
Abstract
Sulfides have been widely acknowledged as one of the most promising solid electrolytes (SEs) for all-solid-state batteries (ASSBs) due to their superior ionic conductivity and favourable mechanical properties. However, the extremely poor air stability of sulfide SEs leads to destroyed structure/performance and release of toxic H2S gas, which greatly limits mass-production/practical application of sulfide SEs and ASSBs. This review is designed to serve as an all-inclusive handbook for studying this critical issue. First, the research history and milestone breakthroughs of this field are reviewed, and this is followed by an in-depth elaboration of the theoretical paradigms that have been developed thus far, including the random network theory of glasses, hard and soft acids and bases (HSAB) theory, thermodynamic analysis and kinetics of interfacial reactions. Moreover, the characterization of air stability is reviewed from the perspectives of H2S generation, morphology evolution, mass change, component/structure variations and electrochemical performance. Furthermore, effective strategies for improving the air stabilities of sulfide SEs are highlighted, including H2S absorbents, elemental substitution, design of new materials, surface engineering and sulfide-polymer composite electrolytes. Finally, future research directions are proposed for benign development of air stability for sulfide SEs and ASSBs.
Graphical Abstract

Similar content being viewed by others

Avoid common mistakes on your manuscript.
1 Introduction
Widespread application of lithium-ion batteries (LIBs) in electronic devices and electric vehicles confirms their great importance in modern society [1]. However, commercialized LIBs encounter the upper limit of energy density and severe safety issues due to leakage, low thermal stability and flammability of organic liquid electrolytes (OLEs) [2, 3]. Replacing OLEs with nonflammable solid electrolytes (SEs) would ultimately solve the safety problem and may potentially enable use of lithium metal anodes for higher energy density and make all-solid-state batteries (ASSBs) the most desirable energy-storage devices and technologies [4,5,6,7,8,9,10,11,12]. However, different types of SEs suffer from various challenges. For example, among these are the large grain boundary impedance and fragile ceramic nature for oxide SEs [13, 14], low room-temperature ionic conductivity and low Li+ transfer number for polymer SEs [15, 16], limited ionic conductivity and expensive manufacturing process for thin film LiPON [17], etc.
Among all kinds of SEs, sulfide SEs have been widely acknowledged as one of the most promising candidates [7,8,9,10,11,12,13,14,15] due to their remarkable ductilities and high ionic conductivities, which are on par with those of OLEs [18, 19]. However, their poor stabilities, including narrow electrochemical windows [20,21,22,23,24], chemical/electrochemical incompatibility with oxide cathodes [25,26,27] and lithium metal anodes [28,29,30,31,32,33,34,35], and poor air stability [36], greatly limit practical application of sulfide-based ASSBs [37]. Among these problems, air stability is a common issue for all or most solid-state batteries [38, 39]. Oxide-based SEs can react slowly with humid air by Li+/H+ exchange and result in the formation of ionic resistive LiOH and Li2CO3, which can increase the interfacial resistance [40,40,41,42,44]. When a halide SE such as Li3InCl6 is exposed to humid air, it first becomes a crystalline hydrate and then decomposes into In2O3, HCl and LiCl [45, 46]. Although polymer SEs such as PEO are chemically stable toward water and air, they are oxidized easily during cycling in a pure O2 environment [6, 47]. In particular, the extremely poor air stability leads to evolutions of toxic H2S gas [48], formation of completely damaged structures [49] and decayed performance [50] and also makes synthesis, storage, transportation and posttreatment of sulfide SEs very complex; they often require an inert atmosphere or dry room, which significantly increases production costs. For these reasons, numerous efforts have been made to unravel the origins of poor air stability [36, 51] and to develop air-stable sulfide SEs exhibiting other satisfactory properties [52].
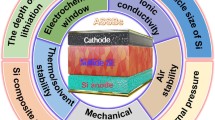
In this comprehensive review, the research history of sulfide SEs and milestone breakthroughs for air stability are summarized. Then, theoretical explanations, including random network theory tailored for glass materials, hard and soft acids and bases (HSAB) theory based on the affinities of chemical species, thermodynamic analyses based on the energy changes for hydrolysis reactions, and kinetics of interfacial reactions based on the reactivity of crystalline planes, are summarized to provide scientific interpretations of the air instability problem. To better understand structure-performance relationships, characterizations of air stability, including the amount of H2S generated, morphological evolution, mass changes, component/structure variations and electrochemical performance, are reviewed. Furthermore, effective strategies for developing air-stable sulfide SEs, including use of H2S absorbents, elemental substitution, design of new materials, surface coatings and sulfide-polymer composite electrolytes, are highlighted. Finally, future research directions and perspectives for the air stability problem of sulfide SEs are proposed. The major contents of this comprehensive review are summarized with the schematic illustration in Fig. 1. This review is designed to provide fundamental understanding and facilitate benign development of air-stable sulfide SEs for mass production and wide practical application of sulfide ASSBs.

Framework for air stability research with sulfide SEs
2 Research History for the Air Stability of Sulfide SEs and ASSBs
The earliest work on this topic involved the random network theory of glass proposed by Zachariasen in 1932 for glassy sulfide SEs, which was regarded as theoretical guidance for modifying glassy electrolytes. Research on the air stability of sulfide SEs attracted little attention until Martin et al. [53] proposed improving the air stability of sulfide SEs by decreasing the proportion of nonbridging sulfurs in 2008. In 2011, Tatsumisago et al. [36] investigated the structural changes undergone by glass and glass-ceramic Li2S-P2S5 sulfides in the atmosphere for the first time. In 2013, Hayashi et al. [48] successfully suppressed H2S gas generation by adding metal oxides into glassy electrolytes and used oxygen substitution to partially replace Li2S with Li2O. Subsequently, a series of studies based on an oxygen substitution strategy (e.g., xLi2O·(75 − x)Li2S·25P2S5 [54], 75Li2S·(25 − x)P2S5·xP2O5 [55] and Li6PS5−xOxBr [56]) were reported. Although these were effective in improving air stability, the loss in ionic conductivity resulting from this strategy was often criticized. In 2019, Xiao et al. [57] performed cosubstitution of Zn and O and obtained a glass-ceramic electrolyte with significantly enhanced air stability and ionic conductivity. Moreover, they calculated the energy change for hydrolysis involving the reactions of cosubstituted sulfide SEs or pristine sulfide with H2O for the first time, which further demonstrated the effectiveness of this strategy. Subsequently, the codoping strategy was also successfully applied to various sulfide SEs (e.g., Li3+3xP1−xZnxS4−xOx, Li6−2xZnxPS5−xOxBr [58] and Li6.988P2.994Nb0.2S10.934O0.6 [59]), which led to comprehensively enhanced properties. In 2014, Liang et al. [51] proposed an innovative use of HSAB theory, which was proposed by Pearson in 1963 [60], as theoretical guidance for designing air-stable sulfide SEs. Moreover, they prepared As-substituted Li4SnS4 (LSS) with the highest known ionic conductivity and air stability among air-stable and recoverable sulfide SEs. Based on HSAB theory, a series of novel air-stable materials were developed. In 2019, Hayashi et al. [61] successfully synthesized Li3SbS4 with higher air stability but reduced ionic conductivity, which was one order of magnitude less than that of LSS. Huang et al. [62] creatively synthesized air-stable Li4Cu8Ge3S12 with an open-framework structure and reversible water adsorption/desorption capability. However, the parent phases of these new materials (i.e., LSS, Li3SbS4, and Li4Cu8Ge3S12) generally exhibited superior air stability but lower ionic conductivity than the well-known thiophosphates. Fortunately, various materials (e.g., Li3.833Sn0.833As0.166S4 [51], 0.4LiI-0.6Li4SnS4 [52], Li3.8Sn0.8Sb0.2S4 [63], Li3.85Sn0.85Sb0.15S4 [64]) from the Li4SnS4 family were developed, and solution-coating was realized, which greatly advanced practical application. In aiming to develop air-stable sulfide SEs suitable for dry-room conditions, Sun et al. performed soft-acid substitution to obtain Sn-substituted Li6PS5I [65] and Sb-substituted Li10GeP2S12 (LGPS) [50] with significantly enhanced air stability and ionic conductivity in 2020. Furthermore, other strategies, such as sulfide-polymer composite electrolytes [49, 66, 67] and surface engineering [68], were proposed to improve the air stabilities of sulfide SEs. In 2020, Mo et al. [69] systematically investigated the moisture stabilities of lithium ternary sulfides (Li-M-S) with different central cations M with thermodynamic analyses, which provided guidance for designing air-stable sulfide SEs. All of this significant progress on the air stabilities of sulfide SEs is summarized in Fig. 2 for a clear demonstration.

Research history and milestone breakthroughs for air stabilities of sulfide SEs and ASSBs. Reprinted with permission from Ref. [36]. Copyright © 2010, Elsevier. Reprinted with permission from Ref. [94]. Copyright © 2012, American Chemical Society. Reprinted with permission from Ref. [119]. Copyright © 2017, Wiley-VCH. Reprinted with permission from Ref. [57]. Copyright © 2018, Elsevier. Reprinted with permission from Ref. [66]. Copyright © 2019, American Chemical Society. Reprinted with permission from Ref. [62]. Copyright © 2019, Wiley-VCH. Reprinted with permission from Ref. [61]. Copyright © 2019, Elsevier. Reprinted with permission from Ref. [69]. Copyright © 2020, Wiley-VCH. Reprinted with permission from Ref. [50]. Copyright © 2020, American Chemical Society. Reprinted with permission from Ref. [65]. Copyright © 2020, Wiley-VCH
3 Theoretical Explanations
As the understanding of the air stabilities of sulfide SEs is deepening, various theoretical explanations have been proposed from different perspectives, including the random network theory of glass, HSAB theory, thermodynamic analysis and kinetics of interfacial reactions.
3.1 Random Network Theory of Glass
In 1932, Zachariasen proposed the random network theory of glass (RNTG) to explain glass construction from ionic polyhedra (e.g., tetrahedra and octahedra) arrangements exhibiting short-range order and long-range disorder (Fig. 3a). Since early-stage SE research started from glass and glass-ceramic sulfide SEs [70], the RNTG turned out to provide theoretical guidance. In 2008, Martin et al. [53] pointed out that nonbridging sulfur anions were unstable sites vulnerable to be attacked by water molecules. Therefore, nonbridging sulfur units were bridged by introducing trivalent ions such as Ga3+ and La3+ to eliminate some nonbridging sulfurs and improve the air stabilities of glasses. However, convincing experimental data, such as the amount of H2S gas generated, XRD patterns and other direct evidence, were lacking. In 2011, Muramatsu et al. [36] found that the air stabilities of L2S-P2S5 sulfide SEs were related to their local structures. The 75Li2S·25P2S5 glass and glass-ceramic sulfides containing PS43− groups showed the highest air stabilities compared with those of 67Li2S·33P2S5 glass and Li2S crystals that contain P2S74− and Li–S–Li groups, respectively. They concluded that the bridging sulfurs in P2S74− and Li–S–Li were first attacked by water molecules and transformed into –SH groups, which subsequently reacted with water molecules and were finally transformed into H2S gas. In this case, bridging sulfurs seemed to be less stable than nonbridging sulfurs based on experimental results. Fukushima et al. [71] found that 60Li2S·25P2S5·10Li3N glass-ceramic sulfides exhibited high ionic conductivity and high stability against moisture, which was attributed to formation of crosslinks in the glass network due to nitrogen addition. In addition to comparisons of air stabilities for various materials in glass systems, air stability differences among glasses, glass-ceramic and crystalline sulfide SEs must be studied further [72].

Theoretical explanations. a Schematic illustration of the network structure of glass materials. Reprinted with permission from Ref. [150]. Copyright © 2005, Elsevier. Schematic illustration of HSAB theory for b “soft-soft” and c “hard-hard” reactions. Reprinted with permission from Ref. [74]. Copyright © 1968, American Chemical Society. d Energy for hydrolysis reactions of sulfides with different central cations. Reprinted with permission from Ref. [69]. Copyright © 2020, Wiley-VCH. e. Exposed crystalline planes (predicted) of β-Li3PS4. f Linear relationship between \(\left( {\Delta E_{\text{p}} } \right)\) and \(\left( {E_{{{\text{VS}},\;\mathop {{\text{seg}}}\limits^{\cdot\cdot} }} } \right)\). Reprinted with permission from Ref. [76]. Copyright © 2020, Elsevier. g Schematic illustration of anion polymerizations induced by atmospheric deterioration and electrochemical deterioration. Reprinted with permission from Ref. [78]. Copyright © 2021, American Chemical Society
3.2 Hard and Soft Acids and Bases Theory
In 1963, Pearson [60] proposed the hard and soft acids and bases (HSAB) theory on the basis of Lewis acid-base theory. According to the binding abilities of atoms, ions and molecules to electrons (from strong to weak), chemical species can be classified into three categories: hard, boundary and soft. Pearson [73] then defined absolute hardness to calculate and quantify the hardness of various chemical species. Subsequently, Klopman [74] further elaborated the HSAB theory with molecular orbital theory. As shown in Fig. 3b, soft bases and acids exhibit higher energy for the highest occupied molecular orbitals (HOMOs) and lower energy for the lowest unoccupied molecular orbitals (LUMOs), respectively. The energy difference between them is small and favors electron migration, which results in a low-energy hybrid orbital and a strong covalent bond. In contrast, hard bases and acids have lower HOMO energy and higher LUMO energy (Fig. 3c), respectively. The energy difference is large and unfavourable for electron migration. Therefore, they are more inclined toward ionization, thus inducing Coulomb interactions and forming strong ionic bonds. A combination of hard and soft chemical species generally induces a weak ionic bond and leads to an unstable product. In conclusion, the HSAB theory can be summarized in one sentence: soft acids/bases have high affinities for soft bases/acids, and hard acids/bases have high affinities for hard bases/acids, which is confirmed by the high stability of their products. In 2014, Sahu et al. [51] first proposed that the HSAB theory might serve as a guideline for designing air-stable sulfide SEs. With this guidance, Sn4+ [65], As5+ [51, 75], Sb5+ [50], and other soft acids [62], which are inclined to bind tightly with the soft base S2− and impede attack by H2O, have been used to improve the air stabilities of sulfide SEs.
3.3 Thermodynamic Analysis
Thermodynamic analyses based on changes in Gibbs energy for hydrolysis reactions between sulfide SEs and H2O facilitate evaluation of the air stabilities of various sulfide SEs and screening of potential air-stable candidates. Liu et al. [57] first calculated the energy change to demonstrate the improved air stability of ZnO-doped β-Li3PS4. Recently, Zhu et al. [69] performed thermodynamic analysis to systematically investigate the moisture stability of lithium ternary sulfides Li-M-S with different central cations M. As shown in Fig. 3d, the overall stability trend with respect to the central cations M is consistent with the empirical HSAB theory and previous experiments. More specifically, sulfide SEs with central cations Sn4+, Sb5+ and As5+ showed significantly better moisture stability than those containing B3+, Al3+, Si4+ and P5+.
3.4 Kinetics of Interfacial Reactions
Chemical reactions between sulfide SEs and air or moisture first occur at their interfaces, so it is necessary to study the kinetics of these interfacial reactions. Kim et al. [76] defined the p-band centre of the S-ion \(\left( {\Delta E_{\text{p}} } \right)\) and segregation energy of the S vacancy \(\left( {E_{{{\text{VS}},\;\mathop {{\text{seg}}}\limits^{\cdot\cdot} }} } \right)\) as electronic and structural descriptors with which to evaluate the atmospheric instability of β-Li3PS4 based on density functional theory. Among the four exposed surfaces (Fig. 3e), i.e., (111), (101), (001) and (100), the (110) and (111) surfaces were subsequently identified to be highly unstable due to their positive \({\Delta E_{\text{p}} }\) and negative \({E_{{{\text{VS}},\;\mathop {{\text{seg}}}\limits^{\cdot\cdot} }} }\), which were observed to present a linear relationship, as shown in Fig. 3f. Thus, formation and growth of surfaces with high surface reactivities should be suppressed during syntheses of sulfide SEs by controlling the exposed crystalline planes. However, most of the currently synthesized sulfide SEs are polycrystalline powders with randomly exposed crystalline planes that deviate from equilibrium crystal shapes following the Gibbs-Wulff theorem. Despite the successful growth of single crystals of LGPS [77], further development of a method for controlling exposed crystalline planes with high chemical stabilities and thus improving the air stabilities of sulfide SEs is required.
Subsequently, Kim et al. [78] revealed that H2O adsorption-dissociation reactions of surface Li ions constituted the initial stage for deteriorative hydrolysis reactions of sulfide SEs, as indicated by AIMD simulation results for the Li7P3S11 (100) surface. Then, they found and verified a correlation between atmospheric and electrochemical deterioration based on XPS results for air-exposed and delithiated Li7P3S11 samples. More specifically, as shown in Fig. 3g, Li-ion loss from the particle surface during delithiation caused by electrochemical charging and formation of lithium hydrates during air exposure promoted polymerizations of anions from PS43− or P2S74− to large PaSb(5a−2b) anionic clusters, which corresponded to transformations from P–S bonds to P–Sn–P bonds. Unfortunately, the polymerized anionic structures lowered the energy barriers for H2S generation and facilitated hydrolysis reactions instead of stimulating passivation.
Recently, Xu et al. [79] investigated the influence of crystallinity on air stability experimentally. They prepared two representative samples with the same Li9P3S9O3 compositions and LGPS-type structures via mechanochemical and melt quenching methods (denoted as M-Li9P3S9O3 and Q-Li9P3S9O3, respectively). Q-Li9P3S9O3 exhibited higher crystallinity, even at the particle surface, than M-Li9P3S9O3, which was confirmed by both XRD patterns and TEM images. In this case, these two samples were first exposed to air as powders and then subjected to temperature-programmed desorption-mass spectrometry (TPD-MS) to quantify the amounts of moisture and H2S gas adsorbed by the powder or to measure the ionic conductivity retained in the pellet state. Q-Li9P3S9O3 exhibited a much higher ionic conduction retention rate (> 70%) than M-Li9P3S9O3 (19%). In addition, the exposed Q-Li9P3S9O3 released H2O with a relatively lower evolution rate (1.8 ppm s−1 at 358 K, 1 ppm = 1 μmol mol−1) and generated minor amounts of H2O and H2S gas, whereas M-Li9P3S9O3 emitted H2O at a faster rate (45 ppm s−1) and generated large amounts of H2O and H2S gas. Given that the chemical compositions and LGPS-type structures were the same and there was only a small difference in BET specific surface areas for Q-Li9P3S9O3 (0.98 m2 g−1) and M-Li9P3S9O3 (0.64 m2 g−1), Xu et al. speculated that the highly crystalline particle surfaces of Q-Li9P3S9O3 provided a limited number of defect sites for H2O molecules to adsorb on, thus slowing propagation of the hydrolysis reaction from the surface to bulk regions and consequently generating minor amounts of H2S gas.
In summary, research involving the random network theory of glass and kinetics of sulfide-air interfacial reactions is limited to glassy materials and specific crystalline planes, respectively. Nevertheless, recent studies starting from the perspective of interfacial reaction kinetics still generate some interesting discoveries and deepen our fundamental understanding of the mechanisms for hydrolysis reactions. The HSAB theory based on binding of electrons to chemical species contained in sulfide SEs and thermodynamic analyses of the energy changes for chemical reactions are more widely used.
4 Characterization of Air Stability
Air stability reflects the chemical stabilities of SE materials in an air environment. Air-stable SEs will not react with any air components, such as nitrogen, oxygen, carbon dioxide and moisture, and maintain their physicochemical properties. Therefore, characterizations of the air stabilities of sulfide SEs can be based on macroscopic chemical reaction phenomena (i.e., amount of H2S gas generated [36, 80], morphology [59, 66] and mass [65] changes with exposure time), microscopic chemical components and structures, and electrochemical properties and performance before and after exposure to air, which are summarized in Fig. 4. The amount of H2S gas generated is calculated from the concentration detected by a H2S sensor after exposing sulfide SE samples to a controlled atmosphere. Morphological changes can be captured with optical photos or scanning electron microscopy (SEM) images. Furthermore, mass changes with exposure time can be recorded with thermogravimetric analysis (TGA) apparatus [65]. Characterization of the chemical components and structures of sulfide samples before and after exposure to air is beneficial for identifying the air stabilities of various sulfide SEs and understanding the mechanisms of structural degradation and chemical reactions. In addition to the common characterization methods universally adopted for materials research (e.g., X-ray diffraction (XRD), Raman and X-ray photoelectron spectroscopy (XPS)), advanced characterization methods, such as solid-state magic-angle-spinning nuclear magnetic resonance (MAS-NMR) and synchrotron radiation source (e.g., X-ray absorption near-edge spectra (XANES) and extended X-ray absorption fine structure (EXAFS)), have been applied to determining air stabilities for sulfide SEs. Finally, analyses of the electrochemical properties of sulfide SEs and electrochemical performance of sulfide ASSBs with air-exposed sulfide SEs contribute to the establishment of structure-performance relationships.

