
Abstract
Hematopoietic stem and progenitor cells (HSPC) express at their surface an array of cell adhesion receptors that enables them to interact with extracellular matrix components and transmembrane cell adhesion molecules expressed within stem cell niches in the bone marrow. Herein, we review the different cell adhesive interactions taking place within the bone marrow between HSPC and their niches (via integrins, CD44, selectin ligands, and Eph receptors) and how they support HSPC anchoring in the bone marrow and regulate HSPC fate during hematopoiesis. We review how malignant hematopoietic cells utilize adhesive interactions to resist chemotherapy treatments. Finally, we review how these adhesive interactions can be therapeutically targeted to increase HSPC mobilization for transplantation and to chemosensitize malignant hematopoietic cells to chemotherapy.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Hematopoietic stem cells (HSC) are not randomly distributed in the bone marrow (BM) [1] but located in specific functional microenvironmental units called niches [2–5, 6••, 7••]. In adult humans and mice, HSC are mostly dormant in steady-state, dividing approximately once or twice a year, respectively [8, 9]. However in response to stress [9] and infections [10, 11], these HSC can be rapidly stimulated to divide to generate a burst of highly proliferative hematopoietic progenitor cells (HPC) that will expend and differentiate to reconstitute different blood and immune lineages. HPC have very limited self-renewal capacity and HSC with high self-renewal potential must be preserved lifelong. To ensure HSC longevity, HSC return to dormancy once the physiological challenge is resolved and then can serve as a back-up until the next challenge [9]. HSC and HPC are capable of dividing either symmetrically to self-renew, or asymmetrically to generate a lineage committed progenitor cell while preserving a functional original HSC or HPC [12]. Specific HSC niches can exquisitely regulate these different states and functions of HSC and HPC via their specific cellular components, extracellular matrix, cell-cell interactions, soluble mediators, and perfusion gradients. It has also been proposed that several types of HSC niches may be necessary to instruct different fates to HSC [5, 6••, 7••, 13, 14]. Indeed, individual HSC can choose one of several fates, such as to stay quiescent, or to proliferate, to either self-renew or differentiate into committed hematopoietic progenitor cells (HPC). These decisions are thought to be largely controlled by microenvironmental cues from the niches in which HSC reside. Dormant HSC (those HSC in prolonged quiescence) have been reported to associate specifically with small arterioles near the BM—bone interface that are ensheathed by NG2+ pericytes (a subset of mesenchymal stromal cell: MSC) [6••, 7••]. In contrast proliferative or “active” HSC are found adjacent to sinusoidal endothelium [6••, 7••]. Critical factors regulating HSC survival and self-renewal at these niches are under intense investigation. Cell adhesion molecules such as selectins and integrin-ligand adhesion molecules are involved in maintaining homeostasis at the HSC and HPC BM niche.
In order to find (home to) and stay in their specific niches (retention), HSC and HPC express a range of cell adhesion receptors which interact with extracellular matrix proteins and cell adhesion molecules expressed by niche cells. In addition to their role in ensuring HSC and HPC are retained in their niches, there is mounting evidence that these cell adhesion receptors also signal and influence HSC and HPC regulation. An important aspect of such adhesive interactions is that the affinity of these receptors to their adhesive ligands is regulated by post post-translational mechanisms in order to adapt the niche and HSC state to a particular demand. Herein, we will review some of these adhesive interactions between HSC and their niche and how they regulate HSC fate in normal and pathological situations.
Leukocyte Adhesion Receptors Expressed by HSC
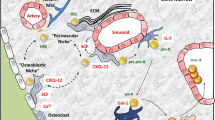
HSC express a range of cell adhesion receptors belonging to several structural families with best known examples P-selectin glycoprotein ligand -1 (PSGL-1 of CD162), integrins α4β1 (CD49d/CD29 complex also cells VLA-4) and α5β1 (CD49d/CD29 complex or VLA-5) and CD44 which were identified in the 1990s and more recently the Eph-ephrin axis. We have chosen to limit this review to these well-characterized cell adhesion molecules as summarized in Fig. 1.

