
Abstract
Purpose
Heterozygous loss-of-function mutations in the glucokinase (GCK) gene cause MODY 2, which is characterized by asymptomatic fasting hyperglycemia and does not require insulin treatment. Conversely, homozygous loss-of-function mutations in the same gene give rise to permanent neonatal diabetes mellitus (DM) that appears in the first 6–9 months of life and necessitates lifelong insulin treatment. We aimed to present the genotypic and phenotypic features of a 13-year-old patient diagnosed with DM at the age of 3 years due to a homozygous variant in the GCK gene.
Methods
The patient’s clinical and laboratory findings at follow-up were not consistent with the initial diagnosis of type 1 DM; thus, next-generation sequencing of MODY genes (GCK, HNF1A, HNF1B, and HNF4A genes) was performed to identify monogenic causes of DM.
Results
A novel homozygous variant c.1222 G > T in the GCK gene was revealed. In silico analysis identified it as a pathogenic variant. His mother, father, and brother had the same heterozygous variant in the GCK gene and were diagnosed with MODY 2 (mild fasting hyperglycemia and elevated HbA1c) after genetic counseling.
Conclusion
In this case report, a patient with a homozygous variant in the GCK gene, who was diagnosed with DM after the infantile period, was presented, highlighting the fact that cases with homozygous variants in the GCK gene can, though rarely, present at a later age with a milder phenotype.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Background
Glucokinase (GCK) has been called “the glucose sensor” of pancreatic beta cells because of its kinetics, allowing pancreatic beta cells to change glucose phosphorylation rate over a range of physiological glucose concentrations [1, 2]. As a result, it controls glucose-mediated insulin secretion and glucose homeostasis. Gain-of-function mutations in the GCK gene cause inappropriate hypersecretion of insulin despite hypoglycemia, known as hyperinsulinemic hypoglycemia [3]. Homozygous [4] or compound heterozygous [5] loss-of-function mutations in the GCK gene give rise to permanent neonatal diabetes mellitus (PNDM). GCK-PNDM is diagnosed during the first 6 months of life with severe hyperglycemia, and patients usually have intrauterine growth retardation [4]. It necessitates lifelong insulin treatment. Heterozygous loss-of-function mutations in the same gene cause a monogenic form of DM, maturity-onset diabetes of the young (MODY), specifically GCK-MODY or MODY 2. MODY, the most common monogenic form of diabetes, is characterized by autosomal dominant inheritance and the appearance of non-insulin-dependent diabetes before the age of 25 years [6]. It can often be misdiagnosed as type 1 diabetes mellitus (T1DM), as it typically presents in adolescence or young adulthood, or even type 2 diabetes mellitus (T2DM) [7]. Although it is present from birth, it is diagnosed during routine investigations in later life as mild subclinical fasting hyperglycemia (96–140 mg/dL) without any symptoms [2]. Family history may not be easily detected in GCK-MODY because affected parents or family members may be undiagnosed or have been mistakenly diagnosed with T2DM. Insulin treatment is unnecessary due to deterioration in the dose–response relationship between plasma glucose concentration and insulin secretion. Consequently, GCK-MODY requires no specific treatment; dietary intervention is usually sufficient [8]. Previously, three patients diagnosed with DM outside the infantile period and with homozygous GCK mutations were reported [9, 10]. We aimed to present the genotypic and phenotypic features of a 13-year-old patient diagnosed with DM at the age of 3 years due to a homozygous variant in the GCK gene.
Written informed consent was obtained from the patient’s parents for publication of this case report.
