
Abstract
This study is related to the effect of sintering on a commercially synthetic hydroxyapatite powder (CSHAp) uniaxially pelleted and sintered at various temperatures. A series of thermal analysis methods and tests were used to evaluate both phase changes that occurred during sintering and physical and mechanical properties of sintered samples. Bioactivity property of CSHA was investigated in SBF solution during 3, 7, 15, and 30 days. The results show that changes in colors occur in sintered samples at 1200 and 1300 °C. Phase changes occur in HA β-TCP at 1200 °C and HA β-TCP, α-TCP, and CaO phases at 1300 °C at less than 5% of each phases. The hardness and density values attain to maximum value of 4.87 ± 0.15 GPa and 3.06 ± 0.02 g/cm3 with 98.35 ± 0.17% relative density at 1300 °C, respectively. However, its compressive and flexural strengths increase by increasing sintering temperature until 1100 °C (130.20 ± 6.22 MPa for σcompressive and 60.27 ± 9.93 MPa for σflexural) and then drastically decrease at elevated temperatures. Its fracture toughness and average grain size values change between 0.68 ± 0.05 and 0.96 ± 0.05 MPam1/2 and 0.162 ± 0.019 and 17.167 ± 2.156 μm. Apatite crystals occur on the surface of CSHAp as needle at 3 days and they grow by ascending dwelling time.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
The replacement or restoration of troubled, damaged, or lost bone is a major clinical problem worldwide. Present healing methods to treat bone loss or damage include use of bone grafts such as autograft and allograft [1]. Autograft sources show an excellent osteoinductivity, but they are not available in unlimited amounts, which leads to some anatomical and structural problems, inadequate resorption rate during healing, and also require an additional surgery. In the case of allografts or xenografts, there are concerns about a possible contaminations with infectious material (e.g., HIV, hepatitis, or BSE), a potential immune response, and they may induce the loss of osteoinduction [2, 3]. In order to overcome these problems, artificial materials which can repair the bony defects are needed [4]. Metallic and polymeric materials are used in healthcare industry, but there are shortcomings of these materials. Metallic materials such as stainless steel, Co-based alloys, and Ti and Ti-alloys are commonly used for biomedical applications, though having good mechanical strength, but one of the major problems is about metals that are often prone to corrosion in the body environment [5]. Polymers have lower hard tissue compatibility and they release acidic degradation products which lead to inflammatory responses [6]. On the other hand, ceramic materials such as hydroxyapatite do not exhibit any cytotoxic effects, and it shows excellent biocompatibility with hard tissues and also with skin and muscle tissues, which is the most appropriate ceramic material for hard tissue replacement [7]. HA has been synthesized using several methods including wet-chemical method in aqueous solutions, sol–gel method, hydrothermal method, thermal deposition, conversion of coastal corals, and continuous precipitation [8], or it is commercially available from some companies such as Merck, Sigma Aldrich, and etc. There have been numerous publications on the determination of sinterability and bioactivity properties of CSHA powders in literature [9–18].
In the present study, not only thermal behavior but also physical and mechanical properties of a commercially synthetic HA obtained from Across Organics Company were investigated by a series of analysis methods and tests. The results showed that decomposition at elevated temperatures leads to reduction in mechanical properties before it attained higher densification. Samples with higher compressive strength were also tested to evaluate their in vitro bioactivity in a simulated body fluid. SBF test shows that it has in vitro bioactivity property.
Experimental procedure
Sample preparation
As said by M. Knepper et al. [19], bioactive synthetic HAp is commercially available in different forms depending on the material application, e.g., as fine powder (particle size <20 μm) or as spray-dried agglomerates (particle size up to 165 μm). In the present study, a commercially synthetic hydroxyapatite was purchased from Across Organics Company (CAS Number: 1306-06-5), and it was used in SEM image of CSHA powder showing that it has a broad size distribution as shown in Fig. 1.

a SEM image of b particle size distribution of CSHA powder
The composition of HA powder is listed in Table 1. As shown in Table 1, it can be used for biomedical applications due to the rates of impurities containing Hap that are compatible with ASTM F1185-88 standard [20]. The data which is determined by supply company show that Ca/P weight ratio of 2.156 and molar ratio of 1.667, respectively, are the same as stoichometric HAp [21].