Schematic representation of main cell adhesion receptors and molecules expressed by HSC and niche cells or in the niche extracellular matrix. Ribbon structure from X-ray crystallography data has been shown when available
Selectins
Selectins are a family of three transmembrane calcium-dependent lectins (L-selectin, E-selectin, and P-selectin) mediating cell-cell adhesion. Selectins are responsible for recruitment and homing of leukocytes to sites of inflammation, lymphoid organs, and the BM [15]. They specifically bind to fucosylated and sialylated tetrasaccharide motifs called sialyl LewisX (sLex) that decorates glycoproteins and glycolipids on the surface of many leukocytes. The three selectins mediate rolling and tethering of fast passing leukocytes onto activated (as in the case of inflammation) or specialized endothelial cells (such as in lymph nodes or BM). This step of rolling is mediated by a weak adhesive interaction, enabling leukocytes in the circulation to slow down and tether to these endothelial cells, a preliminary step to further strong attachment mediated by integrins. These steps of rolling and attachment are necessary for leukocyte extravasation across the endothelium and migration into the extravascular compartment of the tissue.
L-selectin (CD62L) is expressed by most leukocytes and best known to mediate the recruitment of leukocytes (lymphocytes and monocytes) to the high endothelial venules in lymph nodes via its interaction with glycosylation-dependent cell adhesion molecule (GlyCAM)-1 and to the endothelium of gut-associated lymphoid organs via mucosal vascular address in cell adhesion molecule (MadCAM)-1. Although expressed on HSC and HPC, the role of L-selectin on these cells remains unclear. Sell −/− mice in which the L-selectin gene has been deleted have defective lymphocyte migration into the lymph nodes, Peyer’s patches and spleen, and defective granulocyte recruitment to inflamed peritoneum [16].
P-selectin (CD62P), expressed by megakaryocytes, platelets, inflamed endothelial cells, and a subset of BM endothelial cells, mediates the rolling of leukocytes expressing its transmembrane ligand PSGL-1. P-selectin in concert with E-selectin plays an important role in leucocyte recruitment to inflamed tissues and homing of HSPC to the BM. P-selectin knock-out mice (Selp −/−) display a subtle phenotype. Megakaryocytes have recently been identified as critical HSC niche component [17] that secrete transforming growth factor (TGF)-β which maintain HSC quiescence [18••, 19••]. As megakaryocyte express P-selectin and human/mouse HSC express a functional P-selectin ligand PSGL-1 [20, 21], P-selectin may play an important role in anchoring HSC in close association with megakaryocytes in the BM. This remains, however, to demonstrate.
E-selectin (CD62E) is expressed by endothelial cells during inflammation as well as in specialized domains of the BM endothelium [22]. Unlike P-selectin, E-selectin has many potential ligands [23] such as PSGL-1 [24], a particular glycoform of CD44 called HCELL [25], E-selectin ligand (ESL)-1 [26•], and poorly defined fucosylated cell surface glycosphingolipids [27]. The combination of both E-selectin and P-selectin is critical to granulocyte recruitment into inflamed tissue and HSPC homing to the BM. Indeed, P- and E-selectin are not normally expressed by naive endothelial cells; however, following inflammation, endothelial selectins become rapidly expressed, and leukocytes with counter-receptors begin to roll on the activated endothelium. P-selectin is stored as a pre-form protein in endothelial cell Weibel-Palade bodies and translocated to cell surfaces within minutes of endothelial cell activation while expression of E-selectin begins hours later following de novo synthesis. Initial “fast” rolling (shear-resistant adhesion) of leukocytes on the endothelium is mediated by P-selectin, and rolling begins to “slow” when E-selectin is also expressed. Final immobilization of leukocytes occurs with the activation of integrins followed by extravasation to the site of tissue damage/inflammation (Fig. 1). Consistent with this role, leukocytes in mice deficient for both E- and P-selectins (Selp −/− Sele −/−) are unable to migrate properly to sites of tissue injury or inflammation [28]. Although these mice are more susceptible to bacterial infections, they are also less likely to develop hypersensitivity (allergic) or other inflammatory reactions. HSC homing and engraftment upon transplantation of wild-type HSC is defective in lethally irradiated Selp −/− Sele −/− mice [29]. This function of E- and P-selectin in HSC homing and engraftment is somewhat redundant as HSC homing and engraftment can still occur in recipient mice in which either selectin gene [29].
Unique Role of E-Selectin in Regulating HSC
Unlike P- and L-selectin, it has recently emerged that E-selectin plays a unique role in HSC regulation. E-selectin, which is expressed on the surface of specialized endothelial cells in HSC perivascular niches, promotes HSC proliferation. In steady-state, only ~3–5 % of BM vascular endothelial cells express E-selectin, which this rises dramatically following hematopoietic stress, such as the recovery phase following irradiation, where >90 % of BM endothelial cells express E-selectin at a time when the surviving HSC pool must cycle to replenish the depleted blood and immune system [30••]. When E-selectin is therapeutically blocked with the small molecule antagonist GMI-1070 or is absent in Sele −/− E-selectin gene-deleted mice, a large proportion of HSC returns to quiescence with heightened self-renewal potential as shown by increased potential for serial-transplantation in mice [30••]. Another consequence of slower HSC cycling is their increased resistance to cytotoxic chemotherapy or radiation in the absence of E-selectin or following administration of the E-selectin antagonist [30••]. Surprisingly, these effects are mediated solely by E-selectin, not P-selectin as Selp −/− mice do not display increase in HSC dormancy.