Case
The male proband was first admitted to the hospital when he was 3 years old with polyuria and polydipsia for 2 weeks and weight loss throughout the previous month. He was born at term with a birth weight of 2500 g. His medical history was unremarkable, and mental-motor development and auxological measurements were normal. His parents were first-degree cousins. There was no history of DM in the family except for the grandmother, who had T2DM. On physical examination, weight was 15.5 kg (0.28 SDS), height was 99 cm (0.43 SDS), and BMI was 19.6 (− 0.16 SDS); he was prepubertal according to Tanner staging, while no pathological features existed. Laboratory findings were the following: fasting serum glucose 172 mg/dL (N, 60–100); C-peptide 1.1 ng/mL (N, 0.9–7.1); insulin < 2 mIU/mL (N, 1.9–23); and HbA1c 7.1% (54.1 mmol/mol, N, 4–5.7). There was no acidosis in the blood gas analysis and no ketone in the urine. Only two diabetes autoantibodies (anti-glutamic acid decarboxylase and anti-insulin) could be performed, and they were negative. As blood glucose monitoring showed mild fasting and postprandial hyperglycemia, a diabetes diet was recommended and a basal-bolus insulin regimen at a total daily dose of 0.5 units/kg was initiated. After offering a DM self-management education program to the parents, he was discharged from the hospital. Bolus insulin was discontinued after 3 years due to normal postprandial glucose levels. In nearly 10 years of outpatient follow-up, he had good metabolic control (HbA1c 6–7.1%, 42.1–54.1 mmol/mol) and no ketoacidosis events since diagnosis under treatment with a single daily dose (0.2 units/kg) of insulin glargine. At his last examination at 13 years of age, weight was 53.2 kg (0.08 SDS), height was 164.5 cm (0.5 SDS), and BMI was 19.6 (− 0.16 SDS). Testicular volume was 8 mL bilaterally, pubic hair development was compatible with Tanner stage 2, and the rest of the physical examination was normal. Owing to the inappropriately low insulin requirements beyond the honeymoon period and negative diabetes autoantibodies that were not compatible with T1DM, he was re-evaluated via laboratory testing, which indicated preserved beta cell function: C-peptide 1.08 ng/mL (N, 0.9–7.1). Despite the absence of strong family history for the inherited forms of diabetes, next-generation sequencing of MODY genes (GCK, HNF1A, HNF1B, and HNF4A genes) was performed.
Methods
For the molecular genetic evaluation, a Custom Target Amplicon MODY NGS Panel (Celemics, Inc., Seoul, Korea), designed for the most frequent MODY types, was used. This panel covered all the coding regions and the exon–intron boundaries of the four most frequent MODY-associated genes, namely, GCK, HNF1A, HNF1B, and HNF4A. Target amplicon NGS was performed on an Illumina MiSeq NGS System (Illumina, Inc., San Diego, CA, USA) using the MiSeq Reagent Nano Kit v3 (Illumina, Inc., San Diego, CA, USA). FASTQ sequencing files were collected and transferred to “SEQ” variant analysis software (Genomize, Istanbul, Turkey). The variant interpretation was made according to the standards and guidelines released by the American College of Medical Genetics (ACMG) [11]. The Varsome platform was used for in silico analysis. The variant detected in the index case was studied in other family members using the Sanger sequence method by designing region-specific primers. DUET server was used to predict protein stability change due to detected mutation [12].
Results and clinical follow-up
Next-generation sequencing analyses of the HNF1A, HNF1B, and HNF4A genes did not identify any mutations. However, the novel homozygous missense variant c.1222 G > T was revealed in exon 10 of the GCK gene which has never been previously reported. This nucleotide change c.1222 G > T leads to substitution of valine by lysine at residue 408 (p.V408L). In silico analysis indicated that the variant is disease-causing (from mutation taster) and likely pathogenic (from Varsome). This variant was found neither in the Exome Aggregation Consortium (ExAC) nor in the 1000 Genomes Project databases. The c.1222 G > T variant was defined as pathogenic by 21 computational prediction methods, namely, BayesDel_addAF, BayesDel noAF, DANN, DEOGEN2, EIGEN, EIGEN PC, FATHMM, FATHMM-MKL, FATHMM-XF, LIST-S2, LRT, MVP, MetaLR, MetaSVM, MutPred, MutationAssessor, MutationTaster, PROVEAN, REVEL, SIFT, and SIFT4G vs. one benign prediction from PrimateAI. As a result of clinical findings and in silico analysis, the variant was evaluated as likely pathogenic according to ACMG criteria (PM1, PM2, PP2, PP3) (11). According to the DUET server’s prediction, the detected variant (p.V408L) causes the GCK protein to be destabilized.
