
Abstract
Currently, Fe-N-C materials are considered to be among the most important oxygen reduction reaction (ORR) catalysts, because they are potential substitutes for Pt-based catalysts and are therefore promising in the development of non-noble metal-based catalysts. However, challenges such as electron transfer kinetics still exist and need to be improved upon. From a chemical stand point, improvements can be made through the better understanding of mechanisms in Fe-N-C-based ORR catalysis along with a deeper understanding of the chemical origin of active sites on Fe-N-C catalyst surfaces. Based on these, this comprehensive review will focus on the energy conversion, transformation kinetics and electron transfer of the ORR process as catalyzed by Fe-N-C catalysts. And by taking these and other relevant analytical results for Fe-N-C materials into consideration, primary strategies in the improvement in Fe-N-C catalyst activity will be presented.
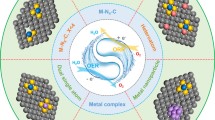
Graphical Abstract
As the promising Pt substrate for oxygen reduction catalysis, the Fe-N-C materials are active toward the four-electron reduction of O2 to H2O. This review focuses on the profound understanding of heterogeneous oxygen reduction reaction on Fe-N-C materials from the following aspects: (1) thermodynamics of energy conversion in ORR processes, (2) kinetics of ORR processes based on Fe-N-C catalysts, (3) the textural features of Fe-N-C and analytic results known as far, (4) fundamental principle for Fe-N-C materials synthesis and (5) practical application for fuel cell and metal–air batteries.

Similar content being viewed by others


Avoid common mistakes on your manuscript.
1 Introduction
Environmentally friendly energy storage through the reversible conversion of chemical energy to electrical energy through devices such as fuel cells and metal–air batteries is vital to the abatement of pollution and greenhouse effects in which a shift away from carbon and fossil fuel (coal, oil and gas)-driven economies is required globally [1]. However, a major bottleneck in the commercialization of these devices is the requirement for oxygen reduction reactions (ORR) in their operation, in which high overpotential for ORR reactions due to sluggish kinetics at the cathode exists [2]. Here, platinum (Pt) and its alloys, possessing appropriate oxygen binding energies (Fig. 1a), have been demonstrated to enhance ORR performances [3, 4]. However, the use of Pt possesses substantial economic challenges due to its scarcity and cost (Fig. 1b), making Pt-based applications significantly more costly than earth-abundant first-row (3d) transition metals (Fe, Co, Ni). There is an additional problem, which has to do with the surface chemistry of Pt. Catalysts based on Pt tend to gets deactivated rather quickly due to the inevitable phenomena of methanol crossover (the diffusion of the methanol fuel from the anode to the cathode) [5]. In fact, (1) methanol electro-oxidation of the crossed-over methanol at the cathode and (2) the poisoning effect by CO due to incomplete methanol oxidation are two factors that would reduce the efficiency of ORR reactions. This is known to degrade the performance of the fuel cell in which Pt catalysts are employed. Therefore, significant efforts have been devoted to the identification of cost-effective and methanol/CO-tolerant non-noble metal electrocatalysts, including transition metal and nitrogen co-doped carbon-based catalysts (M-N-C) [6, 7], as well as metal-free heteroatom-doped carbon catalysts [8,9,10].

(Part a adapted with permission from Ref. [3]; Copyright 2004 American Chemical Society. Part b adapted with permission from Ref. [11]; Copyright 2012 Royal Society of Chemistry.)
a Volcano curve of activity as a function of oxygen binding energy in which Pt is the most active metal for ORR. b Annual production-dependent market prices of VIII metals
Owing to the unique structural and surface stability of Fe-N-C catalysts and their robustness against methanol/CO poisoning [12], these materials have garnered the attention of researchers in ORR applications and there has been an exponential increase in the articles being published on Fe-N-C materials as ORR catalysts (Fig. 2a) based on publication and citation statistics from the Web of Science, along with an exponentially growing citation trend (Fig. 2b), suggesting that this research area is progressively growing. One may note that from the stand point of a larger impact, Fe-N-C ORR materials are also relevant to other areas, such as selective oxidation reaction [13,14,15,16], CO2 reduction [17, 18] and hydrogenation [19, 20]. Therefore, a timely comprehensive review on this rapidly growing category of Fe-N-C for ORR catalysis is of great significance to a vast body of readers.

Articles based on Fe-N-C catalysts for oxygen reduction reactions: a histogram of the number of articles and b citations in recent years. Statistics data are based on indexing keywords of oxygen reduction reaction and Fe-N-C on the Web of Science
Till now, several review articles [2, 21,22,23,24,25,26], related or referred to the preparation and electrochemical performance of Fe-N-C materials for ORR catalysis, have been published. A comprehensive review specifically focusing on the structure of Fe-N-C and the activity determined factor as principle for this kind catalysts’ design are definitely necessary to facilitate future research and development in this area. In this review, we engage to the profound understanding of heterogeneous oxygen reduction reaction on Fe-N-C materials from the following aspects: (1) the thermodynamics of energy conversion in the ORR process, (2) the kinetics of the ORR process based on Fe-N-C catalysts, (3) the textural features of Fe-N-C and analytic results known as far, (4) the fundamental principles for Fe-N-C materials synthesis and (5) the practical applications in fuel cells and metal-air batteries. In each of these cases, open challenges and the way forward will be described.
2 Thermodynamic Description of the ORR Process
Thermodynamics, the study of energy transformation, enables the quantitative discussion and prediction of chemical reaction energetics [27]. In fuel cells, the high efficiency of energy conversion (i.e., from chemical to electrical) in terms of the theoretical thermodynamics makes it be a promising technique for an energy-sufficient in future.
For example, in hydrogen fuel cells:
Here the product (water) is in liquid form. At standard conditions of 25 °C (298 K) and 1 atm, the thermal energy (ΔH) of the hydrogen/oxygen reaction is 285.8 kJ mol−1, and the free energy available for useful work is 237.1 kJ mol−1. Based on this, the thermal efficiency of ideal fuel cells operating reversibly on pure hydrogen and oxygen at standard conditions is:
Then, an actual cell thermal efficiency is:
At standard state, in which Eideal = 1.23 V, and hence:
In a fuel cell, oxygen (O2) is electrochemically reduced at the cathode through the mediation of ORR. Taking the overpotential (Eop) into consideration for a real fuel cell, the actual cell efficiency is found to be:
Therefore, a high EORR with low Eop is anticipated, where the EORR depends on the process of electron transfer pathway and the Eop is up to the cathodic electrode materials and the electrolyte.
The complete electrochemical ORR involves four net coupled proton and electron transfers. And in general, the mechanisms of ORR follow two pathways (Fig. 3), i.e., the (1) direct 4-electron reduction pathway from O2 to H2O (in acidic media) or to OH− (in alkaline media); (2) 2-electron pathway which involves the formation of an intermediate of hydrogen peroxide molecule, depending on the electrode material chosen and pH of the electrolyte [3, 28]. Thermodynamically, the EORR for the 4e− reduction pathway is 1.23 V, and the 2e− pathway is 0.68 V, meaning that the 4e− pathway is more efficient and hence desirable.

ORR pathways in acidic and alkaline solution, in which a 4-electron reduction pathway with high potential is desirable
3 Kinetic Mechanisms of ORR Processes
A catalyst is defined by the IUPAC as a substance that can increase the rate of a reaction without modifying the overall standard Gibbs energy change occurring through the process. That is, the kinetic mechanism of reaction can be changed by a catalyst, and doing so would not affect the thermodynamics at all. As a promising non-precious metal catalysis, studying the kinetics of ORR on Fe-N-C is very useful in understanding the mechanism of ORR and for further improving the catalytic performance. For electrochemical catalysis, the kinetic mechanisms of substance transformation and electron transfer can in fact be investigated individually.
3.1 Substance Transformation
Typically, in acidic solutions, the substance transformation of ORR is best described by using the Damjanovic model [30] in Fig. 4a. The ORR is considered to follow one of the two different reduction pathways; at times both of them can occur during the reduction of O2 to water. Taking advantage of Damjanovic model and using the method developed by Hsueh et al., the Ohsaka group [31, 32] calculated and compared the reaction rate constants of k1, k2 and k3 by hydrodynamic (rotating ring–disk electrode) voltammetry. It is found that the Fe-N-C formed by Fe introduction is active toward the four-electron reduction of O2 and small amounts of Fe species leads to a rather large increase in k1 than in k2 [32]. A suitably chosen cationic surfactant can modulate the ORR selectivity on a Fe-N-C catalyst by kinetic promotion through Coulombic interaction with the peroxo-Fe complexes [33]. As the first step during ORR, the adsorption and activation of O2 on catalysts involves associative or dissociative pathways, or a combination of these. Using in situ IR spectroscopy, the associative pathway has been demonstrated. The primary signature of this is the observation of surface-adsorbed superoxide (OOHad) and hydroperoxide (HOOHad). It may be noted that the breaking of O–O bond only occurs at the peroxo level [34]. The Ohsaka group’s work has addressed the adsorption equilibrium between the adsorbed and desorbed H2O2 near the disk surface. And a Wroblowa model (Fig. 4b) is employed to distinct the parallel-sequential path and the sequential paths of the ORR on Fe-N-C materials. The compared results (Fig. 4c) suggest that the Damjanovic model always overestimates the k1 values and underestimates the k2 values [29].

a Damjanovic and b Wroblowa model for ORR. c Variations of the rate constants k1 and k2 on Fe-N-C catalysts using Damjanovic (solid lines) and Wroblowa models (dotted lines). (Part a, b and c adapted with permission from Ref. [29]; Copyright 2015 American Chemical Society.)
3.2 Electron Transfer
Mukerjee’s research [35] reveals that there exists two-electron transfer pathway for ORR: (1) the inner-sphere electron transfer (ISET) mechanism (Fig. 5a, inset (1) involves chemisorption of desolvated O2 onto an active site leading to a direct/series 4e− ORR pathway without the desorption of reaction intermediates, (2) an outer-sphere electron transfer (OSET) mechanism [Fig. 5a, inset (2)], wherein the noncovalent hydrogen bonding forces between specifically adsorbed hydroxyl species (OHads acting as an outer-sphere bridge) and solvated O2 (localized in the outer-Helmholtz plane) promote a 2e− reduction step forming intermediates.

(Part a, b and c adapted with permission from Ref. [35]; Copyright 2013 American Chemical Society.)
a Schematic illustration of the inner-sphere (inset i) and outer-sphere (inset ii) electron transfer mechanisms during ORR. b ORR activity (ID) and concomitant ring current (IR) due to hydrogen peroxide oxidation. c Proposed ORR mechanism on Fe-N-C in alkaline solution
As shown in Fig. 5b for ORR in alkaline media (pH > 12), the characteristic ring–electrode signature at ~ 0.8 V for peroxide-intermediate formation through the outer-sphere process is clearly absent on Fe-N-C. This is because Lewis basic anionic hydrogen peroxide intermediates (HO2−, pKa ≈ 11.6) can be stably adsorbed and immediately reduced to the 4e− product on Lewis acidic Fe2+ active sites via the formation of stabilized Lewis acid–base adduct [35]. This ensures that the catalytic cycle in alkaline medium undergoes complete 4e− transfer (Fig. 5c) to regenerate the active site via the formation of ferric-hydroxyl species. The ORR activity is essentially governed by the dynamic structure associated with the Fe2+/3+ redox transition (Fig. 5c), rather than the static structure of bare sites [36]. The neutral intermediate H2O2 negates its Lewis basic character and undermines its stabilization on Fe2+ active sites, facilitating the 2e− ORR pathway and generating H2O2 (as in Fig. 5b).
From above, it may be noted that the kinetics of ORR is determined to a large extent by the choice of electrode materials and the electrolyte chosen. Evidently a clear understanding of the kinetic mechanisms involved in ORR on Fe-N-C would facilitate further research to design, synthesize and investigate ORR catalysts.
4 Fe-N-C Structure
The Fe atom in Fe-N-C materials could be coordinated by the pyridine/pyrrole nitrogen, carbon or a combination of C and N [37]. The different coordination environments give rise to the different property and hence lead to varied ORR activities. For the sake of the effective catalyst design, it is hence reasonable to identify and ideally, quantify the Fe-N-C structure–ORR catalytic activity correlations [38, 39].
4.1 Identification
The synthesis conditions used for making Fe-N-C materials, for instance pyrolysis temperature [40, 41] and protected atmosphere [42], result in different models of the Fe-N-C configurations with two- to six-coordinated nitrogen (pyridinic or pyrrolic nitrogen atoms) [43,44,45]. As shown in Fig. 6, it has been reported that ideally in a hypothetical graphene sheet (parent system); the Fe-N-C structure would likely include Fe-N2 at the edge defects (Fig. 6a); Fe-N4 in-plane defects of graphene sheet (Fig. 6b–d), Fe-N5 and Fe-N6 with one or two additional pyridinic N perpendicular to graphene sheet is to be expected as well (Fig. 6e–f), respectively [44, 46]. And based on the form of the surrounding carbon atoms, Fe-N configurations can be divided into several sub-categories such as in the case of pyridinic N4-Fe, sites embedded in an intact graphitic layer and surrounded by ten carbon atoms can form FeN4C10 in Fig. 6b, or in the case of 8 surrounding carbon atoms, FeN4C8 can exist on the edge of micropores to bridge two adjacent armchair-like graphitic edges (Fig. 6c) [38].