Selectin Ligands on HSC
Most of the physiological selectin ligands are mucin-type glycoproteins decorated with sialylated, fucosylated glycan chains which are presumed to interact with the C-type lectin domain of the selectins (for example PSGL-1, CD34, CD44, GlyCAM-1, MADCAM-1) [15]. Mucins are large rod-like proteins with extensive O- and N-linked glycosylations that extend away from the cells outer surface. Selectins appear to recognize the “sialyl Lewisx”(sLex) or similar tetrasaccharide structures, displayed at the ends of these long glycan chains or glycolipids. However, purified sLex tetrasaccharide only partially blocks selectin-mediated cell adhesion [31]. Therefore, although sLex is part of the recognition element for selectins, it must be correctly presented on the appropriate polypeptide or glycolipid backbone to mediate the shear-resistant adhesion that occurs in vivo. PSGL-1 (CD162) is a well-characterized selectin receptor on leukocytes and binds to all three selectins with varying affinities depending on adequate glycosylations and tyrosine sulfation. Although most hematopoietic cells express PSGL-1, they cannot adhere to selectins without the co-expression of five different glycosyl-transferases required to generate the sLex tetrasaccharide. In particular, fucosyl-transferase (FucT)-VII is absolutely essential to generate functional selectin receptors. Leukocytes from Fut7 −/− mice can no longer adhere to P- and E-selectin even though they express PSGL-1 [32].
Although mouse and human HSC express active selectin ligand PSGL-1 [20, 21], the effect of E-selectin on HSC cycling is not mediated by PSGL-1 nor CD44, the alternative canonical E-selectin ligand [25], as the anti-proliferative effect of E-selectin gene deletion is not replicated in mice lacking these two genes (Selplg −/− Cd44 −/−). Furthermore, HSC from these mice still bind to E-selectin [30••]. Candidate receptors for this proliferative effect of E-selectin are not definitively identified but may include membrane glycosphingolipids [30••]. ESL-1 has also been recently shown to promote HSC quiescence; however, this effect was independent of E-selectin involving instead decreased release of anti-proliferative TGF-β [33••].
Integrins
Integrins form a vast and diverse array of cell adhesion receptors. They are formed by a heterodimeric complex between one out of 18 possible integrin α subunits and one out of 8 possible integrin β subunits all encoded by a unique gene. Not all combination of αβ complexes exist but the numbers of existing integrins remains mind boggling. They are classified according to the β subunit involved (e.g., β1 integrins, β2 integrins, etc). Each type of integrin αβ dimer has a unique repertoire of ligands that can be either extracellular matrix molecules or transmembrane cell adhesion molecules. Each integrin has unique function. For the sake of simplicity, we will review those known to be expressed on HSC which include several β1, β2 [34•], β3, and β7 integrins [35].
The β1 integrin chain (CD29) dimerizes with integrins α1 to α11 or integrin αv. Mouse and human HSC are known to express only integrin α2 (CD49b), integrin α4 (CD49d), integrin α5 (CD49e), integrin α6 (CD49f), and integrin α9 [36–39]. Each of these integrins binds to several extracellular matrix proteins such as collagens (integrin α2β1), fibronectin (α4β1 and α5β1), laminin (α6β1), cleaved osteopontin (α4β1, α9β1) [39], and a key transmembrane cell adhesion molecule called vascular cell adhesion molecule-1 (VCAM-1 or CD106) that binds to integrins α4β1, α9β1, and α4β7 all present on HSC [35, 39]. The distribution of these extracellular matrix proteins in the BM can thus act as an address code. For instance collagens I and IV are abundant at the endosteum [40]. Fibronectin is distributed throughout the BM and particularly abundant at the endosteum and in and around megakaryocytes [40], which are both associated with HSC niches. Laminin is abundant in the wall of arteries and sinusoids in the BM [40], again two sites where niches for more primitive HSC have been described [6••, 7••]. Osteopontin has been reported at the endosteum [41] where it is cleaved by thrombin that has been activated by megakaryocytes [42] to become a ligand for integrins α4β1 and α9β1 [39]. Osteopontin after cleavage by thrombin is an important regulator of HSC within their niche as it inhibits HSC proliferation in vitro and mice knocked out for the osteopontin gene (Opn −/−) have increased number of HSC in the BM. Furthermore, HSC lodgment into their Opn −/− endosteal niche is perturbed after transplantation [41]. Finally, the integrin-ligand VCAM-1 is expressed by most cells known to participate in HSC niches, particularly BM endothelial cells [43], mesenchymal stem cells and osteoprogenitors [44], osteoblasts, and a subset of BM-resident macrophages while it is absent from lymphocytes, HSPC, and monocytes [45]. Conditional deletion of the VCAM-1 gene in hematopoietic and BM stromal cells, or conditional deletion of integrin α4 gene in hematopoietic cells both results in spontaneous HSPC mobilization into the blood and spleen with dramatic expansion of splenic hematopoiesis [45, 46] proving that the adhesive interaction between VCAM-1 and α4 integrins (which include α4β1 and α4β7 both expressed by HSPC [35]) is key to anchor HSC in their niche.