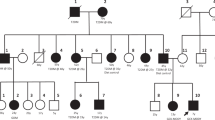
The patient’s normal weight parents and brother also had the same heterozygous variant in the GCK gene (Fig. 1). After genetic counseling, they all underwent laboratory evaluation; the mother’s fasting serum glucose was 108 mg/dL and HbA1c was 6.2% (44.3 mmol/mol), while the father’s fasting serum glucose was 130 mg/dL and HbA1c was 6.2% (44.3 mmol/mol); the brother had a fasting serum glucose level of 102 mg/dL and HbA1c was 6.0% (42.1 mmol/mol) (Table 1). The pedigree of the family is presented in Fig. 2. They were all diagnosed with GCK-MODY and dietary intervention was recommended. The insulin therapy of the index case was discontinued after genetic analysis; however, blood glucose levels increased up to 212 mg/dL, and insulin glargine was re-initiated. In the nearly 10 years of clinical follow-up, the patient’s HbA1c levels ranged between 6.5 (47.5 mmol/mol) and 7.9% (62.8 mmol/mol), with an insulin treatment of 0.3 unit/kg/day, and DM-related microvascular and macrovascular complications did not develop.

Electropherogram of family members. The electropherograms show that the proband was homozygous, and other family members were heterozygous for the variant (c.1222G > T)

Pedigree of the family with the novel variant in the glucokinase (GCK) gene. Arrow indicates the proband
Discussion
Heterozygous loss-of-function mutations in the GCK gene cause GCK-MODY. It is one of the most common subtypes. Autosomal dominant inheritance occurs in MODY types, and three generations can exhibit diabetes. More generations can exhibit diabetes, but diagnosis is further supported if a diagnosis is present in at least > 2 generations. Many studies have shown that the frequency of GCK-MODY varies between countries [13,14,15,16], with, for example, approximately one in four MODY cases having a GCK mutation in the Turkish pediatric cohort [17]. As a result of decreased GCK caused by GCK gene mutation, glucose phosphorylation and glucose sensitivity decrease in pancreatic beta cells, leading to a shift to the right in the dose–response relationship between plasma glucose concentration and insulin secretion [18]. For this reason, glucose-lowering therapy is ineffective, and it is not recommended outside pregnancy. Postprandial glucose levels are generally normal in patients with GCK-MODY, and their HbA1c is usually below 7.5% [19]. During a 50-year follow-up period, individuals with GCK-MODY had a similar prevalence of microvascular and macrovascular complications (nephropathy, retinopathy, peripheral neuropathy, peripheral vascular disease, and cardiovascular disease) to controls [20].
Homozygous loss-of-function mutations in the GCK gene generally cause PNDM. Today, over 50 patients with homozygous GCK mutations have been reported thanks to the increasing accessibility of molecular genetic analysis [4, 9, 21,22,23,24,25,26,27,28]. PNDM is an extremely rare hereditary condition, its incidence being estimated at one case per 260,000 live births in Europe [29]. However, the annual incidence of PNDM in the South-East Anatolian region of Turkey was found to be one in 48, 000 live births, possibly as a consequence of consanguineous marriages [30]. Unlike GCK-MODY, GCK-PNDM becomes symptomatic with insulin-requiring severe hyperglycemia in the first 6–9 months of life [4]. Intrauterine growth retardation is often present [4, 9, 24]. It is suggested that fetal growth is affected by the mutation in the GCK gene, with supporting evidence in studies on animals and humans [31, 32]. Patients with homozygous GCK mutations usually need insulin therapy; however, sulfonylurea treatment was reported to reduce the insulin dose in one case [22]. In a case series of 19 patients with homozygous GCK mutations, all patients with neonatal diabetes required insulin treatment, with the median dose being 1 unit/kg/day (range 0.7–1.3 units) [9].