Atomic structure of Fe-N-C sites supported on graphene with different coordination environments. Gray ball: carbon; Red ball: iron; Blue ball: nitrogen
To unravel the nature of Fe-N-C structure, the techniques of X-ray photoelectron spectroscopic (XPS), electron microscopy, Mössbauer spectroscopic and X-ray absorption fine structure (XAFS) are mostly available. XPS is effective to elucidate the chemical composition and nitrogen bonding configuration for Fe-N-C materials. In Fig. 7a, five differentiate peaks in the deconvoluted N1s spectra of UK 65, a Fe-N-C material pyrolyzed from iron(III) chloride 5, 10, 15, 20-tetrakis(4-methoxyphenyl) porphyrin (FeClTMPP) as the precursor for Fe, N and C in an inert gas, is assigned to pyrrolic (Pyr-N), Fe coordinated (Fe-N), pyrrolic (Pyr-N), graphitic-like (G-like) and oxidized type (Py-N-O) nitrogen, respectively [47]. Drawing from this, Shen et al. [48] have studied the evolution of the N1s spectrum for single-atom dispersed Fe-N-C samples prepared via template casting methods with different amounts of Fe [48]. For iron phthalocyanine (FePc)-derived Fe-N-C materials, the pyrrolic N keeps associating with Fe, resulting in the presence of two peaks for N1s (pyrrolic N with Fe bonding—denoted as Nα and the pyridinic N with carbon bonding—denoted as Nβ). Compared with FePc, Deng et al. [14] found that part of the pyridinic species on the outside macrocycle was destroyed during ball milling whereas Fe-N4 (pyrrolic nitrogen) was well retained. This peak differentiation principle is widely used in the research on structure of the Fe-N-C catalysts and their correlations with the electrochemical performance for ORR [49]. Though the XPS is reasonable to study the Fe–N bonding, it is found to be insufficient to delineate the details of nitrogen coordination which is relevant for ORR catalytic activity.

(Part a adapted with permission from Ref. [47]; Copyright 2009 American Chemical Society. Part b, c and d adapted with permission from Ref. [50]; Copyright 2018 John Wiley and Sons.)
a N1s narrow scan spectrum of UK-65 (a Fe-N-C material) and its deconvolution. b Fe K-edge, c XANES and d EXAFS spectra of materials with corresponding EXAFS fitting curves
As a sensitive characterization method to identify non-crystalline species, X-ray absorption spectroscopy (XAS) including X-ray absorption near edge structure (XANES) and extended X-ray absorption fine structure (EXAFS) spectrum recorded at the Fe K-edge is developed to explore the coordination environment of iron. For example, the absorption edge of the XANES of Fe in the Fe-N-C sample (SA-Fe-N-1.5-800) is close to but exhibits a slightly negative shift as compared with reference Fe2O3 samples (Fig. 7b), suggesting that the average valence state of Fe in SA-Fe-N-1.5-800 is slightly lower than + 3 [50]. Correspondingly, the Fourier transform (FT) of the Fe K-edge EXAFS spectra, as shown in Fig. 7c is more intuitive and the coordination number for Fe could be obtained through data fitting (Fig. 7d). Theoretically, the bond length of Fe–N is shorter than that of Fe–C and Fe–Fe, and slightly longer than that of Fe–O. The absence of M–M bond as evaluated by XAS indicates the complete dispersion of metal in materials is isolated single atom [51]. Confirmed by EXAFS, as listed in Table 1, the Fe-N-C materials with atomically dispersed Fe-Nx configuration is successfully synthesized via carbonizing Fe, N, C involves precursors (the mixture, polymer or Fe-N4 contained organic molecule) under 600–1000 °C at inert atmosphere or ammonia along with/without post-acidic treatment.
Commonly, pure FeO, Fe2O3 or iron phthalocyanine are used as references to clarify that Fe in Fe-N-C materials is coordinated with nitrogen but not oxygen [49, 50, 54]. However, XAS/EXAFS is censorable to distinguish the Fe–O and Fe–N, since their bond length is similar. And therefore the 57Fe Mössbauer spectroscopy, based on the recoil-free absorption of γ rays by Fe57 nuclei [46], is a powerful tool for the identification of iron species [38, 57], in which different coordination environments result in the change of isomer shift (IS) and a quadrupole splitting (QS). By taking advantage of 57Fe Mössbauer spectra, Dodelet group and cooperators achieved a number of instructive results on Fe-N-C structure [42, 58,59,60]. For example, a catalyst containing the highest concentration of FeN4 sites can be achieved through the purification of porphyrin-based catalysts with a second heat treatment in forming gas (10% H2 in N2) followed by a secondary acid-leaching step. Mössbauer spectra in Fig. 8 reveal that the acid-unstable inorganic metal species can be dramatically reduced in some cases, even down to zero through the purification treatment. In this case, only three doublet peaks (Fig. 8c) are observed which are assigned to the electronically different FeN4 sites (Table 2). And without the interference from inorganic metal species, the ORR activity is tremendously increased in Fig. 8e, f [60]. Unexpectedly, a new doublet with an IS of 0.30 mm s−1 and a QS of 0.63 mm s−1 appears and can be attributed to the Fe2N or –Fe2+xN with x = 0.1 [38]. And the new active Fe-N-C site with an IS of 0.60 mm s−1 and a QS of 3.13 mm s−1 is reported. These peaks are assigned to Fe-N2 moiety and correspond to an average coordination number of 2 for N (obtained from EXAFS fitting) [48].

(Part a, b, c, d, e and f adapted with permission from Ref. [60]; Copyright 2016 American Chemical Society.)
57Fe Mössbauer spectra of (Fe,Fe) and (Fe, Co) catalysts before a, b and after c, d purification treatment. e, f The corresponding ORR current densities at a rotation rate of 900 rpm in 0.5 MH2SO4
Electron microscopy and associated techniques have contributed significantly to the characterization of heterogeneous catalysts and can provide nano- or atomic-scale information on the structure, morphology, composition and electronic state of areas of interest [61]. With advancement in aberration corrections to achieve an image resolution below 0.1 nm and with the rapid development of in situ techniques, advanced electron microscopy is poised to probe fundamental questions of Fe-N-C catalysts [54, 62]. As shown in Fig. 9a–c, the atomic structure of FeN4 centers in graphene is revealed for the first time through a combining high-resolution transmission electron microscopy/high-angle annular dark-field scanning transmission electron microscopy with low-temperature scanning tunneling microscopy (STM) [14]. The Fe-N-C centers embedded in the plane of a graphene matrix are a desired model for STM. However, for most Fe-N-C materials obtained through high-temperature pyrolysis, possessing complex textural property with rough surfaces is not appropriate for STM. In most of the published work, high-resolution transmission electron microscopy (HRTEM) with elemental mapping is employed for preliminary estimation of dispersion of Fe and N in Fe-N-C materials [16, 52, 56, 63,64,65,66]. In situ electrochemical scanning tunneling microscopy (ECSTM) has been carried out to investigate the iron–phthalocyanine (FePc)-catalyzed oxygen reduction reaction on Au(111) terrace [66]. A tetragonal structure with a bright spot at the center and four additional spots at the corners (as seen in Fig. 9d) is observed, corresponding to iron and the benzene rings, respectively. The transformation between the FePc molecules (the dim spots) and the FePc-O2 complex (the bright spots) is reversible under applied potential, as it varies from 350 to 50 mV in oxygen-saturated 0.1 M HClO4. In addition, theoretical calculations were conducted in this study based on the results and indicated that the lowest unoccupied molecular orbital (LUMO) of FePc-O2 was largely localized at the center of the phthalocyanine framework and further extended to the O-O part (Fig. 9e), while it is homogeneous distributed on the Pc framework (Fig. 9f).

(Part a, b and c adapted with permission from Ref. [14]; Copyright 2015 American Association for the Advancement of Science. Part d, e and f adapted with permission from Ref. [67]; Copyright 2016 John Wiley and Sons.)
Structural analysis of a FeN4 (FeN4/GN) catalyst. a High-resolution transmission electron microscopy (HRTEM) image. b High-angle annular dark-field scanning transmission electron microscopy (HAADF-STEM) image. Inset is the electron energy loss spectroscopy (EELS) atomic spectra of elemental Fe and N from the bright dots. c Low-temperature scanning tunneling microscopy (LS-STM) image. d High-resolution STM image of a FePc monolayer on Au (111) surface. Top views of the electron density distribution in e FePc-O2 and f FePc
It is to be noted that hitherto the complex Fe-N-C structure is not completely identified; it is a result that ought to be corroborated further. This hence presents a rather interesting puzzle for both catalyst researchers and those who work on advanced characterization techniques.
4.2 Quantification
Turnover frequency (TOF), calculated as the derivative of the number of turnovers of the catalytic cycle with respect to the time per active site, is a direct evaluation parameter for intrinsic activity comparisons on different active sites [68], in which active site quantifications are key to TOF calculations. And unlike precious metal (Pt, Pd, Ru) [69,70,71]-based catalysts, titration by under potential deposition (UPD) of copper, H2 or CO stripping titration is inactive for Fe-N-C materials. However recently, significant improvements have been made for Fe-N-C quantification, making it relevant for the further development and rational design of Fe-N-C catalysts.
Ideally, without interference of inorganic metal sites, the densities of pristine Fe-N-C moieties on materials can be quantified from the information of either Fe or N. Through peak differentiation on deconvolution of N1s XPS spectra (Fig. 10a), the amount N in the form of Fe-N is thus determined by Shen et al. [48, 73]. As the average coordination number of N is 2.0 confirmed by EXAFS, the amount Fe-N-C therefore is half of that of N at. % present in Fe-N. Since low Fe concentrations are not detected by routine XPS, Jason et al. [74] digested the sample using a mixture of nitric acid and hydrochloric acid in 5:1 ratio and measured the Fe content by inductively coupled plasma optical emission spectroscopy (ICP-OES). The total reflection X-ray fluorescence spectroscopy (TXRF) is also a reliable means for iron content determination in the Fe-N-C catalysts [72]. Another relevant approach would be neutron activation analysis (NAA) as is introduced by Kramm [60], to determine the concentrations of mononuclear Fe-N4 moieties. However, we cannot regard all the Fe in the catalyst as active sites for a large proportion is likely to be inactive phases or in-accessably buried in the carbon skeleton [42, 75]. Similar to CO adsorbed on platinum, Daniel [39, 72] found that nitrite anion can strongly interact with Fe-N-C to form a stable poisoned catalyst adduct 3, as shown in Fig. 10b. The as-formed adduct 3 can be reduced and stripped at very low potential (Fig. 10c), along with complete recovery of ORR activity (Fig. 10d). This stripping process makes the in situ quantification of Fe-N-C sites for ORR reasonable. So that the gravimetric site density (MSD) of active sites can be calculated as:
where Qstrip is the excess Coulometric charge associated with the stripping peak (Fig. 10c), nstrip is the number of electrons associated with the reduction in one adsorbed nitrosyl per site, F is the Faraday constant. On the other hand, if one is to simply count the amount of Fe using the nitrite stripping, it would result in an underestimation of active sites in view of that the ORR activity of poisoned Fe-N-C material is still much higher than metal-free N-C material (Fig. 10d).