The β2 integrin subunit (CD18) can dimerize with four integrin α subunits, namely αL (CD11a), αM (CD11b), αX (CD11c), and αD (CD11d). Only β2 integrin αLβ2 (previously called LFA-1) has been reported on primitive HSPC. It binds to intercellular adhesion molecules (ICAM)-1,-2 and -3, transmembrane adhesion molecules expressed by endothelial cells. Integrin αLβ2 has been found expressed on short-term reconstituting multipotent progenitors but absent from long-term reconstituting HSC in adult mice [47•] and in mouse embryos [48]. Therefore, integrin αLβ2 and its ligands are not regulator of HSC but could play an important role in regulating multipotent progenitors in their respective niches. This would be consistent with the recent observation that mice expressing an hypomorphic variant of integrin β2 have increased numbers of multipotent progenitors in the BM [34•].
The β3 integrin subunit (CD61) can associate with either integrin αIIb (CD41) or αV (CD51). Integrin αIIbβ3 is expressed by quiescent HSC in aged mice [49•] and has been known to be expressed by embryonic HSC in the yolk sac [50]. Integrin αIIbβ3 is the classic platelet glycoprotein IIb-IIIa complex that mediates platelet aggregation during blood clotting. It binds fibrinogen, fibronectin, vitronectin, and von Willebrand factor (vWF or CD146) following its activation by ADP or thrombin [51]. Integrin αIIbβ3 plays an important role in adult hematopoiesis as integrin αIIb deficient mice (Itga2b −/−) display a pancytopenia while their HSC display increased apoptosis and proliferation, with a transplantation defect [49•]. As vWF, an integrin αIIbβ3 ligand, is produced by megakaryocytes and endothelial cells which are necessary to maintain HSC quiescent [18••, 19••, 52••], it may be that vWF plays a key role in maintaining HSC in contact with megakaryocytes and endothelial cells at their niche. Interestingly, only a small proportion of HSC express αIIbβ3 in the young adult mouse suggesting that the state of HSC quiescence may vary with age. The function of αIIbβ3 in yolk sac HSC is probably different as these cells need to expand to populate the growing embryo with primitive hematopoietic cells (mostly fetal erythrocytes and macrophages).
Integrin Activation States—Key to Find the Good Address
The spectrum of integrins expressed by HSC suggests that they play a key role in HSC anchoring in the BM, as well as homing to the BM during development or following transplantation. A key aspect of integrin biology is that unlike other cell adhesion receptors, integrins require prior conformational activation by extracellular soluble mediators to become capable of binding their ligands. This is because integrin αβ complexes can adopt several conformational states with different levels of affinity or avidity for their ligands. As many integrin ligands are expressed in many different tissues, this enables them to lock-in cells at the right time at the right place provided that the integrin activating molecule is present. Integrins require metallic divalent cations such as Mg2+ and Ca2+ to complex together in the plasma membrane. On hematopoietic cells, β1 integrins can be activated by inside-out signaling by cytokine receptors such as the interleukin-3 receptor or c-Kit [36, 53]. Integrins have also divalent cation binding sites near the ligand binding site. Mg2+ at concentrations over 1 mM is sufficient to initiate β1 integrin activation without cytokine. In contrast, Ca2+ in excess of 10 mM (concentrations found physiologically in the periphery of osteoclasts resorbing bones) reduces β1 integrin activation by cytokines [54]. Interestingly, Mn2+ is a “super-activator” of β1 integrins as between 0.03 and 0.1 mM it locks β1 integrins in an activated state [54]. The recent finding that hematopoietic cells in the endosteal region of the BM have high level of Mn2+ associated with their plasma membrane [55••] opens new perspectives. Indeed, Mn2+ is necessary to proper bone development and at 1.4 ppm is relatively abundant in the compact bone [56] compared to the blood (1.5-2.0 μg /L) [57]. Mn2+ release under the effect of osteoclasts remodeling the bone, could play a major role in maintaining β1 integrins activated at the surface of HSC to lock them in at their niches.