Methods for characterizing the air stabilities of sulfide SEs
4.1 Macroscopic Chemical Reaction Phenomena
Sulfide SEs show hygroscopic properties and generation of toxic H2S gas when exposed to humid air, leading to evolutions in morphology and mass changes.
4.1.1 Amount of H2S Gas Generated
The amount of H2S gas generated has generally been regarded as a critical index for evaluating air stability and the possibility of practical applications of sulfide SEs. As shown in Fig. 5a, the conventional method for measuring the amount of H2S gas generated is to place a sulfide sample into a closed desiccator/container with a certain volume, expose the sulfide sample to a precontrolled atmosphere with temperature and relative humidity (RH) within certain ranges and record H2S gas concentrations in real time with a gas sensor [81, 82]. The total amount of H2S gas generated (V) can be calculated with Eq. (1) [80]:
where V denotes the total amount of H2S generated (cm3 g−1) normalized by the weight m of the sulfide electrolyte sample (g), C denotes the value recorded for the H2S concentration (ppm), and L is the volume of the desiccator (cm3).

Measurement of H2S gas amounts. a Conventional detecting system. Reprinted with permission from Ref. [66]. Copyright © 2019, American Chemical Society. b, c Improved detecting systems. Reprinted with permission from Ref. [83]. Copyright © 2021, American Chemical Society. d Our designed detecting system. Reprinted with permission from Ref. [84]. Copyright © 2021, Wiley-VCH
However, accurate measurement of the H2S gas amount generated still encounters three major challenges, including position-related and time-delayed detection of the H2S gas concentrations and fluctuations of RH as the hydrolysis reaction occurs. Due to point detection by the gas sensor and the heterogeneous distribution of H2S gas in the whole space of the desiccator, the detected concentration is position-related. Specifically, the large density and slow diffusion rate of H2S gas hinder uniform diffusion and instant detection, which leads to accumulation of H2S gas around the sulfide sample as the hydrolysis reaction occurs at the interface between the sulfide SE and air/moisture. In other words, the closer the gas sensor is to the sulfide sample, the higher the concentration of H2S gas detected. Therefore, a small electric fan was additionally placed in the desiccator to circulate the atmosphere (Fig. 5b and c) and create an approximately uniform distribution of H2S gas [83], which made the detected value of H2S concentration more representative. However, the possibility of time-delayed detection still exists because instantly generated H2S gas circulates with air flow until it is detected by the gas sensor. Moreover, the fluctuations of RH during the continuous hydrolysis reaction probably influence the rate for generation of H2S gas. Subsequently, Kimura et al. [61] overcame this problem by exposing sulfide samples to flowing air with a constant RH and flow rate. Inspired by this idea, a pump suction-type gas sensor was used instead of a point detection sensor based on gas diffusion, and the whole setup was connected to a pipeline to achieve unidirectionally flowing gas, instant detection of H2S gas and a constant RH for the exposed atmosphere [84], as shown in Fig. 5d. Therefore, this design overcame the aforementioned challenges for accurate measurements of H2S gas. The total amount of H2S generated was calculated with Eq. (2).
where \(A\) denotes the total/accumulated amount of H2S generated normalized by the weight \(\left( M \right)\) of S atoms in the sulfide electrolyte sample, \(C_{{{N}}}\) denotes the \(N\)th recorded value for the H2S concentration, \(v\) is the air-flow velocity and \(\Delta t\) is the time interval of recording.
In this review, reported data obtained from air stability tests of various sulfide SEs are summarized in Table 1. The improved air stabilities of modified sulfide SEs can be confirmed by the lower amounts of H2S gas generated and minor structural degradations. Two obvious conclusions can be drawn from the data in Table 1. The larger the exposed surface area between the sulfide sample and the atmosphere is, the intenser the hydrolysis reaction is, and the larger the amount of H2S gas generated. The higher the RH used for exposure of the sulfide sample is, the more H2S gas generated. However, it is difficult to quantitively identify air stability differences among various sulfide SEs based on data collected from numerous reports due to the differences in detecting systems, exposure conditions (e.g., volume of the airtight container, temperature, relative humidity and exposure time, specific surface area, morphology and crystallinity of sulfide samples) and evaluation methods. Therefore, it is necessary to establish a unified standard for measuring the amount of H2S gas generated to achieve comparable data from different labs.
4.1.2 Changes in Morphology and Mass
In addition to generation of H2S gas, morphology and mass changes also occur during hydrolysis reactions. Morphological changes in sulfide SE powders, pellets and membranes before and after exposure to humid air were investigated. Recently, Lu et al. [84] used optical imaging to record the morphological changes of Li9.54Si1.74P1.44S11.7Cl0.3 (LSPSC), Li3PS4 (LPS), LSS and Li3.875Sn0.875As0.125S4 (LSAS) powders upon exposure to a humid atmosphere (100% RH). While LSPSC and LPS suffered from large volume changes and obvious colour changes, LSS and LSAS only absorbed H2O molecules and turned into transparent solutions without hydrolysis reactions, as shown in Fig. 6a. Tufail et al. [85] performed ex situ SEM characterizations for particles of the glass-ceramic Li7P3S11 and the Li6.95Zr0.05P2.9S10.8O0.1I0.4 electrolyte. The morphology of Li7P3S11 became porous when it was exposed to humid air, while the Li6.95Zr0.05P2.9S10.8O0.1I0.4 electrolyte with enhanced air stability retained its morphology and showed no discernible variation. Tufail et al. further investigated ex situ SEM images of the cross-sections and surfaces of Li7P3S11 and Li7Sb0.05P2.95S10.5I0.5 pellets. While the morphologies of the Li7Sb0.05P2.95S10.5I0.5 pellets were similar before and after exposure to moist air with a 40%–47% humidity for 20 min, the morphology of pristine Li7P3S11 showed more cracks (Fig. 6b). Optical images captured by Li et al. [86] indicated that undoped Li7P3S11 reacted with H2O immediately, while Li7Sn0.1P2.8S10.5O0.2 remained stable for 60 min. Although Na3SbS4 is stable in dry air, the surface morphology of the Na3SbS4 pellet became rough and uneven with pulverization due to hydration in humid air, in contrast to the smooth and flat surfaces of the bare sample [87]. Tan et al. [66] recorded the morphology changes of the pristine Li7P3S11 film and the SEBS-Li7P3S11 composite SE film after flooding in water. The pristine Li7P3S11 film was completely hydrolyzed and disappeared once it came into contact with water, while the SEBS-Li7P3S11 film retained its original shape despite being immersed in water. Xu et al. [88] designed a protective layer consisting of fluorinated polysiloxane (F-POS)-coated Li1.4Al0.4Ti1.6(PO4)3 (LATP) nanoparticles on the surface of a Li6PS5Cl membrane. Even during continuous dripping of water, the Li6PS5Cl membrane with the superhydrophobic layer repelled the water droplets and maintained its original morphology, whereas the bare Li6PS5Cl membrane presented a colour change and a violent reaction, as shown in Fig. 6c.

Morphological evolution of sulfide SEs. a LSPS, LPS, LSS and LSAS particles. Reprinted with permission from Ref. [84]. Copyright © 2021, Wiley-VCH. b Li7P3S11 and Li7Sb0.05P2.95S10.5I0.5 pellets. Reprinted with permission from Ref. [85]. Copyright © 2021, Elsevier. c Spray-coated and bare Li6PS5Cl membranes. Reprinted with permission from Ref. [88]. Copyright © 2021, Wiley-VCH
Zhao et al. [65] investigated the reactivities of Li6PS5I (LPSI) and Sn-substituted LPSI-20Sn with O2 by exposing these two SEs to a pure oxygen atmosphere and monitored the mass changes with exposure times in a TGA apparatus. The mass of LPSI increased by 1.12% after exposure to pure oxygen for 10 h, while that of LPSI-20Sn increased by 0.35% after exposure to the same atmosphere for 20 h.
4.2 Microscopic Components and Structures
The structural changes and stabilities of Li2S-based sulfide SEs exposed to air were first studied by Muramatsu et al. [36]. They found that the air stabilities of L2S-P2S5 glass and glass-ceramic SEs were related to their local structures, which changed with changing molar proportions of Li2S. A series of SEs with different molar proportions of Li2S (i.e., 67%, 70%, 75%, 80% and 100%) were pressed into pellets and then exposed to humid air in the desiccator to determine their air stabilities from the amount of H2S generated. As shown in Fig. 7a, the amount of H2S generated first decreased, reached a minimum at a Li2S molar proportion of 75% and then increased. Raman spectra (Fig. 7b and c) of 67L2S-33P2S5 glass and Li2S crystals before and after exposure for 4 min, 90 min and one day showed evolution of local structures or chemical groups according to exposure time. The P2S74− peak at 407 cm−1 gradually weakened and disappeared with longer exposure, and this was accompanied by the emergence of new peaks at 2 560 and 3 300 cm−1, which corresponded to S–H and O–H groups, respectively. Similarly, Li–S groups were indicated by a peak 376 cm−1, and this gradually weakened and disappeared as new peaks emerged at 2 560, 3 300 and 3 600 cm−1; these corresponded to S–H, O–H and Li–O–H groups, respectively. Based on the aforementioned changes in local structures, they speculated that both the P2S74− group in 67Li2S-33P2S5 glass and the Li–S group in crystalline Li2S underwent two-step hydrolysis reactions (Fig. 7f and g) and ended up generating H2S gas after breaking of bridging sulfur bonds. In contrast, Raman spectra for the 75Li2S-25P2S5 glass and glass-ceramic electrolytes before and after exposure to air for one day showed no additional peaks except for those corresponding to the PS43− group (Fig. 7d and e). Furthermore, the ionic conductivity of the 75Li2S-25P2S5 glass-ceramic pellet decreased slightly from 1.9 × 10−4 S cm−1 to 1.5 × 10−4 S cm−1 after exposure to air for 7 h (Fig. 7h). Therefore, 75Li2S-25P2S5 glass and glass-ceramic electrolytes with local structures based on PS43− groups exhibited the highest air stabilities among Li2S-P2S5 sulfide SEs, which was corroborated by the amounts of H2S generated, local structures and ionic conductivities. In addition, X-ray diffraction (XRD) is generally used to identify changes in the overall crystalline structures of various sulfide SEs after exposure to air and generation of hydrolysis reaction products. Moreover, the local structures and chemical components of sulfide SEs before and after exposure to air have been characterized by MAS-NMR, XANES, EXAFS and XPS, which will be described in subsequent sections.

Evolution of the microscopic components and structures. Humid-air exposure of pelletized Li2S-P2S5 glasses with different Li2S contents. a Amount of H2S gas generated. Raman spectra for b 67%Li2S-33%P2S5 glass and c Li2S crystal before and after exposure to humid air for 4 min, 90 min and 1 day. Raman spectra of 75%Li2S-25%P2S5 glass and glass-ceramic d before and e after exposure to humid air. Structural changes of f P2S74− in 67%Li2S-33%P2S5 glass and g crystalline Li2S in humid air. h Ionic conductivity of pelletized 75%Li2S-25%P2S5 glass-ceramic as a function of exposure time. Reprinted with permission from Ref. [36]. Copyright © 2010, Elsevier
4.3 Electrochemical Properties and Performance
The air instabilities of sulfide SEs lead to structural damage and side product formation, which greatly degrades the electrochemical properties of sulfide SEs/ASSBs. At the material level, the ionic conductivities of sulfide SEs after exposure to humid air exhibit different degrees of diminution according to their different air stabilities. After exposure to humid air, the ionic conductivities of air-instable sulfide SEs (e.g., Li3PS4, Li6PS5Cl and LGPS) decrease dramatically due to hydrolysis reactions and undesired products. For air-sensitive sulfide SEs (e.g., due to oxygen or soft-acid substitution) that exhibit stability toward water, the ionic conductivity also decreased due to slow but irreversible hydrolysis reactions. In contrast, the ionic conductivities of air-stable sulfide SEs (e.g., LSS, Li3SbS4) were only reduced slightly due to absorption of water, and they can be recovered by removing the water. It is worth noting that some moisture-exposed materials, such as Li4Cu8Ge3S12 [62] and Li2Sn2S5[89], even exhibited elevated ionic conductivity, which may have originated from proton conduction or acceleration induced by coupling of Li+ and water molecules, which will be discussed in subsequent sections. However, the electronic conductivities and electrochemical stabilities of air-exposed SEs have not been studied deeply.
The electrochemical performance of a full cell is expected to worsen due to reduced ionic conductivity and undesired products. Cho et al. [90] incorporated zeolite into Li6PS5Cl as a scavenger for H2O and toxic H2S gas and to mitigate the hydrolysis reaction of Li6PS5Cl. As shown in Fig. 8a, an ASSB with zeolite-incorporated Li6PS5Cl (Z-Li6PS5Cl) showed a much lower overpotential than pristine Li6PS5Cl (P-Li6PS5Cl). While the ASSB with P-Li6PS5Cl exhibited a low discharge capacity and a rapid loss of capacity, the ASSB with Z-Li6PS5Cl showed a much higher discharge capacity and superior cyclability (Fig. 8b). Park et al. [52] assembled ASSBs from a Li3PS4/LiCoO2 mixture and a 0.4LiI-0.6Li4SnS4-coated LiCoO2 cathode before and after exposure to dry air for 24 h, respectively. While the Li3PS4/LiCoO2 mixture cathode exhibited a significantly increased overpotential and decreased capacity after exposure to dry air (Fig. 8c), the capacity of the 0.4LiI-0.6Li4SnS4-coated LiCoO2 cathode was slightly reduced after exposure to dry air, which indicated potential compatibility with practical applications. Liang et al. [50] reported that the reversible capacity (Fig. 8d) of an ASSB constructed with air-exposed Li10Ge(P1−xSbx)2S12 at 0.1 C was only slightly reduced compared with that containing fresh Li10Ge(P1−xSbx)2S12, which originated from the enhanced air stability caused by Sb substitution. Interestingly, Ye et al. [91] found that an ASSB with Li9.54Si1.74(P1−xSbx)1.44S11.7Cl0.3 (x = 9.7%) showed a slightly improved specific capacity of 182.4 mAh g−1 after one hour of air exposure and better long-term cycling stability than even pristine Li9.54Si1.74P1.44S11.7Cl0.3 (Fig. 8e and f). The mechanisms behind these phenomena are still unknown. Lu et al. [84] reported that ASSBs with air-exposed and heat-treated Li3.875Sn0.875As0.125S4 (LSAS) exhibited superior performance for the heat-treated sample, as indicated by a high discharge capacity and long-term cycling stability, whereas that with the air-exposed sample showed merely negligible discharge capacity (Fig. 8g and h). The enhanced performance may be attributed to reversible water adsorption/desorption by LSAS and favorable interfacial products generated from trace amounts of water. Xu et al. [88] designed a superhydrophobic layer for the Li6PS5Cl membrane without sacrificing the electrochemical properties even after water exposure. While the ASSB with the bare Li6PS5Cl membrane exhibited no capacity (Fig. 8i), the ASSB with the Li6PS5Cl membrane delivered a discharge capacity comparable to that seen with an unexposed membrane (Fig. 8j). Li et al. [67] prepared an air-stable membrane comprising a Li7PS6-embedded composite electrolyte in an ambient environment. The ASSB with this air-exposed membrane exhibited a high rate capacity (Fig. 8k) because the poly(vinylidenefluoride-co-hexafluoropropylene) (PVDF-HFP) polymer matrix protected the Li7PS6 sulfide SE from air. Therefore, it is crucial to investigate the interfacial stabilities of sulfide SEs with electrodes, including identification of reaction products and degradation and stabilization mechanisms for ASSBs with pristine and air-exposed sulfide SEs. Tian et al. [87] found that a Na3SbS4⋅8H2O hydrate coating layer was beneficial for interfacial stability with Na metal, which was formed in situ on the surface of the Na3SbS4 SE after exposure to humid air (68% RH) for 10 min. Due to formation of passivating products (i.e., NaH and Na2O) and the reduced fraction of the electronically conducting phase (i.e., Na3Sb) at the interface, decomposition of Na3SbS4 SEs and impedance growth upon cycling were limited. This reactivity-guided interface design ingeniously takes advantage of the instabilities of sulfide SEs toward humid air and constructs an effective passivation or protection layer, which will inspire further studies on the relationships between the air stabilities of sulfide SEs and the electrochemical performance of sulfide ASSBs.

Electrochemical performance of ASSBs with air-exposed sulfide SEs. a Charge-discharge profiles of NMC811/SE/Li-In ASSBs with P-Li6PS5Cl and Z-Li6PS5Cl before (the solid line) and after exposure (the dotted line). b Cycling performance of ASSBs with air-exposed P-Li6PS5Cl and Z-Li6PS5Cl. Reprinted with permission from Ref. [90]. Copyright © 2020, The Electrochemical Society. c Voltage profiles for LiCoO2/LPS/Li-In cells in which the LiCoO2 composite electrodes are the LPS/LiCoO2 mixed electrodes or 0.4LiI-0.6Li4SnS4-coated LiCoO2 electrodes before and after exposure of LPS powders and the 0.4LiI-0.6Li4SnS4-coated LiCoO2 powders to dry air for 24 h. Reprinted with permission from Ref. [52]. Copyright © 2015, Wiley-VCH. d Charge-discharge curves of In/Li10Ge(P0.925Sb0.075)2S12/LiCoO2 cells with Li10Ge(P0.925Sb0.075)2S12 before and after air exposure. Reprinted with permission from Ref. [50]. Copyright © 2020, American Chemical Society. Charge and discharge profiles at 0.5 C and 55 °C for ASSBs using the NMC811 cathode, the graphite-protected Li metal anode and Li9.54Si1.74(P1−xSbx)1.44S11.7Cl0.3 as the solid electrolyte before and after air exposure at e x = 9.7% and f x = 0. Reprinted with permission from Ref. [91]. Copyright © 2021, Elsevier. Charge and discharge profiles of g LCO@LNO/LSAS/LTO ASSB fabricated in humid air (70% RH, 28.9 °C) and h LCO@LNO/SE/LTO ASSB with air-exposed LSAS after heat treatment at 280 °C. Reprinted with permission from Ref. [84]. Copyright © 2021, Wiley-VCH. Charge-discharge profiles of LCO@LNO/SE/LTO ASSBs using i a bare Li6PS5Cl membrane after exposure to water and j an LATP@F-POS/Li6PS5Cl/LATP@F-POS membrane. 1 mL in−2 = 0.155 mL cm−2. Reprinted with permission from Ref. [88]. Copyright © 2021, Wiley-VCH. k Comparison of rate performance for cells with SCE and PVDF-HFP/LiTFSI polymer electrolytes. Reprinted with permission from Ref. [67]. Copyright © 2020, American Chemical Society
In summary, various characterization methods have been developed to investigate the air stabilities of sulfide SEs from different perspectives, including macroscopic chemical reaction phenomena, microscopic components/structures and electrochemical properties. Among these characterization methods, it is advisable that basic and universal methods, including measurements of H2S generation upon exposure, XRD of crystal structures, Raman spectra of local structures and measurements of ionic conductivity before and after exposure, should be performed to construct a unified system for evaluation of data from different labs. Advanced characterization methods, such as XAS, NMR and in situ TEM, can be alternative approaches providing further proof and deepening our understanding.
5 Strategies to Improve Air Stability
After decades of research, five promising strategies (Fig. 9) have been proposed to improve the air stabilities of sulfide SEs: (1) using additives to absorb H2S gas [48, 92], (2) partial substitution of S2− by oxygen anions [54, 56, 57] or the hard base P5+ by soft acids [50, 65, 93], (3) designing new materials with soft acids as the central cations [61, 94], (4) surface coating or passivation [68] and (5) construction of sulfide-polymer composite electrolytes [49, 66]. All of these strategies were shown to be effective in reducing the amount of H2S gas generated (Table 1). Among them, substitution with oxygen or soft acids and designing new materials are currently considered optimal solutions for improving the inherent air stabilities of sulfide SEs. The strategy of constructing composite SEs is promising for integrating the advantages of sulfide and polymer SEs [95], thus potentially satisfying all prerequisite properties of SEs and accelerating commercialization of ASSBs. However, as H2S absorbents, metal-oxide additives decrease the ionic conductivities of sulfide SEs and consume only H2S via chemical reactions instead of improving the inherent air stability. It is noteworthy that strategies tailored toward improving air stability generally induce changes in other critical properties (e.g., ionic conductivity and compatibility with Li metal) of sulfide SEs, as summarized in Table. 2.