(Part a adapted with permission from Ref. [48]; Copyright 2017 Elsevier. Part b adapted with permission from Ref. [72]; Copyright 2016 American Chemical Society. Parts c and d adapted with permission from Ref. [39]; Copyright 2016 Nature Publishing Group.)
a Deconvoluted N1s of XPS. b Proposed reaction mechanism of Fe-N-C active sites after treatment with a nitrite containing solution and subsequent treatment under different pH conditions for the hybrid catalysts. c Homogeneous reduction in aqueous nitrite and excess current associated with the reductive stripping of intermediates. d ORR performance of catalyst layers before, during and after nitrite adsorption
5 Activity of Fe-N-C
High activities can be achieved for Fe-N-C catalysts in alkaline media, and the nature of the active sites has been progressively investigated in recent studies [16, 54, 76]. However, in acidic medium, Fe-N-C catalysts still exhibit higher overpotentials and lower stabilities, despite significant improvements. This is because as has been discussed in Sect. 3.2, in acidic medium, the neutral intermediate H2O2 negates the Lewis basic character and undermines stabilization on Fe2+ active sites, driving the 2e− ORR pathway and generating H2O2 (as in Fig. 5b) [35]. For ORR on Fe-N-C materials, to improve the activity (i.e., for facilitating substance transformation and to enhance electron transfer rate), similar to the traditional heterogeneous catalyst, there are two strategies: (1) increasing the intrinsic activity of Fe-N-C or (2) increasing the number of active site on a given electrode (e.g., through increased loading or tuned catalyst structure to expose more active sites per gram) [77]. Besides, the activity of electrochemical catalyst can also be enhanced by the improvement in conductivity for fast electron transport.
5.1 Improve Intrinsic Activity of Fe-N-C
Since the adsorption of O2 on carbon and nitrogen atoms is rather weak, the O2 molecule is mainly activated on the Fe atom of Fe-N-C catalysts. The spin-polarized DFT computations reveal that the 2p orbitals of O2 are hybridized with the 3d orbitals of Fe in both spin-up and spin-down channels and that the O2–2π* state can be partially occupied in the spin-down channel due to the charge transfer from the Fe-N-C monolayer to the O2 molecule [78]. The d 2z orbital of Fe, which can be controlled through the incorporation of functional groups, would split into bonding and anti-bonding states before and after the adsorption of O2. Subsequently the relative energy position of the d 2z orbital with respect to the Fermi level can induce an Fe redox couple potential shift and modulate the catalytic activity toward ORR [79]. Hence, the intrinsic activity of Fe-N-C relies on the coordination environments toward Fe, including the coordination number of N, the property of the coordinated N and any other invasive factors.
As a stable structure, Fe-N4 with different states of N exhibits varied performance for ORR. One important attempt to correlate bonding in the solid with observed ORR activity has come through the work by Xu et al. [37]. They took the number of valence electrons in the d orbital (θd) and electronegativity into consideration together, to arrive at a corresponding descriptor (φ) for activity prediction:
where θd is the number of valence electrons in the occupied d orbital of the metal element, EM represents the electronegativity of metal element, EN and EC represent the electronegativity of nitrogen and carbon elements, nN and nC represent the number of nearest-neighbor N and C atoms, and α is the correction coefficient.
A volcano relationship (Fig. 11a) between onset potentials for ORR and the descriptor φ is observed in both theoretical and corresponding experimental verification [80]. The volcano curve indicates that the intrinsic activity of Fe-N4C10 with four pyridinic nitrogen atoms is higher than that of Fe-N4C12 (Fig. 6d) with four pyrrolic nitrogen atoms. Furthermore for the Fe-N4 moiety with four pyridinic nitrogens, Liu et al. [36, 80] took advantage of DFT formalism and conducted calculations to reveal that the activation energy for O–O bond breakage in FeN4C8 is about 0.20 eV, less than that for FeN4C10 of 0.56 eV (Fig. 11b). This suggests that the intrinsic catalytic activity for ORR on FeN4C8 is higher than FeN4C10 [36, 81]. And Koslowski et al. show that a C-Fe-N2 center where iron is bonded to two nitrogen atoms and one carbon atom, favor a 2-electron ORR that yields hydrogen peroxide and that it is less active than an in-plane Fe-N4 center [50, 82]. Compared with Fe-N4 in Fig. 11c, the edge site Fe-N2 (iron is not directly bonded to carbon) achieves superior intrinsic activity due to a lower interaction with *O2 and *OH intermediates and enhanced electron transport [48, 83,84,85]. In the work of Dodelet and co-workers, both Fe-N2 and Fe-N4 are shown to coexist in the carbon-supported materials with a ratio determined by preparation conditions [43, 84].

(Part a adapted with permission from Ref. [37]; Copyright 2018 Nature Publishing Group. Part b adapted with permission from Ref. [80]; Copyright 2017 American Chemical Society. Parts c and d adapted with permission from Ref. [48]; Copyright 2017 Elsevier.)
a Theoretical and corresponding experimental onset potentials for ORR versus the descriptor φ. b Calculated free energy evolution diagrams for O2 reduction at the active sites of FeN4-C10, FeN4-C12 and FeN4-C8. c Free energy diagram of the ORR on FeN2 and FeN4 sites. d Synthesis procedure for the Fe-N2 catalyst (FeN2/NOMC)
To summarize, the intrinsic activity of different active sites on Fe-N-C can be listed in the order of: Fe-N2 > Fe-N4C8 > Fe-N4C10 > Fe-N4C12 > C-Fe-N2. And hence, a catalyst consisting of abundant Fe-N-C site with high intrinsic activity is desirable; in particular there would be advantages to have sufficient Fe-N2 species on the edge of carbon support. For example in Fig. 11d, a nominal Fe-N2-based catalyst (FeN2/NOMC) is synthesized using an intriguing template casting strategy. In the sample, 57Fe Mössbauer spectroscopy confirmed that Fe is only coordinated by N atoms, and EXAFS at Fe K-edge X-ray absorption spectroscopy reveals that the average coordination number of N is 2.0. The well-defined structure exhibits superior performance with a more positive half-wave potential for ORR compared to Pt/C catalyst [48].
In addition to the above pointers to improve the ORR activity of Fe-N-C, it may be noted that there is scope for further improvement through the tuning of electronic structure of Fe in Fe-N-C configuration. This in fact may be possible via use of a suitable catalysts supporting either carbon with heteroatom doping or other nanoparticles. Among them, it is reported that S-doped Fe-N-C catalyst can exhibit better ORR activity than undoped Fe-N-C in both alkaline and acidic electrolyte [86,87,88,89,90,91]. This is consistent with another report [73] wherein thiophene-like structure (C-S-C) formed by S incorporation reduces the electron localization around Fe centers and improves interaction with oxygenated species, thus lower ORR activation barriers on Fe-N-C and boosts the ORR activity. In addition, Sun et al. [92] demonstrate that embedding of oxygen into the Fe-N-C structure is favorable for boosting durability and methanol endurance of the catalyst through the elongation of the inter-atomic atom distance (from the active core) [92]. Not only electronegative elements S and O, the electropositive element B [93] has also been introduced to modify the property of Fe-N-C site. As reported by Kai et al., boric acid is selected to be mixed with other precursors [94]. Then after direct pyrolysis and acid leaching, B-decorated porous carbon frameworks with atomically dispersed Fe-N-C species (denoted as FeBNC) are obtained. And the B atom around the Fe-N-C structure is more favorable to the adsorption of chemical species and catalytic activity for ORR [95]. The electropositive effect also exists on the Fe-N4 treated by NH3 and with higher coordinated N to enhance the intrinsic ORR activity [38, 44]. Factually, without post-acid leaching, the magnetic Fe/Fe3C nanoparticle is ubiquitous as a by-product in the preparation of Fe-N-C catalysts through high-temperature pyrolysis [73, 96,97,98]. Although the Fe/Fe3C is also active toward ORR [99,100,101], the activity is much less than Fe-N-C. The vicinity of Fe/Fe3C nanocrystals is not required per se [60, 72, 97, 102], but it could enable the enhancement of catalystic performance prepared via some specific preparation routes. For instance, an Fe-based material, synthesized by pyrolysis of thiourea and agarose containing α-Fe2O3 nanoplate as Fe precursor, displays excellent catalytic performance for ORR with more positive onset potential (1.1 V vs. the reversible hydrogen electrode) [103]. Likewise Jiang et al. [49] reported a facile and easily scalable method to prepare Fe-N-C materials via pyrolysis of the mixture of carbon nanotube, glucose, sodium dodecyl sulfate and nitrate nonahydrate. The resulting sample contains Fe-N-C configurations and graphene-encapsulated Fe/Fe3C nanocrystals. It is proven that the metallic iron nanoparticles reduces the charge density of central Fe atoms in neighboring Fe-N-C configurations and favors the adsorption of oxygen molecule to deliver high ORR catalytic performance.
5.2 Increase the Number of Fe-N-C Sites
Catalytic activity relies on the number of accessible active Fe-N-C sites on the surface. Here, our previous study found that ORR activity is linearly proportional to the concentration of Fe-N-C sites (Fig. 12a). Furthermore as is deduced from Butler–Volmer formulation, the half-wave potential (E1/2), a characteristic of ORR activity, is independent of the substrate concentration and is directly related to the number of active sites:
where A is the coefficient constant of current to ORR reaction rate, S is the number of active site, Eθ is equilibrium potential, F is the Faraday constant, k0 is the standard rate constant, and α is the transfer coefficient.