CD44
CD44 is a highly polymorphic and multifunctional transmembrane proteoglycan expressed on most leukocytes and HSPC. CD44 is expressed with many splicing variants, some of them characteristic of leukemic transformation. Appropriately decorated with sLex fucosylated polysaccharides, CD44 can be a receptor for L-selectin and E-selectin [25, 58] as mentioned above. CD44 is also a receptor for high molecular weight hyaluronic acid. Hyaluronic acid is a highly hydrophilic polysaccharide that is produced in many tissues to form a gel once hydrated. Hyaluronic acid is present in the BM at the endosteum and around blood vessels [59]. It is also synthesized by HSPC themselves via the expression of hyaluronate synthase 3 [60]. CD44 expressed by human and mouse HSPC binds to hyaluronic acid [60, 61]. Similar to β1 integrins, the binding of CD44 to hyaluronic acid is activated by interleukin-3 receptor and c-Kit by an undefined inside-out signaling mechanism [61]. HSC from mice knocked out for the hyaluronate synthase-3 gene (Has3 −/−) have reduced lodgment in the metaphyseal endosteal region of the BM following transplantation [59]. Therefore, hyaluronic acid is likely to guide HSC to their niche via CD44.
Interestingly, the monoclonal anti-human CD44 antibody H90 activates CD44 on CD34+ HSPC to bind to hyaluronic acid [61]. Most interestingly, this antibody reverses the differentiation blockage in acute myeloid leukemia (AML) cells in vitro which then progress towards a more mature monocytic morphology [62]. Likewise in immune-deficient mice transplanted with primary human AML cells, administration of H90 antibody to the leukemic mice reduced leukemia progression, induced AML cell differentiation with induction the monocytic antigen CD14 expression and inhibition of engraftment in secondary transplants. This suggests that this anti-CD44 antibody directly targets leukemia stem cells [63]. A6 peptide is a capped octopeptide with the sequence Ac-KPSSPPEE-NH2 derived from urokinase plasminogen activator [64]. A6 peptide has been shown to bind and activate CD44 with downstream phosphorylation of focal adhesion kinase (FAK) and increase adhesion to hyaluronic acid [65]. A6-peptide selectively kills primary chronic lymphoblastic leukemia cells in vitro and in vivo in xenograft in immunodeficient mice [66].
Eph and Ephrins
Transmembrane cell-cell adhesion receptors Eph and their ligands ephrins form a unique family of cell adhesion molecules. Eph receptors are integral tyrosine kinases belonging to two different classes A and B with 10 different EphA and 6 different EphB all encoded by different genes. In general, EphA receptors can bind to 5 different glycophosphoinositide-anchored ephrin A, while EphB receptors can bind to 3 different transmembrane ephrin B, with the exception of EphB2, ephrin B2, and EphA4 which can bind across both A and B classes of Eph and ephrins [67]. Binding of ephrins to Eph receptors initiates both forward signaling via the tyrosine kinase domain of Eph receptors and reverse signaling in the cell presenting ephrins [68•]. Eph and ephrins are best known for their role in axonal guidance and remodeling of the nervous system during development [68•]. Their expression is also induced in a number of tissues following systemic inflammation or injury [67].