CSHA powders were not calcinated before sintering treatments as calcination treatment leads to formation of hard agglomerates, and it has negative effect on sinterability of HA powders as indicated by Yeong K.C.B. et al. [22]. The samples were uniaxially pressed at 350 MPa according to our previous study [23]. For lubrication, the interior surface of the die was coated with a suspension of stearic acid in ethylene alcohol. Before sintering treatments, the green bodies were dried at 105 °C for 1 day. The compacted samples were sintered in air condition at temperatures ranging from 900 to 1300 °C, at furnace ramp rate of 5 °C, and soaking time of 4 h and then cooled to room temperature ramp rate of 5 °C at air atmosphere.
Characterization of sintered bodies
The X-ray diffraction (XRD) patterns were obtained at room temperature in an X’Pert MPD Philips diffractometer using Cu–Kα radiation at 40 kV and 30 mA. Intensity data were collected by step counting method (step 0.02° and a time per step of 0.6 s) in the range of 2θ (between 10° and 90°). The microstructures of the samples were determined by scanning electron microscope (SEM, JOEL Ltd., JSM-5910 LV), equipped with energy dispersive spectroscopy (EDS, Oxford Industries INCAx-Sight 7274, 133-Ev resolution 5.9 keV) for elemental analysis after gold coating. Differential thermal analysis (DTA) was carried out at the heating rate of 5 °C/min between room temperature and 1300 °C (Perkin Elmer PE-7 Series, USA). α-Alumina was used as a reference material. Thermogravimetric analyzer (TGA) was used in order to find the weight loss during heating at the same device. The average grain size of sintered samples was evaluated using the mean linear intercept method from scanning electron micrographs [24].
Determination of physical and mechanical properties
The shrinkage rates of samples were calculated by the comparison of the lengths of green bodies and sintered bodies using a digital caliper as shown in Eq. 1.
where L1 and L2 are the lengths before and after drying or sintering process, respectively [25]. The green densities were measured from the weight and size of the samples as shown in Eq. 2.
where m is the weight (gr), r is the radius (mm), and h is the height (mm) of the pelleted samples [25]. The bulk densities and apperant porosity rates of sintered samples were determined by Archimedes method using the Eqs. 3–5.
where D is the dry weight of the samples, W is the wet weight of the samples, and S is the wet weight suspended in the water of the samples [26]. Relative density of green bodies and sintered samples were calculated by dividing the green and sintered densities by the theoretical density of HAp (3.156 g/cm3). Hardness of the sintered samples was measured using Shimadzu HMV2 microindentor at a load of 1.962 N for 20 s. Prior to indentation, the surface of samples was polished to 3 μm surface finish. Average hardness and standard deviation values were taken from 20 indents. For fracture toughness measurements, the samples were polished by 10, 5, 3, and 1 μm diamond paste until obtaining of mirror like surface after grinding with 800, 1200, and 2500 mesh SiC papers. Niihara equation [27] was used for fracture toughness measurements as shown in Eq. 6. At least ten indentations were taken using 300 g load for 10 s.
where Hv is the Vickers hardness, a the half diagonal of the indentation, c the radial crack dimension measured from the center of the indent impression, i.e., c = L + a, and L is the crack length. Compressive strength of sintered samples were measured using Devotrans universal testing device (Istanbul-Turkey) at speed of 2 mm/min. The bending strength was determined by 3-point bending method according to ASTM C1161-94 standard [28] on a universal testing machine (Zwick) which applied force with cross head speed of 1 mm/min to the specimens until failure and recorded the strength as shown in Eq. 7. The inner ring had a diameter of 10 mm and the outer support ring had a diameter of 20 mm. The sintered test bars with dimensions 40 × 4 × 4 mm3 were ground (1200 grit) on both sides before testing. The distance between inner spans was 20 mm. Four specimens were tested for a single point.
where P is the break load (N), L is the distance between inner spans (mm), b is the specimen width (mm), and h is the specimen thickness (mm).