Methods for improving the air stabilities of sulfide SEs
5.1 H2S Absorbents
Given that ZnO reacts with H2S gas to function as an absorbent, Hayashi et al. [48] speculated that the chemical reaction between metal oxide MxOy and H2S gas follows Eq. (3).
As the change in Gibbs free energy should be large and negative to absorb H2S gas effectively, three metal oxides (Fe2O3, ZnO and Bi2O3) were screened due to their large Gibbs free energy (∆G) (−43.9, −78.0 and −232.0 kJ mol−1, respectively). Fe2O3, ZnO and Bi2O3 nanoparticles were physically mixed with Li3PS4 glass powders by ball milling at 230 r min−1 for 2 h. As shown in Fig. 10a and b, both the ZnO and LPS glasses remained after ball milling, and 31P MAS-NMR spectra taken before and after ZnO addition did not differ, suggesting that there was no chemical interaction between ZnO and LPS glass. Fe2O3, ZnO and Bi2O3 in composite electrolytes absorb H2S gas effectively. Among them, Bi2O3 showed the optimal absorption effect because it has the most negative ∆G, and almost no H2S gas was detected by the gas sensor (Fig. 10c). Furthermore, formation of ZnS (Fig. 10d) after air exposure of composite electrolytes with ZnO and LPS glass indicated that the reaction between ZnO and H2S followed Eq. (3). Unfortunately, the ionic conductivity decreased monotonically with increasing mole percentage of ZnO, as shown in Fig. 10e. Therefore, it is necessary to find the optimal mole percentage of ZnO to strike a balance between air stability and ionic conductivity.

Air stability enhanced by H2S absorbents. a XRD pattern for the composite consisting of 90 mol% Li3PS4 glass and 10 mol% ZnO (mol% means the molar fraction). b 31P MAS-NMR spectra of the composite and Li3PS4 glass. c Amount of H2S generated as a function of exposure time to air. d XRD patterns of the 90Li3PS4·10ZnO composite and Li3PS4 glass after exposure to air for one day. e Ionic conductivity at 25 °C for (100 − x)Li3PS4·xZnO (mol%) composite electrolytes as a function of ZnO content. Reprinted with permission from Ref. [48]. Copyright © 2013, Royal Society of Chemistry. f XRD patterns for the 70(0.75Li2S·0.25P2S5)·30MxOy (MxOy: Li2O, MgO, CaO and CuO) composites, the 75Li2S·25P2S5 glass, and the 70(0.75Li2S·0.25P2S5)·30FeS composite. g Exposure time dependence of H2S gas amount generated from the 70(0.75Li2S·0.25P2S5)·30MxOy (MxOy: Li2O, MgO, CaO and CuO) composites, the 75Li2S·25P2S5 glass, and the 70(0.75Li2S·0.25P2S5)·30FeS composite. Reprinted with permission from Ref. [92]. Copyright © 2013, Springer Nature. h Schematic illustration of the composite electrode prepared with the zeolite-incorporated Li6PS5Cl SE. i Quantities of H2S gas produced by P-Li6PS5Cl and Z-Li6PS5Cl when exposed to humid air. Reprinted with permission from Ref. [98]. Copyright © 2021, Royal Society of Chemistry
Subsequently, Hayashi et al. [92] added FeS and basic metal oxides (e.g., Li2O, MgO, CaO and CuO) into 75Li2S·25P2S5 glass to catalyze the decomposition of H2S and react with acidic H2S gas. It was obvious from the XRD patterns that the additives and 75Li2S·25P2S5 glass existed independently without any chemical interactions (Fig. 10f). As shown in Fig. 10g, these additives suppressed H2S gas generation, and the inhibition effects decreased in the order FeS > CuO > CaO > Li2O > MgO. However, the ionic conductivities of these composite electrolytes decreased due to addition of nonionic conductors. Fortunately, FeS showed optimal inhibition of H2S gas generation and induced minimal loss of ionic conductivity.
Apart from some specific metal oxides that react with H2S or even H2O, porous materials, such as zeolites, absorb H2O molecules and target harmful gases [96, 97] due to their three-dimensional porous structures. Lee et al. [98] first incorporated calcined ZSM-5 zeolite into Li6PS5Cl (Z-Li6PS5Cl) powder to scavenge H2S gas and H2O molecules surrounded by Li6PS5Cl particles (Fig. 10h), thus mitigating hydrolysis reactions and associated irreversible degradation of structure and performance. While the concentration of H2S for pristine Li6PS5Cl underwent a dramatic increase and reached ~ 120 ppm after exposure to humid air (50% RH) for 1 h, that of Z-Li6PS5Cl first rose slowly and then dropped from ~ 50 ppm to 40 ppm, as shown in Fig. 10i. This indicated that H2O molecules and H2S gas were effectively adsorbed into the pores of zeolites, which resulted in a low H2S generation rate and a subsequent decrease in the H2S concentration. However, introduction of lithium-ion-insulated zeolites will inevitably lower the ionic conductivity.
In conclusion, introduction of ion-insulated H2S absorbents by physical mixing with sulfide SEs inevitably impedes ion conduction. Using additives to absorb H2S or catalyze its decomposition suppresses H2S gas generation but does not enhance air stability, since separation of S atoms from sulfide SEs is always accompanied by structural degradation/collapse. Given that sulfide SEs exhibit inherent hydroscopic properties and a tendency to hydrolyze, only modification methods tailored to overcome these two challenges will improve the air stabilities of sulfide SEs.
5.2 Element Substitution
Element substitution processes can be classified into three types. The first is oxygen substitution, which potentially combines the high chemical stabilities of oxide SEs and the high ionic conductivities of sulfide SEs by forming oxysulfides. Soft acid substitution based on HSAB theory, which generates robust M–S bonds to resist attack by O2 and H2O, is the second type. Other element substitutions with complicated mechanisms belong to the last type.
5.2.1 Oxygen Substitution
Ohtomo et al. [54] synthesized xLi2O·(75 − x)Li2S·25P2S5 by replacing Li2S with Li2O with certain proportions x. As shown in Fig. 11a, the structure of 75Li2S·25P2S5 glass was retained from x = 4% to x = 17% without Li2O diffraction peaks, which did not indicate physical mixing of oxide additives. When x = 4%, the amount of H2S gas generated began to decrease significantly (Fig. 11b). When x \(\geqslant\) 7%, the generated H2S gas concentration reached the lower limit of the gas sensor. Ohtomo et al. speculated that introduction of Li2O reduced the residual amount of Li2S in 75Li2S·25P2S5 glass, thus suppressing H2S gas generation and improving air stability. Unfortunately, the ionic conductivity of 75Li2S·25P2S5 glass decreased monotonically with increases in the proportion x for Li2O (Fig. 11c). Therefore, x = 7% was regarded as the optimal proportion allowing xLi2O·(75 − x)Li2S·25P2S5 to exhibit both decent air stability and ionic conductivity. Recently, Ren et al. [99] obtained a (70 − x)Li2S·30P2S5·xLi2O glass ceramic by replacing Li2S with different amounts of Li2O. While the highest ionic conductivity of 1.2 × 10−3 S cm−1 was achieved with x = 1%, the best moisture stability and a compromised ionic conductivity of 9.9 × 10−4 S cm−1 were displayed for x = 5%.

Air stability enhanced by oxygen substitution. a XRD patterns for xLi2O·(75 − x)Li2S·25P2S5 (x = 4, 7, 11, and 17) samples prepared by two-step mechanical milling. b Amounts of H2S gas generated from xLi2O·(75 − x)Li2S·25P2S5 (x = 0, 4, 7, 11, and 17) glass powders. c Ionic conductivities of pelletized xLi2O·(75 − x)Li2S·25P2S5 (x = 0, 4, 7, 11, and 17) glass powders. Reprinted with permission from Ref. [151]. Copyright © 2013, Springer Nature. d 31P MAS NMR spectra of 75Li2S·(25−x)P2S5·xP2O5 (x = 0 and 10) glasses. e Amounts of H2S gas generated from pelletized 75Li2S·(25−x)P2S5·xP2O5 (x = 0 and 10) glasses after exposure to air. f Composition dependence of conductivity at 25 °C and activation energy (Ea) for conduction by pelletized 75Li2S·(25−x)P2S5·xP2O5 glasses. Reprinted with permission from Ref. [55]. Copyright © 2013, Elsevier. g XRD patterns for Li3+3xP1−xZnxS4−xOx (x = 0, 0.02, 0.06) and 0.98Li3PS4·0.02ZnO. h Amount of H2S generated from Li3.06P0.98Zn0.02S3.98O0.02 after exposure to humid air for different durations. i Arrhenius plots for Li3+3xP1−xZnxS4−xOx (x = 0, 0.01, 0.02, 0.03, 0.04, 0.05, 0.06). Reprinted with permission from Ref. [57]. Copyright © 2018, Elsevier
By following the same strategy for oxygen substitution, Hayashi et al. [55] synthesized 75Li2S·(25 − x)P2S5·xP2O5 by replacing P2S5 with P2O5 with a certain proportion x. 31P NMR spectra (Fig. 11d) indicated that the coordination environment of P was changed, and a series of oxysulfide units (e.g., PS3O, PS2O2, PSO3, PO4) were formed by oxygen doping with P2O5; this was significantly different from simple generation of POS3 by oxygen doping with Li2O. Although the total amount of H2S generated (Fig. 11e) for 75Li2S·(25 − x)P2S5·xP2O5 with x = 10 was almost identical to that of pristine 75Li2S·25P2S5 glass, emergence of the maximum concentration of H2S was delayed. Therefore, oxygen doping with P2O5 significantly suppressed the generation rate rather than the total amount of H2S generated. Unfortunately, the ionic conductivity of 75Li2S·(25 − x)P2S5·xP2O5 glass (Fig. 11f) also decreased monotonously with increasing P2O5 content in the range 0 \(\leqslant\) x \(\leqslant\) 10, for which nonbridging oxygens in the newly formed oxysulfide units served as strong traps for lithium-ion conduction. However, Xu et al. [100] reported that the ionic conductivity of 75Li2S·(25 − x)P2S5·xP2O5 with low-concentration doping (x = 1) reached a maximum value of 8 × 10−4 S cm−1. By considering the experimental results reported by Hayashi et al., they concluded that nonbridging sulfur atoms were replaced directly by bridging oxygen atoms that served as weak traps for lithium ions when the substitution proportion x = 1, which led to the increased ionic conductivity. However, a larger proportion for substitution of sulfur atoms by oxygen atoms resulted in formation of a series of oxysulfide units with nonbridging oxygen atoms serving as strong traps for lithium-ion conduction, which led to reduced ionic conductivity.
Furthermore, Zhang et al. [56] found that O-doped Li6PS5−xOxBr (0 \(\leqslant\) x \(\leqslant\) 1) exhibited comprehensively but not significantly enhanced properties, including good air stability, excellent dendrite suppression capability and superior electrochemical/chemical stability against Li metal and high-voltage oxide cathodes. In contrast to other O-incorporated sulfides, the O atoms were more inclined to substitute S atoms at free S2− sites instead of at PS43− tetrahedral sites, which was confirmed by the unchanged relative intensities and symmetries of Raman peaks with the increased O content. Although impurity phases appeared after exposing the samples to humid (35% RH) air for 10 min, the amounts reached a minimum at x = 0.3. However, due to the strong electrostatic attraction between O2− and Li+ and increases in the levels of impurities, the ionic conductivity decreased with increasing O content. Due to the enhanced shear modulus and oxide-containing interfacial layer (e.g., Li3OBr), O-doped Li6PS5−xOxBr achieved a high critical current density (CCD) close to 0.9 mA cm−2. Since CCD is defined as the lowest current density at which battery shorting occurs due to Li dendrite penetration [101, 102], the high CCD value reveals excellent dendrite suppression capability. Peng et al. [103] synthesized O-substituted Li5.5PS4.5−xOxCl1.5 (x = 0, 0.075, 0.175, 0.25) SEs by partially replacing the raw material P2S5 with P2O5. The air stability of Li5.5PS4.5−xOxCl1.5 determined from the total amount of H2S produced increased monotonically with increasing O content, whereas the ionic conductivity dropped monotonically with increasing O content. Ultimately, Li5.5PS4.425O0.075Cl1.5 with compromised properties was selected. Recently, Xu et al. [104] synthesized Li6.25PS4O1.25Cl0.75 exhibiting high stability toward moist air and an enhanced ionic conductivity of 2.8 mS cm−1 caused by partial oxygen substitution at both S and Cl sites. After exposure to humid air (53% RH), the argyrodite structure of Li5.5PS4.425O0.075Cl1.5 was well maintained, whereas the XRD patterns for Li6PS5Cl showed numerous unknown peaks. Air-exposed Li6.25PS4O1.25Cl0.75 after 180 °C postannealing still maintained an argyrodite structure with minor Li2S impurities, in stark contrast with the collapsed structure of Li6PS5Cl. In addition, the H2S sensing response curve for Li6.25PS4O1.25Cl0.75 was much weaker than that for undoped Li6PS5Cl. Xu et al. [79] performed oxygen substitution on Li9P3S12 and obtained Li9P3S9O3 with an LGPS-type structure by a melt quenching method. Li9P3S9O3 showed ionic conductivity retention as high as 70%, which was much higher than that for undoped Li9P3S12 (10%), after exposure to dry air for 6 h, which confirmed the effectiveness of oxygen substitution for improving air stability. However, the highest ionic conductivity of > 1 × 10−3 S cm−1 was obtained for a low oxygen content. The opposite trends seen for variations of ionic conductivity and air stability with O substitution amount inevitably hinder the development of O-substituted sulfide SEs with optimal comprehensive properties.
In addition to single oxygen substitution for sulfur to enhance air stability, dual substitution of either Li+, P5+ or S2− sites has been performed recently to improve properties, including air stability, ionic conductivity and interfacial stability, toward Li metal. Liu et al. [57] selected ZnO as the dopant based on theoretical calculations and synthesized a Li3+3xP1−xZnxS4−xOx (0 \(\leqslant\) x \(\leqslant\) 0.06) glass-ceramic electrolyte by partially substituting P5+ and S2 with Zn2+ and O2−, respectively. Only diffraction peaks of β-Li3PS4 were observed in the XRD pattern (Fig. 11g) when x = 0.02, which confirmed successful dual doping of ZnO into the crystalline lattice instead of physical mixing. It was noted that the crystal structure of β-Li3PS4 was changed when x = 0.06, consistent with theoretical calculations showing that dissolution of ZnO into crystalline β-Li3PS4 was energetically unfavourable. Subsequently, the energy changes (∆Es) for hydrolyses of the Li3+3xP1−xZnxS4−xOx electrolytes with x = 0 and x = 0.02 were calculated. ∆E was found to increase from −912.15 to −882.3 J mol−1 after doping, indicating the improved air stability of the ZnO-doped electrolyte. The improved air stability was also supported by the low amount of H2S gas generation (0.017 5 cm3 g−1) after exposing Li3.06P0.98Zn0.02S3.98O0.02 to humid air for three hours (Fig. 11h). The ionic conductivity reached a highest value of 1.12 mS cm−1 when x = 0.02 (Fig. 11i). Theoretical calculations showed a synergistic effect in which the O atom tended to locate next to a Zn atom and enlarge the migration channel for Li+ when x = 0.021 (thus increasing the ionic conductivity), while doping with Zn alone hindered migration of Li+. No apparent changes were observed in a symmetric cell during galvanostatic charge-discharge testing at a current density of 0.5 mA cm−2, which indicated good interfacial stability for Li3.06P0.98Zn0.02S3.98O0.02 and Li metal. Chen et al. [58] also adopted the ZnO codoping strategy and realized comprehensively enhanced properties for Li6−2xZnxPS5−xOxBr (0 ≤ x ≤ 1.5). However, Li sites rather than P sites were substituted by Zn atoms in Li6PS5Br. Due to substitution of Zn atoms by Li, which increased the Li content, the high ionic conductivity was maintained despite the negative impacts of oxygen substitution of sulfur and impurity formation. After exposure to air with a humidity of ~ 10% for 10 min, an impurity phase comprising LiBr·H2O quickly appeared for Li6PS5Br, but no impurity phase was observed for Li6−2xZnxPS5−xOxBr (x = 0.15); this demonstrated the enhanced air stability of ZnO-codoped Li6PS5Br. Furthermore, when x = 0.15, the polarization voltages of symmetric cells were lower and more stable than that for cells with x = 0. This enhancement in interfacial stability and suppression of Li dendrites was attributable to formation of Li-Zn alloy and Li3OBr at the Li/Li6−2xZnxPS5−xOxBr interface, as well as to reduced electronic conductivity resulting from ZnO doping.
Ahmad et al. [59] developed a Nb and O codoping strategy to improve the chemical/electrochemical stability of glass-ceramic Li7P3S11. The ionic conductivity reached a maximum at x = 0.2, which was attributed to precipitation of a highly conductive crystalline phase with PS43− and P2S74− units. However, this was compromised when the dopant level was further increased (x ≥ 0.4) due to formation of a low-conductivity phase Li4P2S6. With increasing content of the LiNbO3 dopant, the amount and rate of H2S generation from glass-ceramic sulfide SEs decreased monotonically. In particular, a sharp decrease appeared when x = 0.2, at which point bridging sulfurs with poor air stability in the P2S74− units were substituted by oxygen to give P2OS64− units that were difficult to hydrolyze. Moreover, the symmetric cell with a Nb- and O-codoped Li7P3S11 SE displayed a lower overpotential and flat and stable stripping/plating behaviour compared with pristine Li7P3S11. Recently, Li et al. [86] designed a Sn and O cosubstitution strategy and obtained Li7Sn0.5xP3−xS11−2.5xOx (x = 0.2) with enhanced stability against moisture and Li metal. After exposure to humid air (40%–45% RH) for 250 s, the amount of H2S generated by Li7Sn0.1P2.8S10.5O0.2 was 6.7 times lower than that of pristine Li7P3S11. Moreover, the symmetric cell with Li7Sn0.1P2.8S10.5O0.2 exhibited a flatter and more stable Li plating/stripping curve and a higher CCD of 0.4 mA cm−2 than pristine Li7P3S11. Although Sb and O cosubstitution was first performed on β-Li3PS4 by Xie et al. [105] and resulted in a high ionic conductivity of 1.08 mS cm−1 and excellent stability against lithium even at a current density of 1 mA cm−2, an investigation of air stability was not performed. Fortunately, Zhao et al. [106] demonstrated the effectiveness of congener substitution of Sb and O for P and S in improving the air stability of Li7P3S11. Due to formation of the oxysulfide units POS33− and P2OS64−, Li7P3−xSbxS11−2.5xO2.5x (x = 0.1) showed an enhanced ionic conductivity of 1.61 mS cm−1 and air stability. The amount of H2S it generated after exposure to humid air was nearly 2.8 times lower than that of pristine Li3P3S11. Recently, Tufail et al. [85] even performed triple substitution of Li3P3S11 by ZrO2 and LiI dopants and obtained a Li6.95Zr0.05P2.9S10.8O0.1I0.4 (LZPSOI) SE with a high ionic conductivity of 3.01 mS cm−1. While the Zr dopant was regarded as the cause, introduction of a small amount of oxygen promoted the motion of Li+ and stability against moist air, based on previous reports. In addition, the introduction of larger and more polarizable I− anions enhanced the ionic conductivity, lithium-metal compatibility [107] and utilization of Li2S-based active materials. After exposure to humid air (41%–43% RH) for 50 min, the amount of H2S generated by LZPSOI was five times lower than that of pristine Li7P3S11. The enhanced air stability was attributed to formation of oxysulfide units (i.e., POS33− and P2OS64−). However, it is inevitable that numerous attempts will be required to achieve improvements in comprehensive properties with multisubstitution due to the variability of atom sites and contents upon substitution.
Therefore, oxygen substitution effectively improved the air stability of sulfide SEs due to changes in the coordination environment (i.e., oxygen atoms partially replaced sulfur atoms and were bound tightly with the hard acid P5+). However, to obtain both satisfactory ionic conductivity and high air stability, more effort is required to optimize oxygen substitution at a relatively low content level and within a narrow range to form favourable oxysulfide units, since a large oxygen substitution level dramatically reduces ionic conductivity. Fortunately, the codoping strategy with introduction of another cation results in enhanced properties of sulfide SEs and promotes practical application.
5.2.2 Soft Acid Substitution
Based on HSAB theory, Liang et al. [50] proposed that reducing the P content would improve the air stability of Li10GeP2S12 (LGPS), since the hard acid P5+ tends to react with the hard base O and form P to O bonds instead of maintaining only P–S bonds. Inspired by the presence of Na3SbS4·xH2O in the natural environment rather than decomposed products of Na3SbS4, they predicted that Sb-substituted LGPS would exhibit both improved air stability and ionic conductivity, because the large Sb5+ ion would broaden Li+ diffusion pathways. No broadening or additional diffraction peaks (Fig. 12a) appeared for Li10Ge(P0.925Sb0.075)2S12 after exposure to a dry-room environment with a relative humidity of 1%–3% for 24 h. In contrast, broadening and split diffraction peaks (Fig. 12b) as well as impurity phases emerged for undoped LGPS under the same conditions. Furthermore, the amount of H2S generated (Fig. 12c) decreased continuously with increasing Sb content (i.e., decreasing P content). The ionic conductivity (Fig. 12d) of undoped LGPS decreased by 54% from 10.9 to 5 mS cm−1 after exposure to dry air, while that of Sb-doped Li10Ge(P1−xSbx)2S12 only decreased by 5%–18%. Ye et al. [91] subsequently performed Sb substitution and obtained Li9.54Si1.74(P1−xSbx)1.44S11.7Cl0.3 (x = 0.097) and an increase in ionic conductivity from 5.4 to 8.8 mS cm−1. Although enhanced air stability was demonstrated by XRD patterns in which no obvious changes were observed, the ionic conductivity still dropped from 8.8 to 5.8 mS cm−1 because the exposure conditions (15% RH and 35 °C) were harsher than those of Sb-substituted LGPS. Tufail et al. [82] obtained a Li7Sb0.05P2.95S10.5I0.5 SE by dual substitution of Sb and I into Li7P3S11. The ionic conductivity of Li7Sb0.05P2.95S10.5I0.5 was increased from 1.40 to 2.55 mS cm−1 after doping. The amount of H2S generated (Fig. 12e) by Li7P3S11 was 1.32 cm3 g−1 (92 ppm) after exposure to humid air (40%–47% RH) for 35 min, while that of Li7Sb0.05P2.95S10.5I0.5 was only 0.37 cm3 g−1 (26 ppm). The intensities of the diffraction peaks for Li7Sb0.05P2.95S10.5I0.5 (Fig. 12f) remained unchanged after exposure, in contrast to the peak broadening and diminished intensity seen for Li7P3S11; this indicated improved air stability for Li7Sb0.05P2.95S10.5I0.5.

