Transcriptome analysis revealed highly expressed genes encoding secondary metabolite pathways and small cysteine-rich proteins in the sclerotium of Lignosus rhinocerotis. PLoS One 10(11):e0143549
Yin Z, Liu H, Li Z, Ke X, Dou D, Gao X, Song N, Dai Q, Wu Y, Xu JR, Kang Z, Huang L (2015) Genome sequence of Valsa canker pathogens uncovers a potential adaptation of colonization of woody bark. New Phytol 208(4):1202–1216
Young MD, Wakefield MJ, Smyth GK, Oshlack A (2010) Gene ontology analysis for RNA-seq: accounting for selection bias. Genome Biol 11:R14
Zhang Y, Zhang K, Fang A, Han Y, Yang J, Xue M, Bao J, Hu D, Zhou B, Sun X, Li S (2014) Specific adaptation of Ustilaginoidea virens in occupying host florets revealed by comparative and functional genomics. Nat Commun 5:3849
Zhao Z, Liu H, Wang C, Xu JR (2013) Comparative analysis of fungal genomes reveals different plant cell wall degrading capacity in fungi. BMC Genomics 14:274
Zhou SM, Chen LM, Liu SQ, Wang XF, Sun XD (2015) De novo assembly and annotation of the Chinese chive (Allium tuberosum Rottler ex Spr.) transcriptome using the Illumina platform. PLoS One 10(5):e0133312
Zhu XM, Li L, Wu M, Liang S, Shi HB, Liu XH, et al (2018) Current opinious on autophagy in pathogenicity of fungi. Virulence
Acknowledgements
We thank the scientific and technological research program of the Chongqing Municipal Education Commission (KJ15012017) and the National Natural Science Foundation (31400130) for supporting this research.
Funding
This study was funded by the scientific and technological research program of the Chongqing Municipal Education Commission (KJ15012017) and the National Natural Science Foundation (31400130).
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
All the authors declare that they have no conflict of interest.
Ethical approval
This article does not contain any studies with human participants or animals performed by any of the authors.
Informed consent
Informed consent was obtained from all individual participants included in the study.
Additional information
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
ESM 1
(XLSX 27 kb)
Rights and permissions
About this article

Cite this article
Wang, A., Shu, X., Niu, X. et al. Transcriptome analysis and whole genome re-sequencing provide insights on rice kernel smut (Tilletia horrida) pathogenicity. J Plant Pathol 102, 155–167 (2020). https://doi.org/10.1007/s42161-019-00401-8
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s42161-019-00401-8