(Part a adapted with permission from Ref. [48]; Copyright 2017 Elsevier. Part b adapted with permission from Ref. [41]; Copyright 2014 American Chemical Society. Part c adapted with permission from Ref. [6]; Copyright 2016 American Chemical Society. Part d adapted with permission from Ref. [104]; Copyright 2015 American Chemical Society.)
a Correlation between ORR current density and Fe-N-C concentration at potentials of 0.89, 0.84 and 0.79 V. b Dependence of H2O2 yield at 0.70 V on Fe-N-C catalyst loading at the electrode surface. Synthesis scheme for c the “silica-protective-layer-assisted” approach and d the “shape fixing via salt recrystallization” method
Thus, a high content of Fe-N-C active sites is essential to deliver high ORR catalytic performance. So far, several advanced strategies have been established to improve Fe-N-C sites during catalyst preparation and electrochemical measurement.
The most straightforward method for Fe-N-C sites enhancement is to increase the materials loading during testing. As presented in Fig. 12b, Wang et al. [41] found that the H2O2 yields on Fe-N-C are inversely proportional to the catalyst loading. Once the catalyst loading increase from 0.06 to 0.6 mg cm−2, the intermediate H2O2 yield drops from 16 to < 1%. The estimated rate constants based on the Wroblowa model (Fig. 4b) reveal that at high loading density (> ca. 200 μg cm−2), k3/k5 is larger than unity, k1 ≫ k2 and k6 value decreases. These indicate that the intermediate H2O2 undergoes further reduction to H2O rather than desorption. Whereas, the lower loading density leads to the 2-electron reduction of O2, which corresponds to k1 < k2 and k3/k5 is less than unity [29]. However, the higher loading of catalysts, the thicker of modified layer, brings about the more intensive normal diffusion, especially for the three-dimensional porous materials [8].
On the other hand, significant efforts have been devoted to improve Fe-N-C active sites, either by increasing the density of Fe-N-C or enhancing the accessible area of materials. It is generally recognized that the avoidance of carbide formation during pyrolysis represents a promising way to enhance the density of ORR active sites on Fe-N-C catalysts [97]. A general “silica-protective-layer-assisted” approach in Fig. 12c is developed to preferentially generate catalytically active Fe-N-C sites in catalysts by suppressing the formation of large Fe-based particles [6]. Yang et al. propose a surfactant-assisted method to increase the active site with a small amount of iron [105]. In addition, the Fe-based particles are more often removed by post-acid etching, since iron, iron carbides or iron oxides are acid-unstable [96, 106]. The inorganic metal species in Fe-N-C catalysts can be drastically reduced by performing a second heat treatment of as-synthesized catalysts in forming gas or NH3 with a subsequent acid leaching [38, 59]. The method is very efficient since the inorganic Fe nanoparticle covered or buried in the carbon can be exposed to acid after the carbon removed via methanation under forming gas or NH3 treatment [42, 107]. Using a semi-embed strategy by pre-anchoring Fe precursor on the surface of template (SBA-15), the strong anchoring effect of SBA-15 prevents the Fe-based species to penetrate into the carbon skeleton and make the formed Fe species be accessible on the surface. In this way, different amounts of Fe-N-C sites are introduced by simply regulating the amount of impregnated Fe precursor [47]. However, it is noteworthy here that it is impossible to continuously increase Fe-N-C content by monolithically adding Fe precursor, since the transition metals such as Ni, Co and Fe are common catalysts to enhance the graphitization of carbon [108, 109] and hence lead to the evolution of doped nitrogen as well as lower specific area. Therefore, keeping balance of the invasive Fe precursor and doped nitrogen is a director to optimize the Fe-N-C materials. Interestingly, a “shape fixing via salt recrystallization” method in Fig. 12d is employed to efficiently synthesize N-doped carbon nanomaterials with a high density of Fe-N-C sites as an ORR catalyst, where the NaCl crystal functions as a fully sealed nanoreactor and facilitates the N incorporation and graphitization [104].
Currently large surface carbon with a mass of defect is used as support for Fe-N-C materials preparation. Benefiting from pore-forming technology, in addition to the typical nanocarbons such as carbon nanotube- [49, 82], nanofiber- [110] or graphene [14, 88, 111]-based catalysts, numerous novel Fe-N-C materials with tunable porous structure can be fabricated by bottom-up strategy [112]. Using the method of hard template sacrifice, the sample after carbonization can inversely replicate the porous structure of template and maintain its morphology, where the surface area is significantly determined by the selected template. For examples, silica nanoparticles, ordered mesoporous silica and montmorillonite are used as templates for achieving mesoporous structures, which reveals a strong correlation between the activity and the apparent BET surface area [113, 114]. Song et al. introduced ultrathin tellurium nanowires as template with biomass-derived d(+)-glucosamine hydrochloride and ferrous gluconate as precursors to synthesize Fe-N-C carbon nanofibers aerogels as efficient non-precious metal catalysts for ORR (Fig. 13a) [115]. In Fig. 13b [116], Wang et al. [116] report a 3D simple cubic carbon frameworks (SCCFs) with the surface decorated homogeneously with Fe-N-C moieties. It is synthesized via in situ carbonization of the surface-coating ligands on Fe3O4 3D simple cubic superlattices along with acid etching and NH3 activation. Benefiting from the homogeneously dispersed and fully accessible Fe-N-C sites, highly graphitic nature and enhanced mass transport achieved through the porous nature of the structure, these SCCFs outperformed Pt/C for ORR. It is observed that the specific surface area of N-doped carbon spheres doubles through CO2 activation treatment. It hence improves the availability of accessible N-functional groups to form high-density and homogeneous Fe-N-C moieties for boosting ORR electrocatalysis [117]. Appealingly, metal–organic frameworks (MOFs), as a novel porous material, have received wide attention in recent years and have been brought in Fe-N-C materials fabrication [53]. After rational carbonation, MOFs derived or decorated materials inherit the porous structure of corresponding MOFs with high specific surface area and uniform active sites, particularly with the zeolitic imidazolium framework (ZIFs), which has transition metal ions and nitrogen-containing organic ligands, such as ZIF-7 [76] and ZIF-8 [56, 118, 119], as the most applicable for self-sacrificed precursor/template for the preparation of nanoporous carbons. As shown in Fig. 13c [76], the 3D structure of ZIF-7 is transformed to 2D by surface functionalization with AFC, and after pyrolysis, a 2D mesoporous carbon doped with Fe-N-C active sites can be realized with interconnected and mesopore-dominated pore structure for high performance toward ORR in alkaline medium. Figure 13d [56] represents a highly stable isolated Fe-N-C catalyst with excellent ORR reactivity which is prepared via a cage encapsulated precursor pyrolysis strategy using ZIF-8 with cavity diameter of 11.6 nm and pore diameter of 3.4 nm as molecular-scale cages. Due to the confinement effect, the trapped Fe precursor in the molecular-scale cages turns into isolated single iron atoms anchored on nitrogen species during carbonization directly without acid treatment. Furthermore, a “semi-closed system” pyrolysis strategy using ZnCl2/KCl eutectic salts is adopted to control the precursor carbonization and pore-making process [120]. The molten eutectic salts create an ionic liquid-confined space for preventing the fast decomposition, cracking, shrinking and sintering of precursors. This approach also suppresses the large weight loss and prevents N evaporation of the precursor during pyrolysis. It thus contributes to the unique three-dimensional porous graphene-like structure with a high surface area and graphitization degree, as well as high density of the Fe-N-C sites. Accordingly, the as-prepared Fe-N-C catalyst exhibits superior performance with E1/2 of 0.803 V and 0.918 V in acidic and alkaline media.

(Part a adapted with permission from Ref. [115]; Copyright 2016 John Wiley and Sons. Part b adapted with permission from Ref. [116]; Copyright 2017 American Chemical Society. Part c adapted with permission from Ref. [76]; Copyright 2017 American Chemical Society. Part d adapted with permission from Ref. [56]; Copyright 2017 John Wiley and Sons.)
Schematic illustration of the synthesis of Fe-N-C catalysts using templates of a ultrathin tellurium nanowires, b Fe3O4 cubic super lattices, c ZIF-7 and d ZIF-8
5.3 Introduce Secondary Active Sites
A Fe-N-C catalyst with less H2O2 yield during ORR is crucial for displacing Pt [121]. The fundamental understanding of ORR mechanisms on Fe-N-C catalysts in acid medium is as shown in Fig. 14a. Choi et al. proposed that the desirable 4-electron reduction pathway for ORR can proceed via a direct 4e- or a consequential 2e− × 2e− mechanism on a single active site. Likewise a bifunctional 2e− + 2e− mechanism on two active sites is plausible too [57]. Thus, the introduction of a secondary active sites to catalyze the peroxide reduction reaction (PRR) is a rational approach to alleviate the issue associated with H2O2. Since the Fe particles encapsulated in N-doped carbon layers (Fe@N-C) are active toward the intermediate H2O2 in acidic solution. However, in moderation, it can play the role of secondary active sites to lower the yield of H2O2 during ORR through 2e− + 2e− mechanism. Therefore, a material containing Fe-N-C moieties and Fe@N-C exhibits high O2 reduction performance with enhanced selectivity and durability [57].

(Part a adapted with permission from Ref. [57]; Copyright 2017 John Wiley and Sons. Part b adapted with permission from Ref. [126]; Copyright 2018 Royal Society of Chemistry. Part c adapted with permission from Ref. [73]; Copyright 2017 John Wiley and Sons.)
a ORR mechanisms of Fe-N-C catalysts in acidic media and the promotion of Fe particles encapsulated in N-doped carbon layers (Fe@N-C) toward Fe-N-C catalysis through the 2e−+2e −pathway. b Relationship between the ratio of the amount of pyridinic and Fe-N-C to the amount of pyrrolic N and the ORR half-wave potential of the catalysts. c Kinetic current densities of Fe-N-C catalysts with and without S-doping in O2-saturated 0.1 M KOH at a scan rate of 10 mV s−1
To bring in extra active sites is also effective to further enhance the activity of Fe-N-C materials. As the member of M-N-C materials, the single-atom dispersed Co-N-C catalyst also shows excellent performance for O2 reduction [122,123,124]. An optimized Fe + Co-N-C sample is prepared by one-step pyrolysis of Fe/Co bimetal zeolitic imidazolate framework (Fe,Co-ZIF). It exhibits a positive half-wave potential (E1/2) of 0.875 V and 0.764 V in the alkaline and acidic electrolyte with superior methanol tolerance and electrochemical stability [125]. Actually, the site with low activity, such as remainder pyridinic, pyrrolic nitrogen or others in Fe-N-C sample, can also contribute to the ORR activity. For example, it is confirmed that the ORR activity is proportional to the ratio of the amount of pyridinic and Fe-N-C to the amount of pyrrolic N [126], because the carbon atoms with Lewis basicity next to pyridinic N are the main active sites in N-doped carbon materials [127]. As shown in Fig. 14b [126], the sample with higher ratio of the amount of pyridinic and Fe-N-C to the amount of pyrrolic N present results in a more positive E1/2. On the other hand, compared with S free Fe-N-C sample (Fe/NC), an improved ORR kinetic currents density (Jk) from C-S-C site on support is observed for the S-doped Fe-N-C catalyst (Fe/SNC) in alkaline solution (Fig. 14c) [73].
5.4 Enhance the Conductivity of Fe-N-C Materials
A material with high conductivity is approved for electrochemical catalysis. For carbon materials, the higher degree of graphitization, the better of its conductivity. Previous studies reveal that the pyrrolic N is the most unstable nitrogen, which is mainly formed at low temperatures (200–350 °C) in the nitrogen-doped carbons. When temperature reaches about 550–750 °C, the pyrollic N would transform to pyridinic N and further to graphitic-N (above 750 °C) [129,130,131,132,133,134,135,136,137]. It is hence inferred that a higher temperature gives rise to higher degree of graphitization. Besides, the introduction of non-3d high-valency Mo can not only prevent Fe species from aggregation through the dilution effect of Mo, but also enhance the graphitization degree of carbon skeleton to resulting in a high electrical conductivity of Fe-N-C catalysts [138]. However, it is indeed a double-edged sword since the graphitizing process gives rise to the sacrifice of surface area, evolution of heteroatom and destroys the active Fe-N-C structure. Otherwise, the conductivity could also be improved by structural combination. Such as in Fig. 15a [128], a porous Fe-N-C hybrid material composed of hierarchically ordered porous carbon (OPC) microblocks interlinked with CNTs is developed for catalyzing ORR. The abundant graphitic CNTs maximize the conductivity and results in the outstanding catalytic activity. Lately, a high surface area of 1380 m2 g−1 and homogeneous distribution of Fe, N-embedded hierarchical carbon framework wired onto 1D porous carbon nanotubes is rationally designed from a nucleation and growth of ZIF-8 on the surface of porous tellurium nanotubes (Fig. 15b) [65]. The 1D structures enhance the electron transport and the high surface supply enabling host for abundant access to Fe-N-C sites, contributing to a E1/2 of 0.867 and 0.818 V in alkaline and acidic media, respectively.

(Part a adapted with permission from Ref. [128]; Copyright 2014 John Wiley and Sons. Part b adapted with permission from Ref. [65]; Copyright 2017 John Wiley and Sons.)
Schematic illustration of the synthesis of a Fe-N-C NT-OPC catalysts and b Fe,N-embedded interconnected MOF-derived porous carbon nanotubes using tellurium nanotubes as a sacrificial template
6 Application for Energy Conversion Systems
6.1 Proton Exchange Membrane Fuel Cells
Membrane electrode assembly fuel cells (MEAFC, Fig. 16a) are able to efficiently convert hydrogen/oxygen chemical energy into electricity at low temperatures (normally < 90 °C) and are ideal clean-energy systems for sustainable economic development. MEAFCs can be classified into two categories based on the type of polymer electrolyte membrane (PEM) used in which one is the alkaline polymer electrolyte fuel cell (APEFC) in which the PEM conducts hydroxide ions and the other is the proton exchange membrane fuel cell (PEMFC) in which the PEM conducts protons in acidic environments [139]. Of these two, APEFCs possess several advantages over PEMFCs, including faster oxygen reduction reactions, less corrosive environments, decreased fuel crossover rates and mitigated CO poisoning [140]. However, disadvantages of APEFCs include system complexities due to water removal requirements at the cathode, the occasional need to replenish KOH electrolytes and the requirement for pure H2 and O2 gas (APEFCs cannot tolerate even atmospheric levels of CO2). And as a result, PEMFCs are currently considered to be the most suitable candidates for application in portable and automotive applications [141]. And currently, the dominant limiting factor in PEMFCs, unlike APEFCs, which is the PEM, is the electrocatalyst [140], and therefore, the focus of this review will be on the application of Fe-N-C catalysts for PEMFCs.