We present a 13-year-old boy diagnosed with diabetes at the age of 3 years and with reasonable glycemic control with a single low dose of insulin glargine during follow-up despite having a homozygous variant in the GCK gene. To the best of our knowledge, three patients have been reported in the literature to date who were diagnosed beyond infancy with monogenic diabetes with homozygous GCK mutations [9, 10]. In the first report, Raimondo et al. [9] evaluated the clinical phenotype of 19 patients with homozygous GCK mutations and studied their molecular mechanisms of GCK dysfunctions. Two of these cases were diagnosed with homozygous GCK mutations. The first patient was born at term with a birth weight of 3285 g. She was diagnosed at 9 years of age with diabetes owing to high fasting blood glucose and mildly elevated postprandial glucose levels. A homozygous mutation, c.478G.A, in the GCK gene was detected, and she did not require insulin treatment. The second patient had been treated with multiple daily insulin injections due to a diagnosis of T1DM at 15 years of age. She was born at term with a birth weight of 3500 g. Next-generation sequencing was performed due to negative anti-insulin antibodies and revealed the c.676G > A homozygous GCK mutation. The third patient with a homozygous GCK mutation (c.1116G > C) was a 51-year-old Italian woman whose 3-year-old grandson was being investigated for diabetes [10]. She had been diagnosed with diabetes at 22 years of age, during pregnancy, when she had mildly elevated fasting glucose and HbA1c levels under metformin treatment. After the genetic diagnosis, her treatment was discontinued. The presence of long-term complications of diabetes in the patient was not mentioned in the report [30]. These three cases presented with mild, youth-onset diabetes and had average birth weight. Moreover, only one of them was treated with insulin [25, 30]. Similarly, there was no history of low birth weight, hyperglycemia, or hospitalization in the neonatal period in our patient. After the genetic diagnosis and owing to the low insulin requirement, we tried to discontinue insulin treatment but failed. We did not detect any complications in our patient during about 10 years of medical monitoring. Our patient’s clinical phenotype was compatible with that of the previous cases that were reported to be diagnosed with diabetes outside the infantile period and had homozygous GCK mutations. When we evaluate the clinical features of all four patients, including ours, we can conclude that some homozygous GCK mutations cause mild or childhood-onset diabetes resembling GCK-MODY in many aspects.
Variability in the clinical phenotype of GCK mutations can be influenced by the characteristics of mutant enzymes and many factors involved in GCK homeostasis. GCK is called “the glucose sensor” due to its kinetic properties that alter glucose phosphorylation in beta cells to maintain physiological glucose levels [1]. In other words, glucose-stimulated insulin release (GSIR) is dependent on its kinetic properties. A mathematical model has been developed for predicting the threshold for GSIR in GCK mutations [1]. Inactivating mutations in the GCK gene decrease the phosphorylating potential of the GCK enzyme [33]. According to this model, the glucose threshold for GSIR in PNDM-GCK is extraordinarily high, and GCK-MODY is moderately high [1]. While the kinetics of the enzyme are generally useful in predicting the clinical phenotype, in some cases, they may be inadequate to predict other phenotypes. Different regulatory molecules (PFKFBI, BAD, and purified free penta-ubiquitin chains) are involved in the regulation and interaction of glucokinase, and their defects can alter glycemic control [2]. A mutation in the GCK gene, affecting protein stability possibly due to a defect in the protein structure though not greatly changing the kinetics of GCK, has been also hypothesized [34]. The thermolability properties of mutant GCK enzymes can play a role in predicting the severity of the phenotype [9, 35].