In relation to the hematopoietic system, EphA2 and EphB2 were found expressed on mobilized human CD34+ or CD133+ HSPC [69] while EphA3 has been reported on erythroid progenitors [70]. Human BM stromal cells express EphB1 and EphB2, EphB4, ephrin-B1 and ephrin-B2 [71]. In the mouse EphA 1–5, receptors were found expressed at the surface of mouse HPSC [72]. Mouse HSPC were also found to express ephrins ligand using EhpA1-Fc and EhpA3-Fc probes; however, their precise identity could not be confirmed unambiguously as monoclonal antibodies specific for mouse ephrins were not available. Likewise, primary mouse BM stromal cells were also found to express EphA2 and EphA3 as well as ephrins binding to the EphA3-Fc and EphA1-Fc probes [72]. In other reports, EphA2, EphB4, ephrin B1, and ephrin B2 were detected on mouse BM stromal cells [71]. Interestingly, stimulation of the mouse myeloid cell line FDCP1 with EphA3-Fc construct increased cell adhesion to VCAM-1 while in vivo administration of EphA3-Fc into mice caused a rapid and transient mobilization of colony-forming units (CFU) and more primitive CFU-S12 and cobblestone area forming cells (CAFC) into the peripheral blood [72]. Therefore, both HSPC and stromal cells express an array of EphA receptors and ephrin ligands suggesting that this system may establish a cross-talk between HSC and their niche stromal components. However, this may be difficult to demonstrate genetically as Eph and ephrins are redundant with promiscuity in their receptor/ligand preference. To illustrate this, Epha2−/− mice did not show any obvious hematopoietic abnormality [73]. Eph receptors have been found expressed on AML [74], chronic lymphoblastic leukemia [75] and a variety of B [76] and T leukemia cell lines [77] and may have a role in the biology of these hematopoietic malignancies.
Clinical Application: HSC Mobilization and Transplantation
Until the early 1990s, HSPC harvested for use in transplantation were collected by multiple BM aspirations under general anesthesia. This, however, changed rapidly when it was discovered that daily injection of the cytokine granulocyte colony-stimulating factor (G-CSF) promoted the egress of HSPC from the BM into the blood in mice [78], a phenomenon called mobilization. Subsequent studies rapidly confirmed G-CSF-induced mobilization in humans [79] leading to progressive replacement of BM aspirates by peripheral blood CD34+ cells as the source of HSC for transplantation [80]. Owing to this success, over one million HSC transplantations have been performed with mobilized blood CD34+ cells worldwide to date.
For many years, the mechanisms by which G-CSF promotes HSPC mobilization remained obscure. However, intense research since the early 2000s has identified several mechanisms working in parallel. G-CSF causes a complete remodeling of the BM landscape and HSPC niches [81]. The expression of VCAM-1 cell adhesion protein and the chemokine CXCL12 (also called stromal-derived factor-1; SDF-1) which both work in concert to retain HSPC within their niches are both strikingly down-regulated in response to G-CSF in vivo [82, 83]. This is due to a combination of mechanisms such as (1) proteolytic cleavage by neutrophil proteases released in the BM as a consequence of neutrophil expansion [82–84] and (2) direct down-regulation of Vcam1 and Cxcl12 transcripts in BM stromal cells [85, 86]. This latter effect is in part mediated by BM-resident macrophages which express the G-CSF receptor [86–88].
While in healthy donors G-CSF treatment is sufficient to elicit mobilization of sufficient numbers of CD34+ HSPC to ensure safe engraftment in an allogeneic recipient, G-CSF fails to mobilize adequate numbers of CD34+ HSPC in a proportion of heavily pre-treated multiple myeloma and non-Hodgkin’s lymphoma patients eligible for autologous HSC transplantation which adversely affect their prognosis of survival [89]. A number of therapeutic strategies have been devised to overcome HSC mobilization failure in these patients. The only drug which is currently approved to rescue mobilization in patients who previously failed to mobilize in response to G-CSF is the small synthetic CXCR4 antagonist Plerixafor (previously called AMD3100). By inhibiting chemotaxis in response to CXCL12, Plerixafor synergizes with G-CSF in eliciting HSPC mobilization [90] enabling mobilization rescue in an important proportion of these poor mobilizers [91, 92]. Alternative CXCR4 antagonists are being developed to elicit mobilization in autologous transplantation patients [93]. An alternative strategy to CXCR4 antagonism is to directly target integrins (particularly α4β1 and α9β1) responsible for HSPC anchoring to VCAM-1 within their niche. The humanized anti-integrin α4 antibody natalizumab is used to treat relapsing multiple sclerosis and Crohn’s disease by blocking lymphocyte recruitment in chronically inflamed nervous system and gut. Natalizumab is a potent HSC mobilizing agent [94, 95]. Interestingly, CD34+ HSPC mobilized with natalizumab have higher level of CXCR4 expression and enhanced chemotaxis to CXCL12 compared G-CSF-mobilized HSPC [95]. BIO5192, a small synthetic antagonist of integrin α4 also causes weak mobilization of HSPC alone and synergizes with G-CSF to levels similar to Plerixafor [96]. As VCAM-1 binds to both integrins α4β1 and α9β1 on HSPC, N-(benzenesulfonyl)-L-prolyl-L-O-(1-pyrrolidinylcarbonyl)tyrosine (BOP) a small synthetic molecules that antagonizes both integrins has been developed. Interestingly, a single dose of BOP rapidly mobilized HSC within 30–60 min in mice and showed strong synergism with G-CSF [55••]. Most interestingly, transplantation of blood mobilized with the combination of BOP and Plerixafor led to higher reconstitution and engraftment than blood mobilized with G-CSF alone [55••]. Therefore, targeting both the CXCL12/CXCR4 axis and the VCAM-1/integrin α4β1 α9β1 axis by combining CXCR4 antagonist and BOP may represent a superior alternative to mobilize HSC in patients that mobilize poorly in response to G-CSF. However, at present only Plerixafor has been approved by the FDA to enhance HSC mobilization.