(Part a adapted with permission from Ref. [144]. Part b adapted with permission from Ref. [107]; Copyright 2009 American Association for the Advancement of Science. Part c adapted with permission from Ref. [145]; Copyright 2018 John Wiley and Sons. Part d adapted with permission from Ref. [146]; Copyright 2011 Nature Publishing Group.)
a Schematic of a membrane electrode assembly fuel cell (MEAFC). b Comparison of the polarization curves from H2-O2 fuel cells tested (pO = pH2 = 1.5 bar, 80 °C) using Fe-N-C catalysts with different mass loading and a Pt-based catalyst. c Polarization and power density curves (80 °C, absolute pressure = 2.5 bar) of Fe2-Z8-C, Z8-Fe2-C, and Pt/C(20%) with cathode loadings of 2.8, 2.7 and 0.33 mg cm−2, respectively. d Tafel plots of the volumetric activity of cathodes made from Fe-N-C materials
To achieve high-performance O2 electrochemical reduction, Fe-N-C catalysts are currently the most promising substitutes for Pt-based catalysts in membrane cathode fabrication, in which the current density of a MEA using Fe-N-C electrocatalysts of 5.3 mg cm−2 at the cathode is equivalent to that of a Pt-based cathode with a loading of 0.4 mg Pt cm−2 at a cell voltage of ⩾ 0.9 V (Fig. 16b) [107]. And recently, high-power densities of 1.14 and 1.11 W cm−2 at 2.5 and 2.0 bar H2/O2 were achieved using a MEA fabricated with a Fe-N-C catalyst (Fe2-Z8-C, Fig. 16c), in which 280 mW cm−2 at 0.8 V was achieved, which is higher than that of the 2020 target (250 mW cm−2) of the US Department of Energy (DOE) [142]. However, in comparison with Pt/C, the nominally high power density of the reported Fe-N-C catalyst was related to higher mass loadings. Another crucial target for MEAs using non-Pt catalysts, as per DOE guidelines, is a kinetic activity per volume of 300 A cm−3@0.8ViR-free (intersection of the extrapolated Tafel slope with the 0.8 ViR-free axis, Fig. 16d) [142, 143]. Here, ORR volumetric performance is related to catalytic activity because mass transport limitations are related to electrode thicknesses, in which a thick MEA will lower mass transport and decrease power density [107]. Therefore, it is important to attain the same kinetic current density on NPMC cathodes as Pt-based cathodes without exceeding thickness; however, the volumetric current densities of state-of-the-art Fe-N-C catalysts are still far from the DOE target (Table 3). This can be explained by the facts that Fe-N-C catalysts are carbonized at high temperatures (> 700 °C) and are more hydrophobic, and that although the porous structure of Fe-N-C catalysts can contribute to higher surface areas for active site dispersion and mass transport enhancement, it also lightens bulk density and cause these catalysts to be much thicker than Pt/C at same mass loading. Therefore, the fabrication of thin Fe/N/C catalyst layers with fast mass transfer is a key issue in future studies [91].
6.2 Metal-Air Batteries
Metal-air batteries (MABs) are attracting increasing attention from researchers due to advantages such as their cost-effectiveness and environmental compatibility as well as their high energy density [151]. In practical applications, the discharge performance of metal-air battery systems [152, 153] with either two-electrode (Fig. 17a) or tri-electrode configuration (Fig. 17b) relies on the oxygen reduction reaction at the air electrodes. Therefore, a Fe-N-C material that can facilitate ORR through the direct four-electron reduction pathway is highly preferred. There are various MABs according to the metals used [154,155,156,157,158]. Among them, Li-air and Zn-air batteries with theoretical energy density of 5200 and 1084 Wh kg−1 (including oxygen) are the most promising ones [152].