Air stability enhanced by Sb and Sn substitution. XRD patterns for a Li10Ge(P1−xSbx)2S12 (x = 7.5%) and b Li10GeP2S12 samples before and after air exposure. c Amounts of H2S gas generated from commercial Li10GeP2S12 and synthesized Li10Ge(P0.925Sb0.075)2S12 powders. d Room-temperature ionic conductivities of the Li10Ge(P1−xSbx)2S12 (0 ≤ x ≤ 15%) sample before and after air exposure. Reprinted with permission from Ref. [50]. Copyright © 2020, American Chemical Society. e Amounts of H2S gas produced from both Li7Sb0.05P2.95S10.5I0.5 and Li7P3S11 solid-state electrolyte samples when exposed to moist air. f XRD patterns for Li7Sb0.05P2.95S10.5I0.5 and Li7P3S11 before and after exposure to humid air. Reprinted with permission from Ref. [82]. Copyright © 2020, Elsevier. XRD patterns of g LPSI-20Sn and h LPSI electrolytes before and after exposure to air with 10% humidity, as well as after a postheating process. i XANES of the P K-edge in LPSI-20Sn after exposure to 10% humidity. j Arrhenius plots for the LPSI-20Sn electrolyte before and after exposure to air with 10% humidity, as well as after the postheating process. k Li plating/stripping polarization of the Li//LPSI-20Sn//Li symmetric cell tested under a high current density of 1.26 mA cm−2 and cut-off capacity of 1 mAh cm−2. Reprinted with permission from Ref. [65]. Copyright © 2020, Wiley-VCH. l Synchrotron-based XRD patterns for gc-Li3.2P0.8Sn0.2S4 SSEs before and after exposure to air with 5% humidity. Arrhenius plots for m gc-Li3.2P0.8Sn0.2S4 and n gc-Li3PS4 SSEs before and after exposure to air with 5% humidity. Reprinted with permission from Ref. [93]. Copyright © 2021, Wiley-VCH
Based on HSAB theory, Zhao et al. [65] used another soft acid, Sn4+, to partially replace the hard acid P5+ in Li6PS5I (LPSI) and synthesized Sn-doped LPSI (LPSI-20Sn) with superior air stability demonstrated by various characterization methods. The XRD spectra (Fig. 12g) and P K-edge X-ray absorption spectra (Fig. 12i) showed little difference for LPSI-20Sn before and after exposure to an atmosphere with 10% humidity for 12 h. However, LiI and other impurity phases formed after exposure of LPSI to humid air (Fig. 12h). Furthermore, the ionic conductivity (Fig. 12j) of LPSI-20Sn dropped slightly from 3.5 × 10−4 to 2.2 × 10−4 S cm−1 after exposure to humid air and recovered to 3.1 × 10−4 S cm−1 after a postheating process (180 °C in the vacuum oven). To reveal the mechanism of enhanced air stability for LPSI-20Sn SE, density functional theory (DFT) calculations of the oxygen replacement reaction energy (ΔE) were conducted. The higher ΔE of LPSI-20Sn indicated that the bonding energy for (P/Sn)–S in (P/Sn)S4 tetrahedra was much higher than that of P–S in PS4 tetrahedra after replacing S with O. Therefore, the crystalline structure of Sn-doped LPSI-20Sn was more stable against ambient air. In addition, a Li//LPSI-20Sn//Li symmetric cell demonstrated very stable Li plating and stripping behaviours (Fig. 12k) for ≈200 h (125 cycles) at RT, even with a high current density of 1.26 mA cm−2 and a cut-off capacity of 1 mAh cm−2. The LiI formed at the Li anode interface served as a vital component and created a uniform electron/ion distribution pathway and suppressed Li dendrite formation, thus resulting in good Li metal compatibility. Zhao et al. [93] also synthesized a Sn-substituted glass-ceramic Li3PS4 (gc-Li3.2P0.8Sn0.2S4) with high ionic conductivity, improved air stability and good Li-metal compatibility. gc-Li3.2P0.8Sn0.2S4 exhibited a 6.2-fold increase in ionic conductivity compared with pristine glass-ceramic Li3PS4 due to an enlarged lattice and higher Li+ ion concentration. Benefiting from the strength of the Sn–S bond, gc-Li3.2P0.8Sn0.2S4 showed excellent overnight air stability in humid air (5% RH), as corroborated by the unchanged crystal structure (Fig. 12l), negligible reduction in ionic conductivity (Fig. 12m) and unchanged P, S K-edge and Sn L3-edge XANES data. In contrast, a significant reduction in ionic conductivity resulted for gc-Li3PS4 exposed to humid air (Fig. 12n). Moreover, a symmetric cell with gc-Li3.2P0.8Sn0.2S4 demonstrated stable Li plating/stripping behaviour for over 600 h at a current density of 0.1 mA cm−2, and the lifetime was 4 times longer than that of pristine glass-ceramic Li3PS4. This is attributable to regulation of Li deposition at the Li/sulfide SSE interface by Li-Sn alloys. Rajagopal et al. [108] prepared a Sn-doped Li7P2S8I0.75Br0.25 solid electrolyte with an improved ionic conductivity of 7.78 mS cm−2 and high air stability. After exposure to humid air (5%–10% RH), the ionic conductivity retention for Sn-doped Li7P2S8I0.75Br0.25 was as high as 71%, and no structural changes or decomposition were observed.
Cu+ is a soft acid with an ionic radius (74 pm) similar [109] to that of Li+ (73 pm) and is rarely used for substitution of the hard-acid P5+ with a smaller ionic radius (31 pm). Recently, Taklu et al. [110] successfully obtained argyrodite Li6+3xP1−xCuxS5−xClx (x = 0.1, LPSC-1) via dual substitution of P5+ and S2− by Cu+ and Cl−, respectively. The highest ionic conductivity of 4.34 mS cm−2 was obtained at x = 0.1 and was attributed to synergetic effects of multiple factors, including an increased charge carrier density from extra Li+, enhanced anion disorder from added Cl−, and a smaller electronegativity difference between the Cu+ dopant and S2−. The relatively low amount of H2S generated (Fig. 13a) suggested the improved air stability of LPSC-1. The strong Cu–S bond resulting from incorporation of the soft acid Cu+ stabilized the localized structure of PS43−, which was demonstrated by the side product Cu3PS4 (Fig. 13b). Apart from the enhanced ionic conductivity, the lowest electronic conductivity and improved interfacial compatibility of LPSC-1 brought about by Li metal jointly contributed to superior suppression of dendrite formation with critical current densities as high as 3 mA cm−2 and stable Li plating and stripping processes for more than 200 h at the same current density. In contrast, pristine Li6PS5Cl (LPSC-P) with the highest electronic conductivity showed a low critical current density of 0.75 mA cm−2 (Fig. 13c), unflattened plating/striping profiles and fast short circuit at 3 mA cm−2, which indicated the lowest dendrite suppression capability and severe Li incompatibility, respectively. Ce3+, a rare earth element, was regarded as a soft acid and was first verified to enhance air stability through strong bonding to the soft acid S2− by Zhou et al. [111]. They prepared a Li7P2.9Ce0.2S10.9Cl0.3 glass-ceramic with an enhanced ionic conductivity of 3.2 mS cm−1 and improved air stability via dual substitution of Ce3+ and Cl− for P5+ and S2−, respectively. After exposure to humid air (40% RH), Li7P2.9Ce0.2S10.9Cl0.3 exhibited a lower amount H2S generation (Fig. 13d) compared with those of pristine and Ce-substituted Li7P3S11. XPS results showed that the introduction of Ce3+ facilitated the formation of Ce–S bonds without impacting the bridging sulfurs in Li7P3S11. The greater strengths of Ce–S or P = S bonds in Li7P2.9Ce0.2S10.9Cl0.3 compared with those of the other two air-exposed samples (Fig. 13e and f) indicated high chemical resistance of the Ce–S bond toward moist air. Yu et al. [75, 112] synthesized Na3P0.62As0.38S4 by substitution with the soft-acid As, and it exhibited an increase in ionic conductivity from 0.46 to 1.46 mS cm−1 and high moisture stability. The XRD pattern for Na3P0.62As0.38S4 was well maintained even after exposure to humid air (15% RH) for 100 h.

Air stability enhanced by substitution with Cu, Ce and In. a H2S amount generated under a controlled humidity of 55%–60%. b XRD patterns for LPSC-1 before and after exposure to ambient air (66% RH) for 1 h followed by sintering for 1 h at 550 °C. c Critical current density with increasing step size current density of LPSC-1. Reprinted with permission from Ref. [110]. Copyright © 2021, Elsevier. d Concentration of H2S within 60 min when exposed to humid air (40% RH). High-resolution S 2p spectrum and peak-deconvolution results for e Li7P3S11 and f Li7P2.9Ce0.2S10.9Cl0.3. Reprinted with permission from Ref. [111]. Copyright © 2021, American Chemical Society. g Quantities of H2S gas produced from Li6.5In0.25P0.75S5I and Li6PS5I electrolytes when exposed to moist air. h XRD patterns and i Raman spectra for Li6.5In0.25P0.75S5I and Li6PS5I electrolytes before and after exposure to moist air. Reprinted with permission from Ref. [113]. Copyright © 2021, Wiley-VCH
According to the thermodynamic analytical results of Mo et al., [69] In3+ is a good candidate dopant for enhancing the air stabilities of sulfide SEs. Jiang et al. [113] enhanced the air stability and ionic conductivity of Li6PS5I (LPSI) through incorporation of In3+. As shown in Fig. 13g, the amount of H2S generated by In-doped LPSI decreased dramatically from almost 1.2 to 0.18 cm3 g−1 after exposure to dry-room air (10% RH) for 60 min. While some unknown peaks appeared in the XRD pattern for LPSI, that of In-doped LPSI remained unchanged (Fig. 13h) after air exposure. In addition, Raman spectroscopy also confirmed the unchanged structure of air-exposed In-doped LPSI, whereas the enlarged Raman spectra of pristine LPSI showed two additional peaks (Fig. 13i).
5.2.3 Substitution with Other Elements
In addition to substitutions with oxygen and soft acids to enhance the air stabilities of sulfide SEs, substitutions with other elements were also investigated and showed positive effects, despite the complicated controlling mechanisms. Fukushima et al. [71] synthesized (75–1.5x)Li2S·25P2S5·xLi3N glass-ceramics by partial substitution of Li2S with Li3N. The 60i2S·25P2S5·10Li3N glass-ceramic achieved the highest ionic conductivity of 1.4 mS cm−2 (Fig. 14a) and enhanced moisture stability with less H2S generation (Fig. 14b), which was attributed to formation of crosslinked P and N in the glass network (Fig. 14c). In addition to the S2− bonded in PS43−, nonbonded S2− was considered another site in the argyrodite structure that was vulnerable to oxygen and moisture. Subramanian et al. [114] substituted nonbonded S2− with Br− and reported Li5.6PS4.6ClBr0.4 SEs with both enhanced air stability and improved ionic conductivity. After exposure to low-humidity air (10% RH), Li5.6PS4.6ClBr0.4 exhibited a lower rate for generation of H2S (Fig. 14d) and a higher ionic conductivity retention of 61.5% (Fig. 14e) than Li6PS5Cl. However, it was difficult to identify the minor structural changes for both pristine and Br-substituted Li6PS5Cl from XRD patterns and Raman spectra (Fig. 14f), which may be ascribed to the low humidity. Min et al. [115] synthesized Li6+2xAlxP1−xS5Cl (x = 0, 0.025, 0.05, 0.075) by partial substitution of Al2S3 for P2S5. As shown in Fig. 14g, H2S generation was suppressed as the Al3+ content was increased from x = 0 to x = 0.075, which verified the enhanced air stability resulting from Al2S3 substitution. Moreover, the XRD pattern (Fig. 14h) for undoped Li6PS5Cl presented stronger peaks for the side-product Li3PO4 compared with its Al2S3-substituted counterparts, again revealing the positive effects of Al3+. Although the ionic conductivity decreased monotonically as the amount of Al2S3 incorporation was increased (Fig. 14i), the decrease was relatively small compared to that of sulfide SEs substituted with oxygen alone.

Air stability enhanced by substation with other elements. a Room-temperature ionic conductivities of (75 − 1.5x)Li2S·25P2S5·xLi3N glasses and glass-ceramics. The Li2S crystal was precipitated from composition with 25 \(\leqslant\) x \(\leqslant\) 40. b Amounts of H2S gas generated from 60Li2S·25P2S5·10Li3N and 75Li2S·25P2S5 glass-ceramic powders. c N 1 s XPS spectrum of the 60Li2S·25P2S5·10Li3N glass-ceramic. Reprinted with permission from Ref. [71]. Copyright © 2017, Elsevier. d H2S amount, e bar diagram of the ionic conductivity values before and after air stability testing and f Raman spectra after air stability testing of Li6PS5Cl and Li5.6PS4.6Cl1.0Br0.4 solid electrolytes. Reprinted with permission from Ref. [114]. Copyright © 2021, Elsevier. g Amount of H2S generated by Li6+2xAlxP1−xS5Cl (x = 0, 0.025, 0.05, 0.075) in air. h XRD patterns for Li6+2xAlxP1−xS5Cl (where x = 0, 0.075) after reacting with moisture for 300 s. i Ionic conductivity of Li6+2xAlxP1−xS5Cl (x = 0, 0.025, 0.05, 0.075). Reprinted with permission from Ref. [115]. Copyright © 2021, The Electrochemical Society
In summary, elemental substitution is a common strategy used to tune atom sites/contents with high degrees of freedom and achieve homogenous properties for modified sulfide SEs. However, partial substitution of hard acids may result in irreversible structural degradation due to the presence of unstable P–S bonds. Moreover, rich experience and more experimental attempts are needed to achieve satisfactory properties for sulfide SEs.
5.3 Design of New Materials
Based on HSAB theory, Li/Na-M-S ternary or Li/Na-M-M´-S quaternary sulfide SEs obtained by complete substitution of hard acids with soft acids M/M´ (e.g., As, Sn and Sb) may provide the ideal configuration for optimal air stability. Since all S atoms in these completely substituted SEs are covalently bonded with soft-acid atoms M, their air stabilities should be enhanced significantly compared with those of partially substituted analogues.
5.3.1 Li/Na-Sn-S System
A series of fast ionic conductors based on Li3x[LixSn1−xS2] have been identified, among which Li[Li0.33Sn0.67S2] (x = 0.33, i.e., Li2SnS3) and Li0.6[Li0.2Sn0.8S2] (x = 0.2, i.e., Li2Sn2S5) are two representative examples with similar layered structures (Fig. 15a) [116]. In 2014, Kuhn et al. [117] first synthesized Li2SnS3 with the monoclinic space group C2/c (No. 15) via a facile wet chemistry approach. In 2015, Brant et al. [118] obtained Li2SnS3 through a solid-phase method, and it exhibited outstanding thermal stability up to ~ 750 °C and superior air stability. No additional diffraction peaks (Fig. 15b) or crystalline decomposition products appeared in Li2SnS3 after exposure to humid air (60% RH) for one week, so its structure is stable. Fast ion conduction in Li2SnS3 is expected from the high mobilities of Li(1) and Li(3) sites that reside in lithium sulfide layers between the honeycomblike [SnS3]2− layers, despite the disappointing ionic conductivity of 1.5 × 10−5 S cm−1 originating from a low pellet density of 56%. In 2016, Holzmann et al. [116] reported a lithium-poor phase Li2Sn2S5 with the monoclinic space group C2/m (No. 12). Partial occupation (38%) of interlayered Li+ sites was beneficial for facile Li-ion diffusion between the covalent Sn(Li)S2 layers, which was consistent with the superior bulk ionic conductivity of 1.5 × 10−2 S cm−1. However, the high grain boundary impedance and hydration in air hindered the application of Li2Sn2S5 in ASSBs. Fortunately, Joos et al. [89] found that the total ionic conductivity of the hydrated or water-intercalated phase Li2Sn2S5·xH2O (0 < x \(\leqslant\) 10) was as high as 10−2 S cm−1. Upon exposure to humid air, pristine Li2Sn2S5 underwent a two-step phase transition from the anhydrous phase (x = 0) to the first hydrated phase (x ≈ 2–4) and the second hydrated phase (x ≈ 8–10), which resulted in a two-step increase in the layer distance. In addition, it is interesting that the ionic conductivity increased with increasing intercalated water content, as shown in Fig. 15c. After excluding the operation of protonic conduction and internal Li+-H+ exchange, the authors speculated that coupling of Li+ ions and interlayer water molecules may have effectively accelerated Li+ motion.