Evaluation of the genotypic features of patients who had homozygous GCK mutations diagnosed with diabetes beyond infancy could be critical in assessing the clinical picture. In the report by Raimondo et al. [9], examining 19 patients with homozygous GCK mutations, no correlation was found between the severity of the clinical picture and the kinetics of mutant GCK proteins. Mutations of the two cases who were diagnosed with homozygous GCK mutations at 9 and 15 years showed inactivating kinetics indistinguishable from the neonatal-onset mutations; however, interestingly, they exhibited increased thermostability characteristics. The investigators suggested that increased thermostability leads to a more stable protein and may reduce the clinical severity of the disease. They concluded that protein instability is more highly correlated with phenotypic severity than kinetic dysfunction [9]. The in silico analysis of the third patient with a novel E372D mutation showed an increased protein flexibility compared with the GCK wild type, also leading to the conclusion that protein stability can affect clinical severity [10].
In our patient, a c.1222 G > T missense novel homozygous variant was revealed in the GCK gene. In silico analysis indicated that the variant was disease-causing (from mutation taster) and likely pathogenic (from Varsome), and as a result caused destabilization of the GCK protein. Functional analyses for the mutant GCK activity could not be performed. However, the patient’s father, mother, and brother were heterozygous for this variant, and their clinical and laboratory findings were consistent with GCK-MODY.
Our patient is so far the fourth reported case with a homozygous GCK mutation presenting with mild hyperglycemia. The detected variant is a novel mutation that has never been previously reported. This report presents the disease-causing novel variant c.1222 G > T in the GCK gene and its rare clinical presentation of mild hyperglycemia, in contrast to the usual phenotype of permanent neonatal DM. In conclusion, the present report emphasizes the fact that, although infrequent, homozygous mutations in the GCK gene may result in childhood-onset mild diabetes. In cases where the type of diabetes cannot be distinguished on the basis of clinical and laboratory findings, molecular analysis of monogenic diabetes genes may provide exact diagnosis, reveal novel variants, and offer a better understanding of the relationship between genotype and phenotype.
Data Availability
Data and material are stated in the text.
Code availability
Not applicable.
References
Matschinsky FM (2002) Regulation of pancreatic beta-cell glucokinase: from basics to therapeutics. Diabetes 51(Suppl 3):S394-404
Osbak KK, Colclough K, Saint-Martin C, Beer NL, Bellanné-Chantelot C, Ellard S et al (2009) Update on mutations in glucokinase (GCK), which cause maturity-onset diabetes of the young, permanent neonatal diabetes, and hyperinsulinemic hypoglycemia. Hum Mutat 30(11):1512–1526
Glaser B, Kesavan P, Heyman M, Davis E, Cuesta A, Buchs A et al (1998) Familial hyperinsulinism caused by an activating glucokinase mutation. N Engl J Med 338(4):226–230
Njølstad PR, Sagen JV, Bjørkhaug L, Odili S, Shehadeh N, Bakry D et al (2003) Permanent neonatal diabetes caused by glucokinase deficiency: inborn error of the glucose-insulin signaling pathway. Diabetes 52(11):2854–2860
Esquiaveto-Aun AM, De Mello MP, Paulino MF, Minicucci WJ, Guerra-Júnior G, De Lemos-Marini SH (2015) A new compound heterozygosis for inactivating mutations in the glucokinase gene as cause of permanent neonatal diabetes mellitus (PNDM) in double-first cousins. Diabetol Metab Syndr 7:101