In vitro bioactivity test
In vitro bioactivity test was only performed for having higher compressive strength sample. Before in vitro test, totally, four samples were pelleted at 350 MPa with a size of ∅11 and 4 mm2. Sintering treatment was carried out at 1100 °C with same sintering regime. The samples were grind by sand paper up to 1200 mesh and then they were dried in an oven at 105 °C for 1 day. Simulated body fluid (SBF) solution was prepared according to Kokuba’s formulation [29] as follows. The solution was prepared by dissolving reagent grade sodium chloride (NaCl), potassium chloride (KCl), calcium chloride dihydrate (CaCl2·2H2O), magnesium chloride hexahydrate (MgCl2·6H2O), sodium hydrogen carbonate (NaHCO3), dipotassium hydrogen phosphate trihydrate (K2HPO4·3H2O), sodium sulfate (Na2SO4) in deionized water. Then, the solution was buffered to physiological pH 7.4 at 37.5 °C by both hydrochloric acid (HCl) and tris (hydroxymethyl)-aminomethane ((CH2OH)3CNH2). The ion concentration of the SBF is shown in Table 2. The sintered samples were immersed in sealed test tubes containing 20 ml of SBF for 3, 7, 15, and 30 days. The experiment was performed in a laboratory water bath that was maintained at a constant temperature of 36.5 °C. The solution was agitated daily to help maintain uniform ion concentrations. After immersion in SBF for various periods, the sintered samples were taken out, gently rinsed with distilled water, and dried at room temperature. Apatite formations were analyzed by SEM analysis after immersed samples were coated by gold.
Results and discussion
Microstructural properties
The structural properties of unsintered CSHAp was determined by XRD analysis and it is shown in Fig. 2. The XRD peaks are found at 2θ of 25,9 (002), 28,9 (120), 31,7 (121), 32,1 (112), 32,9 (030), 34,0 (022), 39,7 (130), 46,6 (222), 48,0 (132), 49,4 (123), 50,4 (231), 51,2 (140), 52,0 (042), and 53,2 (004) which agree well with the JCPDS card number of 98-004-0618 of X’Pert Philips Software, and it has the hexagonal space group of P6/3 m with a = b = 9.415 and c = 6.879. It also does not include any other calcium phosphates such as β and/or α-TCP and CaO. As indicated in literature, lattice parameters of HA change as a = 9.432–9.418 and c = 6.881–6.884 [30]. The lattice parameter values of CSHAp used in this study are noted to be compatible with stoichometric HA.

XRD result of HA at RT
The DTA thermograms and TGA curve are shown in Fig. 3. A relative weight loss was determined between 25 and 100 °C and is attributed to adsorbed water [31], and the second weight loss that occurred between 200 and 400 °C is due to the removal of lattice water [32]. Further weight loss at high temperatures (800–1100 °C) can be due to partial dehydroxylation of hydroxyapatite [33]. A total of 11.8% loss in weight from room temperature to 1300 °C is also seen.

DTA thermograms and TGA curve of CSHAp
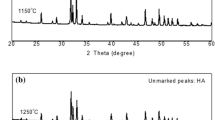
The results of phase analysis by XRD of the sintered samples are presented in Fig. 4. The XRD patterns of the sintered samples below 1100 °C include only HA peaks at same 2θ angles for pure CSHAp, but their intensities and sharpness increase by increasing sintering temperatures. However, β-TCP peaks were detected at 2θ of 27.784, 31.013, and 34.348 which are compatible with the JCPDS card number of 98-007-6896 when samples were sintered at 1200 °C, in addition to the HA. This means that HA starts to decompose at this temperature. The highest β-TCP peak was detected at 2θ of 31.013. Moreover, at 1300 °C, α-TCP and CaO phases were detected in addition to HA and β-TCP. The α-TCP peaks were detected at 2θ of 30.699, 30.705, and 30.711 which are compatible with the JCPDS card number of 98-007-8499. CaO phases were detected at 2θ of 32.242, 37.401, and 53.928 which are compatible with the JCPDS card number of 98-003-4977.

XRD analysis of sintered CSHA samples
Quantitative values of occurred phases during sintering determined by Rietvield analysis are given in Fig. 5. As shown in Fig. 5, β or α-TCP and CaO begin to form at elevated temperatures less than 5%, but major phase is HA at each sintering temperatures. Monolithic HA transformed to biphasic calcium phosphate (BCP) at elevated temperatures. BCP can be used as scaffolding material in some biomedical applications [34] due to the fact that BCPs easily react with human bone, but free calcium oxide constitutes a very undesirable impurity of the HA ceramics, and in general, it excludes the possibility to use such material in biological applications due to its high reactivity against water as indicated by Ślósarczyk A. et al. and Evis Z. et al. [35, 36]. Dehydroxylation and decomposition of CSHA can be explained as shown in Eqs. 8 and 9 below, respectively.