(Parts a and b adapted with permission from Ref. [152]; Copyright 2018 American Association for the Advancement of Science. Parts c and d adapted with permission from Ref. [161]; Copyright 2018 Elsevier. Part e adapted with permission from Ref. [94]; Copyright 2018 American Chemical Society. Part f adapted with permission from Ref. [162]; Copyright 2018 Royal Society of Chemistry.)
a Tri-electrode and b two-electrode systems of rechargeable metal-air batteries. c Comparison of the polarization and power density of Zn-air batteries (ZAB) between Fe-N-C and commercial Pt/C at a catalyst loading of 2.0 mg cm−2. d ORR activity of Fe-N-C and commercial Pt/C measured using rotating disk electrode (RDE) at 1600 rpm. Polarization curves and corresponding power density plots of ZABs using Fe-N-C doped with e boron and f sulfur as ORR catalysts
In Shui et al. work, atomically dispersed Fe-N-C sample is for the first time used as the cathode catalyst to improved performance of the rechargeable Li-air battery (LABs). Compared with the benchmark, which is metal oxide catalyst (α-MnO2/XC-72); LABs with Fe-N-C as the catalyst exhibit high cyclability and low overpotential during both discharge and charge processes [159]. However, the reversibility is achieved only at the smaller current densities of 0.045 mA cm−2 (making it far away from the practical applications). Using the air cathode with Fe-N-C which is uniformly dispersed on the graphene sheets, a high discharge capacity of 6762 mAh g−1 and 50 discharge–charge cycles is also achieved for LABs [160].
Accordingly, the Fe-N-C materials show promising applications for the booming activities in Zn-air battery (ZABs). An efficient ORR catalyst constructed by anchoring Fe-N4 moieties on the carbon matrix with high Fe loading (up to 3.8 wt %) exhibits 121.8 mW cm−2 for ZABs at catalyst loading of 2.0 mg cm−2, comparable to that of the Pt/C-based air cathode (Fig. 17c) [161]. It is worth noting that the ZABs performance is assessed at a static station, and the diffusion process is the rate determining step and can impact the ORR activity (Fig. 17d) during rotating disk electrode (RDE) testing under forced convection. Although the ORR activity of Fe-N-C materials is much higher than Pt/C assessed on RDE in alkaline solution, there still exists the problem of mass transport limitations for ZABs. It is confirmed that the incorporation of heteroatom can adjust the surface polarities and electronic properties of Fe-N-C materials to elevate ORR activity. In comparison with boron-free Fe-N-C catalyst in Fig. 17e, the B doped one shows 19.1% increment in peak power density during ZABs discharge process [94]. Using S-promoted Fe-N-C catalyst as air cathode for ZABs, power densities of 250 mW cm−2 (Fig. 17f) are achieved at 5 mA cm−2, double that of the untreated Fe-N-C materials [162]. In this sample, sulfur is employed as a “promoter” to increase the specific surface area via affording interpenetrating holes, enhance the dispersion of Fe to form evenly dispersed Fe-N-C site and maintain a litchi-like structure of carbon framework.
Promisingly, the Fe-N-C materials are also adopted to the rechargable ZABs in two-electrode system as bifunctional catalyst in the reports [96, 163]. However, the fraught deactivation is observed during charge and discharge cycles. It is due to the damage/oxidation on ORR activity site of Fe-N-C during charge process as Fe-N are thermodynamically less stable and can be in situ oxidized at high potential [164].
7 Challenges and the Way Forward: a Perspective
In recent years, significant progress has been made in developing Fe-N-C materials for oxygen reduction reactions in both alkaline and acidic media. Due to the developments in characterization techniques at high resolution, as well as theoretical simulations, reasonable progress has been made to elucidate these complex real catalytic systems. The potential of Fe-N-C materials is evident; however, there remain many scientific and technological challenges.
Most of the work relating to Fe-N-C for ORR being reported recently is focused on novel methods for material preparation; however, the activity seems to be stagnate [102, 165]. Especially in acidic media, the activity of Fe-N-C materials is much less than Pt/C with ~ 50 mV lower of E1/2. Is it the extreme value for actual Fe-N-C materials, although the activity of Fe-N-C materials can be further theoretically enhanced by the structural optimization?
The verified Fe-N-C-based catalysts have activities that are primarily due to their Fe-N4 active centers. However, this is not the most active site according to theoretical predictions [73, 83]. Fe-N2 sites, the most active sites for ORR still unconfirmed experimentally with solid evidence. Also fundamental questions remain, such as the coordination state of Fe in these systems remains unanswered.
A key limitation for further activity would be alleviating the rapid decay in fuel cell performance, which is usually observed with Fe-N-C catalysts at the initial testing due to the loss of iron located in the open micropore network or radical oxygen species produced by Fenton reactions [122, 166, 167].
Despite the many challenges, there is reason to be hopeful about Fe-N-C for ORR since advancements in situ and ex situ characterization techniques will continue to give insights of relevance to progressively move toward champion catalysts. The gap between theoretical and experimental value is narrowing over time and mechanistic insights are certainly getting more robust. The key reason to continue this activity may eventually be to do for its sweeping relevance for ORR, and perhaps beyond!
References
Wang, Y., Qiu, W., Song, E., et al.: Adsorption-energy-based activity descriptors for electrocatalysts in energy storage applications. National Sci. Rev. 5, 327–341 (2018)
Shao, M., Chang, Q., Dodelet, J.P., et al.: Recent advances in electrocatalysts for oxygen reduction reaction. Chem. Rev. 116, 3594–3657 (2016)
Nørskov, J.K., Rossmeisl, J., Logadottir, A., et al.: Origin of the overpotential for oxygen reduction at a fuel-cell cathode. J. Phys. Chem. B 108, 17886–17892 (2004)
Wang, Y.J., Zhao, N., Fang, B., et al.: Carbon-supported Pt-based alloy electrocatalysts for the oxygen reduction reaction in polymer electrolyte membrane fuel cells: particle size, shape, and composition manipulation and their impact to activity. Chem. Rev. 115, 3433–3467 (2015)
Li, Y., Zhou, W., Wang, H., et al.: An oxygen reduction electrocatalyst based on carbon nanotube-graphene complexes. Nat. Nanotech. 7, 394–400 (2012)
Sa, Y.J., Seo, D.J., Woo, J., et al.: A general approach to preferential formation of active Fe-Nx sites in Fe-N/C electrocatalysts for efficient oxygen reduction reaction. J. Am. Chem. Soc. 138, 15046 (2016)
Liu, Y., Jiang, H., Zhu, Y., et al.: Transition metals (Fe Co, and Ni) encapsulated in nitrogen-doped carbon nanotubes as bi-functional catalysts for oxygen electrode reactions. J. Mater. Chem. A 4, 1694–1701 (2016)
Li, J., Hou, P., Zhao, S., et al.: A 3D bi-functional porous N-doped carbon microtube sponge electrocatalyst for oxygen reduction and oxygen evolution reactions. Energy Environ. Sci. 9, 3079–3084 (2016)
Zhang, J., Dai, L.: Heteroatom-doped graphitic carbon catalysts for efficient electrocatalysis of oxygen reduction reaction. ACS Catal. 5, 7244–7253 (2015)
Tang, C., Zhang, Q.: Nanocarbon for oxygen reduction electrocatalysis: dopants, edges, and defects. Adv Mater 29(13), 1703185 (2017)
Vesborg, P.C.K., Jaramillo, T.F.: Addressing the terawatt challenge: scalability in the supply of chemical elements for renewable energy. RSC Adv. 2, 7933–7947 (2012)
Ren, G., Lu, X., Li, Y., Zhu, Y., et al.: Porous core-shell Fe3C embedded N-doped carbon nanofibers as an effective electrocatalysts for oxygen reduction reaction. ACS Appl. Mater. Interfaces 8, 4118–4125 (2016)
Saha, B., Gupta, D., Abu-Omar, M.M., et al.: Porphyrin-based porous organic polymer-supported iron(III) catalyst for efficient aerobic oxidation of 5-hydroxymethyl-furfural into 2,5-furandicarboxylic acid. J. Catal. 299, 316–320 (2013)
Deng, D., Chen, X., Yu, L., et al.: A single iron site confined in a graphene matrix for the catalytic oxidation of benzene at room temperature. Sci. Adv. 1, 1500462 (2015)
Zhang, P., Chen, X.F., Lian, J.S., et al.: Structural selectivity of CO oxidation on Fe/N/C catalysts. J. Phys. Chem. C 116, 17572–17579 (2012)
Liu, W., Zhang, L., Liu, X., et al.: Discriminating catalytically active FeNx species of atomically dispersed Fe–N–C catalyst for selective oxidation of the C–H bond. J. Am. Chem. Soc. 139, 10790–10798 (2017)
Ju, W., Bagger, A., Hao, G.P., et al.: Understanding activity and selectivity of metal-nitrogen-doped carbon catalysts for electrochemical reduction of CO2. Nat. Commun. 8, 944 (2017)
Huan, T.N., Ranjbar, N., Rousse, G., et al.: Electrochemical reduction of CO2 catalyzed by Fe–N–C materials: a structure-selectivity study. ACS Catal. 7, 1520–1525 (2017)
Xu, S., Yu, D., Liao, S., et al.: Nitrogen-doped carbon supported iron oxide as efficient catalysts for chemoselective hydrogenation of nitroarenes. RSC Adv. 6, 96431–96435 (2016)
Li, J., Jj, Zhang, Liu, H., et al.: Graphitic carbon nitride (g–C3N4)-derived Fe–N–C catalysts for selective hydrodeoxygenation of 5-hydroxymethylfurfural to 2,5-dimethylfuran. ChemistrySelect 2, 11062–11070 (2017)
Nie, Y., Li, L., Wei, Z.: Recent advancements in Pt and Pt-free catalysts for oxygen reduction reaction. Chem. Soc. Rev. 44, 2168–2201 (2015)
Scofield, M.E., Liu, H., Wong, S.S.: A concise guide to sustainable PEMFCs: recent advances in improving both oxygen reduction catalysts and proton exchange membranes. Chem. Soc. Rev. 44, 5836–5860 (2015)
Raj, C.R., Samanta, A., Noh, S.H., et al.: Emerging new generation electrocatalysts for the oxygen reduction reaction. J. Mater. Chem. A 4, 11156–11178 (2016)
Xia, W., Mahmood, A., Liang, Z., et al.: Earth-abundant nanomaterials for oxygen reduction. Angew. Chem. Int. Ed. 55, 2650–2676 (2015)
Xia, Z., An, L., Chen, P., et al.: Non-Pt nanostructured catalysts for oxygen reduction reaction: synthesis, catalytic activity and its key factors. Adv. Energy Mater. 6, 1600458 (2016)
Wenling, G., Liuyong, H., Jing, L., et al.: Recent advancements in transition metal-nitrogen–carbon catalysts for oxygen reduction reaction. Electroanalysis 30, 1217–1228 (2018)
Atkins, P.W., De Paula, J.: Atkins’ Physical Chemistry. Oxford University Press, Oxford (2006)
Wan, K., Yu, Z., Li, X., et al.: pH Effect on electrochemistry of nitrogen-doped carbon catalyst for oxygen reduction reaction. ACS Catal. 5, 4325–4332 (2015)
Muthukrishnan, A., Nabae, Y., Okajima, T., et al.: Kinetic approach to investigate the mechanistic pathways of oxygen reduction reaction on Fe-containing N-doped carbon catalysts. ACS Catal. 5, 5194–5202 (2015)
Damjanovic, A., Genshaw, M.A., Bockris, J.O.M.: Distinction between intermediates produced in main and side electrodic reactions. J. Chem. Phys. 45, 4057–4059 (1966)
Muthukrishnan, A., Nabae, Y., Hayakawa, T., et al.: Fe-containing polyimide-based high-performance ORR catalysts in acidic medium: a kinetic approach to study the durability of catalysts. Catal. Sci. Technol. 5, 475–483 (2015)
Wu, J., Zhang, D., Niwa, H., et al.: Enhancement in kinetics of the oxygen reduction reaction on a nitrogen-doped carbon catalyst by introduction of iron via electrochemical methods. Langmuir 31, 5529–5536 (2015)
Wu, K.H., Shi, W., Wang, D., et al.: In situ electrostatic modulation of path selectivity for the oxygen reduction reaction on Fe–N doped carbon catalyst. Chem. Mater. 29, 4649–4653 (2017)
Nayak, S., McPherson, I.J., Vincent, K.A.: Adsorbed intermediates in oxygen reduction on platinum nanoparticles observed by in situ IR spectroscopy. Angew. Chem. Int. Ed. 130, 1–5 (2018)
Ramaswamy, N., Tylus, U., Jia, Q., et al.: Activity descriptor identification for oxygen reduction on nonprecious electrocatalysts: linking surface science to coordination chemistry. J. Am. Chem. Soc. 135, 15443–15449 (2013)
Jia, Q., Ramaswamy, N., Hafiz, H., et al.: Experimental observation of redox-induced Fe–N switching behavior as a determinant role for oxygen reduction activity. ACS Nano 9, 12496–12505 (2015)
Xu, H., Cheng, D., Cao, D., et al.: A universal principle for a rational design of single-atom electrocatalysts. Nat. Catal. 1, 339–348 (2018)
Zitolo, A., Goellner, V., Armel, V., et al.: Identification of catalytic sites for oxygen reduction in iron-and nitrogen-doped graphene materials. Nat. Mater. 14, 937–942 (2015)
Malko, D., Kucernak, A., Lopes, T.: In situ electrochemical quantification of active sites in Fe–N/C non-precious metal catalysts. Nat. Commun. 7, 13285 (2016)
Kong, A., Zhu, X., Han, Z., et al.: Ordered hierarchically micro- and mesoporous Fe–Nx-embedded graphitic architectures as efficient electrocatalysts for oxygen reduction reaction. ACS Catal. 4, 1793–1800 (2014)
Wang, Q., Zhou, Z., Lai, Y., et al.: Phenylenediamine-based FeNx/C catalyst with high activity for oxygen reduction in acid medium and its active-site probing. J. Am. Chem. Soc. 136, 10882–10885 (2014)