Anik A, Çatli G, Abaci A, Böber E (2015) Maturity-onset diabetes of the young (MODY): an update. J Pediatr Endocrinol Metab 28(3–4):251–263
Shields BM, Hicks S, Shepherd MH, Colclough K, Hattersley AT, Ellard S (2010) Maturity-onset diabetes of the young (MODY): how many cases are we missing? Diabetologia 53(12):2504–2508
Hattersley AT, Greeley SAW, Polak M, Rubio-Cabezas O, Njølstad PR, Mlynarski W et al (2018) ISPAD Clinical Practice Consensus Guidelines 2018: the diagnosis and management of monogenic diabetes in children and adolescents. Pediatr Diabetes 19(Suppl 27):47–63
Raimondo A, Chakera AJ, Thomsen SK, Colclough K, Barrett A, De Franco E et al (2014) Phenotypic severity of homozygous GCK mutations causing neonatal or childhood-onset diabetes is primarily mediated through effects on protein stability. Hum Mol Genet 23(24):6432–6440
Marucci A, Biagini T, Di Paola R, Menzaghi C, Fini G, Castellana S et al (2019) Association of a homozygous GCK missense mutation with mild diabetes. Mol Genet Genomic Med 7(7):e00728
Richards S, Aziz N, Bale S, Bick D, Das S, Gastier-Foster J et al (2015) Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet Med 17(5):405–424
Pires DE, Ascher DB, Blundell TL (2014) DUET: a server for predicting effects of mutations on protein stability using an integrated computational approach. Nucleic Acids Res 42(Web Server issue):314–9
Fendler W, Borowiec M, Baranowska-Jazwiecka A, Szadkowska A, Skala-Zamorowska E, Deja G et al (2012) Prevalence of monogenic diabetes amongst Polish children after a nationwide genetic screening campaign. Diabetologia 55(10):2631–2635
Barrio R, Bellanne-Chantelot C, Moreno JC, Morel V, Calle H, Alonso M et al (2002) Nine novel mutations in maturity-onset diabetes of the young (MODY) candidate genes in 22 Spanish families. J Clin Endocrinol Metab 87(6):2532–2539
Yorifuji T, Fujimaru R, Hosokawa Y, Tamagawa N, Shiozaki M, Aizu K et al (2012) Comprehensive molecular analysis of Japanese patients with pediatric-onset MODY-type diabetes mellitus. Pediatr Diabetes 13(1):26–32
Feigerlova E, Pruhova S, Dittertova L, Lebl J, Pinterova D, Kolostova K et al (2006) Aetiological heterogeneity of asymptomatic hyperglycaemia in children and adolescents. Eur J Pediatr 165(7):446–452
Haliloglu B, Hysenaj G, Atay Z, Guran T, Abali S, Turan S et al (2016) GCK gene mutations are a common cause of childhood-onset MODY (maturity-onset diabetes of the young) in Turkey. Clin Endocrinol 85(3):393–399
Fajans SS, Bell GI, Polonsky KS (2001) Molecular mechanisms and clinical pathophysiology of maturity-onset diabetes of the young. N Engl J Med 345(13):971–980
Steele AM, Wensley KJ, Ellard S, Murphy R, Shepherd M, Colclough K et al (2013) Use of HbA1c in the identification of patients with hyperglycaemia caused by a glucokinase mutation: observational case control studies. PloS one 8(6):e65326
Steele AM, Shields BM, Wensley KJ, Colclough K, Ellard S, Hattersley AT (2014) Prevalence of vascular complications among patients with glucokinase mutations and prolonged, mild hyperglycemia. JAMA 311(3):279–286
Porter JR, Shaw NJ, Barrett TG, Hattersley AT, Ellard S, Gloyn AL (2005) Permanent neonatal diabetes in an Asian infant. J Pediatr 146(1):131–133
Turkkahraman D, Bircan I, Tribble ND, Akçurin S, Ellard S, Gloyn AL (2008) Permanent neonatal diabetes mellitus caused by a novel homozygous (T168A) glucokinase (GCK) mutation: initial response to oral sulphonylurea therapy. J Pediatr 153(1):122–126
Rubio-Cabezas O, Díaz González F, Aragonés A, Argente J, Campos-Barros A (2008) Permanent neonatal diabetes caused by a homozygous nonsense mutation in the glucokinase gene. Pediatr Diabetes 9(3 Pt 1):245–249
Bennett K, James C, Mutair A, Al-Shaikh H, Sinani A, Hussain K (2011) Four novel cases of permanent neonatal diabetes mellitus caused by homozygous mutations in the glucokinase gene. Pediatr Diabetes 12(3 Pt 1):192–196