Phase compositions of sintered samples at various temperatures
Sintering of hydroxyapatite is complicated by the fact that HAp is hydrated phase that decomposes to anyhdrous calcium phosphates such as TCP at approximately 1200–1450°C. Decomposition results from dehydroxylation beyond a critical point. For temperatures below the critical point, the HAp crystal structure is retained despite dehydroxylation and the HAp rehydrates on cooling. If the critical point is exceeded, complete and irreversible dehydroxylation occurs, resulting in collapse of the HAp structure and decomposition [37]. In literature, the initial decomposition temperature is variously specified at 1323 [38], 1573 [39], or even 1750 K [40]. In the present study, it is seen that CSHAp starts to decompose at 1473 K (1200 °C). This may be related to impurities in CSHAp, history of manufacturing process, and also water vapor pressure in the furnace atmosphere [41].
As shown in Fig. 6, the necks are formed at lower rate between interconnected set of rounded HA particulates with average grain size of 0.162 ± 0.019 μm and high degree of porosity sintered sample at 900 °C. Increment in sintering temperatures contributed to the increase in not only average grain size but also length of necks between CSHA particulates and the formation of denser microstructures. The average grain size was determined to be 0.186 ± 0.020 and 0.473 ± 0.035 μm for sintered samples at 1000 and 1100 °C, respectively. For sintered samples both at 1200 and 1300 °C, formation of microcracks was observed with increase in average grain size of 7.955 ± 1.616 and 17.167 ± 2.156 μm. It is seen that transgranular microcracks occurred at these temperatures, but the rate and also length of microcracks are higher for sintered samples at 1300 °C. This situation is related to the formation of second (β-TCP), third (α-TCP), and fourth (CaO) phases as we detailed in XRD analyis and their difference in thermal shock behaviors and also thermal expansion coefficient compared to HA [42].

SEM images of sintered CSHA samples at a 900 °C, b 1000 °C, c 1100 °C, d 1200 °C, and e 1300 °C
Physical and mechanical properties of CSHA
The measured shrinkage rates% of the sintered samples with a photograph showing the changes in color depending on the sintering temperatures between 900 and 1300 °C are given in Fig. 7. It is seen that the shrinkage rates of the sintered samples increase from 5.33 ± 0.57 to 18.20 ± 0.62% with an increment rate of 241.3% by increasing sintering temperatures and they become light blue and then dark blue in color at 1200 and 1300 °C, respectively. Change in color is due to the presence of Mn5+ or MnO4 3− ions at the PO4 3− sites in the apatite crystal structure reported by G. De With et al. and Yubao et al. Especially explained by Yubao et al., sintering at high temperature not only increases the intensity of oxidation in the oxidizing atmosphere, but also provides enough energy for the oxidized manganese ion (Mn2+ to Mn5+) to migrate within the crystal lattice [43, 44]. It is also determined that shrinkage rates% in height of CSHAp are compatible with a review which has been prepared by Champion E. [45] and his referred studies.

a Colors and b %shrinkage rates of sintered samples between 900 and 1300 °C
Maximum open porosity rate of 36.99 ± 0.50% is obtained for CSHA sintered at 900 °C. Increment in sintering temperature leads to the decrease of open porosity content, and it attains the lowest value of 2.91 ± 0.76% at 1300 °C as shown in Fig. 8a. On the other hand, sintered and also relative density of the samples increase by increasing sintering temperatures. Sintered density was 1.95 ± 0.01 g/cm3 with a relative density of 61.9 ± 0.37 for sintered samples at 900 °C. These values increase linearly until 1100 °C as 2.31 ± 0.02 g/cm3 with a relative density of 75.3 ± 0.28%. After this temperature, sintered density increase by a factor of 130% and the density of sintered samples at 1200 °C attained 3.02 ± 0.01 g/cm3 with a relative density of 97.81 ± 0.46%. However, at 1300 °C, a negligible increment in density was observed compared to sintered samples at 1200 °C. This means that densification has finished at approximately 1200 °C. As indicated by Ruys. A.J. et al., the sigmoidal behavior shows the density of HA ceramics and it plateaus at elevated temperatures as limiting densification is related to the grain growth in lower rate [37]. It is also seen that relative density of green bodies can be improved to maximum value of 98.35 ± 0.17% for pelleted samples at 350 MPa via sintering treatment. However, the relative density of sintered samples is higher than the other studies in which British 7253 standard had used [46–48]. This can be related to average grain size and morphology of HA powder.

