
Abstract
With the development of smart materials, stimulus-responsive self-repairing materials attract more and more research attention. Although self-healing materials including concretes, rubbers, and hydrogels have been extensively investigated in the past decades, novel functionalities or properties such as shape memory, sol–gel transition, adhesion, anti-biofouling, and electronic/magnetic property have been introduced to self-repairing systems in recent years in order to broaden the scope of their applications in energy, drug delivery, tissue adhesion, cell culture, etc. In this paper, we first present an overview of the general strategies to prepare self-healing materials and the characterization methods to assess their healing performance. Then, we mainly focus on reviewing recent progress in novel self-healing materials possessing unique functionalities and their potential applications in biomedical engineering. Finally, the challenges and scope for future development are also discussed.

ᅟ
Similar content being viewed by others

Explore related subjects
Discover the latest articles, news and stories from top researchers in related subjects.Avoid common mistakes on your manuscript.
1 Introduction
As the degradation, damage, and deformation of traditional materials is an unavoidable process, a new class of materials with autonomic structure/functionality recovery are highly demanded in various applications. In the past decades, spontaneous healing of wound or injury in biological systems inspired researchers to develop synthetic materials able to mimic such elegant self-recovery process after damage. Up to date, numerous self-healing synthetic materials have been developed with the advancements of novel synthetic technologies, thus introducing a new chapter into material applications.
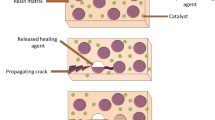
Self-healing materials have been used by Ancient Egyptians, Indians, and Romans for building purposes; however, insightful understanding of the self-recovery mechanism and rationale design of self-healing materials remained elusive until the end of the 1990s [1, 2]. Since then, there have been numerous self-healing materials developed, which could be simply classified into either extrinsic or intrinsic self-healing materials based on the resource of self-recovery. In general, structure and functionality recovery of extrinsic self-healing materials after being damaged heavily relies on the release and reaction of sequestered healing agents in the damaged region [3, 4]. Based on different approaches to realize the sequestration of healing agents, singular or multiple local self-healing performance could be observed. For example, capsule-based sequestration methods lead to singular local healing event due to the depletion of healing agents after one-time damage, while multiple healing events could occur based on vascular sequestration of healing agents as replenishment from the network or external supply is allowed. Different from extrinsic self-healing materials, inherent reversibility of bonding in the material’s matrix typically accounts for the healing performances in intrinsic self-healing materials; thus, repetitive local self-healing performance can be achieved without healing agents. Reversible non-covalent interactions and dynamic covalent bonds are the two most common self-healing strategies employed in intrinsic self-healing materials (Table 1). Reversible physical interactions such as hydrogen bonds, hydrophobic interactions, ionic interactions, π–π stacking, and host–guest interactions have been used to play the major role in the stimulus-responsive healing process of self-healable rubber, film, and hydrogels [11, 26, 33, 35,36,37,38,39,40,41,42,43,44,45,46,47]. Compared to non-covalent interactions, relatively strong dynamic covalent bonds introduce both structural stability and higher mechanical strength into intrinsic self-healing materials. For this reason, acylhydrazone bonds, imine bonds, disulfide bonds, and urea bonds have been introduced into polymers for self-recovery after damage [23,24,25, 31, 48,49,50,51].
In recent years, smart materials possessing stimulus-responsive property further boosted the research wave of self-healing materials. With the capability to respond to certain external stimuli, rationally designed smart materials with self-healing properties hold great potential for applications in the fields of which specific environmental conditions are inevitable. For example, low pH-stimulated self-repairing materials are desired for tissue adhesion in the stomach which contains corrosive acid solution for digestion [37, 52]. In addition, stimulus-responsive properties bring other functionalities into the same system beyond self-healing function, broadening the scope of applications in which multiple functionalities are required simultaneously.
We have witnessed that functional self-healing materials have been becoming the new trend in material design and development in the past decade. Recently, there are several excellent reviews on the topic of self-healing materials (including rubbers, hydrogels, metals, and polymers) in terms of healing strategies and mechanisms [53,54,55,56]. However, functionalities, other than self-healing, were barely reviewed and discussed. The purpose of this review is to introduce novel self-healing materials with other unique functionalities and their potential applications in biomedical engineering. This review article starts with a brief presentation of the general strategies to prepare self-healing materials and the characterization methods to assess self-healing performance in Sect. 2. Then, it concentrates on a number of unique functional self-healing materials in Sect. 3 and their potential applications in biomedical fields in Sect. 4. Finally, this review is closed with the discussion on current challenges and the future perspective for the development of functional self-healing materials.
2 Assessment of self-healing performance
The self-healing process includes both structure and functionality healing. Qualitative evaluation of self-repairing is usually through observation of structure recovery which is the first step in assessing self-healing performance. Multiple advanced characterization techniques employing optical microscope, laser microscope, scanning electron microscope (SEM), and atomic force microscope (AFM) have been utilized to record the healing process of structure [14, 21, 37, 41, 57,58,59,60]. Disappearance of the damage or coalescence of the cutting surface is frequently used as a general criterion to validate the occurrence of self-recovery. Due to different healing abilities, various degrees of structure recovery are observed. However, it is difficult to quantitatively determine the healing extent by optical observation.
To quantitatively evaluate the self-healing performance, the first step is to choose a desired property of function. It is worth noting that self-healing does not simply mean the restoration of all properties or functions to the original level. It is similar as what is observed in biological systems; sometimes, a scar is left after spontaneous healing of a wound [61]. Furthermore, restoration of each functionality may differ in degree. Thus, before quantitative evaluation of the self-healing extent, a certain property or function needs to be designated first. The healing efficiency of the self-healing material can be obtained by comparing the difference between original and healed materials, as the percent value indicates the extent of property restoration. Up to date, mechanical properties such as tensile strength and rheological property are mostly used to represent a certain function in the calculation of the healing efficiency. Thus, either a tensile test or a rheological measurement was widely accepted as the characterization method to quantify the healing extent in the literature reports. Healing efficiency relying on a tensile test can be defined as the ratio of the stress of the healed and original material [52, 62]. This method is more suitable for self-healing materials which are able to stand the stress of clamps [7, 63, 64]. Rheological measurement is also widely used to investigate the internal recovery of materials from 3D versions. Damage of material is initiated by a large strain, while the recovery of both storage modulus (G′) and loss modulus (G″) is recorded [34, 38, 65]. By comparing the stabilized modulus to its original value before deformation, the healing extent is obtained. Such characterization method is more applicable for soft materials which can be destructed by a large strain.
3 Functional self-healing materials
Based on the functionality, the self-healing materials can possess different properties including electronic/magnetic properties, adhesion, fluorescence, sol–gel transition, pH sensitivity, light sensitivity, and shape memory, which will be discussed in detail in the subsequent section.
3.1 Self-healing materials with electronic/magnetic properties
Electrical properties are generally required for the components of most electronic devices to support their normal operation. Failure of key components may lead to eventual dysfunction of electronic devices. Thus, self-healing materials with electronic conducting properties, serving as the self-recovery components, could bring a new concept to elongate the lifetime of electrical devices, which are especially important for implanted medical devices and electronic skin. Consequently, it stimulates the research in the design and development of conducting self-healing materials. Recently, a neural compatible graphene–poly(N,N-dimethylacrylamide) (PDMAA) hydrogel with high conductivity and low impedance was fabricated by Hou et al. [66]. Self-healing of the conducting hydrogel was trigged by a thermal stimulus, thus re-establishing the equilibrium of disrupted hydrogen bonding between PDMMA chains and graphene walls. Sixty percent of initial electrical conductivity was restored via healing at 37 °C over a period of 12 h. Near-infrared laser was also used as photothermal stimulus to initiate the recovery of the developed material, and healing was observed after 2 h by maintaining the temperature of the laser spot below 50 °C. Moreover, self-healing stretchable (SHS) wires with metallic conductivity were fabricated [67]. Liquid metal–eutectic gallium indium was injected into microchannels composed of the self-healing polymer Reverlink® to form stretchable conducting wires. After being broken completely and realigned in sequence, reintegration of liquid metal within the self-healing microchannels leads to the recovery of its conductivity (Fig. 1). The results show that both mechanical and electrical properties were restored simultaneously. Although the resistance increased slightly after the healing process and only 40% of its original tensile strain was recovered, the friendly healing process occurred within 10 min at ambient conditions, which is impressive. The self-healing conducting materials provide potential substitutes for conventional electronic components with an improved durability and lifespan and also offer the opportunity for the development of stretchable electronic devices with self-healing properties [68, 69].