Clinical Application: Blocking Cell Adhesion to Overcome Chemoresistance
It has known for nearly two decades that adhesion of multiple myeloma [97], B-lymphoma [98], B cell acute lymphoblastic leukemia (B-ALL) [99], B-CLL [100], AML [101] and more recently chronic myeloblastic leukemia (CML) cells [102] to BM stroma cells or immobilized cell adhesion molecules increases malignant cell survival to cytotoxic drugs or in some cases to tyrosine-kinase inhibitors and immunomodulators such as lenalidomide. This phenomenon called cell adhesion-mediated drug resistance (CAM-DR) has been shown in many cases to involve in part adhesive interaction between α4 integrins and VCAM-1 [97, 98, 103–107, 108•].
In addition to integrin-mediated CAM-DR, the interaction between CD44 and hyaluronic acid has also been reported to mediate CAM-DR in multiple myeloma [109]. L-selectin has also recently been reported to mediate CAM-DR in B-CLL cells by promoting B-CLL cell escape in lymph nodes and protection once adhered to BM stromal cells [110, 111•]. Finally, we have recently found in an immune competent mouse model of human AML, that E-selectin but not P-selectin promotes potent CAM-DR in vitro and in vivo as deletion of the E-selectin receptor in mice or administration of the small E-selectin antagonist GMI-1271 sensitized leukemia stem cells to chemotherapy [112]. Likewise, E-selectin has been found to induce CAM-DR to the tyrosine-kinase inhibitor imatinib in an immune competent mouse model of human CML an effect reversed in vivo by GMI-1271 [113].
The mechanisms of CAM-DR have not been fully elucidated and may be disease dependent. Activation of focal adhesion kinase [101], which is activated by integrin clustering, NF-κB/RelB [108•, 114], Wnt/β-catenin signaling [115], or by activating drug efflux by ATP-binding cassette transporters [106] may play a role.
These findings have prompted a few clinical trials to test safety and efficacy of disrupting cell adhesion as an adjuvant therapy to conventional therapy (Table 1). The most common approach has been to mobilize hematopoietic malignant cells from their protective niches to sensitize them to chemotherapy. This approach has been successful in mouse models of AML [116, 117]. There are currently five clinical trials registered in the USA to test chemosensitizing effect of various CXCR4 antagonists. Of note a number of phase I/II trials to test Plerixafor safety and efficacy in treatment of AML have been completed with only one with published results [118].
While the anti-α4 integrin natalizumab is in many clinical trials to test efficacy in treating multiple sclerosis, there was only one trail on its effect on multiple myeloma, a trial that was terminated in 2014 due to low enrolment and technical problems with measurements.
There is currently one phase II clinical trial recruiting to test efficacy and tolerability of CD44-activating A6-peptide in CLL and small lymphocytic lymphoma patients.
The efficacy of small E-selectin antagonist GMI-1271 in combination with conventional chemotherapy is being tested in a phase I/II trial on refractory and relapsed AML patients. In view of promising initial results, the FDA granted Fast Track status to this clinical trial in June 2016 to progress to phase II trial on newly diagnosed AML patients.
Finally, the efficacy of blocking EphA3 receptor with the humanized antibody KB004 was trialed on myelodysplasia and myelofibrosis patients; however, the trial was suspended in November 2015 after the company Kalobios making KB004 filed for bankruptcy.