Changes in a porosity-sintered density and b sintered density and relative density of samples
It can be seen in Fig. 9 that the lowest microhardness value of 0.76 ± 0.01 GPa was observed for sintered samples at 900 °C. A slight increase in microhardness was observed until 1100 °C, and it started to increase sharply between 1100 and 1200°C and then attained the maximum value of 4.87 ± 0.15 GPa at 1300 °C. The relatively low microhardness values for sintered samples <1200 °C are related to lower sintered densities of CSHA.

Microhardness values of CSHA
As shown in Fig. 10, compressive strength of sintered samples increases by increasing temperature until 1100 °C, but CSHA samples containing second and/or third phases (β- or α-TCp and CaO) showed poor compressive strength. Figure 10 also clearly reveals that the maximum compressive strength did not correspond to the maximum sintered density. As indicated by H. Zhou and J. Lee, for conventional sintering, a higher temperature is necessary to attain high-density parts with superior mechanical properties. However, HA tends to transform to tricalcium phosphate (TCP, Ca3(PO4)2) and/or CaO phases, which is named as decomposition of HA, during sintering, particularly at 1200 °C and above [49]. It is worth mentioning that decomposition of HA suppresses densification and will be accompanied by a decrease in mechanical properties and also effect biological properties [50].

Compressive strengths of CSHA with sintering temperature
The CSHAp sintered at 1100 °C shows a maximum three point bending strength value of 60.27 ± 9.93 MPa, and at higher sintering temperatures, the value decreases as shown in Fig. 11. For specimens sintered above 1100 °C, the flexural strength drops slightly because of considerable grain growth [51].

Three point bending strength of the sintered CSHAp with sintering temperature
Figure 12 shows the Vickers indentations for the measurements of fracture toughness obtained on the surface of sintered samples with an optical microscopy photograph. As shown in optical microscopy photographs, the cracks emanating from the four vertices and diagonals of indentations are longer for sintered samples at 1200 and 1300 °C than the others. This is due to lower densifications of sintered samples <1200 °C. It is also seen that Kıc values of sintered samples increase by increasing sintering temperature until 1100 °C (0.96 ± 0.05 MPam1/2) and then drastically decrease at elevated temperatures as seen from the results of compressive and flexural strength measurements. A decrease of KIC with increasing grain size is usually observed in ceramics where fracture mechanism is transgranular because the major contribution to crack resistance is related to the crossing of the grain boundaries [52]. The fracture toughness of HAp is less than 1 MPam1/2, which is well known to be one of the biggest obstacles for wider application of this material [53]. The results on Kıc measurements in the present study are the same as others commercially, biologically, or synthetically prepared HAp.

Fracture toughness of CSHA with sintering temperatures
In vitro bioactivity of HAp
To determine the in vitro bioactivity of CSHA by owning only higher compressive strength sample, it was immersed in simulated body fluid for 3, 7, 15, and 30 days. As shown in Fig. 13a, the plurality of apatite crystals formed in cubic shape after 3 days of immersion. In the case of 7 days, a change occurred in shapes of apatite crystals formed on the surface of sample from cubic to dendritic structure. The size and thickness of calcium phosphate layers increased with increasing SBF immersion time. After 30 days of immersion, the surface of the samples showed highly dense calcium phosphate layers.

Surface morphology of SBF tested samples with immersion time of a 3, b 7, c 15, and d 30 days
The essential requirement for an artificial material to bond with a living bone is the formation of this calcium phosphate layer on the surfaces. This layer is similar to the bone mineral in composition and structure; hence, the bone-producing cell is called as osteoblast, which can proliferate preferentially on this surface [54–56]. The presence of apatites shows that CSHA powder used in this study has bioactivity property.