Kramm, U.I., Lefèvre, M., Larouche, N., et al.: Correlations between Mass Activity and physicochemical properties of Fe/N/C catalysts for the ORR in PEM fuel cell via 57Fe Mössbauer spectroscopy and other techniques. J. Am. Chem. Soc. 136, 978–985 (2014)
Lefèvre, M., Dodelet, J.P., Bertrand, P.: Molecular oxygen reduction in PEM Fuel Cells: evidence for the simultaneous presence of two active sites in Fe-based catalysts. J. Phys. Chem. B 106, 8705–8713 (2002)
Lai, Q., Zheng, L., Liang, Y., et al.: Metal-organic-framework-derived Fe-N/C electrocatalyst with five-coordinated Fe-Nx sites for advanced oxygen reduction in acid media. ACS Catal. 7, 1655–1663 (2017)
Li, Q., Li, X., Zhang, G., et al.: Cooperative spin transition of monodispersed FeN3 sites within graphene induced by CO adsorption. J. Am. Chem. Soc. 140, 15149–15152 (2018)
Zhu, Y., Zhang, B., Liu, X., et al.: Unravelling the structure of electrocatalytically active Fe-N complexes in carbon for the oxygen reduction reaction. Angew. Chem. Int. Ed. 53, 10673–10677 (2014)
Jaouen, F., Herranz, J., Lefèvre, M., et al.: Cross-laboratory experimental study of non-noble-metal electrocatalysts for the oxygen reduction reaction. ACS Appl. Mater. Interfaces 1, 1623–1639 (2009)
Shen, H., Gracia-Espino, E., Ma, J., et al.: Atomically FeN2 moieties dispersed on mesoporous carbon: a new atomic catalyst for efficient oxygen reduction catalysis. Nano Energy 35, 9–16 (2017)
Jiang, W., Gu, L., Li, L., et al.: Understanding the high activity of Fe–N–C electrocatalysts in oxygen reduction: fe/Fe3C nanoparticles boost the activity of Fe-Nx. J. Am. Chem. Soc. 138, 3570–3578 (2016)
Miao, Z., Wang, X., Tsai, M., et al.: Atomically dispersed Fe-Nx/C electrocatalyst boosts oxygen catalysis via a new metal-organic polymer supramolecule strategy. Adv Energy Mater 8(24), 1801226 (2018)
Qiao, B., Wang, A., Yang, X., et al.: Single-atom catalysis of CO oxidation using Pt1/FeOx. Nat. Chem. 3, 634–641 (2011)
Yi, J., Xu, R., Wu, Q., et al.: Atomically dispersed iron–nitrogen active sites within porphyrinic triazine-based frameworks for oxygen reduction reaction in both alkaline and acidic media. ACS Energy Lett. 3, 883–889 (2018)
Long, J., Gang, W., Rui, Z., et al.: From metal-organic frameworks to single-atom Fe implanted N-doped porous carbons: efficient oxygen reduction in both alkaline and acidic media. Angew. Chem. Int. Ed. 130, 8661–8665 (2018)
Fei, H., Dong, J., Feng, Y., et al.: General synthesis and definitive structural identification of MN4C4 single-atom catalysts with tunable electrocatalytic activities. Nat. Catal. 1, 63–72 (2018)
Zhang, H., Hwang, S., Wang, M., et al.: Single atomic iron catalysts for oxygen reduction in acidic media: particle size control and thermal activation. J. Am. Chem. Soc. 139, 14143–14149 (2017)
Chen, Y., Ji, S., Wang, Y., et al.: Isolated single iron atoms anchored on N-doped porous carbon as an efficient electrocatalyst for the oxygen reduction reaction. Angew. Chem. Int. Ed. 129, 7041–7045 (2017)
Choi, C.H., Choi, W.S., Kasian, O., et al.: Unraveling the nature of sites active toward hydrogen peroxide reduction in Fe–N–C catalysts. Angew. Chem. Int. Ed. 56, 8809–8812 (2017)
Herranz, J., Jaouen, F., Lefèvre, M., et al.: Unveiling N-protonation and anion-binding effects on Fe/N/C catalysts for O2 reduction in proton-exchange-membrane fuel cells. J. Phys. Chem. C 115, 16087–16097 (2011)
Kramm, U.I., Herranz, J., Larouche, N., et al.: Structure of the catalytic sites in Fe/N/C-catalysts for O2-reduction in PEM fuel cells. Phys. Chem. Chem. Phys. 14, 11673–11688 (2012)
Kramm, U.I., Herrmann-Geppert, I., Behrends, J., et al.: On an easy way to prepare metal-nitrogen doped carbon with exclusive presence of MeN4-type sites active for the ORR. J. Am. Chem. Soc. 138, 635–640 (2016)
Liu, J.J.: Advanced electron microscopy of metal-support interactions in supported metal catalysts. Chemcatchem 3, 934–948 (2011)
Gu, J., Cai, Z., Wang, D., et al.: Single-molecule imaging of iron-phthalocyanine-catalyzed oxygen reduction reaction by in situ scanning tunneling microscopy. ACS Nano 10, 8746–8750 (2016)
Zhong, W., Chen, J., Zhang, P., et al.: Air plasma etching towards rich active sites in Fe/N-porous carbon for the oxygen reduction reaction with superior catalytic performance. J. Mater. Chem. A 5, 16605–16610 (2017)
Tan, H., Li, Y., Jiang, X., et al.: Perfectly ordered mesoporous iron–itrogen doped carbon as highly efficient catalyst for oxygen reduction reaction in both alkaline and acidic electrolytes. Nano Energy 36, 286–294 (2017)
Ahn, S.H., Yu, X., Manthiram, A.: “Wiring” Fe-Nx-embedded porous carbon framework onto 1D nanotubes for efficient oxygen reduction reaction in alkaline and acidic media. Adv Mater 29(26), 1606534 (2017)
Chung, D.Y., Kim, M.J., Kang, N., et al.: Low-temperature and gram-scale synthesis of two-dimensional Fe–N–C carbon sheets for robust electrochemical oxygen reduction reaction. Chem. Mater. 29, 2890–2898 (2017)
Ding, Y., Niu, Y., Yang, J., et al.: A metal-amino acid complex-derived bifunctional oxygen electrocatalyst for rechargeable Zinc–air batteries. Small 12, 5414–5421 (2016)
Kozuch, S., Martin, J.M.L.: Turning over definitions in catalytic cycles. ACS Catal. 2, 2787–2794 (2012)
Cui, C., Gan, L., Heggen, M., et al.: Compositional segregation in shaped Pt alloy nanoparticles and their structural behaviour during electrocatalysis. Nat. Mater. 12, 765–771 (2013)
Li, M., Zhao, Z., Cheng, T., et al.: Ultrafine jagged platinum nanowires enable ultrahigh mass activity for the oxygen reduction reaction. Science 354, 1414–1419 (2016)
Mahmood, J., Li, F., Jung, S.M., Okyay, M.S., et al.: An efficient and pH-universal ruthenium-based catalyst for the hydrogen evolution reaction. Nat. Nanotech. 12, 441–446 (2017)
Malko, D., Kucernak, A.R.J., Lopes, T.: Performance of Fe–N/C oxygen reduction electrocatalysts towards NO2 −, NO, and NH2OH electroreduction-from fundamental insights into the active center to a new method for environmental nitrite destruction. J. Am. Chem. Soc. 138, 16056–16068 (2016)
Shen, H., Gracia-Espino, E., Ma, J., et al.: Synergistic effects between atomically dispersed Fe–N–C and C–S–C for the oxygen reduction reaction in acidic media. Angew. Chem. Int. Ed. 129, 13988–13992 (2017)
Varnell, J.A., Sotiropoulos, J.S., Brown, T.M., et al.: Revealing the role of the metal in non-precious-metal catalysts for oxygen reduction via selective removal of Fe. ACS Energy Lett. 3, 823–828 (2018)
Wu, G., More, K.L., Johnston, C.M., et al.: High-performance electrocatalysts for oxygen reduction derived from polyaniline, iron, and cobalt. Science 332, 443–447 (2011)
Ye, Y., Li, H., Cai, F., et al.: Two-dimensional mesoporous carbon doped with Fe–N active sites for efficient oxygen reduction. ACS Catal. 7, 7638–7646 (2017)
Seh, Z.W., Kibsgaard, J., Dickens, C.F., et al.: Combining theory and experiment in electrocatalysis: insights into materials design. Science 355, 146–157 (2017)
Wang, Y., Yuan, H., Li, Y., et al.: Two-dimensional iron-phthalocyanine (Fe–Pc) monolayer as a promising single-atom-catalyst for oxygen reduction reaction: a computational study. Nanoscale 7, 11633–11641 (2015)
Seo, M.H., Higgins, D., Jiang, G., et al.: Theoretical insight into highly durable iron phthalocyanine derived non-precious catalysts for oxygen reduction reactions. J. Mater. Chem. A 2, 19707–19716 (2014)
Titov, A., Zapol, P., Kral, P., et al.: Catalytic Fe-xN sites in carbon nanotubes. J. Phys. Chem. C 113, 21629–21634 (2009)
Liu, K., Wu, G., Wang, G.: Role of local carbon structure surrounding FeN4 Sites in boosting catalytic activity for oxygen reduction. J. Phys. Chem. C 121, 11319–11324 (2017)
Yasuda, S., Furuya, A., Uchibori, Y., et al.: Iron–nitrogen-doped vertically aligned carbon nanotube electrocatalyst for the oxygen reduction reaction. Adv. Funct. Mater. 26, 738–744 (2016)
Song, P., Wang, Y., Pan, J., et al.: Structure-activity relationship in high-performance iron-based electrocatalysts for oxygen reduction reaction. J. Power Sources 300, 279–284 (2015)
Jaouen, F., Marcotte, S., Dodelet, J.P., et al.: Oxygen reduction catalysts for polymer electrolyte fuel cells from the pyrolysis of iron acetate adsorbed on various carbon supports. J. Phys. Chem. B 107, 1376–1386 (2003)
Zhu, C., Shi, Q., Xu, B.Z., et al.: Hierarchically porous M–N–C (M = Co and Fe) single-atom electrocatalysts with robust MNx active moieties enable enhanced ORR performance. Adv Energy Mater 8(29), 1801956 (2018)
Hu, K., Tao, L., Liu, D., et al.: Sulfur-doped Fe/N/C nanosheets as highly efficient electrocatalysts for oxygen reduction reaction. ACS Appl. Mater. Interfaces 8, 19379–19385 (2016)
Kone, I., Xie, A., Tang, Y., et al.: Hierarchical porous carbon doped with iron–nitrogen–sulfur for efficient oxygen reduction reaction. ACS Appl. Mater. Interfaces 9, 20963–20973 (2017)
Men, B., Sun, Y., Liu, J., et al.: Synergistically enhanced electrocatalytic activity of sandwich-like N-doped graphene/carbon nanosheets decorated by Fe and S for oxygen reduction reaction. ACS Appl. Mater. Interfaces 8, 19533–19541 (2016)
Sasan, K., Kong, A., Wang, Y., et al.: From hemoglobin to porous N–S–Fe–doped carbon for efficient oxygen electroreduction. J. Phys. Chem. C 119, 13545–13550 (2015)
Kwak, D.H., Han, S.B., Lee, Y.W., et al.: Fe/N/S-doped mesoporous carbon nanostructures as electrocatalysts for oxygen reduction reaction in acid medium. Appl. Catal. B Environ. 203, 889–898 (2017)
Wang, Y.C., Lai, Y.J., Song, L., et al.: S-doping of an Fe/N/C ORR catalyst for polymer electrolyte membrane fuel cells with high power density. Angew. Chem. Int. Ed. 127, 10045–10048 (2015)
Sun, M., Davenport, D., Liu, H., et al.: Highly efficient and sustainable non-precious-metal Fe–N–C electrocatalysts for the oxygen reduction reaction. J. Mater. Chem. A 6, 2527–2539 (2018)
Zhang, J., Byeon, A., Lee, J.W.: Boron-doped carbon-iron nanocomposites as efficient oxygen reduction electrocatalysts derived from carbon dioxide. Chem. Commun. 50, 6349–6352 (2014)
Yuan, K., Sfaelou, S., Qiu, M., et al.: Synergetic contribution of boron and Fe-N-x species in porous carbons toward efficient electrocatalysts for oxygen reduction reaction. Acs Energy Lett. 3, 252–260 (2018)
Fajrial, A.K., Saputro, A.G., Agusta, M.K., et al.: First principles study of oxygen molecule interaction with the graphitic active sites of a boron-doped pyrolyzed Fe–N–C catalyst. Phy. Chem. Chem. Phy. 19, 23497–23504 (2017)
Chen, P., Zhou, T., Xing, L., et al.: Atomically dispersed iron-nitrogen species as electrocatalysts for bifunctional oxygen evolution and reduction reactions. Angew. Chem. Int. Ed. 56, 610–614 (2017)
Kramm, U.I., Herrmann-Geppert, I., Fiechter, S., et al.: Effect of iron-carbide formation on the number of active sites in Fe–N–C catalysts for the oxygen reduction reaction in acidic media. J. Mater. Chem. A 2, 2663–2670 (2014)
He, Z., Maurice, J.L., Gohier, A., et al.: Iron catalysts for the growth of carbon nanofibers: Fe, Fe3C or both? Chem. Mater. 23, 5379–5387 (2011)
Fan, X., Peng, Z., Ye, R., et al.: M3C (M: Fe Co, Ni) nanocrystals encased in graphene nanoribbons: an active and stable bifunctional electrocatalyst for oxygen reduction and hydrogen evolution reactions. ACS Nano 9, 7407–7418 (2015)
Hou, Y., Huang, T., Wen, Z., et al.: Metal-organic framework-derived nitrogen-doped core-shell-structured porous Fe/Fe3C@C nanoboxes supported on graphene sheets for efficient oxygen reduction reactions. Adv. Energy Mater. 4, 1220–1225 (2014)
Hu, Y., Jensen, J.O., Zhang, W., et al.: Hollow spheres of iron carbide nanoparticles encased in graphitic layers as oxygen reduction catalysts. Angew. Chem. Int. Ed. 53, 3675–3679 (2014)
Zhang, Z., Sun, J., Wang, F., et al.: Efficient oxygen reduction reaction (ORR) catalysts based on single iron atoms dispersed on a hierarchically structured porous carbon framework. Angew. Chem. Int. Ed. 130, 9176–9181 (2018)
Han, S., Hu, X., Wang, J., et al.: Novel route to Fe-based cathode as an efficient bifunctional catalysts for rechargeable Zn–air battery. Adv. Energy Mater. 8, 1800955 (2018)
Ding, W., Li, L., Xiong, K., et al.: Shape fixing via salt recrystallization: a morphology-controlled approach to convert nanostructured polymer to carbon nanomaterial as a highly active catalyst for oxygen reduction reaction. J. Am. Chem. Soc. 137, 5414–5420 (2015)
Yang, L., Cheng, D., Xu, H., et al.: Unveiling the high-activity origin of single-atom iron catalysts for oxygen reduction reaction. PNAS 115, 6626–6631 (2018)
Serov, A., Artyushkova, K., Atanassov, P.: Fe–N–C oxygen reduction fuel cell catalyst derived from carbendazim: synthesis, structure, and reactivity. Adv. Energy Mater. 4, 1301735 (2014)
Lefevre, M., Proietti, E., Jaouen, F., et al.: Iron-based catalysts with improved oxygen reduction activity in polymer electrolyte fuel cells. Science 324, 71–74 (2009)
Kim, J., Lee, J., Choi, Y., et al.: Synthesis of hierarchical linearly assembled graphitic carbon nanoparticles via catalytic graphitization in SBA-15. Carbon 75, 95–103 (2014)