Air stability of the Li-Sn-S system. a Crystal structures of Li[Li0.33Sn0.67S2] and Li0.6[Li0.2Sn0.8S2]. Reprinted with permission from Ref. [116]. Copyright © 2016, Royal Society of Chemistry. b X-ray powder diffraction data for Li2SnS3 before and after humid-air exposure. Reprinted with permission from Ref. [118]. Copyright © 2014, American Chemical Society. c Conductivity at 30 °C as a function of the average water content in Li2Sn2S5·xH2O. Reprinted with permission from Ref. [89]. Copyright © 2021, Royal Society of Chemistry. Fragment of the crystal structure of d orthorhombic LSS with the [SnS4]4− anionic units highlighted and e cubic Li4SnS4·13H2O. f TGA/DSC data for dehydration of Li4SnS4·13H2O. Reprinted with permission from Ref. [94]. Copyright © 2012, American Chemical Society. g XRD patterns of aqueous-solution processed LSS after heat-treatment at various temperatures. h Ionic conductivities at 30 °C for aqueous-solution processed LSS as a function of heat-treatment temperature. i H2S amount generated as a function of time for an aqueous solution of LSS. Reprinted with permission from Ref. [119]. Copyright © 2017, Wiley-VCH. j XRD patterns of Li4SnS4 heated to 260 and 390 °C for 2 h compared to that of orthorhombic Li4SnS4. k H2S gas generation from hexagonal Li4SnS4 and Li3PS4 glass-ceramic powders upon exposure to humid air. Reprinted with permission from Ref. [80]. Copyright © 2018, American Chemical Society. l Schematic graph of the ion exchange process. Reprinted with permission from Ref. [152]. Copyright © 2016, Elsevier. NSS stands for Na4SnS4 and LSS stands for Li4SnS4. m XRD patterns of the obtained powder before and after atmospheric exposure and reheating at 240 °C. Reprinted with permission from Ref. [120]. Copyright © 2019, Elsevier
In 2012, Kaib et al. [94] first synthesized a Li4SnS4 (LSS) SE with an ionic conductivity of 7 × 10−5 S cm−1 (20 °C). They found that (1) LSS was soluble in polar solvents (e.g., methanol or water), (2) Li4SnS4·13MeOH or Li4SnS4·13H2O hydrates formed after slow evaporation of the solvent, and (3) LSS could be completely recovered by consecutive heating above 320 °C. As shown in Figs. 15d and 15e, LSS converted from the orthorhombic (space group Pnma, No. 62) to the cubic (space group P213, No. 198) phase after absorbing 13 water molecules, while [SnS4]4− tetrahedra structure were still maintained. Based on the results of thermogravimetric analysis (TGA) and differential scanning calorimetry (DSC) (Fig. 15f), they further speculated that 13 water molecules were completely removed by thermal treatment above 300 °C, while 0.5 water molecules remained in the crystalline structure after mild thermal treatment at 150–300 °C. Subsequently, Choi et al. [119] investigated the evolution of the LSS crystalline structure by subjecting aqueous solutions to different heat-treatment temperatures. XRD patterns (Fig. 15g) for LSS recovered with heat treatments in the range 360–450 °C corresponded well with that of orthorhombic LSS synthesized by the solid-phase method. However, another set of “unknown” XRD patterns appeared and showed amorphous features when the heating temperature was in the range of 200–320 °C. Interestingly, the ionic conductivity (Fig. 15h) first increased and then decreased as the heat-treatment temperature was increased from 200 to 450 °C and reached the maximum value of 1.4 × 10−4 S cm−1 at 320 °C. Although the crystallinity of LSS increased upon increasing the heating temperature from 360 to 450 °C, its ionic conductivity monotonously decreased. As shown in Fig. 15i, the amount of H2S generated by LSS was almost negligible compared with that of LGPS containing P due to the superior stability of the SnS44− structure toward water. Unfortunately, the “unknown” XRD pattern with amorphous features was not identified, and no explanation was found for the dependence of ionic conductivity for recovered LSS on the heat-treatment temperature. Subsequently, Kanazawa et al. [80] treated ball-milled precursors to LSS at 260 °C and 390 °C. The XRD pattern (Fig. 15j) for LSS synthesized by ball milling and a postheating treatment at 260 °C was similar to the “unknown” XRD pattern (Fig. 15g) seen with recovered LSS, and the ionic conductivities of these two SEs were almost the same (1.1 × 10−4 S cm−1 and 1.4 × 10−4 S cm−1, respectively). Finally, Rietveld refinement analyses of synchrotron XRD data for LSS heated to 260 °C enabled identification of the “unknown” XRD pattern as hexagonal LSS in the space group P63/mmc (No. 194). The higher ionic conductivity of hexagonal LSS compared with that of orthorhombic LSS was ascribed to its larger free volume, which was potentially more favourable for ion conduction. As shown in Fig. 15k, hexagonal LSS generated only little H2S gas after exposure to humid air (70% RH), in contrast to the LPS glass-ceramic SE. Matsuda et al. [120] took advantage of the moisture stability of LSS and prepared hexagonal LSS from a Na4SnS4 aqueous solution by ion exchange (Fig. 15l) and realized, for the first time, an air-stable LSS SE. This novel synthetic method was beneficial for low-cost and large-scale preparation of sulfide SEs. The synthesized LSS also exhibited good air stability after exposure to humid air (50% RH) for one day and then was recovered by heat treatment at 240 °C, based on the XRD patterns (Fig. 15m) obtained for the LSS before and after exposure to humid air.
Numerous attempts have been made to improve the ionic conductivities of LSS (3.1 × 10−4 S cm−1 for orthorhombic LSS and 1.1 × 10−4 S cm−1 for hexagonal LSS). Sahu et al. [51] synthesized Li3.833Sn0.833As0.166S4 with an ionic conductivity of 1.39 × 10−3 S cm−1 by substituting Sn with As to create interstitial vacancies that accounted for the enhanced ionic conductivity. Only the diffraction peak at ~16° was broadened (Fig. 16a) due to absorbed moisture after exposure to the laboratory environment (64 °F and 80% RH; °F = °C × 1.8 + 32 °C) for 48 h. After heating the air-exposed Li3.833Sn0.833As0.166S4 at 140 °C for one hour, the intensity of the originally widened diffraction peak was significantly reduced, while the other diffraction peaks remained similar to those of pristine Li3.833Sn0.833As0.166S4. In contrast, the XRD pattern (Fig. 16b) for β-Li3PS4 after treatment under the same conditions changed completely, indicating destruction of the crystalline structure. Moreover, the ionic conductivity (Fig. 16c) of air-exposed Li3.833Sn0.833As0.166S4 decreased slightly from 1.39 × 10−3 to 9.95 × 10−4 S cm−1, while that of air-exposed β-Li3PS4 decreased by more than one order of magnitude. Therefore, the outstanding air stability of Li3.833Sn0.833As0.166S4 limited moisture absorption from humid air and minimized the impacts on the crystalline structure and ionic conductivity (which was recovered by moderate heat treatment), potentially enabling practical application. Recently, Lu et al. [84] prepared a similar As-substituted Li4SnS4 analogue, Li3.875Sn0.875As0.125S4 (LSAS), with the highest room-temperature ionic conductivity value (2.45 mS cm−1) among all reported lithium-ion sulfide SEs stable to moist air; this material was realized, for the first time, with a one-step gas-phase synthetic method in an ambient environment. After immersion in water, the crystalline structure (Fig. 16d) and localized structure (Fig. 16e) of LSAS were completely recovered by heat treatment above 350 °C. In addition, the superior moisture stability of LSAS compared to LPS and LSPSC was proven by minimal generation of H2S (Fig. 16f).

Air stability of As- and Sb-substituted Li4SnS4. a, b XRD patterns for Li3.833Sn0.833As0.166S4 and β-Li3PS4 before and after exposure to humid-air. c Arrhenius plots for Li3.833Sn0.833As0.166S4 and β-Li3PS4 before and after air exposure. Reprinted with permission from Ref. [51]. Copyright © 2014, Royal Society of Chemistry. d XRD patterns and e Raman spectra of LSAS before and after water immersion and heat treatment. f Amounts of H2S generated by LSAS, LSS, LPS and LSPSC. Reprinted with permission from Ref. [84]. Copyright © 2021, Wiley-VCH. g Calculated total ionic conductivities of cold-pressed Li4−xSbxSn1−xS4 (0 \(\leqslant\) x \(\leqslant\) 0.8) powders at room temperature. h XRD patterns of Li3.8Sb0.2Sn0.8S4 before and after air exposure for 12 h. i H2S gas generation from Li3.8Sb0.2Sn0.8S4 and LGPS powders upon exposure to humid air. Reprinted with permission from Ref. [63]. Copyright © 2019, Elsevier. j Li+ conductivities at 30 °C for Li4−xSn1−xSbxS4 [64]. k XRD patterns for Sb-doped Li4SnS4 (Li4−xSn1−xSbxS4 (0 \(\leqslant\) x \(\leqslant\) 0.30)). l XRD patterns of Li3.85Sn0.85Sb0.15S4 before and after air exposure. m H2S amount as a function of time in atmospheric air for Li3.85Sn0.85Sb0.15S4 compared with Li6PS5Cl or Li4SnS4. Reprinted with permission from Ref. [64]. Copyright © 2019, Elsevier
Considering the toxicity of arsenic and its use in LSAS, Zhang et al. [63] synthesized an air-stable and environmentally friendly SE, Li4−xSn1−xSbxS4 (LSSS), by substituting Sn with Sb. As shown in Fig. 16g, its ionic conductivity reached the maximum value of 3.5 × 10−4 S cm−1 at x = 0.2. The XRD patterns (Fig. 16h) obtained before and after exposure of LSSS to humid air (60% RH, 22 °C) for 12 h were almost identical. As shown in Fig. 16i, the amount of H2S generated by air-exposed LSSS was negligible compared with that of LGPS. Kwak et al. [64] also performed Sb substitution for Sn to obtain Li3.85Sn0.85Sb0.15S4 with ionic conductivities of 4.6 × 10−4 S cm−1 and 8.5 × 10−4 S cm−1 (Fig. 16j) for cold-pressed and hot-sintered pellets by further optimizing the substitution proportion. As shown in Fig. 16k, the diffraction peaks (e.g., 25.8°) shifted to the left with increasing Sb substitution proportion from x = 0 to x = 0.30, and an impurity phase appeared at ~ 17° when x ≥ 0.15. Therefore, the upper limit for Sb dissolution was determined to be within the range 0.10 < x < 0.15. The crystalline structure (Fig. 16l) of Li3.85Sn0.85Sb0.15S4 barely changed after exposure to dry air for 12 h, and the ionic conductivity was slightly reduced to 4.1 × 10−4 S cm−1. The amount of H2S generated by Li6PS5Cl after exposure to humid air (50% RH) was as high as 89 ppm (Fig. 16m), while that of Li3.85Sn0.85Sb0.15S4 without P was extremely low (6 ppm). This may have been caused by trace amounts of impurities, such as Li2S.
The LSS SE also exhibited good solubility and compatibility with the wet-coating process [121]. Park et al. [52] dissolved LiI and LSS SEs into anhydrous methanol to obtain 0.4LiI-0.6Li4SnS4 SEs and tuned the proportion of these two components to optimize the ionic conductivity at 4.1 × 10−4 S cm−1. xLiI-(1 − x)Li4SnS4 SEs (x < 0.5) showed an amorphous feature, and the added LiI was not present in crystalline form but dissolved into LSS and acted as a glass forming agent to reduce the crystallinity of LSS. In contrast, LSS synthesized at 450 °C without the addition of LiI was highly crystalline. Furthermore, it is noteworthy that [SnS4]4− anions were always stable and unaffected by heating and LiI introduction. Therefore, the proportion of LiI and the crystallinity (which depended on the heating temperature) jointly affected the ionic conductivity of xLiI-(1 − x)Li4SnS4 SEs. The ionic conductivity of 0.4LiI-0.6Li4SnS4 after exposure to dry-air flow for 24 h dropped from 4.1 × 10−4 to 2.6 × 10−4 S cm−1, while that of Li3PS4 decreased by two orders of magnitude from 1.0 × 10−3 to 8.0 × 10−6 S cm−1.
Na4SnS4 and Na3SbS4 are analogues of Li4SnS4 and Li3SbS4, respectively, and they also exhibited outstanding moisture stability. Although a series of Na4SnS4 hydrates was reported by Schiwy et al. [122] in 1973, the solid-state ionic conductor Na4SnS4 was first reported by Heo et al. [123] in 2018. Heo et al. discovered tetragonal Na4−xSn1−xSbxS4 (0.02 \(\leqslant\) x \(\leqslant\) 0.33) with space group I41/acd, which was distinctly different from Na4SnS4 or Na3SbS4. In addition to a high ionic conductivity of 0.51 mS cm−1, Na3.75Sn0.75Sb0.25S4 exhibited excellent dry-air stability and recoverability after complete dissolution into water without releasing H2S gas. Jia et al. [124] designed Na3.7[Sn0.67Si0.33]0.7P0.3S4 based on the structural template for Na4Sn0.67Si0.33S4. Although the “hard acid” P5+ was introduced to improve the ionic conductivity, Na3.7[Sn0.67Si0.33]0.7P0.3S4 still showed excellent humid-air stability since no impurity peaks were observed, even in the enlarged XRD patterns, for samples exposed to humid air (35% RH) for 24 h without or with a 150 ℃ drying process. Xiong et al. [125] substituted Sb5+ and Cl− for Sn4+ and S2−, respectively, and obtained Na3.7Sn0.8Sb0.2S3.9Cl0.1 with an improved ionic conductivity of 0.61 mS cm−1. However, an investigation of the air stability was not reported.
5.3.2 Li/Na-Sb-S System
Considering the superior moisture stability of the [SbS4]3− group (e.g., the sodium ion conductor Na3SbS4 is stable in the natural environment in the hydrated form Na3SbS4⋅9H2O), Kimura et al. [61] designed and synthesized the air-stable lithium ion conductor Li3SbS4 (Fig. 17a) by ball milling and postheating. The ionic conductivity of Li3SbS4 glass (1.5 × 10−6 S cm−1) was much higher than that of glass-ceramic Li3SbS4 due to their different structures (Fig. 17b). The amounts of H2S produced by LPS and LSS after exposure to humid air (70% RH) for 1 000 min were 38 cm3 g−1 and 5 cm3 g−1 (Fig. 17c), respectively, while that for Li3SbS4 was less than 1 cm3 g−1; this indicated air stability better than that of LSS. Although Li3SbS4 glass exhibited outstanding air stability, its ionic conductivity only reached ~ 10−6 S cm−1, far below 10−3 S cm−1. More work is needed to improve its ionic conductivity with various substitution strategies.

Air stability of the Li/Na-Sb-S system. a XRD patterns for prepared Li3SbS4 glass and the glass ceramic heated at 200 °C (HT 200 °C) and 500 °C (HT 500 °C). b Temperature dependence of the conductivities of the Li3SbS4 glass and glass ceramic heated at 200 °C. c Amounts of H2S gas generated from the Li3SbS4 glass, Li3PS4 glass, and Li4SnS4 milled powders. Reprinted with permission from Ref. [61]. Copyright © 2019, Elsevier. d Raman spectra and e XRD patterns of pristine Na3SbS4·9H2O, as-synthesized Na3SbS4, air-exposed Na3SbS4 and reheated air-exposed Na3SbS4. Reprinted with permission from Ref. [126]. Copyright © 2016, Wiley-VCH. f XRD patterns of Na3SbS4 powders before and after air exposure for 5 h and heating at 100 °C after air exposure. Reprinted with permission from Ref. [127]. Copyright © 2016, Wiley-VCH. g H2S gas generated from Na2.88Sb0.88W0.12S4 as a function of exposure time to humid air (70% RH). Reprinted with permission from Ref. [130]. Copyright © 2019, Springer Nature. h H2S gas amounts generated from a Na3PS4 pellet and Na2.937 5SbS3.937 5Cl0.062 5 powder after exposure to air. i XRD patterns for the Na2.937 5SbS3.937 5Cl0.062 5 sample before and after air exposure and after the sample was heated at 170 °C for 1 h after air exposure. Reprinted with permission from Ref. [133]. Copyright © 2020, The Ceramic Society of Japan
In 2016, Wang et al. [126] synthesized a Na3SbS4 solid electrolyte by heating and completely removing the waters of crystallization from the hydrate Na3SbS4·9H2O. Since Na3SbS4·9H2O is stable in the natural environment, SbS43− groups with robust Sb–S bonds should be moisture-stable based on HSAB theory. Raman spectra (Fig. 17d) and XRD patterns (Fig. 17e) indicating the structural evolution of Na3SbS4 upon air exposure and reheating further demonstrated reversible H2O adsorption/desorption and the moisture stability of Na3SbS4. In addition, the ionic conductivity was improved from 5 × 10−7 to 1.05 × 10−3 S cm−1 via transformation from Na3SbS4·9H2O to Na3SbS4. Almost at the same time, Zhang et al. [127] also synthesized Na3SbS4 by the solid-phase method, and it exhibited the same crystal structure (space group \(P\overline{4}2_{1} c\), No. 114). In addition to reversible H2O adsorption/desorption and moisture stability (Fig. 17f), the vacancy-rich Na3SbS4 exhibited a much higher ionic conductivity of 3 mS cm−1. Subsequently, Banerjee et al. [128] reported solution processing of Na3SbS4, which enabled preparation of a Na3SbS4-coated cathode for all-solid-state sodium-ion batteries. In 2018, Kim et al. [129] developed a scalable synthetic method for preparation of Na3SbS4 from aqueous solution. These inspiring results indicated that Na3SbS4 is a promising solid electrolyte for applications with ASSBs.
In 2019, Hayashi et al. [130] increased the ionic conductivity of Na3SbS4 from 2.1 to 32 mS cm−1 via partial substitution of Sb5+ with W6+. The introduction of high-valent W6+ effectively created Na vacancies and facilitated a structural transformation from the tetragonal to cubic phase, which enabled isotropic three-dimensional fast-ion conduction. In addition to the outstanding ionic conductivity, the resulting Na2.88Sb0.88W0.12S4 also exhibited excellent moisture stability indicated by negligible H2S generation (0.1 cm3 g−1, Fig. 17g) and Na3SbS4·9H2O hydrate formation after exposure to humid air (70% RH). Fuchs et al. [131] reported an even higher ionic conductivity of (42 ± 8) mS cm−1 for the Na2.9Sb0.9W0.1S4 analogue, which represented the highest value measured in sulfide SEs to date. Subsequently, the aqueous-solution synthetic protocol was developed for Na3−xSb1−xWxS4 by Yubuchi et al. [132], and this showed significant promise for realization of ASSBs despite a compromised ionic conductivity of 4.28 mS cm−1. Inspired by the vacancy introduction strategy, Tsuji et al. [133] developed Na3−xSbS4−xClx electrolytes by partially substituting S2− with Cl−. The resulting Na2.937 5SbS3.937 5Cl0.062 5 showed a higher ionic conductivity (2.9 mS cm−1) than Na3SbS4. Although the exposure conditions were more demanding, Na2.937 5SbS3.937 5Cl0.062 5 showed better moisture stability than Na3PS4 and minimal H2S generation (Fig. 17h) and reversible H2O adsorption/desorption (Fig. 17i).
5.3.3 Novel Quaternary-Ion Conductors
To improve the inherent air stabilities of sulfide SEs with well-known crystalline structures, much effort has been devoted to exploring sulfide SEs with various compositions and novel structures. Lithium oxysulfide superionic conductors, including the aforementioned oxygen-substituted sulfide SEs, were proposed [100] very early to overcome the moisture sensitivities of sulfide SEs. Inspired by predictions for layered LiAlSO offered by Wang et al. [134] based on first-principles calculations, Kuo et al. [135] successfully synthesized LiSnOS oxysulfide with a layered structure, and it was expected to combine the high chemical stabilities of oxide SEs and the high ionic conductivities of sulfide SEs. The preparation of LiSnOS powder utilized a new synthetic route that differed from conventional solid-/liquid-phase methods and involved thermal precipitation of SnOS and a calcination/sulfurization step. LiSnOS exhibited an ionic conductivity of 1.92 × 10−4 S cm−1 and remained stable in air for at least two weeks without phase decomposition. However, LiAlSO and possible analogues in Li-Al-S-O phases have not yet been synthesized despite the numerous experimental attempts by Gamon et al. [136].
Li4PS4I, which was synthesized by a solvent-based approach, was discovered by Sedlmaier et al. [137] in 2017. It has a novel crystalline structure with the tetragonal space group P4/nmm (No. 129). Although a three-dimensional migration pathway for Li+ is predicted based on topostructural analyses of the PS4I4− substructure, the total ionic conductivities fell within the range 6.4 × 10−5–1.2 × 10−4 S cm−1. Subsequently, the air stability was systematically investigated by Calpa et al. [81] in 2021. Even after exposure to humid air (40% RH) for 60 min, no H2S gas was detected for the Li4PS4I SE. While H2S gas generation from the Li3PS4 sample reached a maximum value of 8.3 cm3 g−1 after exposure for ~ 540 min, that of Li4PS4I merely reached 0.96 cm3 g−1. XRD results showed that the side products LiI·H2O and LiI·3H2O were formed after exposure for 60 and 1 800 min, and they acted as a protective layer between PS43− units in the SE and H2O molecules in the air. After simple drying at 180 °C, the structure of air-exposed Li4PS4I was recovered, and a slightly decreased ionic conductivity of ~ 1 × 10−4 S cm−1 was regained. For the first time, a reversible structure was observed for thiophosphate SEs, which are notorious for their poor air stabilities and irreversible structural losses after exposure to humid air.
Wang et al. [62] completely substituted P5+ in LGPS with Cu+ by a urothermal synthesis method and obtained Li4Cu8Ge3S12 (LCGS) with a novel crystalline structure in the space group \(Fm\overline{3}c\) (No. 262). According to HSAB theory, Ge4+ and Cu+, which are soft acids, tend to bond tightly with the soft base S2− and form covalent bonds that are stronger than the P–S bond. As shown in Fig. 18a, the diffraction peaks obtained for LCGS after exposure to humid air (15% RH) or 2 M LiOH aqueous solution (1 M = 1 mol L−1) and sequential heating at 60 °C for 4 h corresponded well with those of pristine LCGS, indicating the high stability of the crystalline structure. Although water molecules were absorbed into the pores of LCGS after exposure to air, the stable skeleton and weak coordination of water molecules by Li+ facilitated the removal of water molecules without destroying the original structure. Interestingly, the ionic conductivities (Fig. 18b) of LCGS after exposure to humid air and LiOH aqueous solution were even higher that of the pristine material; this was closely related to the amount of water absorbed because the impedance gradually increased with increasing time under flowing argon gas (Fig. 18c). Therefore, the abnormal increase in ionic conductivity was attributed to proton conduction in the LCGS after absorption of water molecules.