Conclusion:
In the present study, the effect of sintering temperature on the microstructural, mechanical, and in vitro bioactivity properties of a commercially available HA was investigated. The following conclusions can be drawn from the present study:
-
1-
This study showed that compressive and three point bending strength and also fracture toughness of CSHA attained maximum values just before the occurrence of higher densifications of 98.35%. While the maximum compressive and three point bending strength is 130.20 ± 6.22 and 60.27 ± 9.93 MPa, the fracture toughness is 0.96 ± 0.05 MPam1/2, respectively.
-
2-
At elevated temperatures, it transformed to biphasic HA due to decomposition, but the rates of β and/or α-TCP and CaO are not more than 5%.
-
3-
The result of SBF investigations showed that it has bioactivity property due to apatite layers deposited on the surface of investigated sample.
-
4-
Because of the low mechanical properties compared to human cortical bone, it may be used for temporary biomedical applications that do not require heavy load resistance in human body.
-
5-
If the low mechanical properties can be improved via doping with the oxide ceramics and/or other materials, it may also be a candidate as a bioceramic for load bearing applications.
References
Kailasanathan, C., Selvakumar, N., Naidu, V.: Structure and properties of titania reinforced nano-hydroxyapatite/gelatin bio-composites for bone graft materials. Ceram Int. 38, 571–579 (2012)
Rodrigues, C.V.M., Serricella, P., Linhares, A.B.R., Guerdes, R.M.R.: Borojevic, Rossi, M.A., Duarte, M.E.L., Farina, M.: Characterization of a bovine collagen–hydroxyapatite composite scaffold for bone tissue engineering. Biomaterials. 24, 4987–4997 (2003)
Tadic, D., Epple, M.: Mechanically stable implants of synthetic bone mineral by cold isostatic pressing. Biomaterials. 24, 4565–4571 (2003)
Ohtsuki, C., Miyazaki, T., Kamitakahara, M., Tanihara, M.: Design of novel bioactive materials through organic modification of calcium silicate. J Eur Ceram Soc. 27, 1527–1533 (2007)
Karanjai, M., Sundaresan, R., Mohan, T.R.R., Kashyap, B.P.: Evaluation of growth of calcium phosphate ceramics on sintered Ti–Ca–P composites. Mater Sci Eng C. 28, 1401–1407 (2008)
Li, N., Jie, Q., Zhu, S., Wang, R.: A new route to prepare macroporous bioactive sol–gel glasses with high mechanical strength. Mater Lett. 58, 2747–2750 (2004)
Zhang, J., Tanaka, H., Ye, F., Jiang, D., Iwasa, M.: Colloidal processing and sintering of hydroxyapatite. Mater Chem Phys. 101, 69–76 (2007)
Pramanik, S., Agarwal, A.K., Rai, K.N., Garg, A.: Development of high strength hydroxyapatite by solid-state-sintering process. Ceram Int. 33, 419–426 (2007)
Afonso, A., Santos, S.J.D., Vasconcelos, M., Branco, R., Cavalheiro, J.: Granules of osteoapatite and glass-reinforced hydroxyapatite implanted in rabbit tibiae. J Mater Sci Mater Med. 7, 507–510 (1996)
Georgiou, G., Knowles, J.C.: Glass reinforced hydroxyapatite for hard tissue surgery-part 1: mechanical properties. Biomaterials. 22, 2811–2815 (2001)
Ambrosio, A.M., Sahota, J.S., Khan, Y., Laurencin, C.T.: A novel amorphous calcium phosphate polymer ceramic for bone repair: I. Synthesis and characterization. J Biomed Mater Res. 58, 295–301 (2001)
Saiz, E., Gremillard, L., Menendez, G., Miranda, P., Gryn, K., Tomsia, A.P.: Preparation of porous hydroxyapatite scaffolds. Mater Sci Eng C. 27, 546–550 (2007)
Guo, X., Xiao, P., Liu, J., Shen, Z.: Fabrication of nanostructured hydroxyapatite via hydrothermal synthesis and spark plasma sintering. Journal of American Ceramic Society. 88, 1026–1029 (2005)