Jiao, Y., Han, D., Liu, L., et al.: Highly ordered mesoporous few-layer graphene frameworks enabled by Fe3O4 nanocrystal superlattices. Angew. Chem. Int. Ed. 54, 5727–5731 (2015)
Wu, Z., Xu, X., Hu, B., et al.: Iron carbide nanoparticles encapsulated in mesoporous Fe–N-doped carbon nanofibers for efficient electrocatalysis. Angew. Chem. Int. Ed. 54, 8179–8183 (2015)
Liu, D., Long, Y.: Superior catalytic activity of electrochemically reduced graphene oxide supported iron phthalocyanines toward oxygen reduction reaction. ACS Appl. Mater. Interfaces 7, 24063–24068 (2015)
Zhou, T., Zhou, Y., Ma, R., et al.: Achieving excellent activity and stability for oxygen reduction electrocatalysis by hollow mesoporous iron-nitrogen-doped graphitic carbon spheres. J. Mater. Chem. A 5, 12243–12251 (2017)
Liang, H.W., Wei, W., Wu, Z.-S., et al.: Mesoporous metal-nitrogen-doped carbon electrocatalysts for highly efficient oxygen reduction reaction. J. Am. Chem. Soc. 135, 16002–16005 (2013)
Kong, A., Dong, B., Zhu, X., et al.: Ordered mesoporous Fe-porphyrin-like architectures as excellent cathode materials for the oxygen reduction reaction in both alkaline and acidic media. Chemistry 19, 16170–16175 (2013)
Song, L., Wu, Z., Zhou, F., et al.: Sustainable hydrothermal carbonization synthesis of iron/nitrogen-doped carbon nanofiber aerogels as electrocatalysts for oxygen reduction. Small 12, 6398–6406 (2016)
Wang, B., Wang, X., Zou, J., et al.: Simple-cubic carbon frameworks with atomically dispersed iron dopants toward high-efficiency oxygen reduction. Nano Lett. 17, 2003–2009 (2017)
Ma, R., Zhou, Y., Hu, C., et al.: Post iron-doping of activated nitrogen-doped carbon spheres as a high-activity oxygen reduction electrocatalyst. Energy Storage Mater. 13, 142–150 (2018)
Deng, Y., Dong, Y., Wang, G., et al.: Well-defined ZIF-derived Fe–N co-doped carbon nanoframes as efficient oxygen reduction catalysts. ACS Appl. Mater. Interfaces 9, 9699–9709 (2017)
Liu, T., Li, M., Jiao, C., et al.: Design and synthesis of integrally structured Ni3N nanosheets/carbon microfibers/Ni3 N nanosheets for efficient full water splitting catalysis. J. Mater. Chem. A 5, 9377–9390 (2017)
Li, J., Chen, S., Li, W., et al.: A eutectic salt-assisted semi-closed pyrolysis route to fabricate high-density active-site hierarchically porous Fe/N/C catalysts for the oxygen reduction reaction. J. Mater. Chem. A 6, 15504–15509 (2018)
Gewirth, A.A., Varnell, J.A., DiAscro, A.M.: Nonprecious metal catalysts for oxygen reduction in heterogeneous aqueous systems. Chem. Rev. 118, 2313–2339 (2018)
Wang, X.X., Cullen, D.A., Pan, Y.T., et al.: Nitrogen-coordinated single cobalt atom catalysts for oxygen reduction in proton exchange membrane fuel cells. Adv. Mater. 30, 1706758 (2018)
Zitolo, A., Ranjbar-Sahraie, N., Mineva, T., et al.: Identification of catalytic sites in cobalt-nitrogen-carbon materials for the oxygen reduction reaction. Nat. Commun. 8, 957 (2017)
Yin, P., Yao, T., Wu, Y., et al.: Single cobalt atoms with precise n-coordination as superior oxygen reduction reaction catalysts. Angew. Chem. Int. Ed. 55, 10800–10805 (2016)
Hu, Z., Guo, Z., Zhang, Z., et al.: Bimetal zeolitic imidazolite framework-derived iron-, cobalt- and nitrogen-codoped carbon nanopolyhedra electrocatalyst for efficient oxygen reduction. ACS Appl. Mater. Interfaces 10, 12651–12658 (2018)
Guan, Z., Zhang, X., Chen, W., et al.: Mesoporous S doped Fe–N–C materials as highly active oxygen reduction reaction catalyst. Chem. Commun. (2018). https://doi.org/10.1039/c8cc05273e
Guo, D., Shibuya, R., Akiba, C., et al.: Active sites of nitrogen-doped carbon materials for oxygen reduction reaction clarified using model catalysts. Science 351, 361–365 (2016)
Liang, J., Zhou, R.F., Chen, X.M., et al.: Fe-N decorated hybrids of CNTs grown on hierarchically porous carbon for high-performance oxygen reduction. Adv. Mater. 26, 6074–6079 (2014)
Yang, Z.X., Xia, Y.D., Mokaya, R.: Aligned N-doped carbon nanotube bundles prepared via CVD using zeolite substrates. Chem. Mater. 17, 4502–4508 (2005)
Sheng, Z.H., Shao, L., Chen, J.J., et al.: Catalyst-free synthesis of nitrogen-doped graphene via thermal annealing graphite oxide with melamine and its excellent electrocatalysis. ACS Nano 5, 4350–4358 (2011)
Kundu, S., Nagaiah, T.C., Xia, W., et al.: Electrocatalytic activity and stability of nitrogen-containing carbon nanotubes in the oxygen reduction reaction. J. Phys. Chem. C 113, 14302–14310 (2009)
Nagaiah, T.C., Kundu, S., Bron, M., et al.: Nitrogen-doped carbon nanotubes as a cathode catalyst for the oxygen reduction reaction in alkaline medium. Electrochem. Commun. 12, 338–341 (2010)
Liu, R., Wu, D., Feng, X., et al.: Nitrogen-doped ordered mesoporous graphitic arrays with high electrocatalytic activity for oxygen reduction. Angew. Chem. Int. Ed. 49, 2565–2569 (2010)
Matter, P., Wang, E., Ozkan, U.: Preparation of nanostructured nitrogen-containing carbon catalysts for the oxygen reduction reaction from SiO2- and MgO-supported metal particles. J. Catal. 243, 395–403 (2006)
Mo, Z., Liao, S., Zheng, Y., et al.: Preparation of nitrogen-doped carbon nanotube arrays and their catalysis towards cathodic oxygen reduction in acidic and alkaline media. Carbon 50, 2620–2627 (2012)
Webster, S., Maultzsch, J., Thomsen, C., et al.: Raman characterization of nitrogen doped multiwalled carbon nanotubes. Nanotub. Devices 772, 129–134 (2003)
Van Dommele, S., Romero-Izquirdo, A., Brydson, R., et al.: Tuning nitrogen functionalities in catalytically grown nitrogen-containing carbon nanotubes. Carbon 46, 138–148 (2008)
Lin, L., Yang, Z.K., Jiang, F., et al.: Nonprecious bimetallic (Fe, Mo)-N/C catalyst for efficient oxygen reduction reaction. ACS Catal. 6, 4449–4454 (2016)
Wang, J., Qiao, J., Baker, R., et al.: Alkaline polymer electrolyte membranes for fuel cell applications. Chem. Soc. Rev. 42, 5768–5787 (2013)
Zhou, T., Shao, R., Chen, S., et al.: A review of radiation-grafted polymer electrolyte membranes for alkaline polymer electrolyte membrane fuel cells. J. Power Sources 293, 946–975 (2015)
O’Hayre, R., Cha, S.W., Colella, W., et al.: Fuel Cell Fundamentals. Wiley, New York (2016)
Spendelow, J.S., Papageorgopoulos, D.C.: Progress in PEMFC MEA component R&D at the DOE fuel cell technologies program. Fuel Cells 11, 775–786 (2011)
Gasteiger, H.A., Kocha, S.S., Sompalli, B., et al.: Activity benchmarks and requirements for Pt, Pt-alloy, and non-Pt oxygen reduction catalysts for PEMFCs. Appl. Catal. B Environ. 56, 9–35 (2005)
Milliken, J. Hydrogen, fuel cells and infrastructure technologies program, multi-year research, development and demonstration plan. US Department of Energy, Page 3.4–2 (2007). https://doi.org/10.2172/920934
Liu, Q., Liu, X., Zheng, L., et al.: The solid-phase synthesis of an Fe–N–C electrocatalyst for high-power proton-exchange membrane fuel cells. Angew. Chem. Int. Ed. 57, 1204–1208 (2018)
Proietti, E., Jaouen, F., Lefevre, M., et al.: Iron-based cathode catalyst with enhanced power density in polymer electrolyte membrane fuel cells. Nat. Commun. 2, 416 (2011)
Yuan, S., Shui, L., Grabstanowicz, L., et al.: A highly active and support-free oxygen reduction catalyst prepared from ultrahigh-surface-area porous polyporphyrin. Angew. Chem. Int. Ed. 125, 8507–8511 (2013)
Wang, J., Huang, Z., Liu, W., et al.: Design of N-coordinated dual-metal sites: a stable and active Pt-free catalyst for acidic oxygen reduction reaction. J. Am. Chem. Soc. 139, 17281–17284 (2017)
Fu, X., Zamani, P., Choi, J.Y., et al.: In situ polymer graphenization ingrained with nanoporosity in a nitrogenous electrocatalyst boosting the performance of polymer-electrolyte-membrane fuel cells. Adv. Mater. 29, 1604456 (2017)
Wu, Y., Wang, C., Wang, R., et al.: Three-dimensional networks of S-doped Fe/N/C with hierarchical porosity for efficient oxygen reduction in polymer electrolyte membrane fuel cells. ACS Appl. Mater. Interfaces 10, 14602–14613 (2018)
Meng, F., Liu, K., Zhang, Y., et al.: Recent advances toward the rational design of efficient bifunctional air electrodes for rechargeable Zn–air batteries. Small 14, 1703843 (2018)
Ganesan, P., Prabu, M., Sanetuntikul, J., et al.: Cobalt sulfide nanoparticles grown on nitrogen and sulfur codoped graphene oxide: an efficient electrocatalyst for oxygen reduction and evolution reactions. ACS Catal. 5, 3625–3637 (2015)
Shinde, S.S., Lee, C.H., Sami, A., et al.: Scalable 3-D carbon nitride sponge as an efficient metal-free bifunctional oxygen electrocatalyst for rechargeable Zn–air batteries. ACS Nano 11, 347–357 (2017)
Wang, R., Chen, Z., Hu, N., et al.: Nanocarbon-based electrocatalysts for rechargeable aqueous Li/Zn–air batteries. ChemElectroChem 5, 1745–1763 (2018)
Pan, J., Xu, Y.Y., Yang, H., et al.: Advanced architectures and relatives of air electrodes in Zn–air batteries. Adv. Sci. 5, 1700691 (2018)
Ma, L., Yu, T., Tzoganakis, E., et al.: Fundamental understanding and material challenges in rechargeable nonaqueous Li–O2 batteries: recent progress and perspective. Adv. Energy Mater. 8, 1800348 (2018)
Huang, Y., Wang, Y., Tang, C., et al.: Atomic modulation and structure design. Adv. Mater. (2018). https://doi.org/10.1002/adma.201803800
Li, Z., Yang, J., Xu, G., et al.: Non-precious cathode electrocatalyst for magnesium air fuel cells: activity and durability of iron-polyphthalocyanine absorbed on carbon black. J. Power Sources 242, 157–165 (2013)
Shui, J., Karan, N.K., Balasubramanian, M., et al.: Fe/N/C composite in Li–O2 battery: studies of catalytic structure and activity toward oxygen evolution reaction. J. Am. Chem. Soc. 134, 16654–16661 (2012)
Yu, L., Shen, Y., Huang, Y.: Fe–N–C catalyst modified graphene sponge as a cathode material for lithium–oxygen battery. J. Alloys. Compd. 595, 185–191 (2014)
Wei, W., Shi, X., Gao, P., et al.: Well-elaborated, mechanochemically synthesized Fe-TPP subset of ZIF precursors (Fe-TPP = tetraphenylporphine iron) to atomically dispersed iron-nitrogen species for oxygen reduction reaction and Zn–air batteries. Nano Energy 52, 29–37 (2018)
Wei, Q., Zhang, G., Yang, X., et al.: Litchi-like porous Fe/N/C spheres with atomically dispersed FeNx promoted by sulfur as highly efficient oxygen electrocatalysts for Zn–air batteries. J. Mater. Chem. A 6, 4605–4610 (2018)
Pan, Y., Liu, S., Sun, K., et al.: A bimetallic Zn/Fe polyphthalocyanine-derived single-atom Fe-N4 catalytic site: a superior trifunctional catalyst for overall water splitting and Zn–air batteries. Angew. Chem. Int. Ed. 57, 8614–8618 (2018)
Jin, S.: Are metal chalcogenides, nitrides, and phosphides oxygen evolution catalysts or bifunctional catalysts? ACS Energy Lett. 2, 1937–1938 (2017)
Hu, B., Wu, Z., Chu, S., et al.: SiO2-protected shell mediated templating synthesis of Fe–N-doped carbon nanofibers and their enhanced oxygen reduction reaction performance. Energy Environ. Sci. 11, 2208–2215 (2018)
Tian, N., Lu, B., Yang, X., et al.: Rational design and synthesis of low-temperature fuel cell electrocatalysts. Electrochem. Energy Rev. 1, 54–83 (2018)
Chenitz, R., Kramm, U.I., Lefevre, M., et al.: A specific demetalation of Fe-N4 catalytic sites in the micropores of NC_Ar + NH3 is at the origin of the initial activity loss of the highly active Fe/N/C catalyst used for the reduction of oxygen in PEM fuel cells. Energy Environ. Sci. 11, 365–382 (2018)
Acknowledgements
This work is supported by the National Natural Science Foundation of China (21471147) and the National Key Research and Development Program of China (2016YFB0101200). M. Yang appreciates the support from the Ningbo 3315 program. J. Wang would like to thank the financial support from the Science and Technology Commission of Shanghai Municipality (15520720400, 16DZ2260603) and the Equipment Research Program (6140721050215). Tiju Thomas would like to thank the Indian Institute of Technology, Madras and the Department of Science and Technology (DST, Government of India) for their financial support in the form of grants such as the Young Scientist Scheme (YSS/2015/001712) and the Centre Grant CHY1718383DSTXTPRA and SOL1819001DSTXHOCX. Tiju Thomas would also like to thank the Ministry of Electronics & Information Technology for their support and the Centre grant ELE1819353MEITENAK (in which he is a Center member). H. Shen would like to acknowledge the contributions made by Liu Yang at the Beijing Advanced Innovation Center for Soft Matter Science and Engineering at the Beijing University of Chemical Technology.
Author information
Authors and Affiliations
Corresponding authors
Ethics declarations
Conflicts of interest
The authors declare that they have no conflicts of interest.
Rights and permissions
About this article

Cite this article
Shen, H., Thomas, T., Rasaki, S.A. et al. Oxygen Reduction Reactions of Fe-N-C Catalysts: Current Status and the Way Forward. Electrochem. Energ. Rev. 2, 252–276 (2019). https://doi.org/10.1007/s41918-019-00030-w
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s41918-019-00030-w