a Schematics illustrating the disconnection and reconnection of a simple electronic circuit using a self-healing wire. A SHS wire is pictured in the inset (ii). The reconnected wires were misaligned on purpose to clearly identify the cut after self-healing in the inset (iii) and show that only the eGain channel has to be aligned to restore electrical conductivity. b Variation of the resistance of SHS wires during connection/disconnection/reconnection experiments. The error bars indicate the variability across three sets of measurements. c Mechanical characterization of a SHS wire (a slab of Reverlink® polymer with an embedded microchannel) before cut (black line) and after self-healing (red squares). (Reprinted with permission from [60]. Copyright 2013, Wiley-VCH.)
Furthermore, capacitors with self-healing properties were also developed. A mechanically and electrically self-recovery supercapacitor was created by spreading single-walled carbon nanotube (SWCNT) films onto a self-healing substrate (Fig. 2) [70]. The self-healing substrate was synthesized via cross-links between a large amount of hydrogen bond acceptors (urea and DETA) and donors (Empol 1016, 80 wt% diacids and 16% triacids) with TiO2 nanoflowers [70,71,72]. Hydrogen bonds in the supramolecular network are also responsible for the self-healing property of the as-developed capacitor. After the damage by cutting, the recovery of self-healing substrate helps the broken SWCNT film to contact again. Thus, both configuration and conductivity of the capacitor were restored. Up to 85.7% capacitance can be restored after cutting five times. A similar strategy was utilized to prepare self-recovery anodes for rechargeable batteries in order to expand the lifetime of batteries. Self-healing polymers formed via hydrogen bonds which were used to coat lithiated silicon microparticles (SiMP), serving as high-capacity electrodes [73]. Over 80% retention of initial capacity was achieved for the self-healed SiMP electrodes after 90 cycles of charging–discharging, which is more than ten times durable than other SiMP electrodes (< 9 cycles). These results indicate that spontaneous healing of the developed novel capacitor material not only expands the lifetime of the electrodes but also retains the electrical performance of the battery.

a The design and manufacturing process flow of a flexible, electrically and mechanically self-healing supercapacitor. The self-healing composite is composed of hierarchical flower-like TiO2 nanostructures (black spheres) and a supramolecular network (red wires) with a large amount of hydrogen bond acceptors (blue rods) and donors (green rods), which is then compressed under heat to form a self-healing substrate. CNT films are deposited on the self-healing substrates, which are then assembled to form the sandwiched supercapacitors. b Optical image of a flexible self-healing substrate on PET sheet after deposition of the CNT film. c Optical image of an integrated self-healing supercapacitor. (Reprinted with permission from [63]. Copyright 2014, Wiley-VCH.)
Besides aforementioned self-healing materials possessing electronic properties, magnetic self-healing materials were also designed and synthesized. It was recently reported that magnetic Fe3O4 nanoparticles with a carboxyl group on its surface were immobilized in chitosan and then formed hydrogel with telechelic difunctional poly(ethylene glycol) (DF-PEG) via imine bonds [27]. The self-healing performance of the hydrogel relied on the dynamic equilibrium of the bond breaking and reforming in the damaged region. Autonomous recovery of the hydrogel was obtained within 60 s at room temperature with 100% restoration of the storage modulus. The self-healing process of the damaged hydrogel pieces could also be triggered by a magnet due to the induced magnetic force in the hydrogel. Shape transformation was also observed when the hydrogel was driven with a magnet to pass through a channel with a built-in obstacle. These results imply the possibility of remotely controlling the self-healing and functionality of magnetic self-healing materials.
3.2 Self-healing materials with shape memory properties
To completely restore the mechanical performance is a big challenge in the design of self-healing materials. The purpose of utilizing intrinsic reversible chemical bonds or physical interactions for self-healing performance seems to be contradictory with the effort of maintaining structural and mechanical strength of the material simultaneously. This is one of the major hurdles which may impede the application of self-healing materials in a stress-bearing area. To address this challenge, shape memory moieties were introduced into self-healing systems to increase their mechanical performance. Thus, it may open a door for self-healing materials with shape memory properties in the design of novel biomaterials for potential prosthetics and orthotics applications. Recently, a self-healing poly(acrylic acid) (PAAc) hydrogel with enhanced mechanical strength in terms of shape memory behavior was reported [74]. The supramolecular polymer network is formed using hydrophobically modified PAAc with oppositely charged cetyltrimethylammonium (CTA) via hydrophobic and electrostatic interactions. Different from self-healing hydrogels which were formed only through hydrophobic interaction [9], its self-healing behavior still maintained in water due to the electrostatical entrapment of surfactant alkyl chains in the network. Healing efficiency of the hydrogel can be accelerated by heating, treatment of acid, or an addition of surfactant solution. Nearly 100% recovery could be achieved. In addition, the hydrogel exhibits 100% shape recovery and the recovery time could be shortened from 1 min to 10 s by increasing the temperature from 25 to 70 °C. Adapting a different strategy, a supramolecular hydrogel possessing both self-healing and shape memory was also reported via host–guest interactions (Fig. 3) [16]. One host β-cyclodextrin (βCD) and two kinds of guest moieties adamantine (Ad) and ferrocene (Fc) were immobilized to the backbone of the hydrogel, responsible for its self-healing performance. Under wet conditions in the presence of an oxidizing agent, coalescence of the cut hydrogel pieces was observed after 3 h. The extent of self-healing relied on the amounts of host–guest moieties as well as the recovery time. Up to 68% recovery was obtained after 72 h. Shape memory behavior of the hydrogel was also observed after the repetitive oxidation–reduction treatment cycle. Without such treatment, no shape-morphing occurred. The oxidant-sensitive Fc guest moieties are responsible for this stimulus-responsive shape memory effect. The high hydrophilicity of oxidized Fc moieties led to their exclusion from the cavity of βCD. Under the reducing state, those free guest moieties reformed inclusion with the host moieties, thus resulting in the shape memory behavior.

a Properties of the designed supramolecular polymeric hydrogel with two kinds of non-covalent cross-links (βCD-Ad and βCD-Fc): self-healing, expansion–contraction, and shape memory. b Chemical structure of the βCD-Ad-Fc gels; x, y, and z indicate the amount (mol %) of βCD-AAm, Ad-AAm, and Fc-AAm units. c Photograph of the βCD-Ad-Fc pAAm gel (6,3,3). (Reprinted with permission from [10]. Copyright 2015, Wiley-VCH.)
Shape memory function is also used to improve the self-healing performance of materials, which is termed as shape memory assisted self-healing (SMASH) materials. The self-recovery of SMASH materials is initiated with the shape recovery to drive the cracks closer, and then the healing process takes place in the damaged region. Shape memory alloy, polymer fibers, and bulk shape memory material are used in the design of SMASH materials. As shape memory fibers aligned perpendicular to the crack surfaces have the highest crack closing efficiency, alignment of fibers in three dimensions would improve recovery performance than in one dimension. Short shape memory polyurethane fibers were used and dispersed in epoxy with caprolactone-derived thermoplastic particles to form SMASH material by Li and co-workers [75]. Short shape memory polymer fibers enable 3-D alignment for the unpredictable direction of the cracks. However, self-healing of these materials requires specific orientation of the shape memory alloys or polymer fibers to the cracks, which still limits their practical applications. Thermoplastic moieties were also dispersed into bulk shape memory materials. Rodriguez et al. prepared SMASH polymers by entrapping self-healing thermoplastic agents (linear poly(ε-caprolactone) (l-PCL)) into a shape memory cross-linked poly(ε-caprolactone) network (n-PCL) [76]. Upon heating, shape recovery of n-PCL brought cracks closer to each other and then the diffusion of l-PCL chains crossing the cracks led to re-bonding of the cracks. Complete healing could be obtained with over 25 wt% l-PCL in the material. The same strategy was also used to prepare self-healing coatings by electrospinning thermoplastic poly(ε-caprolactone) (PCL) fibers into a shape memory epoxy matrix [77]. As shown in Fig. 4, cracks on this corrosion-resistant coating closed and re-sealed after 10 min of heating at 80 °C and no rust was formed on the coated metal.