Halouani, R., Bernache-Assolant, D., Champion, E., Ababou, A.: Microstructure and related mechanical properties of hot pressed hydroxyapatite ceramics. J Mater Sci Mater Med. 5, 563–568 (1994)
Gibson, I.R., Ke, S., Best, S.M., Bonfield, W.: Effect of powder characteristics on the sinterability of hydroxyapatite powders. Journal of Material Science: Materials In Medicine. 1(2), 163–171 (2001)
Mazaheri, M., Haghighatzadeh, M., Zahedi, A.M., Sadrnezhaad, S.K.: Effect of a novel sintering process on mechanical properties of hydroxyapatite ceramics. J Alloys Compd. 471, 180–184 (2009)
Erkmen, Z.E., Genç, Y., Oktar, F.N.: Microstructural and mechanical properties of hydroxyapatite–zirconia composites. Journal of American Ceramic Society. 90, 2885–2892 (2007)
Ramesh, S., Aw, K.L., Tolouei, R., Amiriyan, M., Tan, C.Y., Hamdi, M., Purbolaksono, J., Hassan, M.A., Teng, W.D.: Sintering properties of hydroxyapatite powders prepared using different methods. Ceram Int. 39, 111–119 (2013)
Knepper, M., Moricca, S., Milthorpe, B.K.: Stability of hydroxyapatite while processing short-fibre reinforced hydroxyapatite ceramics. Biomaterials. 18, 1523–1529 (1997)
Designation: F 1185-88, American Society for Testing and Materials (ASTM), 1988
Pontier, C., Viana, M., Champion, E., Chulia, D.: Apatitic calcium phosphates used in compression: rationalization of the end-use properties. Powder Technol. 130, 436–441 (2003)
Yeong, K.C.B., Wang, J., Ng, S.C.: Fabricating densified hydroxyapatite ceramics from a precipitated precursor. Mater Lett. 38, 208–213 (1999)
Bozkurt, Y., Pazarlioglu, S., Gokce, H., Gurler, I., Salman, S.: Hydroxyapatite lanthanum oxide composites. Acta Phys Pol A. 127, 1407–1409 (2015)
Aktas, B., Tekeli, S., Salman, S.: Improvements in microstructural and mechanical properties of ZrO2 ceramics after addition of BaO. Ceram Int. 42, 3849–3854 (2016)
Ghomi, H., Fathi, M.H., Edris, H.: Effect of the composition of hydroxyapatite/bioactive glass nanocomposite foams on their bioactivity and mechanical properties. Mater Res Bull. 47, 3523–3532 (2012)
Engin, N.O., Tas, A.C.: Manufacture of macroporous calcium hydroxyapatite Bioceramics. J Eur Ceram Soc. 19, 2569–2572 (1999)
Niihara, K., Morena, R., Hasselman, D.: Evaluation of Kıc of brittle solids by the indentation method with low crack-to-indent ratios. J Mater Sci Lett. 1, 13–16 (1982)
Standard Test Method for Flexural Strength of Advanced Ceramics at Ambient Temperature, Annual Book of ASTM standards, (1996)
Kokubo, T., Yamamuro, T., Hench, L.L., Wilson, J.: Bonding mechanism of bioactive glass–ceramic A–W to living bone. CRC Press, United State (1990)
Luo, P., Nieh, T.G.: Synthesis of ultrafine hydroxyapatite particles by a spray dry method. Mater Sci Eng C. 3, 75–78 (1995)
Anee, T.K., Ashok, M., Palanichamy, M., Kalkura, S.N.: A novel technique to synthesize hydroxyapatite at low temperature. Mater Chem Phys. 80, 725–730 (2003)
Manjubala, I., Sivakumar, M.: In-situ synthesis of biphasic calcium phosphate ceramics using microwave irradiation. Materials Chemistry and Pyhsics. 71, 272–278 (2001)
Bahrololoom, M.E., Javidi, M., Javadpour, S., Ma, J.: Characterisation of natural hydroxyapatite extracted from bovine cortical bone ash. J Ceram Process Res. 10, 129–138 (2009)
AbdulQader, S.T., Rahman, I.A., Ismail, H., Kannana, T.P., Mahmood, Z.: Simple pathway in preparation of controlled porosity of biphasic calcium phosphate scaffold for dentin regeneration. Ceram Int. 39, 2375–2381 (2013)