Air stabilities of novel quaternary ion conductors. a XRD patterns for LCGS before and after exposure to 15% moist air and a 2 M LiOH aqueous solution. b Comparative Arrhenius plots showing minor changes in conductivity and activation energy before and after exposure. c Reversible variations in ionic conductivity for LCGS when exposed to moist air and Ar flow. The inset shows the magnified impedance plot. Reprinted with permission from Ref. [62]. Copyright © 2019, Wiley-VCH. d Amount of H2S generated when Li6.6Ge0.6Sb0.4S5I and Li6PS5Cl were exposed to air. Reprinted with permission from Ref. [139]. Copyright © 2020, Wiley-VCH. e Amounts of H2S generated from Li6PS5I (black) and Li6+xSb1−xGexS5I [x = 0 (red), 0.5 (blue), and 0.75 (green)] as a function of exposure time in 15% air humidity. Reprinted with permission from Ref. [83]. Copyright © 2021, American Chemical Society. f Amounts of H2S generated from Li6PS5I (black) and Li6+xSb1−xSixS5I [x = 0 (red), 0.5 (blue), and 0.75 (green)] as a function of exposure time to air with <15% humidity. Galvanostatic cycling of the Li/Li6.75Sb0.25Si0.75S5I/Li symmetric cell g at current densities ranging from 0.2 to 1.7 mA cm−2 and h at 0.3, 0.6, and 1.2 mA cm−2. Reprinted with permission from Ref. [140]. Copyright © 2020, American Chemical Society. i Variations in the ionic conductivities vs. exposure time for Na11Sn2SbS12 and Na11Sn2PS12. Reprinted with permission from Ref. [144]. Copyright © 2018, American Chemical Society. j Amounts of H2S gas generated from Na11Sn2PS12, Na11Sn2SbS12 and Na11.5Sn2Sb0.5Ti0.5S12 as a function of exposure time to humid air (60% RH). Reprinted with permission from Ref. [145]. Copyright © 2021, Elsevier. k Amounts of H2S gas generated from Na10SnSb2S12 when exposed to humid air. Reprinted with permission from Ref. [146]. Copyright © 2020, Elsevier
Li6SbS5I and its derivatives Li6+xMxSb1−xS5I (M = Si, Ge, Sn) were first developed by Zhou et al. in 2019 [138]. Although the ionic conductivity of the sintered pellet was improved to as high as 18.4 and 24 mS cm−1 by substitution of Sb5+ with Ge4+ and Si4+, respectively, there was no air stability investigation reported. Subsequently, Sun et al. [139] found that the air stability of Ge-substituted Li6SbS5I was better than that of the typical sulfide electrolyte Li6PS5Cl; it took a longer time for Li6.6Ge0.6Sb0.4S5I to reach the testing limit of the sensor (Fig. 18d), and its ionic conductivity only dropped from 1.61 to 1.06 mS cm−1 after exposure for 2 h. Lee et al. [83] systematically studied the air stability of Ge-substituted Li6SbS5I by monitoring the amount of H2S produced with in situ Raman spectroscopy and cryo-TEM images. Since the central cations Ge4+ and Sb5+ are soft acids, the air stabilities of Li6SbS5I and Li6.5Ge0.5Sb0.5S5I were much better than that of phosphorus-based LPSI, which was demonstrated by generation of 68% and 84% H2S, respectively (Fig. 18e). Notably, the amount of H2S generated by Li6+xGexSb1−xS5I decreased with increasing Ge content. In contrast to the rapid drop in P–S bond strength for LPSI and the gradual decline in Sb–S bond strength for Li6SbS5I, Ge-substituted Li6SbS5I showed superior air stability due to minor changes in Sb–S and Ge–S bond strengths with exposure time. Air-exposed LPSI was severely damaged and decomposed to Li2S, LiI and Li3PS4, while the other two samples maintained argyrodite structures. Like Ge-substituted Li6SbS5I, Si-substituted Li6SbS5I was also verified to be more stable than LPSI and undoped Li6SbS5I by comparing H2S concentrations (Fig. 18f). [140] However, due to the weak chemical stability of nonbonded S2− ions, structural degradation of Li6+xMxSb1−xS5I (M = Si, Ge, Sn) argyrodites after exposure to moisture/water should be irreversible despite the enhanced resistance to humid air resulting from robust M/Sb–S bonding in [M/SbS4] units or possible protection from LiI·xH2O hydrates. Interestingly, Si-substituted Li6SbS5I even exhibited excellent Li compatibility, despite the high oxidation state of Si4+ and Sb5+ and possible formation of Li-Si or Li-Sb alloys. Zhou et al. [138] reported stable Li+ plating/stripping in a symmetric cell with Li6.7Si0.7Sb0.3S5I at a 0.6 mA cm−2 current density for 400 h, and Lee et al. [140] subsequently measured a critical current density as high as 1.5 mA cm−2 (Fig. 18g) and pushed the stable Li+ plating/stripping current density to 1.2 mA cm−2 (Fig. 18h) for the Li6.75Sb0.25Si0.75S5I analogue. However, the excellent Li compatibility of Si-substituted Li6SbS5I has not been verified in ASSBs using lithium metal instead of a Li-In alloy as the anode, despite the promising results obtained with symmetric cells.
Although promising quaternary superionic conduction by LGPS-type Na10MP2S12 (M = Si, Ge) was predicted by Richards et al. [141] in 2016, little progress has been made to date, except for preparation of an impure Na10SnP2S12 phase with an ionic conductivity of 0.4 mS cm−1. In 2018, Zhang et al. [142] synthesized Na11Sn2PS12 with a crystal structure distinctly different from that of its LGPS counterpart. Shortly after, Duchardt et al. [143] reported an even higher ionic conductivity of 3.7 mS cm−1 for Na11Sn2PS12 with the same tetragonal structure (space group I41/acd, No. 142). Subsequently, Ramos et al. [144] prepared the analogue Na11Sn2SbS12 exhibiting much higher dry-air stability than Na11Sn2PS12, despite a small decrease seen in ionic conductivity after exposure to dry air (Fig. 18i). Weng et al. [145] increased the ionic conductivity of Na11Sn2SbS12 by a factor of three (1.01 mS cm−1) and avoided the NaSbS2 impurity by substituting Sb with Ti. They also verified the outstanding air stability of Na11Sn2SbS12 with negligible H2S generation, as shown in Fig. 18j. However, the resulting Na11.5Sn2Sb0.5Ti0.5S12 with a decreased Sb content showed increased H2S generation when the exposure time was less than 45 min, despite the aforementioned positive effects of Ti substitution. In 2021, Liu et at. [146] reported the preparation of a quaternary Na10SnSb2S12 solid electrolyte, which exhibited an ionic conductivity of 0.52 mS cm−1, by complete replacement of P5+ with Sb5+ in Na10SnP2S12. As a result of the robust Sn–S and Sb–S bonds, Na10SnSb2S12 exhibited excellent air stability with a minor generation of H2S (0.015 cm3 g−1) and a small change in ionic conductivity from 0.52 to 0.51 mS cm−1 after exposure to humid air (55% RH) for 180 min (Fig. 18k).
Although some progress has been made on enhancing the air stabilities of sulfide SEs, more explorations of new materials with novel compositions or structures are required to enrich the variety of sulfide SEs and to overcome air instability and other challenges. Reliable guidelines from theoretical calculations and numerous experimental attempts are crucial for promoting the development of new sulfide SEs.
5.4 Surface Engineering
Since hydrolyses of sulfide SEs first occur at the sulfide surface, it is advisable to construct an inert surface or coating layer to resist chemical attack by O2, water molecules and even organic solvents, as illustrated in Fig. 19a. Jung et al. [68] synthesized oxysulfide-coated Li6PS5Cl with a core-shell structure via environmental mechanical alloying with a controlled oxygen partial pressure. A 50 nm-thick nanolayer with an extremely high oxygen concentration can be identified for the surface microstructure of oxysulfide-coated Li6PS5Cl, which is obviously different from the smooth surface and low oxygen concentration of pristine Li6PS5Cl. Atmospheric stability was evaluated with variations in the XPS O 1 s peak and ionic conductivities. After exposure to humid (35% RH) air, the intensity of the O 1 s peak for pristine Li6PS5Cl increased rapidly due to severe surface oxidation, whereas that of oxysulfide-coated Li6PS5Cl underwent little change (Fig. 19b). Although these two samples exhibited monotonic decreases in ionic conductivities over 120 min, oxysulfide-coated Li6PS5Cl showed slower degradation than pristine Li6PS5Cl, as shown in Fig. 19c. It is well known that typical sulfides are vulnerable to nucleophilic attack in highly polar solvents [147], which results in structural degradation. However, oxysulfide-coated Li6PS5Cl exhibited some resistance to both organic solvents and binders, since the oxysulfide passivation layer effectively suppressed chemical reactions on the surface and maintained a relatively high ionic conductivity. Recently, Xu et al. [88] designed a superhydrophobic surface layer with both water-repellent and Li+-conducting properties at the membrane level, as shown in Fig. 19d. This protective layer was spray-coated onto a Li6PS5Cl membrane, and it consisted of Li1.4Al0.4Ti1.6(PO4)3 (LATP) nanoparticles and fluorinated polysiloxane (F-POS) prepared by hydrolysis and condensation reactions. As shown in Fig. 19e, while water droplets adhered to the surface of the bare membrane, the membrane with the superhydrophobic surface showed distinct water repellency. The outstanding water stability of this designed membrane was further demonstrated by the small variations seen in the XRD patterns, negligible resistance increases and superior electrochemical performance of ASSBs after direct water jetting. It is worth noting that this postprocessing method is applicable to all types of water-sensitive SEs.

Air stability enhanced by surface engineering. a Schematic illustration of the atmospheric stability and chemical resistance of pristine and core-shell sulfide SEs. b O 1 s and C 1 s XPS spectra for pristine and oxysulfide-coated Li6PS5Cl samples, which were tested before (solid lines) and after (dashed lines) exposure to air with 35% RH for 30 min at 25 °C. Reprinted with permission from Ref. [68]. Copyright © 2020, American Chemical Society. d Schematic illustration of the design principles for the superhydrophobic Li+-conducting protective layer on Li6PS5Cl SE membranes. e Serial photographs of the F-POS@LATP/Li6PS5Cl/F-POS@LATP membrane and the bare Li6PS5Cl membrane exposed to extreme conditions with continuous water-drop attack. Reprinted with permission from Ref. [88]. Copyright © 2021, Wiley-VCH
Therefore, this specially designed surface layer is expected to improve air stability toward moisture and oxygen, enhance chemical stability toward organic components during wet casting processes and maintain superior bulk ionic conductivity by preventing structural degradation. In addition, other desirable characteristics, such as superhydrophobic properties and Li-metal compatibility[34], can be easily grafted onto sulfide SEs by surface engineering. Nevertheless, it is noteworthy that ionic conduction from the bulk to the surface should be maintained.
5.5 Sulfide-Polymer Composite Solid Electrolytes
In addition to modification methods for sulfide SEs themselves, combinations of sulfides with polymers have also been proposed. Li et al. [67] prepared a sulfide-incorporated composite electrolyte (SCE) from a combination of Li7PS6 and poly(vinylidene fluoride-co-hexafluoropropylene) (PVDF-HFP) polymer (Fig. 20a) in an ambient environment. XRD (Fig. 20b) and Raman (Fig. 20c) results showed that the main crystalline structure and localized structure (PS43−) of Li7PS6 were well maintained in the SCE without breaking P−S bonds. This SCE delivered a high room-temperature ionic conductivity of 1.1 × 10−4 S cm−1 and the symmetric cells were cycled for up to 1 000 h at 0.2 mA cm−2, since the PVDF-HFP polymer matrix protected the Li7PS6 sulfide SE from humid air. Chen et al. [49] prepared a hybrid SE composed of β-Li3PS4 and poly(glycidyl methacrylate) (PGMA) via a controlled interfacial reaction involving covalent crosslinking, as illustrated in Fig. 20d. The composite SE was obtained with good processability simply by slurry coating. Compared with that of pure β-Li3PS4, the ionic conductivity of this hybrid electrolyte was slightly increased to 1.8 × 10−4 S cm−1. While pure β-Li3PS4 reacted instantly with moisture and generated a series of products, including Li2PS3, LiOH, Li2O and P2O5 (Fig. 15e), the structure of the hybrid electrolyte remained stable in humid air (20% RH) for at least 20 min, as corroborated by in situ environmental XRD results (Fig. 20f). Tan et al. [66] combined the hydrophobic polymer polystyrene-block-poly(ethylene-ran-butylene)-e-polystyrene (SEBS) with Li7P3S11 to obtain the air/moisture-stable composite SEBS-Li7P3S11. The superhydrophobicity of SEBS was comparable to that of well-accepted PTFE based on the contact angle. The amount of H2S generated (Fig. 20g) by the SEBS-Li7P3S11 composite SE after exposure to humid air (50%–55% RH) was significantly lower than that of the pristine Li7P3S11 powder.