Self-healed coatings. The photos of the exposed areas of damaged and self-healed coatings after the linear sweep voltammetry testing are shown: a 10 min and b 15 min electrospinning. The scale bar (bottom right) represents 2 mm. (Reprinted with permission from [70]. Copyright 2013, American Chemical Society.)
3.3 Self-healing materials with sol–gel transition properties
Hydrogels are three-dimensional matrixes with high water content, which typically possess tissue-like physical and mechanical properties [78, 79]. Due to their high biocompatibility, they are widely used in biomedical and pharmaceutical applications such as tissue engineering, drug delivery, and cell encapsulation/immobilization. Reversible sol–gel transition property stimulated by changes in environmental conditions is one of the properties enabling the integration of water-soluble drugs (or cells) and hydrogel precursors through simple solution mixing followed by sol–gel transition. In the design of some self-healing hydrogels, this property was remained for potential programming of hydrogels via stimulus-responsive sol–gel transition. Recently, pH-induced metal ligand-based self-healing hydrogels with sol–gel transition function were reported. Inspired by mussels, this type of hydrogel was designed to mimic the self-repairing mechanism of the creature through which they can survive in mutable destructive conditions under the sea [39, 80]. 3,4-Dihydroxyphenylalanine (DOPA) is a catechol-like amino acid, and it is abundant in the exposed part of mussels which also contains abundant iron and calcium. To mimic the self-healing performance, DOPA-functionalized polymers were prepared and Fe(III) was used to trigger cross-linking in conjunction with pH changes (Fig. 5). In the presence of iron, mono-, bis-, and tris-catechol–Fe3+ complexes formed and varied with environmental pH changes from acidic conditions to basic conditions. This process is fully reversible. In this way, sol–gel transition of the hydrogels could be manipulated by controlling pH. In addition, rapid self-healing performances (less than 60 s) at room temperature without external stimulus were also obtained. Besides mussel-inspired self-healing hydrogels, boronate–catechol complexation-based hydrogel was also reported and possessed both pH responsiveness and self-healing functions [40]. Under the basic aqueous condition, the hydrogel networks are formed via formation of tetrahedral borate esters between boronic acid and catechol groups, while the acidic condition promotes the reversible reaction, thus realizing reversible sol–gel transition.

Fe(III)/DOPA–polyallylamine cross-linking. a Hydrogel formation. In the top photograph, monocomplex formation is evident (green) as a Fe(III) solution is added to the center of the DOPA–polyallylamine solution. The middle photograph illustrates how tris-complexes (red) are formed as a NaOH droplet is added in the center of the Fe(III)/DOPA–polyallylamine solution with bis-complexes (purple/blue) forming at the edge of the base droplet. The bottom photograph shows a hydrogel (pH ∼ 8). b Relative fractions of mono- (green), bis- (blue), and tris-catechol–Fe(III) complexes (red) in solutions of DOPA–polyallylamine and FeCl3 as a function of pH with a DOPA to Fe(III) ratio of 3:1. Note that when no standard deviation can be seen, it is smaller than the size of the points. (Reprinted with permission from [76]. Copyright 2013, American Chemical Society.)
Another strategy to prepare multi-responsive self-healing hydrogel is to utilize the advantage of metal bonding between amino acid-based ligand and ionic metal salt. Recently, a metal bonding hydrogel was fabricated by adding Zn(II) ions into l-valine-based molecules in the presence of sodium perchlorate [13]. Rapid self-recovery was observed in air at room temperature without using any other healing agents. In addition, reversible sol–gel transition could be accomplished by shaking, variation of the temperature or the pH, and the addition of equimolar of Zn(II). In a similar way, a self-healing hydrogel with sol–gel transition function was prepared using amphiphilic tyrosine derivatives and Ni2+ ions [12]. This hydrogel exhibited autonomous recovery upon imposed damages, and the self-healing property could be tuned by controlling the chain length of amphipiles. The increase of the chain length of the amphiphiles resulted in longer healing time. Moreover, complete recovery as rapid as 3 min and 40 s was quantitatively validated by rheology test. In addition, sol–gel transition of this self-healing material could be trigged by low pH (< 7), sonication, and resting, while the reversible process could be triggered by high pH (> 8), heating, resting, and the presence of EDTA or EDA (Fig. 6).

a Chemical structures of various amphiphiles (P1–P4). b Pictures of glass vials containing metallo-hydrogels obtained from different proportions of the P3 and nickel salt (NiCl2). c Multi-stimulus responsiveness shown by the hydrogel obtained from P3. d Illustration of self-healing behavior (i–v) shown by the metallo-hydrogel obtained from P3. (Reprinted with permission from [6]. Copyright 2013, Royal Society of Chemistry.)
Host–guest interactions were also used to prepare stimulus-responsive sol–gel transition hydrogels with self-repairing properties. Recently, a modified PAAc hydrogel with β-CD and Fc as the host and guest molecule was reported by Nakahata et al. [36]. Reversible sol–gel transition occurred in the presence of a reductant or an oxidant. The underlying principle is attributed to the exclusion of the oxidized guest moieties from high hydrophobicity cavities of host molecules and vice versa. Both chemical reagents and electrochemical reactions can be employed to trigger sol–gel transition. Re-adhesion between the cut surfaces of the hydrogel was observed after 24 h at room temperature. Supramolecular networks embedding crown ether-based host–guest moieties were also reported to fabricate this type of hydrogel [33, 81]. Sol–gel transition of this type of self-healing hydrogels can be triggered by pH variation due to pH-dependent host–guest interactions (Fig. 7). By inducing metal–ligand interactions to the backbone of the hydrogel, sol–gel transitions also responded to metallo-stimuli, thermo-stimuli, and cation stimuli.

a The reversible gel–sol transition of supramolecular self-healing gel 4 and b the degradation of self-healing gel 5 upon adding TEA and TFA. (Reprinted with permission from [33]. Copyright 2012, Wiley-VCH.)
Moreover, dynamic covalent bonds were used to fabricate multi-functional hydrogels with both self-repairing and responsive sol–gel transition properties. For example, aldehyde-terminated 3-armed PEO and dithiodipropionic acid dehydrazide were used to prepare self-healing hydrogel in the pH range of 3–9 [23]. This self-healable hydrogel, consisting of two different dynamic covalent bonds (acylhydrazone and disulfide bonds), was responsible for the recovery under acidic/neutral and basic conditions, respectively. In addition, its dual responsive sol–gel transition relied on the aforementioned two dynamic covalent bonds, endowing the hydrogel with the pH-responsive and redox-responsive sol–gel transition property, respectively (Fig. 8). By controlling the sol–gel stimuli, solutions containing functional drugs, active proteins/genes, or even cells can be entrapped in a biocompatible hydrogel matrix through simple mixing, thus providing a facile platform which may be applicable for organ repairing and stimuli-responsive drug delivery.

Sol–gel transitions of HG1G2 and HG1G3 in response to pH and/or redox triggers. The concentration of all of the samples was 20 wt%. (Reprinted with permission from [48]. Copyright 2012, American Chemical Society.)
3.4 Self-healing materials with optical-responsive properties
Photo-induced self-healing via formation of new chemical bonds or reshuffling of polymer chains across damaged regions provides another elegant strategy to develop self-repairing materials [82]. One approach to recognize light-triggered self-healing materials is to disperse immobilized photo-reactive moieties in polymers in which photo-cross-linking reactions are responsible for the healing of cracks. Froimowicz et al. reported a self-healing hydrogel film by exploiting reversible photo-induced dimerization of anthracene on a dendritic polymer scaffold [83]. Recovery of the film was observed overnight after two treatments of UV light (254 nm as the first treatment and 366 nm as the second treatment). In the healing process, a higher-energy UV light was used to cleave the dimer to generate more photo-reactive moieties and a lower-energy UV light was used to trigger the cycloaddition of anthracene to rebuild cross-linking between crack surfaces. Similar strategies using photo-dimerization of coumarin derivatives were also exploited and reported [84, 85]. In brief, a photo-responsive polyurethane network containing coumarin as a photo-sensitive cross-linker was first synthesized by Ling and co-workers. The repair of the film followed a two-step procedure, including the exposure of the fractured surfaces to 254 nm UV light and exposure to 350 nm UV light with fractured surfaces back in contact for 90 min, sequentially. Multiple healings in the same area were both qualitatively and quantitatively investigated, and the tensile test demonstrated that the healing efficiency remained to be 56.6% after three-time repairing. The same group also prepared polyurethane with dihydroxyl coumarin. The healing principle was the same as the previous one, but a higher healing efficiency was obtained (61% after three-time repairing).
Another strategy for the development of light-triggered self-healing material is to use a photo-induced metathesis reaction to restore linkages between cracked surfaces. In this regard, various strategies based on non-covalent interactions or radical catalyzed conversion under UV irradiation were reported [86, 87]. Amamoto et al. reported a self-repairing poly(n-butyl acrylate) (PBA) based on immobilized photo-initiator trithiocarbonate (TTC) as the first example to achieve repeatable healing through a one-step photo-stimulus treatment [88]. Under UV irradiation, a dynamic process relying on covalently reshuffling of TTC units is responsible for the self-healing function. The hemolysis of C–S bonds under photo-stimuli generated radicals to react on the cracked surfaces, and thus, repair is obtained through exchange reactions at the broken surfaces. The synthesized polymer exhibits strong self-recovery ability. Not only were repeatable coalescences of the cut hydrogel rods observed but also the combination of completely discrete polymer particles into one rod was achieved. However, the self-repairing process required UV irradiation in an acetonitrile solvent or under nitrogen atmosphere because of the reactive intermediates of carbon radicals, which may limit its applications. Following a similar principle, the same group designed a self-repairing polyurethane which maintained a similar self-recovery performance but under visible light by exchanging TTC units with thiuram disulfide (TDS) units [28]. TDS units located in the main chain allowed reshuffling reactions in air under visible light due to their low bond dissociation energy and oxygen tolerance. Self-healing of the material was stabilized after 12 h under the light source of a commercial tabletop lamp, and near complete recovery was validated by a tensile test (Fig. 9).