Durmaz E, Flanagan S, Berdeli A, Semiz S, Akcurin S, Ellard S et al (2012) Variability in the age at diagnosis of diabetes in two unrelated patients with a homozygous glucokinase gene mutation. J Pediatr Endocrinol Metab 25(7–8):805–808
Njølstad PR, Søvik O, Cuesta-Muñoz A, Bjørkhaug L, Massa O, Barbetti F et al (2001) Neonatal diabetes mellitus due to complete glucokinase deficiency. N Engl J Med 344(21):1588–1592
Al Senani A, Hamza N, Al Azkawi H, Al Kharusi M, Al Sukaiti N, Al Badi M et al (2018) Genetic mutations associated with neonatal diabetes mellitus in Omani patients. J Pediatr Endocrinol Metab 31(2):195–204
Lin DC, Huang CY, Ting WH, Lo FS, Lin CL, Yang HW et al (2019) Mutations in glucokinase and other genes detected in neonatal and type 1B diabetes patient using whole exome sequencing may lead to disease-causing changes in protein activity. Biochim Biophys Acta 1865(2):428–433
Slingerland AS, Shields BM, Flanagan SE, Bruining GJ, Noordam K, Gach A et al (2009) Referral rates for diagnostic testing support an incidence of permanent neonatal diabetes in three European countries of at least 1 in 260,000 live births. Diabetologia 52(8):1683–1685
Demirbilek H, Arya VB, Ozbek MN, Houghton JA, Baran RT, Akar M et al (2015) Clinical characteristics and molecular genetic analysis of 22 patients with neonatal diabetes from the South-Eastern region of Turkey: predominance of non-KATP channel mutations. Eur J Endocrinol 172(6):697–705
Terauchi Y, Kubota N, Tamemoto H, Sakura H, Nagai R, Akanuma Y et al (2000) Insulin effect during embryogenesis determines fetal growth: a possible molecular link between birth weight and susceptibility to type 2 diabetes. Diabetes 49(1):82–86
Shields BM, Spyer G, Slingerland AS, Knight BA, Ellard S, Clark PM et al (2008) Mutations in the glucokinase gene of the fetus result in reduced placental weight. Diabetes Care 31(4):753–757
Davis EA, Cuesta-Muñoz A, Raoul M, Buettger C, Sweet I, Moates M et al (1999) Mutants of glucokinase cause hypoglycaemia- and hyperglycaemia syndromes and their analysis illuminates fundamental quantitative concepts of glucose homeostasis. Diabetologia 42(10):1175–1186
Galán M, Vincent O, Roncero I, Azriel S, Boix-Pallares P, Delgado-Alvarez E et al (2006) Effects of novel maturity-onset diabetes of the young (MODY)-associated mutations on glucokinase activity and protein stability. Biochem J 393(Pt 1):389–396
Pino MF, Kim KA, Shelton KD, Lindner J, Odili S, Li C et al (2007) Glucokinase thermolability and hepatic regulatory protein binding are essential factors for predicting the blood glucose phenotype of missense mutations. J Biol Chem 282(18):13906–13916
Author information
Authors and Affiliations
Contributions
BEF, İA, and HM collected data and wrote the manuscript, ÖK performed genetic analyses, BND revised the manuscript, and GÇ critically reviewed the manuscript and supervised the entire study process. All authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Ethics approval
The manuscript is a case report; the patient consent statement was obtained.
Consent to participate
Informed consent was obtained from the patient’s parents.
Consent for publication
Not applicable.
Conflict of interest
The authors declare no competing interests.
Additional information
Publisher's note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
About this article

Cite this article
Filibeli, B.E., Çatli, G., Ayranci, İ. et al. Childhood-onset mild diabetes caused by a homozygous novel variant in the glucokinase gene. Hormones 21, 163–169 (2022). https://doi.org/10.1007/s42000-021-00330-1
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s42000-021-00330-1