Air stability enhanced by sulfide-polymer compounding. a Schematic illustration of the Li7PS6-embedded composite electrolyte (Li7PS6/PVDF-HFP/LiTFSI). b XRD patterns and c Raman spectra of Li7PS6/PVDF-HFP/LiTFSI, PVDFHFP/LiTFSI, PVDF-HFP, and pure Li7PS6. Reprinted with permission from Ref. [67]. Copyright © 2020, American Chemical Society. d Schematic illustration of hybrid Li+ conductors with and without covalent interfacial coupling. XRD patterns of e β-Li3PS4 and f the hybrid electrolyte after exposure to air. Reprinted with permission from Ref. [49]. Copyright © 2019, Elsevier. g H2S amount released as a function of exposure time for pristine Li7P3S11 and a composite with the hydrophobic SEBS polymer. Reprinted with permission from Ref. [66]. Copyright © 2019, American Chemical Society
Sulfide-polymer composite SEs potentially combine the advantages of both types of SEs, including the mechanical flexibility and scalable processes of polymer SEs, the high transference number for Li+ and the superior room-temperature ionic conductivity of sulfide SEs, thus providing ultrathin films with high ionic conductivity and other excellent properties [32, 33, 148]. However, as a result of the complex interactions between sulfide SEs and organic solvents and polymers, numerous experimental attempts are needed to tune the ratio of sulfide and polymer SEs and achieve the optimal properties for composite SEs. Due to the heterogenous properties of sulfides and polymers, the ionic conductivity may be compromised.
6 Summary and Perspectives
In summary, the progress of research on the air stability of sulfide SEs and ASSBs was systematically reviewed from the perspectives of theoretical paradigms/mechanisms, characterization techniques, and strategy improvements. In terms of theoretical paradigms/mechanisms, the random network theory of glasses functions well to guide modification of glassy materials, although the improvements in comprehensive properties are still limited. HSAB theory has been generally accepted and proven to be effective for developing air-stable sulfide SEs. Thermodynamic analyses based on energy changes for hydrolysis reactions were performed by Mo et al. [69] to further enrich the selection of alternative cations available for air-stable sulfide SEs. The kinetics of interfacial reactions deepen our understanding of air instability problems originating from the surfaces of sulfide SEs and encourage more research on the growth of sulfide SEs with controlled crystalline orientations. Moreover, characterization techniques for determining air stability have been gradually optimized to study macroscopic chemical reaction phenomena, microscopic chemical components/structures, and electrochemical properties/performance before and after exposure to air. Based on the experimental results, five strategies have been reviewed and demonstrated to be effective for enhancing the air stability of sulfide SEs. In addition, the features, advantages and disadvantages of these five strategies are summarized in Table. 3.
Although significant progress has been made in recent years, practical application and commercialization of sulfide-based ASSBs still have many challenges to be overcome. Based on an in-depth understanding of the air stability problems described herein, several potential directions are proposed here:
-
1)
Developing partial substitution of S/P sites, dual substitution of both S and P sites, and even multiple substitution to trigger synergistic effects and meet the comprehensive prerequisites of sulfide SEs;
-
2)
Developing new materials for Li-M-S (M is soft acid) ternary systems based on theoretical predictions and air-stable sulfide SEs with novel structures (e.g., Li4Cu8Ge3S12 and LiSnOS);
-
3)
Developing sulfide-polymer composite SEs to combine the complementary advantages of these two types of SEs, such as the flexibility and scalable processing of polymer SEs and the high transfer number for Li+ and the superior room-temperature ionic conductivities of sulfide SEs;
-
4)
Developing a surface modification method (e.g., surface oxidation and surface nanostructure design) to maintain the superior bulk ionic conductivity of sulfide SEs and potentially utilize various functionalities of the surface layer, such as passivation of the interface between sulfide SEs and air, suppression of the space-charge layer effect between sulfide SEs and the oxide cathode, improvement in the compatibility between sulfide SEs and the oxide cathode or lithium metal anode, elimination of grain boundaries among particles, etc.;
-
5)
Developing a method to finely control physical properties, such as particle size, specific surface area, crystallinity and exposed crystalline plane, rather than tuning the chemical components.
In addition, the development of innovative synthetic routes/methods tailored for air-stable sulfide SEs, such as the ion exchange method [120], urothermal synthetic method [62] and gas-phase synthetic method [84], will potentially promote mass production of sulfide SEs.
Moreover, accurate measurements of H2S generation should be standardized to achieve comparable data for various sulfide SEs reported by different research groups. The homemade detection system reported in our previous work [84] or advanced gas chromatography-mass spectrometry is recommended. Apart from the standardized detection system, the testing conditions, such as humidity, temperature, sample form and sample mass, should also be standardized. Given the various humidity conditions of current manufacturing processes, including material preparation, storage, transportation, slurry coating, and battery assembly/packaging, and the different air stabilities of various sulfide SEs, it is advisable to measure the H2S gas amounts for all sulfide SEs under mild conditions and build a universal evaluation system for both research and practical applications, such as a dew point of −28 °C corresponding to the dry-room humidity (2% RH) for the slurry coating process. For sulfide SEs with outstanding air stabilities or reversible water absorption/desorption capabilities, durability can be investigated under harsh conditions, such as with high humidity (> 50% RH) or direct water exposure. The recommended testing temperature and sample form are 25 °C and the powder state, respectively. Given the detection range and accuracy of the H2S sensor, the sample mass should correspond to a reasonable range and undergo normalization when calculating the total amount of H2S based on Eq. (2).
Furthermore, it is urgent to improve and enrich the methods available for characterizing the air stabilities of sulfide SEs. Advanced in situ environmental characterization methods will be powerful in uncovering the mechanisms of interfacial reactions between sulfide SEs and air from collected kinetic information. Apart from the in situ XRD method used to analyze the evolution of reaction products during exposure of sulfide SEs to air, Tsukasaki et al. [149] developed a TEM system to evaluate the air stabilities of battery materials, and deterioration of the sulfide-based Li4SnS4 glass-ceramic was observed in situ under flowing air. In addition, basic and universal characterization methods should be used to construct a unified system for evaluating comparable data from different labs, including measurements of H2S generation upon exposure, XRD data for crystal structures, Raman spectra for local structures and measurements of ionic conductivity before and after exposure to air. Advanced characterization methods, such as XAS, NMR and in situ TEM, can be alternative approaches used to provide further proof and deepen our understanding.
Overall, continuous efforts are needed to overcome the aforementioned challenges and to convert laboratory investigations into large-scale application of air-stable sulfide SEs and ASSBs in the future.
References
Armand, M., Tarascon, J.M.: Building better batteries. Nature 451, 652–657 (2008). https://doi.org/10.1038/451652a
Xu, K.: Nonaqueous liquid electrolytes for lithium-based rechargeable batteries. Chem. Rev. 104, 4303–4418 (2004). https://doi.org/10.1021/cr030203g
Hu, Y.S.: Batteries: getting solid. Nat. Energy 1, 16042 (2016). https://doi.org/10.1038/nenergy.2016.42
Manthiram, A., Yu, X.W., Wang, S.F.: Lithium battery chemistries enabled by solid-state electrolytes. Nat. Rev. Mater. 2, 16103 (2017). https://doi.org/10.1038/natrevmats.2016.103
Zhao, Q., Stalin, S., Zhao, C.Z., et al.: Designing solid-state electrolytes for safe, energy-dense batteries. Nat. Rev. Mater. 5, 229–252 (2020). https://doi.org/10.1038/s41578-019-0165-5
Chen, R.S., Li, Q.H., Yu, X.Q., et al.: Approaching practically accessible solid-state batteries: stability issues related to solid electrolytes and interfaces. Chem. Rev. 120, 6820–6877 (2020). https://doi.org/10.1021/acs.chemrev.9b00268
Cheng, X.B., Zhang, R., Zhao, C.Z., et al.: A review of solid electrolyte interphases on lithium metal anode. Adv. Sci. 3, 1500213 (2016). https://doi.org/10.1002/advs.201500213
Sun, C.W., Liu, J., Gong, Y.D., et al.: Recent advances in all-solid-state rechargeable lithium batteries. Nano Energy 33, 363–386 (2017). https://doi.org/10.1016/j.nanoen.2017.01.028
Bachman, J.C., Muy, S.: Inorganic solid-state electrolytes for lithium batteries: mechanisms and properties governing ion conduction. Chem. Rev. 116, 140–162 (2016). https://doi.org/10.1021/acs.chemrev.5b00563
Wu, Y.J., Wang, S., Li, H., et al.: Progress in thermal stability of all-solid-state-Li-ion-batteries. InfoMat 3, 827–853 (2021). https://doi.org/10.1002/inf2.12224
Yan, W.L., Wu, F., Li, H., et al.: Application of Si based anodes in sulfide solid-state batteries. Energy Storage Sci. Technol. 10, 821–835 (2021). http://esst.cip.com.cn/EN/Y2021/V10/I3/821
Wu, F., Liu, L.L., Wang, S., et al.: Solid state ionics-selected topics and new directions. Prog. Mater. Sci. 126, 100921 (2022). https://doi.org/10.1016/j.pmatsci.2022.100921
Zhao, N., Khokhar, W., Bi, Z.J., et al.: Solid garnet batteries. Joule 3, 1190–1199 (2019). https://doi.org/10.1016/j.joule.2019.03.019
Abouali, S., Yim, C.H., Merati, A., et al.: Garnet-based solid-state Li batteries: from materials design to battery architecture. ACS Energy Lett. 6, 1920–1941 (2021). https://doi.org/10.1021/acsenergylett.1c00401
Wang, H.C., Sheng, L., Yasin, G., et al.: Reviewing the current status and development of polymer electrolytes for solid-state lithium batteries. Energy Storage Mater. 33, 188–215 (2020). https://doi.org/10.1016/j.ensm.2020.08.014
Tan, S.J., Zeng, X.X., Ma, Q., et al.: Recent advancements in polymer-based composite electrolytes for rechargeable lithium batteries. Electrochem. Energy Rev. 1, 113–138 (2018). https://doi.org/10.1007/s41918-018-0011-2
López-Aranguren, P., Reynaud, M., Głuchowski, P., et al.: Crystalline LiPON as a bulk-type solid electrolyte. ACS Energy Lett. 6, 445–450 (2021). https://doi.org/10.1021/acsenergylett.0c02336
Kamaya, N., Homma, K., Yamakawa, Y., et al.: A lithium superionic conductor. Nat. Mater. 10, 682–686 (2011). https://doi.org/10.1038/nmat3066
Kato, Y., Hori, S., Saito, T., et al.: High-power all-solid-state batteries using sulfide superionic conductors. Nat. Energy 1, 16030 (2016). https://doi.org/10.1038/nenergy.2016.30
Han, F.D., Gao, T., Zhu, Y.J., et al.: Electrochemical stability of Li10GeP2S12 and Li7La3Zr2O12 solid electrolytes. Adv. Energy Mater. 6, 1501590 (2016). https://doi.org/10.1002/aenm.201501590
Fitzhugh, W., Wu, F., Ye, L.H., et al.: A high-throughput search for functionally stable interfaces in sulfide solid-state lithium ion conductors. Adv. Energy Mater. 9, 1900807 (2019). https://doi.org/10.1002/aenm.201900807
Wu, F., Fitzhugh, W., Ye, L.H., et al.: Advanced sulfide solid electrolyte by core-shell structural design. Nat. Commun. 9, 4037 (2018). https://doi.org/10.1038/s41467-018-06123-2
Liu, L.L., Wu, F., Li, H., et al.: Advances in electrochemical stability of sulfide solid-state electrolyte. J. Chin. Ceram. Soc. 47, 1367–1385 (2019)
Fitzhugh, W., Wu, F., Ye, L.H., et al.: Strain-stabilized ceramic-sulfide electrolytes. Small 15, 1901470 (2019). https://doi.org/10.1002/smll.201901470
Haruyama, J., Sodeyama, K., Han, L.Y., et al.: Space-charge layer effect at interface between oxide cathode and sulfide electrolyte in all-solid-state lithium-ion battery. Chem. Mater. 26, 4248–4255 (2014). https://doi.org/10.1021/cm5016959
Zhu, Y.Z., He, X.F., Mo, Y.F.: Origin of outstanding stability in the lithium solid electrolyte materials: insights from thermodynamic analyses based on first-principles calculations. ACS Appl. Mater. Interfaces 7, 23685–23693 (2015). https://doi.org/10.1021/acsami.5b07517
Wang, Y., Lv, Y., Su, Y.B., et al.: 5V-class sulfurized spinel cathode stable in sulfide all-solid-state batteries. Nano Energy 90, 106589 (2021). https://doi.org/10.1016/j.nanoen.2021.106589
Richards, W.D., Miara, L.J., Wang, Y., et al.: Interface stability in solid-state batteries. Chem. Mater. 28, 266–273 (2016). https://doi.org/10.1021/acs.chemmater.5b04082
Wang, S., Fang, R.Y., Li, Y.T., et al.: Interfacial challenges for all-solid-state batteries based on sulfide solid electrolytes. J. Materiomics 7, 209–218 (2021). https://doi.org/10.1016/j.jmat.2020.09.003
Ye, L.H., Li, X.: A dynamic stability design strategy for lithium metal solid state batteries. Nature 593, 218–222 (2021). https://doi.org/10.1038/s41586-021-03486-3
Xin, S., You, Y., Wang, S.F., et al.: Solid-state lithium metal batteries promoted by nanotechnology: progress and prospects. ACS Energy Lett. 2, 1385–1394 (2017). https://doi.org/10.1021/acsenergylett.7b00175
Zhang, Z.H., Wu, L.P., Zhou, D., et al.: Flexible sulfide electrolyte thin membrane with ultrahigh ionic conductivity for all-solid-state lithium batteries. Nano Lett. 21, 5233–5239 (2021). https://doi.org/10.1021/acs.nanolett.1c01344
Wan, H.L., Liu, S.F., Deng, T., et al.: Bifunctional interphase-enabled Li10GeP2S12 electrolytes for lithium-sulfur battery. ACS Energy Lett. 6, 862–868 (2021). https://doi.org/10.1021/acsenergylett.0c02617
Shi, Y.N., Zhou, D., Li, M.Q., et al.: Surface engineered Li metal anode for all-solid-state lithium metal batteries with high capacity. ChemElectroChem 8, 386–389 (2021). https://doi.org/10.1002/celc.202100010
Peng, J., Wu, D.X., Song, F.M., et al.: High current density and long cycle life enabled by sulfide solid electrolyte and dendrite-free liquid lithium anode. Adv. Funct. Mater. 32, 2105776 (2022). https://doi.org/10.1002/adfm.202105776
Muramatsu, H., Hayashi, A., Ohtomo, T., et al.: Structural change of Li2S-P2S5 sulfide solid electrolytes in the atmosphere. Solid State Ion. 182, 116–119 (2011). https://doi.org/10.1016/j.ssi.2010.10.013
Liu, L.L., Xu, J.R., Wang, S., et al.: Practical evaluation of energy densities for sulfide solid-state batteries. eTransportation 1, 100010 (2019)
Chen, X.F., Guan, Z.Q., Chu, F.L., et al.: Air-stable inorganic solid-state electrolytes for high energy density lithium batteries: challenges, strategies, and prospects. InfoMat 4, e12248 (2022). https://doi.org/10.1002/inf2.12248
Zhao, S., Zhu, X.X., Jiang, W., et al.: Fundamental air stability in solid-state electrolytes: principles and solutions. Mater. Chem. Front. 5, 7452–7466 (2021). https://doi.org/10.1039/d1qm00951f
Galven, C., Dittmer, J., Suard, E., et al.: Instability of lithium garnets against moisture. Structural characterization and dynamics of Li7-xHxLa3Sn2O12 and Li5-xHxLa3Nb2O12. Chem. Mater. 24, 3335–3345 (2012). https://doi.org/10.1021/cm300964k
Yow, Z.F., Oh, Y.L., Gu, W.Y., et al.: Effect of Li+/H+ exchange in water treated Ta-doped Li7La3Zr2O12. Solid State Ion. 292, 122–129 (2016). https://doi.org/10.1016/j.ssi.2016.05.016
Bohnke, O., Lorant, S., Roffat, M., et al.: Fast H+/Li+ ion exchange in Li0.30La0.57TiO3 nanopowder and films in water and in ambient air. Solid State Ion. 262, 563–567 (2014). https://doi.org/10.1016/j.ssi.2013.08.008
Xia, W.H., Xu, B.Y., Duan, H.N., et al.: Reaction mechanisms of lithium garnet pellets in ambient air: the effect of humidity and CO2. J. Am. Ceram. Soc. 100, 2832–2839 (2017). https://doi.org/10.1111/jace.14865
Wang, Y., Wu, Y.J., Wang, Z.X., et al.: Doping strategy and mechanism for oxide and sulfide solid electrolytes with high ionic conductivity. J. Mater. Chem. A 10, 4517–4532 (2022). https://doi.org/10.1039/d1ta10966a
Wang, S.H., Xu, X.W., Cui, C., et al.: Air sensitivity and degradation evolution of halide solid state electrolytes upon exposure. Adv. Funct. Mater. 32, 2108805 (2022). https://doi.org/10.1002/adfm.202108805
Li, W.H., Liang, J.W., Li, M.S., et al.: Unraveling the origin of moisture stability of halide solid-state electrolytes by in situ and operando synchrotron X-ray analytical techniques. Chem. Mater. 32, 7019–7027 (2020). https://doi.org/10.1021/acs.chemmater.0c02419
Harding, J.R., Amanchukwu, C.V., Hammond, P.T., et al.: Instability of poly(ethylene oxide) upon oxidation in lithium-air batteries. J. Phys. Chem. C 119, 6947–6955 (2015). https://doi.org/10.1021/jp511794g
Hayashi, A., Muramatsu, H., Ohtomo, T., et al.: Improvement of chemical stability of Li3PS4 glass electrolytes by adding MxOy (M = Fe, Zn, and Bi) nanoparticles. J. Mater. Chem. A 1, 6320–6326 (2013). https://doi.org/10.1039/c3ta10247e
Li, J., Chen, H.W., Shen, Y.B., et al.: Covalent interfacial coupling for hybrid solid-state Li ion conductor. Energy Stor. Mater. 23, 277–283 (2019). https://doi.org/10.1016/j.ensm.2019.05.002
Liang, J.W., Chen, N., Li, X.N., et al.: Li10Ge(P1–xSbx)2S12 lithium-ion conductors with enhanced atmospheric stability. Chem. Mater. 32, 2664–2672 (2020). https://doi.org/10.1021/acs.chemmater.9b04764
Sahu, G., Lin, Z., Li, J.C., et al.: Air-stable, high-conduction solid electrolytes of arsenic-substituted Li4SnS4. Energy Environ. Sci. 7, 1053–1058 (2014). https://doi.org/10.1039/c3ee43357a
Park, K.H., Oh, D.Y., Choi, Y.E., et al.: Solution-processable glass LiI-Li4SnS4 superionic conductors for all-solid-state Li-ion batteries. Adv. Mater. 28, 1874–1883 (2016). https://doi.org/10.1002/adma.201505008
Saienga, J., Martin, S.W.: The comparative structure, properties, and ionic conductivity of LiI + Li2S + GeS2 glasses doped with Ga2S3 and La2S3. J. Non Cryst. Solids 354, 1475–1486 (2008). https://doi.org/10.1016/j.jnoncrysol.2007.08.058
Ohtomo, T., Hayashi, A., Tatsumisago, M., et al.: Characteristics of the Li2O-Li2S-P2S5 glasses synthesized by the two-step mechanical milling. J. Non Cryst. Solids 364, 57–61 (2013). https://doi.org/10.1016/j.jnoncrysol.2012.12.044
Hayashi, A., Muramatsu, H., Ohtomo, T., et al.: Improved chemical stability and cyclability in Li2S-P2S5-P2O5-ZnO composite electrolytes for all-solid-state rechargeable lithium batteries. J. Alloys Compd. 591, 247–250 (2014). https://doi.org/10.1016/j.jallcom.2013.12.191
Zhang, Z.X., Zhang, L., Yan, X.L., et al.: All-in-one improvement toward Li6PS5Br-based solid electrolytes triggered by compositional tune. J. Power Sources 410(411), 162–170 (2019). https://doi.org/10.1016/j.jpowsour.2018.11.016
Liu, G.Z., Xie, D.J., Wang, X.L., et al.: High air-stability and superior lithium ion conduction of Li3+3xP1−xZnxS4−xOx by aliovalent substitution of ZnO for all-solid-state lithium batteries. Energy Storage Mater. 17, 266–274 (2019). https://doi.org/10.1016/j.ensm.2018.07.008
Chen, T., Zhang, L., Zhang, Z.X., et al.: Argyrodite solid electrolyte with a stable interface and superior dendrite suppression capability realized by ZnO co-doping. ACS Appl. Mater. Interfaces 11, 40808–40816 (2019). https://doi.org/10.1021/acsami.9b13313
Ahmad, N., Zhou, L., Faheem, M., et al.: Enhanced air stability and high Li-ion conductivity of Li6.988P2.994Nb0.2S10.934O0.6 glass-ceramic electrolyte for all-solid-state lithium-sulfur batteries. ACS Appl. Mater. Interfaces 12, 21548–21558 (2020). https://doi.org/10.1021/acsami.0c00393
Pearson, R.G.: Hard and soft acids and bases. J. Am. Chem. Soc. 85, 3533–3539 (1963). https://doi.org/10.1021/ja00905a001
Kimura, T., Kato, A., Hotehama, C., et al.: Preparation and characterization of lithium ion conductive Li3SbS4 glass and glass-ceramic electrolytes. Solid State Ion. 333, 45–49 (2019). https://doi.org/10.1016/j.ssi.2019.01.017
Wang, Y.Q., Lü, X., Zheng, C., et al.: Chemistry design towards a stable sulfide-based superionic conductor Li4Cu8Ge3S12. Angew. Chem. Int. Ed. 58, 7673–7677 (2019). https://doi.org/10.1002/anie.201901739
Zhang, Z.R., Zhang, J.X., Sun, Y.L., et al.: Li4-xSbxSn1−xS4 solid solutions for air-stable solid electrolytes. J. Energy Chem. 41, 171–176 (2020). https://doi.org/10.1016/j.jechem.2019.05.015
Kwak, H., Park, K.H., Han, D., et al.: Li+ conduction in air-stable Sb-Substituted Li4SnS4 for all-solid-state Li-ion batteries. J. Power Sources 446, 227338 (2020). https://doi.org/10.1016/j.jpowsour.2019.227338
Zhao, F.P., Liang, J.W., Yu, C., et al.: A versatile Sn-substituted argyrodite sulfide electrolyte for all-solid-state Li metal batteries. Adv. Energy Mater. 10, 1903422 (2020). https://doi.org/10.1002/aenm.201903422
Tan, D.H.S., Banerjee, A., Deng, Z., et al.: Enabling thin and flexible solid-state composite electrolytes by the scalable solution process. ACS Appl. Energy Mater. 2, 6542–6550 (2019). https://doi.org/10.1021/acsaem.9b01111