Photographs of TDS cross-linked polymer 4a in self-healing reaction. a Before self-healing reaction, b after cutting cross-linked polymer, and c after self-healing reaction for 24 h. d Stress–strain curve and e elongation value at break (%) of cross-linked polymer 4a after irradiation of visible light at room temperature over time. The plots and the error bars show the average values and the standard deviations for five measurements, respectively. The reaction was carried out by exposing 4a to visible light in bulk state and in air at room temperature. (Reprinted with permission from [20]. Copyright 2012, Wiley-VCH.)
Fluorescent hydrogels are another important category in artificial soft materials, which have been widely used in the sensing area and biological imaging [89, 90]. As photo-stimulated luminescent self-healing supramolecular hydrogel could bring two desired properties into one matrix, their appearance would facilitate the development of artificial soft materials. In this regard, a hydrogel with blue fluorescence was first fabricated by pH-induced gelation of amino acid-based ligands [91]. Fluorescence of the hydrogel is ascribed to the pyrene moiety of the low-molecular-weight amino acid-containing gelator. Cross-slinking of the ligand leads to a red shift of fluorescence emission from 417 to 485 nm, which could be used as an indicator for gelation state. Ninety-three percent recovery of the hydrogel was obtained via rheological evaluation of healing efficiency within 3 min. With inclusion of reduced graphene oxide, healing efficiency can be improved to 100% within 100 s. Another kind of self-healing hydrogel with room temperature phosphorescence (RTP) was fabricated by Chen et al. via host–guest interactions (Fig. 10) [17]. The RTP signals were induced by inclusion of a guest α-bromonaphthalene (α-BrNp) moiety in a β-CD host moiety. The RTP emission spectrum of the hydrogels showed a broad peak in the range of 520 to 550 nm under irradiation at 365 or 254 nm. Phosphorescence glowing as long as 80 and 140 s at a photo-stationary state after irradiation of 365 and 254 nm was obtained, respectively. By inducing competitive guest azobenzene polymer to the poly-BrNp/poly-β-CD assembly, RTP signal of the hydrogel can be reversibly tuned. By integrating the self-healing function with fluorescence/luminescence property in one material, it could provide novel substrates for wearable biosensors designs in the future.

RTP spectra of the binary (a) poly-BrNp/β-CD system ([poly-BrNp] = 1 × 10−5 M, [β-CD] = 0, 5 × 10−5, 5 × 10−4, 1 × 10−3 M). b Poly-BrNp/poly-β-CD system ([poly-BrNp] = 1 × 10−5 M, [poly-β-CD] = 0, 1 × 10−6, 5 × 10−6, 1 × 10−5, 1 × 10−4 M). c Poly-BrNp/poly-β-CD hydrogel ([poly-BrNp] = 2 × 10−2 M, [poly-β-CD] = 2 × 10−2 M). Inset: photograph of poly-BrNp/poly-β-CD hydrogel. d RTP lifetime of the binary poly-BrNp/poly-β-CD system ([poly-BrNp] = 1 × 10−5 M, [poly-β-CD] = 1 × 10−4 M). (Reprinted with permission from [12]. Copyright 2014, Wiley-VCH.)
3.5 Self-healing materials with adhesive properties
Underwater adhesion of materials to dissimilar substrates attracts researchers’ attention due to its potential biological, medical, and industrial applications [92,93,94,95]. Excellent adhesion ability could be used not only as sealant for household but also as tissue adhesives for drug delivery. However, the development of adhesive materials remains challenging. The outstanding underwater attachment performance of mussels inspires the development of biomimetic hydrogels composed of adhesive DOPA which are found in abundance in the exposed part of the mussels. DOPA-functionalized polymers were then used to build several self-healing hydrogels with an adhesive property via complex formation with Fe(III) which exhibited underwater attachment to various substrates [39, 80]. However, their relatively low mechanical strength prevented them from any practical applications. To improve the mechanical properties of the hydrogel, a peptide amphiphile with functional DOPA (DOPAK-PA) units was used as the building block for self-repairing hydrogels with increased stiffness. [38] Covalently cross-linked DOPAK-PA hydrogel (without Fe(III)) was also prepared as a control, and both hydrogels displayed a single peak in adhesion histograms recorded by AFM. However, self-healing performance of the covalently cross-linked DOPAK-PA hydrogel only recovered 7.1% while 77.6% recovery was observed for DOPAK-PA/Fe(III) via rheology experiments.
Inspired by the polyionic character of adhesive proteins, self-healing materials with an adhesion property were also fabricated using polyelectrolytes. Huang et al. reported a unique strategy to synthesize adhesive and self-healing gels through ionic cross-linking between polycations poly(allylamine) (PAH) and pyrophosphate (PPi) or tripolyphosphate (TPP) [96]. Both PAH/PPi and PAH/TPP samples exhibited recovery of storage moduli to their original values within 10–30 min and attachment to various substrate under and above water. Most importantly, the adhesion strength (350–450 kPa) was not affected by the hydrophobicity of the substrates. The adhesive attachment was both qualitatively observed by adhesion of the gels on finger or glass vials and quantitatively measured by a tensile test (Fig. 11).

Photographs show how the PAH/PPi self-healing gels adhere a magnetic stir bar to human finger (a) and the bottoms of two glass vials together (b). (Reprinted with permission from [89]. Copyright 2014, American Chemical Society.)
Adhesion through other non-covalent interactions were also exploited and studied. Different from attachment via covalent polymerization and cross-linking reactions, this setting mechanism is more appealing because of its reversible property. Hydrogen bonding groups such as 2-ureido-4[1H]-pyrimidinone quadruple hydrogen bonding groups (UPy) were added to PBA polymer chains to increase adhesion strength and induce the self-repairing performance [6]. Surface force apparatus (SFA) was used to study the adhesion mechanism of the polymer, and self-adhesion strength was chosen as the desired functionality to evaluate healing efficiency (Fig. 12). With strong multiple H-bonding between UPy units, the surface energy of the PBA-UPy polymer increased to about 1.5-fold that of PBA. The adhesion strength was also dependent on the fraction of UPy in the polymer. One hundred percent recovery of the original self-adhesion strength was obtained around 50 h with a slight increase of the temperature to 40 °C. Another acryloyl-6-aminocaproic acid (A6ACA) hydrogel based on hydrogen bonding with high curing efficiency was reported adhesive to both poly(propylene) surface and rabbit gastric mucosa [37].