Ślósarczyk, A., Piekarczyk, J.: Ceramic materials on the basis of hydroxyapatite and tricalcium phosphate. Ceram Int. 25, 561–565 (1999)
Evis, Z., Tahmasebifar, A.: Structural and mechanical characteristics of hydroxyapatite and tricalcium phosphates doped with Al+3 and F− ions. J Ceram Process Res. 14, 549–556 (2013)
Ruys, A.J., Wei, M., Sorrell, C.C., Dickson, M.R., Brandwood, A., Milthorpe, B.K.: Sintering effects on the strength of hydroxyapatite. Biomaterials. 16, 409–415 (1995)
Ravaglioli, A., Krajewski, A.: Bioceramics. Chapman and Hall, London (1992)
Locardi, B., Pazzaglia, V.E., Gabbi, C., Profilo, B.: Thermal behaviour of hydroxyapatite intended for medical applications. Biomaterials. 44, 437–441 (1993)
Legeros, R.Z., Legeros, J.P.: An introduction to bioceramics. World Scientific, Singapore (1993)
R. Jenkins (and others editors): in “Powder Diffraction File, Inorganic Phases” (JCPDS International Centre for Diffraction Data, Swarthomore, (1987)
Case, E.D., Smith, I.O., Baumann, M.J.: Microcracking and porosity in calcium phosphates and the implications for bone tissue engineering. Mater Sci Eng A. 390, 246–254 (2005)
With, G.D., Van Dijk, H.J.A., Hattu, N., Prijs, K.: Preparation. Microstructure and mechanical properties of dense polycrystalline hydroxyapatite. J Mater Sci. 16, 1592–1598 (1981)
Yubao, L., Cpat, K., Xindong, Z., DeGroot, K.: Relationship between the color change of hydroxyapatite and the trace element manganese. Biomaterials. 14, 969–972 (1993)
Champion, E.: Sintering of calcium phosphate bioceramics. Acta Biomater. 9, 5855–5875 (2013)
Oktar, F.N., Göller, G.: Sintering effects on mechanical properties of glass-reinforced hydroxyapatite composites. Ceram Int. 28, 617–621 (2002)
Yelten, A., Yilmaz, S., Oktar, F.N.: Sol–gel derived alumina–hydroxyapatite–tricalcium phosphate porous composite powders. Ceram Int. 38, 2659–2665 (2012)
Göller, G., Oktar, F.N.: Sintering effects on mechanical properties of biologically derived dentine hydroxyapatite. Mater Lett. 56, 142–147 (2002)
Zhou, H., Lee, J.: Nanoscale hydroxyapatite particles for bone tissue engineering. Acta Biomater. 7, 2769–2781 (2011)
Muralithran, G., Ramesh, S.: The effects of sintering temperatures on the properties of hydroxyapatite. Ceram Int. 26, 221–230 (2000)
Liu, H.S., Chin, T.S., Lai, L.S., Chiu, S.Y., Chung, K.H., Chang, C.S., Lui, M.T.: Hydroxyapatite synthesized by a simplified hydrothermal method. Ceram Int. 23, 19–25 (1997)
Thangamani, N., Chinnakali, K., Gnanam, F.D.: The effect of powder processing on densification, microstructure and mechanical properties of hydroxyapatite. Ceram Int. 28, 355–362 (2002)
Zhanga, Y., Tan, S., Yin, Y.: C-fibre reinforced hydroxyapatite bioceramics. Ceram Int. 29, 113–116 (2003)
Hench, L.L.: Bioceramics: from concept to clinic. J Am Ceram Soc. 74, 1487–1510 (1991)
Cao, W., Hench, L.L.: Bioactive materials. Ceram Int. 22, 493–507 (1996)
Best, S.M., Porter, A.E., Thian, E.S., Huang, J.: Bioceramics: past, present and for the future. J Eur Ceram Soc. 28, 1319–1327 (2008)
Acknowledgement
The authors thank the economic support of Scientific Research Centre of Marmara University (Project No: FEN-C-DRP-080212-0026).
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article

Cite this article
Pazarlıoglu, S., Salman, S. Sintering effect on the microstructural, mechanical, and in vitro bioactivity properties of a commercially synthetic hydroxyapatite. J Aust Ceram Soc 53, 391–401 (2017). https://doi.org/10.1007/s41779-017-0048-4
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s41779-017-0048-4