Li, Y., Arnold, W., Thapa, A., et al.: Stable and flexible sulfide composite electrolyte for high-performance solid-state lithium batteries. ACS Appl. Mater. Interfaces 12, 42653–42659 (2020). https://doi.org/10.1021/acsami.0c08261
Jung, W.D., Jeon, M., Shin, S.S., et al.: Functionalized sulfide solid electrolyte with air-stable and chemical-resistant oxysulfide nanolayer for all-solid-state batteries. ACS Omega 5, 26015–26022 (2020). https://doi.org/10.1021/acsomega.0c03453
Zhu, Y.Z., Mo, Y.F.: Materials design principles for air-stable lithium/sodium solid electrolytes. Angew. Chem. Int. Ed. 59, 17472–17476 (2020). https://doi.org/10.1002/anie.202007621
Wang, C.H., Liang, J.W., Zhao, Y., et al.: All-solid-state lithium batteries enabled by sulfide electrolytes: from fundamental research to practical engineering design. Energy Environ. Sci. 14, 2577–2619 (2021). https://doi.org/10.1039/d1ee00551k
Fukushima, A., Hayashi, A., Yamamura, H., et al.: Mechanochemical synthesis of high lithium ion conducting solid electrolytes in a Li2S-P2S5-Li3N system. Solid State Ion. 304, 85–89 (2017). https://doi.org/10.1016/j.ssi.2017.03.010
Park, K.H., Bai, Q., Kim, D.H., et al.: Design strategies, practical considerations, and new solution processes of sulfide solid electrolytes for all-solid-state batteries. Adv. Energy Mater. 8, 1800035 (2018). https://doi.org/10.1002/aenm.201800035
Pearson, R.G.: Absolute electronegativity and hardness correlated with molecular orbital theory. Proc. Natl. Acad. Sci. USA. 83, 8440–8441 (1986). https://doi.org/10.1073/pnas.83.22.8440
Klopman, G.: Chemical reactivity and the concept of charge- and frontier-controlled reactions. J. Am. Chem. Soc. 90, 223–234 (1968). https://doi.org/10.1021/ja01004a00210.1021/ja01004a002
Shang, S.L., Yu, Z.X., Wang, Y., et al.: Origin of outstanding phase and moisture stability in a Na3P1–xAsxS4 superionic conductor. ACS Appl. Mater. Interfaces 9, 16261–16269 (2017). https://doi.org/10.1021/acsami.7b03606
Kim, J.S., Jeon, M., Kim, S., et al.: Structural and electronic descriptors for atmospheric instability of Li-thiophosphate using density functional theory. Solid State Ion. 346, 115225 (2020). https://doi.org/10.1016/j.ssi.2020.115225
Iwasaki, R., Hori, S., Kanno, R., et al.: Weak anisotropic lithium-ion conductivity in single crystals of Li10GeP2S12. Chem. Mater. 31, 3694–3699 (2019). https://doi.org/10.1021/acs.chemmater.9b00420
Kim, J.S., Jung, W.D., Shin, S.S., et al.: Roles of polymerized anionic clusters stimulating for hydrolysis deterioration in Li7P3S11. J. Phys. Chem. C 125, 19509–19516 (2021). https://doi.org/10.1021/acs.jpcc.1c05034
Xu, M., Song, S.B., Daikuhara, S., et al.: Li10GeP2S12-type structured solid solution phases in the Li9+δP3+δ′S12–kOk system: controlling crystallinity by synthesis to improve the air stability. Inorg. Chem. 61, 52–61 (2022). https://doi.org/10.1021/acs.inorgchem.1c01748
Kanazawa, K., Yubuchi, S., Hotehama, C., et al.: Mechanochemical synthesis and characterization of metastable hexagonal Li4SnS4 solid electrolyte. Inorg. Chem. 57, 9925–9930 (2018). https://doi.org/10.1021/acs.inorgchem.8b01049
Calpa, M., Rosero-Navarro, N.C., Miura, A., et al.: Chemical stability of Li4PS4I solid electrolyte against hydrolysis. Appl. Mater. Today 22, 100918 (2021). https://doi.org/10.1016/j.apmt.2020.100918
Tufail, M.K., Zhou, L., Ahmad, N., et al.: A novel air-stable Li7Sb0.05P2.95S10.5I0.5 superionic conductor glass-ceramics electrolyte for all-solid-state lithium-sulfur batteries. Chem. Eng. J. 407, 127149 (2021). https://doi.org/10.1016/j.cej.2020.127149
Lee, Y., Jeong, J., Lee, H.J., et al.: Lithium argyrodite sulfide electrolytes with high ionic conductivity and air stability for all-solid-state Li-ion batteries. ACS Energy Lett. 7, 171–179 (2022). https://doi.org/10.1021/acsenergylett.1c02428
Lu, P.S., Liu, L.L., Wang, S., et al.: Superior all-solid-state batteries enabled by a gas-phase-synthesized sulfide electrolyte with ultrahigh moisture stability and ionic conductivity. Adv. Mater. 33, 2100921 (2021). https://doi.org/10.1002/adma.202100921
Khurram Tufail, M., Ahmad, N., Zhou, L., et al.: Insight on air-induced degradation mechanism of Li7P3S11 to design a chemical-stable solid electrolyte with high Li2S utilization in all-solid-state Li/S batteries. Chem. Eng. J. 425, 130535 (2021). https://doi.org/10.1016/j.cej.2021.130535
Li, Y.Y., Li, J.W., Cheng, J., et al.: Enhanced air and electrochemical stability of Li7P3S11-based solid electrolytes enabled by aliovalent substitution of SnO2. Adv. Mater. Interfaces 8, 2100368 (2021). https://doi.org/10.1002/admi.202100368
Tian, Y.S., Sun, Y.Z., Hannah, D.C., et al.: Reactivity-guided interface design in Na metal solid-state batteries. Joule 3, 1037–1050 (2019). https://doi.org/10.1016/j.joule.2018.12.019
Xu, J.R., Li, Y.X., Lu, P.S., et al.: Water-stable sulfide solid electrolyte membranes directly applicable in all-solid-state batteries enabled by superhydrophobic Li+-conducting protection layer. Adv. Energy Mater. 12, 2102348 (2022). https://doi.org/10.1002/aenm.202102348
Joos, M., Schneider, C., Münchinger, A., et al.: Impact of hydration on ion transport in Li2Sn2S5·xH2O. J. Mater. Chem. A 9, 16532–16544 (2021). https://doi.org/10.1039/d1ta04736a
Cho, W., Kim, W.J., Lee, D.G., et al.: Enhanced air-stability of argyrodite solid electrolyte by introducing zeolite additive as H2S scavenger. Meet. Abstr. MA2020-02, 951 (2020). https://doi.org/10.1149/ma2020-025951mtgabs
Ye, L.H., Gil-González, E., Li, X.: Li9.54Si1.74(P1−xSbx)1.44S11.7Cl0.3: a functionally stable sulfide solid electrolyte in air for solid-state batteries. Electrochem. Commun. 128, 107058 (2021). https://doi.org/10.1016/j.elecom.2021.107058
Ohtomo, T., Hayashi, A., Tatsumisago, M., et al.: Suppression of H2S gas generation from the 75Li2S·25P2S5 glass electrolyte by additives. J. Mater. Sci. 48, 4137–4142 (2013). https://doi.org/10.1007/s10853-013-7226-8
Zhao, F.P., Alahakoon, S.H., Adair, K., et al.: An air-stable and Li-metal-compatible glass-ceramic electrolyte enabling high-performance all-solid-state Li metal batteries. Adv. Mater. 33, 2006577 (2021). https://doi.org/10.1002/adma.202006577
Kaib, T., Haddadpour, S., Kapitein, M., et al.: New lithium chalcogenidotetrelates, LiChT: synthesis and characterization of the Li+-conducting tetralithium ortho-sulfidostannate Li4SnS4. Chem. Mater. 24, 2211–2219 (2012). https://doi.org/10.1021/cm3011315
Zhang, Q., Cao, D.X., Ma, Y., et al.: Sulfide-based solid-state electrolytes: synthesis, stability, and potential for all-solid-state batteries. Adv. Mater. 31, 1901131 (2019). https://doi.org/10.1002/adma.201901131
Ozekmekci, M., Salkic, G., Fellah, M.F.: Use of zeolites for the removal of H2S: a mini-review. Fuel Process. Technol. 139, 49–60 (2015). https://doi.org/10.1016/j.fuproc.2015.08.015
Sigot, L., Ducom, G., Germain, P.: Adsorption of hydrogen sulfide (H2S) on zeolite (Z): retention mechanism. Chem. Eng. J. 287, 47–53 (2016). https://doi.org/10.1016/j.cej.2015.11.010
Lee, D.G., Park, K.H., Kim, S.Y., et al.: Critical role of zeolites as H2S scavengers in argyrodite Li6PS5Cl solid electrolytes for all-solid-state batteries. J. Mater. Chem. A 9, 17311–17316 (2021). https://doi.org/10.1039/d1ta04799j
Ren, H.T., Zhang, Z.Q., Zhang, J.Z., et al.: Improvement of stability and solid-state battery performances of annealed 70Li2S–30P2S5 electrolytes by additives. Rare Met. 41, 106–114 (2022). https://doi.org/10.1007/s12598-021-01804-2
Tao, Y.C., Chen, S.J., Liu, D., et al.: Lithium superionic conducting oxysulfide solid electrolyte with excellent stability against lithium metal for all-solid-state cells. J. Electrochem. Soc. 163, A96–A101 (2015). https://doi.org/10.1149/2.0311602jes
Raj, R., Wolfenstine, J.: Current limit diagrams for dendrite formation in solid-state electrolytes for Li-ion batteries. J. Power Sources 343, 119–126 (2017). https://doi.org/10.1016/j.jpowsour.2017.01.037
Lu, Y., Zhao, C.Z., Yuan, H., et al.: Critical current density in solid-state lithium metal batteries: mechanism, influences, and strategies. Adv. Funct. Mater. 31, 2009925 (2021). https://doi.org/10.1002/adfm.202009925
Peng, L.F., Chen, S.Q., Yu, C., et al.: Enhancing moisture and electrochemical stability of the Li5.5PS4.5Cl1.5 electrolyte by oxygen doping. ACS Appl. Mater. Interfaces 14, 4179–4185 (2022). https://doi.org/10.1021/acsami.1c21561
Xu, H.J., Cao, G.Q., Shen, Y.L., et al.: Enabling argyrodite sulfides as superb solid-state electrolyte with remarkable interfacial stability against electrodes. Energy Environ. Mater (2022). https://doi.org/10.1002/eem2.12282
Xie, D.J., Chen, S.J., Zhang, Z.H., et al.: High ion conductive Sb2O5-doped β-Li3PS4 with excellent stability against Li for all-solid-state lithium batteries. J. Power Sources 389, 140–147 (2018). https://doi.org/10.1016/j.jpowsour.2018.04.021
Zhao, B.S., Wang, L., Chen, P., et al.: Congener substitution reinforced Li7P2.9Sb0.1S10.75O0.25 glass-ceramic electrolytes for all-solid-state lithium-sulfur batteries. ACS Appl. Mater. Interfaces 13, 34477–34485 (2021). https://doi.org/10.1021/acsami.1c10238
Wu, L.P., Liu, G.Z., Wan, H.L., et al.: Superior lithium-stable Li7P2S8I solid electrolyte for all-solid-state lithium batteries. J. Power Sources 491, 229565 (2021). https://doi.org/10.1016/j.jpowsour.2021.229565
Rajagopal, R., Cho, J.U., Subramanian, Y., et al.: Preparation of highly conductive metal doped/substituted Li7P2S8Br(1–x)Ix type lithium superionic conductor for all-solid-state lithium battery applications. Chem. Eng. J. 428, 132155 (2022). https://doi.org/10.1016/j.cej.2021.132155
Deiseroth, H.J., Kong, S.T., Eckert, H., et al.: Li6PS5X: a class of crystalline Li-rich solids with an unusually high li+ mobility. Angew. Chem. Int. Ed. 47, 755–758 (2008). https://doi.org/10.1002/anie.200703900
Taklu, B.W., Su, W.N., Nikodimos, Y., et al.: Dual CuCl doped argyrodite superconductor to boost the interfacial compatibility and air stability for all solid-state lithium metal batteries. Nano Energy 90, 106542 (2021). https://doi.org/10.1016/j.nanoen.2021.106542
Zhou, L., Tufail, M.K., Ahmad, N., et al.: Strong interfacial adhesion between the Li2S cathode and a functional Li7P2.9Ce0.2S10.9Cl0.3 solid-state electrolyte endowed long-term cycle stability to all-solid-state lithium-sulfur batteries. ACS Appl. Mater. Interfaces 13, 28270–28280 (2021). https://doi.org/10.1021/acsami.1c06328
Yu, Z.X., Shang, S.L., Seo, J.H., et al.: Exceptionally high ionic conductivity in Na3P0.62As0.38S4 with improved moisture stability for solid-state sodium-ion batteries. Adv. Mater. 29, 1605561 (2017). https://doi.org/10.1002/adma.201605561
Jiang, Z., Peng, H.L., Liu, Y., et al.: A versatile Li6.5In0.25P0.75S5I sulfide electrolyte triggered by ultimate-energy mechanical alloying for all-solid-state lithium metal batteries. Adv. Energy Mater. 11, 2101521 (2021). https://doi.org/10.1002/aenm.202101521
Subramanian, Y., Rajagopal, R., Ryu, K.S.: Synthesis, air stability and electrochemical investigation of lithium superionic bromine substituted argyrodite (Li6−xPS5−xCl1.0Brx) for all-solid-state lithium batteries. J. Power Sources 520, 230849 (2022). https://doi.org/10.1016/j.jpowsour.2021.230849
Min, S., Park, C., Yoon, I., et al.: Enhanced electrochemical stability and moisture reactivity of Al2S3 doped argyrodite solid electrolyte. J. Electrochem. Soc. 168, 070511 (2021). https://doi.org/10.1149/1945-7111/ac0f5c
Holzmann, T., Schoop, L.M., Ali, M.N., et al.: Li0.6 [Li0.2Sn0.8S2]: a layered lithium superionic conductor. Energy Environ. Sci. 9, 2578–2585 (2016). https://doi.org/10.1039/c6ee00633g
Kuhn, A., Holzmann, T., Nuss, J., et al.: A facile wet chemistry approach towards unilamellar tin sulfide nanosheets from Li4xSn1–xS2 solid solutions. J. Mater. Chem. A 2, 6100–6106 (2014). https://doi.org/10.1039/c3ta14190j
Brant, J.A., Massi, D.M., Holzwarth, N.A.W., et al.: Fast lithium ion conduction in Li2SnS3: synthesis, physicochemical characterization, and electronic structure. Chem. Mater. 27, 189–196 (2015). https://doi.org/10.1021/cm5037524
Choi, Y.E., Park, K.H., Kim, D.H., et al.: Coatable Li4SnS4 solid electrolytes prepared from aqueous solutions for all-solid-state lithium-ion batteries. Chemsuschem 10, 2605–2611 (2017). https://doi.org/10.1002/cssc.201700409
Matsuda, R., Kokubo, T., Phuc, N.H.H., et al.: Preparation of ambient air-stable electrolyte Li4SnS4 by aqueous ion-exchange process. Solid State Ion. 345, 115190 (2020). https://doi.org/10.1016/j.ssi.2019.115190
Xu, J., Liu, L., Yao, N., et al.: Liquid-involved synthesis and processing of sulfide-based solid electrolytes, electrodes, and all-solid-state batteries. Mater. Today Nano 8, 100048 (2019). https://doi.org/10.1016/j.mtnano.2019.100048
Schiwy, W., Pohl, S., Krebs, B.: Darstellung und struktur von Na4SnS4—14H2O. Z. Anorg. Allg. Chem. 402, 77–86 (1973). https://doi.org/10.1002/zaac.19734020110
Heo, J.W., Banerjee, A., Park, K.H., et al.: New Na-ion solid electrolytes Na4−xSn1−xSbxS4 (0.02 ≤ x ≤ 0.33) for all-solid-state Na-ion batteries. Adv. Energy Mater. 8, 1702716 (2018). https://doi.org/10.1002/aenm.201702716
Jia, H.H., Sun, Y.L., Zhang, Z.R., et al.: Group 14 element based sodium chalcogenide Na4Sn0.67Si0.33S4 as structure template for exploring sodium superionic conductors. Energy Storage Mater. 23, 508–513 (2019). https://doi.org/10.1016/j.ensm.2019.04.011
Xiong, S., Liu, Z.T., Yang, L.F., et al.: Anion and cation co-doping of Na4SnS4 as sodium superionic conductors. Mater. Today Phys. 15, 100281 (2020). https://doi.org/10.1016/j.mtphys.2020.100281
Wang, H., Chen, Y., Hood, Z.D., et al.: An air-stable Na3SbS4 superionic conductor prepared by a rapid and economic synthetic procedure. Angew. Chem. Int. Ed. 55, 8551–8555 (2016). https://doi.org/10.1002/anie.201601546
Zhang, L., Zhang, D.C., Yang, K., et al.: Vacancy-contained tetragonal Na3SbS4 superionic conductor. Adv. Sci. 3, 1600089 (2016). https://doi.org/10.1002/advs.201600089
Banerjee, A., Park, K.H., Heo, J.W., et al.: Na3SbS4: a solution processable sodium superionic conductor for all-solid-state sodium-ion batteries. Angew. Chem. Int. Ed. 55, 9634–9638 (2016). https://doi.org/10.1002/anie.201604158
Kim, T.W., Park, K.H., Choi, Y.E., et al.: Aqueous-solution synthesis of Na3SbS4 solid electrolytes for all-solid-state Na-ion batteries. J. Mater. Chem. A 6, 840–844 (2018). https://doi.org/10.1039/c7ta09242c
Hayashi, A., Masuzawa, N., Yubuchi, S., et al.: A sodium-ion sulfide solid electrolyte with unprecedented conductivity at room temperature. Nat. Commun. 10, 5266 (2019). https://doi.org/10.1038/s41467-019-13178-2
Fuchs, T., Culver, S.P., Till, P., et al.: Defect-mediated conductivity enhancements in Na3–xPn1–xWxS4 (Pn = P, Sb) using aliovalent substitutions. ACS Energy Lett. 5, 146–151 (2020). https://doi.org/10.1021/acsenergylett.9b02537
Yubuchi, S., Ito, A., Masuzawa, N., et al.: Aqueous solution synthesis of Na3SbS4–Na2WS4 superionic conductors. J. Mater. Chem. A 8, 1947–1954 (2020). https://doi.org/10.1039/c9ta02246e
Tsuji, F., Yubuchi, S., Sakuda, A., et al.: Preparation of sodium-ion-conductive Na3–xSbS4–xClx solid electrolytes. J. Ceram. Soc. Japan 128, 641–647 (2020). https://doi.org/10.2109/jcersj2.20089
Wang, X.L., Xiao, R.J., Li, H., et al.: Oxysulfide LiAlSO: a lithium superionic conductor from first principles. Phys. Rev. Lett. 118, 195901 (2017). https://doi.org/10.1103/physrevlett.118.195901
Kuo, D.H., Lo, R., Hsueh, T.H., et al.: LiSnOS/gel polymer hybrid electrolyte for the safer and performance-enhanced solid-state LiCoO2/Li lithium-ion battery. J. Power Sources 429, 89–96 (2019). https://doi.org/10.1016/j.jpowsour.2019.05.010
Gamon, J., Duff, B.B., Dyer, M.S., et al.: Computationally guided discovery of the sulfide Li3AlS3 in the Li-Al-S phase field: structure and lithium conductivity. Chem. Mater. 31, 9699–9714 (2019). https://doi.org/10.1021/acs.chemmater.9b03230
Sedlmaier, S.J., Indris, S., Dietrich, C., et al.: Li4PS4I: a Li+ superionic conductor synthesized by a solvent-based soft chemistry approach. Chem. Mater. 29, 1830–1835 (2017). https://doi.org/10.1021/acs.chemmater.7b00013
Zhou, L.D., Assoud, A., Zhang, Q., et al.: New family of argyrodite thioantimonate lithium superionic conductors. J. Am. Chem. Soc. 141, 19002–19013 (2019). https://doi.org/10.1021/jacs.9b08357
Sun, X., Stavola, A.M., Cao, D.X., et al.: Operando EDXRD study of all-solid-state lithium batteries coupling thioantimonate superionic conductors with metal sulfide. Adv. Energy Mater. 11, 2002861 (2021). https://doi.org/10.1002/aenm.202002861
Lee, Y., Jeong, J., Lim, H.D., et al.: Superionic Si-substituted lithium argyrodite sulfide electrolyte Li6+xSb1–xSixS5I for all-solid-state batteries. ACS Sustain. Chem. Eng. 9, 120–128 (2021). https://doi.org/10.1021/acssuschemeng.0c05549
Richards, W.D., Tsujimura, T., Miara, L.J., et al.: Design and synthesis of the superionic conductor Na10SnP2S12. Nat. Commun. 7, 11009 (2016). https://doi.org/10.1038/ncomms11009
Zhang, Z., Ramos, E., Lalère, F., et al.: Na11Sn2PS12: a new solid state sodium superionic conductor. Energy Environ. Sci. 11, 87–93 (2018). https://doi.org/10.1039/c7ee03083e
Duchardt, M., Ruschewitz, U., Adams, S., et al.: Vacancy-controlled Na+ superion conduction in Na11Sn2PS12. Angew. Chem. Int. Ed. 57, 1351–1355 (2018). https://doi.org/10.1002/anie.201712769
Ramos, E.P., Zhang, Z.Z., Assoud, A., et al.: Correlating ion mobility and single crystal structure in sodium-ion chalcogenide-based solid state fast ion conductors: Na11Sn2PnS12 (Pn = Sb, P). Chem. Mater. 30, 7413–7417 (2018). https://doi.org/10.1021/acs.chemmater.8b02077
Weng, W., Liu, G.Z., Shen, L., et al.: High ionic conductivity and stable phase Na11.5Sn2Sb0.5Ti0.5S12 for all-solid-state sodium batteries. J. Power Sources 512, 230485 (2021). https://doi.org/10.1016/j.jpowsour.2021.230485
Liu, G.Z., Sun, X.R., Yu, X.Q., et al.: Na10SnSb2S12: a nanosized air-stable solid electrolyte for all-solid-state sodium batteries. Chem. Eng. J. 420, 127692 (2021). https://doi.org/10.1016/j.cej.2020.127692
Fan, B., Xu, Y.H., Ma, R., et al.: Will sulfide electrolytes be suitable candidates for constructing a stable solid/liquid electrolyte interface? ACS Appl. Mater. Interfaces 12, 52845–52856 (2020). https://doi.org/10.1021/acsami.0c16899
Liu, G.Z., Shi, J.M., Zhu, M.T., et al.: Ultra-thin free-standing sulfide solid electrolyte film for cell-level high energy density all-solid-state lithium batteries. Energy Storage Mater. 38, 249–254 (2021). https://doi.org/10.1016/j.ensm.2021.03.017
Tsukasaki, H., Igarashi, K., Wakui, A., et al.: In situ observation of the deterioration process of sulfide-based solid electrolytes using airtight and air-flow TEM systems. Microscopy 70, 519–525 (2021). https://doi.org/10.1093/jmicro/dfab022
Kim, Y., Saienga, J., Martin, S.W.: Glass formation in and structural investigation of Li2S + GeS2 + GeO2 composition using Raman and IR spectroscopy. J. Non Cryst. Solids 351, 3716–3724 (2005). https://doi.org/10.1016/j.jnoncrysol.2005.09.028
Ohtomo, T., Hayashi, A., Tatsumisago, M., et al.: All-solid-state batteries with Li2O-Li2S-P2S5 glass electrolytes synthesized by two-step mechanical milling. J. Solid State Electrochem. 17, 2551–2557 (2013). https://doi.org/10.1007/s10008-013-2149-5
Yohannan, J.P., Vidyasagar, K.: Syntheses, structural variants and characterization of AInM’S4 (A=alkali metals, Tl; M’ = Ge, Sn) compounds; facile ion-exchange reactions of layered NaInSnS4 and KInSnS4 compounds. J. Solid State Chem. 238, 291–302 (2016). https://doi.org/10.1016/j.jssc.2016.03.045
Ohtomo, T., Hayashi, A., Tatsumisago, M., et al.: Glass electrolytes with high ion conductivity and high chemical stability in the system LiI-Li2O-Li2S-P2S5. Electrochemistry 81, 428–431 (2013). https://doi.org/10.5796/electrochemistry.81.428
Sahu, G., Rangasamy, E., Li, J.C., et al.: A high-conduction Ge substituted Li3AsS4 solid electrolyte with exceptional low activation energy. J. Mater. Chem. A 2, 10396–10403 (2014). https://doi.org/10.1039/c4ta01243g
Acknowledgements
This work is supported by the Key Program-Automobile Joint Fund of the National Natural Science Foundation of China (Grant No. U1964205), the Key R&D Project funded by the Department of Science and Technology of Jiangsu Province (Grant No. BE2020003), the General Program of the National Natural Science Foundation of China (Grant No. 51972334), the General Program of the National Natural Science Foundation of Beijing (Grant No. 2202058), the Cultivation Project of Leading Innovative Experts in Changzhou City (CQ20210003), the National Overseas High-Level Expert Recruitment Program (Grant No. E1JF021E11), the Talent Program of the Chinese Academy of Sciences, “Scientist Studio Program Funding” from the Yangtze River Delta Physics Research Center and the Tianmu Lake Institute of Advanced Energy Storage Technologies (Grant No. TIES-SS0001).
Author information
Authors and Affiliations
Corresponding authors
Ethics declarations
Conflict of interest
The authors declare no competing interests.
Rights and permissions
About this article

Cite this article
Lu, P., Wu, D., Chen, L. et al. Air Stability of Solid-State Sulfide Batteries and Electrolytes. Electrochem. Energy Rev. 5, 3 (2022). https://doi.org/10.1007/s41918-022-00149-3
Received:
Revised:
Accepted:
Published:
DOI: https://doi.org/10.1007/s41918-022-00149-3