FECO fringe patterns (left), corresponding top-view optical microscope images (middle), and illustrations (right) of the contact junction of the PBA–UPy7.2 polymer of thickness 100 nm at T = 40 °C and RH = 100%. (Reprinted with permission from [31]. Copyright 2014, Wiley-VCH.)
4 Self-healing materials for potential biomedical applications
Self-healing materials, especially self-healing hydrogels, have been attracting attention in biomedical applications in the past decade. A number of self-repairing materials have been reported to possess applicability in tissue engineering, cell culture, drug delivery, and so on. For example, PEG-chitosan and chitosan-based hydrogels possess high biocompatibility and were proposed for drug delivery as an injectable hydrogel [26, 27, 97]. A rapid self-healing hydrogel A6ACA was also proposed to be used as a new generation of tissue adhesive due to its excellent adhesion ability [37, 52]. Furthermore, some unique properties of self-healing materials are quite intriguing to in vivo applications. One instance is a bovine serum albumin (BSA) hydrogel regulated by glucose oxidase (GOX) and catalase (CAT), in which recovery is stimulated by the presence of glucose and 100% healing efficiency could be obtained within 5 h at room temperature [25]. Due to the broad applications of self-healing materials, this section mainly focuses on summarizing and discussing various applications of self-healing materials in the biomedical field.
To ensure safety for applications in biomedical fields, biocompatibility of the hydrogel must be evaluated as the first step. Therefore, in vitro cytotoxicity and in vivo biocompatibility studies are generally conducted. For example, BSA-based self-healing hydrogels were demonstrated to possess high biocompatibility by Chen et al. [98]. A thermal stimulus-responsive healing protein hydrogel was fabricated, and its compatibility was demonstrated by both in vitro cytotoxicity test and in vivo injection. In addition, biodegradations of the hydrogels were observed after 97 days of injection. However, to obtain complete recovery, thermal treatment was required which prevents healing of the material for the in vivo environment. To overcome this drawback, the same group designed and synthesized another BSA-based hydrogel which could be self-healed at room temperature under a physiological pH (Fig. 13a, b). The hydrogel was formed by inducing Ca2+ ions into BSA protein nanoparticle solutions. Self-recovery of the hydrogel was observed at ambient conditions without Ca2+, but the presence of Ca2+ greatly accelerated the healing process. According to cytotoxicity results shown in Fig. 13c, the presence of the hydrogel extract solution can facilitate growth of the cells, implying its good biocompatibility which may favor potential in vivo application of the material.

a, b Photographs that demonstrate the self-healing behavior of the protein hydrogel at room temperature: 10% BSA hydrogel with 12.5 mM Ca2+ at original, cut, self-healed, and bend states (a); Self-healing of 10% BSA hydrogels with different Ca2+ concentrations (15, 20, and 30 mM) (b). c Cytotoxicity evaluation of the calcium-induced BSA hydrogel. (Reprinted with permission from [94]. Copyright 2016, Nature Publishing Group.)
4.1 Self-healing hydrogel used as bio-ink for 3D bioprinting
A self-healing hydrogel ink for 3D bioprinting was developed by Highley et al. [15]. Hyaluronic acid (HA) was chosen as the backbone of the hydrogel because of its excellent biocompatibility, and modified with β-CD and Ad as host and guest moieties. To improve the mechanical properties of the hydrogel, methacrylates were introduced into building blocks enabling covalent cross-linking upon UV irradiation. Shear-thin and self-healing structures of CD-Ad-HA and CD-Ad-MeHA can be printed via computer-aided design. As a result, versatility of the printing platform was expanded to printing host–guest interaction-based self-healing hydrogels with or without covalently cross-linking through UV irradiation (Fig. 14). After being stimulated by UV, CD-Ad-HA hydrogel could easily be washed off, leaving the CD-AD-MeHA part due to the strong bonds between the building blocks. In this way, 3D scaffolds with complicated configuration could be prepared through computer-aided design. This new methodology opens new opportunities in preparing structured composites with good biocompatibility via 3D printing.

a Hyaluronic acid modified with both methacrylates (blue) and guest and host molecules (purple). Ad-MeHA and CD-MeHA macromers cross-link by both physical bonding upon mixing and secondary cross-linking of methacrylates with UV light exposure. b The printing of channels, by writing an ink into a support gel that is modified for secondary cross-linking. UV irradiation covalently cross-links the support gel around the ink (epifluorescent image of fluorescein-labeled ink printed in a bifurcating and rejoining design within an unlabeled, covalently cross-linked support). Pressure-driven flow results in the removal of the ink, leaving a channel network (as shown in phase contrast image). Bottom: images of channel perfusion with a red dye solution initially and after 2 s. c The printing of self-supporting structures, by writing an ink that can be covalently cross-linked into a support gel, followed by UV cross-linking, and dissolution of the support with excess β-CD (image of printed 3D tetrahedron composed of six filaments joined at four vertices, imaged from above the top vertex). Scale bars: 500 μm. (Reprinted with permission from [9]. Copyright 2015, Wiley-VCH.)
4.2 Self-healing hydrogels used in cell culture
Cell attachment to self-healing graphene-poly(N,N-dimethylacrylamide) (PDMAA) hydrogels was also observed (Fig. 15) [66]. The hydrogel was fabricated by immersing graphene networks into N,N-dimethylacrylamide (DMAA) monomer solution, followed by free-radical polymerization. Hydrogen bonds are responsible for the recovery of the material, and the healing of wound was obtained at body temperature over a period of 12 h. Furthermore, cell attachment to the hydrogels was demonstrated by growing rat pheochromocytoma (PC-12) cells on a control hydrogel and a graphene–PDMAA hydrogel as shown in Fig. 15a, b, respectively. Compared to the control, most living cells are attached to the dark graphene–PDMAA fragments, indicating good cellular attachment of this self-healing grapheme–PDMAA hydrogel. Additionally, the as-developed self-healing hydrogel also possessed electroactivity due to graphene networks as mentioned in Sect. 3.1, which enabled this hydrogel to be used in soft actuator and robotic device design. Such conductive self-healing hydrogel also has the potential to be used in the development of artificial skin/electronic skin or wearable biosensor.

a, b Optical microscopy images of PC-12 cells cultured in cell culture fluid and graphene–PDMAA hydrogels (black substrates: graphene–PDMAA hydrogels; transparent substrates: the exceeded PDMAA); scale bars are 150 and 200 mm, respectively. c Impedance spectroscopy for graphene and graphene–PDMAA hydrogels. d Temperature curve for graphene–PDMAA exposed to NIR laser; insets show their infrared thermal images. e Self-healing of graphene–PDMAA gels under NIR laser irradiation and their infrared thermal images. (Reprinted with permission from [59]. Copyright 2012, Royal Society of Chemistry.)
Self-healing hydrogels composed of β-sheet-prone C-terminal amyloid β-protein (Aβ42)-based peptides were exploited for cell culture and stem cell differentiation. Hydrogels were prepared by dissolving the peptide powder into sodium phosphate buffer under heating, and hydrogels were obtained after cooling down. It was claimed that the amyloid networks were formed via hydrophobic and π–π interactions and hydrogen bonding. Hydrophobic interactions are highly responsible for self-recovery across the cracked surfaces. Attachments of neuronal and fibroblast cells to the hydrogel were observed, indicating that the amyloid-based hydrogels are suitable for 2D cell culture. Furthermore, culture of human mesenchymal stem cells on the hydrogel indicated that softer gels promote spreading and elongation of cells with βIII-tubulin expression, an indicator for cell differentiation. Taking advantage of the thixotropicity of the hydrogel, the authors also demonstrated its application in constructing 3D cell culture system (Fig. 16) [99].

3D cell culture using amyloid hydrogels. a Schematic depicting entrapment of cells inside thixotropic peptide gels. b 3D cell culture using P5 gel showing cell viability of both SH-SY5Y and L929 cells indicated by calcein AM staining (green) inside the 3D gel matrix. Scale bars are 50 μm. (Reprinted with permission from [93]. Copyright 2015, Elsevier Ltd.)
4.3 Self-healing hydrogels used in anti-biofouling
A dilemma of biofouling films by proteins or microorganisms on implanted hydrogels is due to the fact that it not only triggers inflammation but also prevents implanted drug delivery systems from releasing functional biomolecules. Anti-biofouling hydrogels with self-healing properties were developed to address this dilemma. Recently, an injectable hydrogel based on self-assembly of an ABA tri-block copolymer was reported to possess both rapid self-healing and an anti-biofouling property [100]. The tri-block copolymer DNODN was synthesized by catechol-functionalized poly(N-isopropylacrylamide) (PNIPAM) as A block and poly(ethylene oxide) as B block through reversible additional fragment transfer polymerization. Hydrogen bonds and π–π interactions of mussel-inspired catechol functional moieties are responsible for the rapid self-healing performance. The gelation was obtained at body temperature, and the reversible sol–gel transition could be controlled by varying the temperature. Additionally, exceptional antifouling performance was demonstrated through no cell attachment on the DNODN-coated microwell dishes (Fig. 17).

Representative fluorescence microscopy images of a uncoated and b DNODN hydrogel coated microwell dishes after exposure to Caco-2 cells for 48 h. Alexa Fluor 488 and 4,6-diamidino-2-phenylindole (DAPI) were used to stain the cell membrane and the nuclei, respectively. (Reprinted with permission from [96]. Copyright 2015, Wiley-VCH.)
4.4 Self-healing hydrogels used in drug delivery
Several self-healing materials have been proposed for drug delivery; however, few of them were experimentally investigated. Also, their self-healing property was barely utilized. Recently, a self-healing, drug delivery hydrogel system with enhanced chemotherapy via the stimulation of sonophoresis was developed [58]. The injectable biomaterials were formed by alginate solution in the presence of divalent calcium ions. Repeatable healing was observed in the presence of a physiological level of calcium ions after ultrasound-induced disruption. In such a way, high-dose pulses of drug release were controlled by intermittent ultrasound stimulus. Disruption of the hydrogels led to high-dose release of drugs upon ultrasound stimulus, while the self-healing of the hydrogel structure slowed the drug release when ultrasound was removed. Both in vitro and in vivo tests of the drug delivery system under ultrasound demonstrated that multiple pulses of a drug dose enhanced the effect of chemotherapy, supported by the decreased viability of two cancer cell lines as well as the high survival rate of the mouse models with breast tumors. Even though the application of this pulsed drug dose delivery system would be limited by the disruption method of ultrasound, a potential therapeutic application of a self-healing drug delivery system was demonstrated for the first time and the groundbreaking drug delivery concept can be applied to many other rapid self-healing systems relying on benign disruption/healing strategies.
5 Conclusion and outlook
Instead of simply synthesizing new materials based on trial and error and characterizing their properties, the rationale design of the materials with pre-determined functionality and the exploration of their unique applications have been becoming a new trend in the development of self-healing materials in recent years. Driven by the desire for durable materials with elongated lifespan, the capsule-based self-healing system has been applied to fabricate several commercial self-repairing materials, including self-healing concretes, phone cases, and screen protectors. With the development of advanced science and technologies, the high demand of self-repairable materials in different industrial sectors sprouts the research in seal-healing materials with an emphasis on new applications.
To cater to the need of specific application, additional functionalities or properties should be introduced into self-healing systems. Up to date, various multi-functional self-healing materials have been developed with pre-determined functionality. It has been demonstrated that self-repairing behavior is able to improve the performances of materials in corrosion resistance coating and the supercapacitor. Also, other unique functions besides self-healing property can endow the self-healing materials to be used as new classes of adhesives and shape memory materials. The sol–gel transition property of self-healing hydrogels can also be explored as a facile and friendly method to entrap the targets of interests. Furthermore, biocompatible self-healing hydrogels are elaborately designed and applied to enhance drug delivery, tissue adhesion, and cell culture in biomedical engineering.
Although a number of self-healing materials and their potential applications have been reported and explored in the literature, the practical applications of those self-healing materials are sparse. Relatively weak mechanical properties, harsh self-healing conditions required, and/or poor biocompatibility are a few major hurdles to limit their applications in biomedical fields. The future research should focus on the development of self-healing materials with strong mechanical strength by employing building blocks with high binding but reversible forces, adapting benign self-healing conditions at physiological conditions (e.g., neutral pH, 37 °C, 4–7 mM glucose, 2.6 mM Ca2+), and using biocompatible materials such as natural polymers (chitosan, etc.) and proteins (BSA, etc.) if targeting on biomedical applications. Due to the limited information and understanding of fundamentals in the material structure–property–function relationship, the development of self-healing materials in the past was basically on a basis of trial and error, which is lack of systematic study and cannot generate a full picture. Therefore, another future research trend prioritized is to employ material simulation in the design of multi-functional self-healing materials because material simulation is barely employed in the design/selection of self-healing materials in the past decades. With the development of simulation methods and the establishment of more and more accurate material models to reveal the structure–property–function relationship, a computation method to simulate the properties including a self-healing function of various materials or composites is expected to play a critical role in the screening/selection of specific materials for specific application in the future design of self-healing materials with unique function, good biocompatibility, and benign healing conditions. We believe, with current advances in material science and progress in material simulation, that self-healing materials with unique functionality will have a promising and bright future.
References
Thirumalini P, Ravi R, Sekar S, Nambirajan M (2011) Study on the performance enhancement of lime mortar used in ancient temples and monuments in India. Indian J Sci Technol 4(11):1484–1487
Wayman E (2011) The secrets of Ancient Rome’s buildings. Smithsonian Magazine
Diesendruck CE, Sottos NR, Moore JS, White SR (2015) Biomimetic self-healing. Angew Chem Int Ed 54(36):10428–10447
Blaiszik B, Kramer S, Olugebefola S, Moore JS, Sottos NR, White SR (2010) Self-healing polymers and composites. Annu Rev Mater Res 40:179–211
Yan T, Schröter K, Herbst F, Binder WH, Thurn-Albrecht T (2016) Unveiling the molecular mechanism of self-healing in a telechelic, supramolecular polymer network. Sci Rep 6:32356
Faghihnejad A, Feldman KE, Yu J, Tirrell MV, Israelachvili JN, Hawker CJ, Kramer EJ, Zeng H (2014) Adhesion and surface interactions of a self-healing polymer with multiple hydrogen-bonding groups. Adv Funct Mater 24(16):2322–2333
Wang C, Liu N, Allen R, Tok JBH, Wu Y, Zhang F, Chen Y, Bao Z (2013) A rapid and efficient self-healing thermo-reversible elastomer crosslinked with graphene oxide. Adv Mater 25(40):5785–5790
Tuncaboylu DC, Argun A, Sahin M, Sari M, Okay O (2012) Structure optimization of self-healing hydrogels formed via hydrophobic interactions. Polymer 53(24):5513–5522
Tuncaboylu DC, Sahin M, Argun A, Oppermann W, Okay O (2012) Dynamics and large strain behavior of self-healing hydrogels with and without surfactants. Macromolecules 45(4):1991–2000
Tuncaboylu DC, Sari M, Oppermann W, Okay O (2011) Tough and self-healing hydrogels formed via hydrophobic interactions. Macromolecules 44(12):4997–5005
Luo F, Sun TL, Nakajima T, Kurokawa T, Zhao Y, Sato K, Ihsan AB, Li X, Guo H, Gong JP (2015) Oppositely charged polyelectrolytes form tough, self-healing, and rebuildable hydrogels. Adv Mater 27(17):2722–2727
Basak S, Nanda J, Banerjee A (2014) Multi-stimuli responsive self-healing metallo-hydrogels: tuning of the gel recovery property. Chem Commun 50(18):2356–2359
Saha S, Bachl J, Kundu T, Díaz DD, Banerjee R (2014) Amino acid-based multiresponsive low-molecular weight metallohydrogels with load-bearing and rapid self-healing abilities. Chem Commun 50(23):3004–3006
Bode S, Zedler L, Schacher FH, Dietzek B, Schmitt M, Popp J, Hager MD, Schubert US (2013) Self-healing polymer coatings based on crosslinked metallosupramolecular copolymers. Adv Mater 25(11):1634–1638
Highley CB, Rodell CB, Burdick JA (2015) Direct 3D printing of shear-thinning hydrogels into self-healing hydrogels. Adv Mater 27(34):5075–5079
Miyamae K, Nakahata M, Takashima Y, Harada A (2015) Self-healing, expansion–contraction, and shape-memory properties of a preorganized supramolecular hydrogel through host–guest interactions. Angew Chem Int Ed 54(31):8984–8987
Chen H, Ma X, Wu S, Tian H (2014) A rapidly self-healing supramolecular polymer hydrogel with photostimulated room-temperature phosphorescence responsiveness. Angew Chem Int Ed 53(51):14149–14152
Xu Z, Peng J, Yan N, Yu H, Zhang S, Liu K, Fang Y (2013) Simple design but marvelous performances: molecular gels of superior strength and self-healing properties. Soft Matter 9(4):1091–1099
Burattini S, Greenland BW, Hayes W, Mackay ME, Rowan SJ, Colquhoun HM (2010) A supramolecular polymer based on tweezer-type π− π stacking interactions: molecular design for healability and enhanced toughness. Chem Mater 23(1):6–8
Burattini S, Greenland BW, Merino DH, Weng W, Seppala J, Colquhoun HM, Hayes W, Mackay ME, Hamley IW, Rowan SJ (2010) A healable supramolecular polymer blend based on aromatic π− π stacking and hydrogen-bonding interactions. J Am Chem Soc 132(34):12051–12058
Huang L, Yi N, Wu Y, Zhang Y, Zhang Q, Huang Y, Ma Y, Chen Y (2013) Multichannel and repeatable self-healing of mechanical enhanced graphene-thermoplastic polyurethane composites. Adv Mater 25(15):2224–2228
Deng G, Tang C, Li F, Jiang H, Chen Y (2010) Covalent cross-linked polymer gels with reversible sol− gel transition and self-healing properties. Macromolecules 43(3):1191
Deng G, Li F, Yu H, Liu F, Liu C, Sun W, Jiang H, Chen Y (2012) Dynamic hydrogels with an environmental adaptive self-healing ability and dual responsive sol–gel transitions. ACS Macro Lett 1(2):275–279
Kuhl N, Bode S, Bose RK, Vitz J, Seifert A, Hoeppener S, Garcia SJ, Spange S, van der Zwaag S, Hager MD (2015) Acylhydrazones as reversible covalent crosslinkers for self-healing polymers. Adv Funct Mater 25(22):3295–3301
Gao Y, Luo Q, Qiao S, Wang L, Dong Z, Xu J, Liu J (2014) Enzymetically regulating the self-healing of protein hydrogels with high healing efficiency. Angew Chem 126(35):9497–9500
Yang B, Zhang Y, Zhang X, Tao L, Li S, Wei Y (2012) Facilely prepared inexpensive and biocompatible self-healing hydrogel: a new injectable cell therapy carrier. Polym Chem 3(12):3235–3238
Zhang Y, Yang B, Zhang X, Xu L, Tao L, Li S, Wei Y (2012) A magnetic self-healing hydrogel. Chem Commun 48(74):9305–9307
Amamoto Y, Otsuka H, Takahara A, Matyjaszewski K (2012) Self-healing of covalently cross-linked polymers by reshuffling thiuram disulfide moieties in air under visible light. Adv Mater 24(29):3975–3980
Rekondo A, Martin R, de Luzuriaga AR, Cabañero G, Grande HJ, Odriozola I (2014) Catalyst-free room-temperature self-healing elastomers based on aromatic disulfide metathesis. Mater Horiz 1(2):237–240
Pepels M, Filot I, Klumperman B, Goossens H (2013) Self-healing systems based on disulfide–thiol exchange reactions. Polym Chem 4(18):4955–4965
Ying H, Zhang Y, Cheng J (2014) Dynamic urea bond for the design of reversible and self-healing polymers. Nat Commun 5:3218
Oehlenschlaeger KK, Mueller JO, Brandt J, Hilf S, Lederer A, Wilhelm M, Graf R, Coote ML, Schmidt FG, Barner-Kowollik C (2014) Adaptable hetero Diels–Alder networks for fast self-healing under mild conditions. Adv Mater 26(21):3561–3566
Zhang J, Niu Y, Huang C, Xiao L, Chen Z, Yang K, Wang Y (2012) Self-healable and recyclable triple-shape PPDO–PTMEG co-network constructed through thermoreversible Diels–Alder reaction. Polym Chem 3(6):1390–1393
Wei Z, Yang JH, Du XJ, Xu F, Zrinyi M, Osada Y, Li F, Chen YM (2013) Dextran-based self-healing hydrogels formed by reversible Diels–Alder reaction under physiological conditions. Macromol Rapid Commun 34(18):1464–1470
Herbst F, Döhler D, Michael P, Binder WH (2013) Self-healing polymers via supramolecular forces. Macromol Rapid Commun 34(3):203–220
Nakahata M, Takashima Y, Yamaguchi H, Harada A (2011) Redox-responsive self-healing materials formed from host–guest polymers. Nat Commun 2:511
Phadke A, Zhang C, Arman B, Hsu C-C, Mashelkar RA, Lele AK, Tauber MJ, Arya G, Varghese S (2012) Rapid self-healing hydrogels. Proc Natl Acad Sci 109(12):4383–4388
Ceylan H, Urel M, Erkal TS, Tekinay AB, Dana A, Guler MO (2013) Mussel inspired dynamic cross-linking of self-healing peptide nanofiber network. Adv Funct Mater 23(16):2081–2090
Holten-Andersen N, Harrington MJ, Birkedal H, Lee BP, Messersmith PB, Lee KYC, Waite JH (2011) pH-induced metal-ligand cross-links inspired by mussel yield self-healing polymer networks with near-covalent elastic moduli. Proc Natl Acad Sci 108(7):2651–2655
He L, Fullenkamp DE, Rivera JG, Messersmith PB (2011) pH responsive self-healing hydrogels formed by boronate–catechol complexation. Chem Commun 47(26):7497–7499
South AB, Lyon LA (2010) Autonomic self-healing of hydrogel thin films. Angew Chem 122(4):779–783
Chen Y, Kushner AM, Williams GA, Guan Z (2012) Multiphase design of autonomic self-healing thermoplastic elastomers. Nat Chem 4(6):467–472
Cui J, del Campo A (2012) Multivalent H-bonds for self-healing hydrogels. Chem Commun 48(74):9302–9304
Kakuta T, Takashima Y, Nakahata M, Otsubo M, Yamaguchi H, Harada A (2013) Preorganized hydrogel: self-healing properties of supramolecular hydrogels formed by polymerization of host–guest-monomers that contain cyclodextrins and hydrophobic guest groups. Adv Mater 25(20):2849–2853
Hentschel J, Kushner AM, Ziller J, Guan Z (2012) Self-healing supramolecular block copolymers. Angew Chem 124(42):10713–10717
Ahn BK, Lee DW, Israelachvili JN, Waite JH (2014) Surface-initiated self-healing of polymers in aqueous media. Nat Mater 13(9):867–872
Cao J, Lu C, Zhuang J, Liu M, Zhang X, Yu Y, Tao Q (2017) Multiple hydrogen bonding enables the self-healing of sensors for human-machine interaction. Angew Chem Int Ed 30(56):8795–8800
Imato K, Nishihara M, Kanehara T, Amamoto Y, Takahara A, Otsuka H (2012) Self-healing of chemical gels cross-linked by diarylbibenzofuranone-based trigger-free dynamic covalent bonds at room temperature. Angew Chem Int Ed 51(5):1138–1142
Tseng TC, Tao L, Hsieh FY, Wei Y, Chiu IM, Hsu S (2015) An injectable, self-healing hydrogel to repair the central nervous system. Adv Mater 27(23):3518–3524
Kathan M, Kovaříček P, Jurissek C, Senf A, Dallmann A, Thünemann AF, Hecht S (2016) Control of imine exchange kinetics with Photoswitches to modulate self-healing in polysiloxane networks by light illumination. Angew Chem Int Ed 55(44):13882–13886
Cash JJ, Kubo T, Bapat AP, Sumerlin BS (2015) Room-temperature self-healing polymers based on dynamic-covalent boronic esters. Macromolecules 48(7):2098–2106
Wei Z, Yang JH, Zhou J, Xu F, Zrínyi M, Dussault PH, Osada Y, Chen YM (2014) Self-healing gels based on constitutional dynamic chemistry and their potential applications. Chem Soc Rev 43(23):8114–8131
Billiet S, Hillewaere XK, Teixeira RF, Du Prez FE (2013) Chemistry of crosslinking processes for self-healing polymers. Macromol Rapid Commun 34(4):290–309
Urban MW (2012) Dynamic materials: the chemistry of self-healing. Nat Chem 4(2):80–82
Wool RP (2008) Self-healing materials: a review. Soft Matter 4(3):400–418
Grabowski B, Tasan CC (2016) Self-healing metals. In: Hager MD, Van der Zwaag S, Schubert US (eds) Self-healing materials. Springer, New York
Nakahata M, Takashima Y, Harada A (2016) Highly flexible, tough, and self-healing supramolecular polymeric materials using host–guest interaction. Macromol Rapid Commun 37(1):86–92
Huebsch N, Kearney CJ, Zhao X, Kim J, Cezar CA, Suo Z, Mooney DJ (2014) Ultrasound-triggered disruption and self-healing of reversibly cross-linked hydrogels for drug delivery and enhanced chemotherapy. Proc Natl Acad Sci 111(27):9762–9767
Burattini S, Colquhoun HM, Fox JD, Friedmann D, Greenland BW, Harris PJ, Hayes W, Mackay ME, Rowan SJ (2009) A self-repairing, supramolecular polymer system: healability as a consequence of donor–acceptor π–π stacking interactions. Chem Commun 44:6717–6719
van Gemert GM, Peeters JW, Söntjens SH, Janssen HM, Bosman AW (2012) Self-healing supramolecular polymers in action. Macromol Chem Phys 213(2):234–242
Velnar T, Bailey T, Smrkolj V (2009) The wound healing process: an overview of the cellular and molecular mechanisms. J Int Med Res 37(5):1528–1542
Burattini S, Greenland BW, Chappell D, Colquhoun HM, Hayes W (2010) Healable polymeric materials: a tutorial review. Chem Soc Rev 39(6):1973–1985
Haraguchi K, Uyama K, Tanimoto H (2011) Self-healing in nanocomposite hydrogels. Macromol Rapid Commun 32(16):1253–1258
Liu J, Song G, He C, Wang H (2013) Self-healing in tough graphene oxide composite hydrogels. Macromol Rapid Commun 34(12):1002–1007
Wang Q, Mynar JL, Yoshida M, Lee E, Lee M, Okuro K, Kinbara K, Aida T (2010) High-water-content mouldable hydrogels by mixing clay and a dendritic molecular binder. Nature 463(7279):339–343
Hou C, Duan Y, Zhang Q, Wang H, Li Y (2012) Bio-applicable and electroactive near-infrared laser-triggered self-healing hydrogels based on graphene networks. J Mater Chem 22(30):14991–14996
Palleau E, Reece S, Desai SC, Smith ME, Dickey MD (2013) Self-healing stretchable wires for reconfigurable circuit wiring and 3D microfluidics. Adv Mater 25(11):1589–1592
Kim D-H, Ahn J-H, Choi WM, Kim H-S, Kim T-H, Song J, Huang YY, Liu Z, Lu C, Rogers JA (2008) Stretchable and foldable silicon integrated circuits. Science 320(5875):507–511
Rogers JA, Someya T, Huang Y (2010) Materials and mechanics for stretchable electronics. Science 327(5973):1603–1607
Wang H, Zhu B, Jiang W, Yang Y, Leow WR, Wang H, Chen X (2014) A mechanically and electrically self-healing supercapacitor. Adv Mater 26(22):3638–3643
Tee BC, Wang C, Allen R, Bao Z (2012) An electrically and mechanically self-healing composite with pressure-and flexion-sensitive properties for electronic skin applications. Nat Nanotechnol 7(12):825–832
Cordier P, Tournilhac F, Soulié-Ziakovic C, Leibler L (2008) Self-healing and thermoreversible rubber from supramolecular assembly. Nature 451(7181):977–980
Wang C, Wu H, Chen Z, McDowell MT, Cui Y, Bao Z (2013) Self-healing chemistry enables the stable operation of silicon microparticle anodes for high-energy lithium-ion batteries. Nat Chem 5(12):1042–1048
Gulyuz U, Okay O (2014) Self-healing poly (acrylic acid) hydrogels with shape memory behavior of high mechanical strength. Macromolecules 47(19):6889–6899
Li G, Zhang P (2013) A self-healing particulate composite reinforced with strain hardened short shape memory polymer fibers. Polymer 54(18):5075–5086
Rodriguez ED, Luo X, Mather PT (2011) Linear/network poly (ε-caprolactone) blends exhibiting shape memory assisted self-healing (SMASH). ACS Appl Mater Interfaces 3(2):152–161
Luo X, Mather PT (2013) Shape memory assisted self-healing coating. ACS Macro Lett 2(2):152–156
Nguyen MK, Lee DS (2010) Injectable biodegradable hydrogels. Macromol Biosci 10(6):563–579
Kızılel S, Sawardecker E, Teymour F, Pérez-Luna VH (2006) Sequential formation of covalently bonded hydrogel multilayers through surface initiated photopolymerization. Biomaterials 27(8):1209–1215
Krogsgaard M, Behrens MA, Pedersen JS, Birkedal H (2013) Self-healing mussel-inspired multi-pH-responsive hydrogels. Biomacromolecules 14(2):297–301
Yan X, Xu D, Chen J, Zhang M, Hu B, Yu Y, Huang F (2013) A self-healing supramolecular polymer gel with stimuli-responsiveness constructed by crown ether based molecular recognition. Polym Chem 4(11):3312–3322
Habault D, Zhang H, Zhao Y (2013) Light-triggered self-healing and shape-memory polymers. Chem Soc Rev 42(17):7244–7256
Froimowicz P, Frey H, Landfester K (2011) Towards the generation of self-healing materials by means of a reversible photo-induced approach. Macromol Rapid Commun 32(5):468–473
Ling J, Rong MZ, Zhang MQ (2011) Coumarin imparts repeated photochemical remendability to polyurethane. J Mater Chem 21(45):18373–18380
Ling J, Rong MZ, Zhang MQ (2012) Photo-stimulated self-healing polyurethane containing dihydroxyl coumarin derivatives. Polymer 53(13):2691–2698
Ghosh B, Chellappan KV, Urban MW (2012) UV-initiated self-healing of oxolane–chitosan–polyurethane (OXO–CHI–PUR) networks. J Mater Chem 22(31):16104–16113
Burnworth M, Tang L, Kumpfer JR, Duncan AJ, Beyer FL, Fiore GL, Rowan SJ, Weder C (2011) Optically healable supramolecular polymers. Nature 472(7343):334–337
Amamoto Y, Kamada J, Otsuka H, Takahara A, Matyjaszewski K (2011) Repeatable photoinduced self-healing of covalently cross-linked polymers through reshuffling of trithiocarbonate units. Angew Chem 123(7):1698–1701
Artzi N, Oliva N, Puron C, Shitreet S, Artzi S, Bon Ramos A, Groothuis A, Sahagian G, Edelman ER (2011) In vivo and in vitro tracking of erosion in biodegradable materials using non-invasive fluorescence imaging. Nat Mater 10(9):704–709
Heo YJ, Shibata H, Okitsu T, Kawanishi T, Takeuchi S (2011) Long-term in vivo glucose monitoring using fluorescent hydrogel fibers. Proc Natl Acad Sci 108(33):13399–13403
Roy S, Baral A, Banerjee A (2013) An amino-acid-based self-healing hydrogel: modulation of the self-healing properties by incorporating carbon-based nanomaterials. Chem Eur J 19(44):14950–14957
Ono K, Saito Y, Yura H, Ishikawa K, Kurita A, Akaike T, Ishihara M (2000) Photocrosslinkable chitosan as a biological adhesive. J Biomed Mater Res A 49(2):289–295
Peak CW, Wilker JJ, Schmidt G (2013) A review on tough and sticky hydrogels. Colloid Polym Sci 291(9):2031–2047
Shao H, Stewart RJ (2010) Biomimetic underwater adhesives with environmentally triggered setting mechanisms. Adv Mater 22(6):729–733
Wallace D, Cruise G, Rhee W, Schroeder J, Prior J, Ju J, Maroney M, Duronio J, Ngo M, Estridge T (2001) A tissue sealant based on reactive multifunctional polyethylene glycol. J Biomed Mater Res 58(5):545–555
Huang Y, Lawrence PG, Lapitsky Y (2014) Self-assembly of stiff, adhesive and self-healing gels from common polyelectrolytes. Langmuir 30(26):7771–7777
Chang G, Chen Y, Li Y, Li S, Huang F, Shen Y, Xie A (2015) Self-healable hydrogel on tumor cell as drug delivery system for localized and effective therapy. Carbohydr Polym 122:336–342
Chen J, Ma X, Dong Q, Song D, Hargrove D, Vora SR, Ma AW, Lu X, Lei Y (2016) Self-healing of thermally-induced, biocompatible and biodegradable protein hydrogel. RSC Adv 6(61):56183–56192
Jacob RS, Ghosh D, Singh PK, Basu SK, Jha NN, Das S, Sukul PK, Patil S, Sathaye S, Kumar A (2015) Self healing hydrogels composed of amyloid nano fibrils for cell culture and stem cell differentiation. Biomaterials 54:97–105
Li L, Yan B, Yang J, Chen L, Zeng H (2015) Novel mussel-inspired injectable self-healing hydrogel with anti-biofouling property. Adv Mater 27(7):1294–1299
Acknowledgements
We are grateful for financial support from the National Science Foundation (NSF). Jun Chen acknowledges the financial support from GE Fellowship.
Author information
Authors and Affiliations
Corresponding authors
Rights and permissions
About this article

Cite this article
Chen, J., Huang, Y., Ma, X. et al. Functional self-healing materials and their potential applications in biomedical engineering. Adv Compos Hybrid Mater 1, 94–113 (2018). https://doi.org/10.1007/s42114-017-0009-y
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s42114-017-0009-y