Conclusions
Recent studies with genetically modified mice in which genes encoding cell adhesion molecules are deleted have clearly demonstrated that some of these cell adhesion molecules are key not only to HSPC homing and anchoring within their specific niches in the BM but also regulate HSPC fate within the BM in steady-state such as the balance between quiescence and proliferation. Another essential aspect is how malignant cells utilize these adhesive interactions to resist cytotoxic chemotherapies and targeted therapies, which ultimately causes relapse from resistant clones that accumulate additional mutations during the process causing multi-drug resistance. Understanding these interactions has enabled the design of drugs targeting specific adhesive interactions which can be used to treat chronic inflammatory diseases but also to enhance HSC mobilization for transplantation. Targeting cell adhesive interactions to chemosensitize hematological malignancies is a new direction for treatments. While it is too soon to conclude on the efficacy of this approach, this is an exciting development in the treatment of incurable hematological malignancies such as multiple myeloma and CLL or diseases with very poor prognosis in the absence of allogeneic HSC transplantation such as AML.
References
Papers of the particular interest, published recently, have been highlighted as • Of importance •• Of major importance
Lord BI, Testa NG, Hendry JH. The relative spatial distributions of CFUs and CFUc in the normal mouse femur. Blood. 1975;46:65–72.
Arai F, Hirao A, Ohmura M, Sato H, Matsuoka S, Takubo K, et al. Tie2/angiopoietin-1 signaling regulates hematopoietic stem cell quiescence in the bone marrow niche. Cell. 2004;118:149–61.
Kohler A, Schmithorst V, Filippi M-D, Ryan MA, Daria D, Gunzer M, et al. Altered cellular dynamics and endosteal location of aged early hematopoietic progenitor cells revealed by time-lapse intravital imaging in long bones. Blood. 2009;114:290–8.
Lo Celso C, Fleming HE, Wu JW, Zhao CX, Miake-Lye S, Fujisaki J, et al. Live-animal tracking of individual haematopoietic stem/progenitor cells in their niche. Nature. 2009;457:92–7.
Xie Y, Yin T, Wiegraebe W, He XC, Miller D, Stark D, et al. Detection of functional haematopoietic stem cell niche using real-time imaging. Nature. 2009;457:97–101.
Kunisaki Y, Bruns I, Scheiermann C, Ahmed J, Pinho S, Zhang D, et al. Arteriolar niches maintain haematopoietic stem cell quiescence. Nature. 2013;502:637–43. This paper shows that HSC niche in the proximity of sinusoids and arterioles in the BM and that quiescent HSC reside preferentially near arterioles.
Itkin T, Gur-Cohen S, Spencer JA, Schajnovitz A, Ramasamy SK, Kusumbe AP, et al. Distinct bone marrow blood vessels differentially regulate haematopoiesis. Nature. 2016;532:323–8. This paper shows that HSC that trafick between their niche and the circulatioon are located in perisunisoidal niches.
Catlin SN, Busque L, Gale RE, Guttorp P, Abkowitz JL. The replication rate of human hematopoietic stem cells in vivo. Blood. 2011;117:4460–6.
Wilson A, Laurenti E, Oser G, van der Wath RC, Blanco-Bose W, Jaworski M, et al. Hematopoietic stem cells reversibly switch from dormancy to self-renewal during homeostasis and repair. Cell. 2008;135:1118–29.
Essers MAG, Offner S, Blanco-Bose WE, Waibler Z, Kalinke U, Duchosal MA, et al. IFNα activates dormant haematopoietic stem cells in vivo. Nature. 2009;458:904–8.
Baldridge MT, King KY, Boles NC, Weksberg DC, Goodell MA. Quiescent haematopoietic stem cells are activated by IFN-γ in response to chronic infection. Nature. 2010;465:793–7.
Ting SB, Deneault E, Hope K, Cellot S, Chagraoui J, Mayotte N, et al. Asymmetric segregation and self-renewal of hematopoietic stem and progenitor cells with endocytic Ap2a2. Blood. 2012;119:2510–22.
Winkler IG, Barbier V, Wadley R, Zannettino ACW, Williams S, Levesque J-P. Positioning of bone marrow hematopoietic and stromal cells relative to blood flow in vivo: serially reconstituting hematopoietic stem cells reside in distinct nonperfused niches. Blood. 2010;116:375–85.
Levesque JP, Winkler IG. Hierarchy of immature hematopoietic cells related to blood flow and niche. Curr Opin Hematol. 2011;18:220–5.
Kansas GS. Selectins and their ligands: current concepts and controversies. Blood. 1996;88:3259–87.
Arbones ML, Ord DC, Ley K, Ratech H, Maynard-Curry C, Otten G, et al. Lymphocyte homing and leukocyte rolling and migration are impaired in L-selectin-deficient mice. Immunity. 1994;1:247–60.
Heazlewood SY, Neaves RJ, Williams B, Haylock DN, Adams TE, Nilsson SK. Megakaryocytes co-localise with hemopoietic stem cells and release cytokines that up-regulate stem cell proliferation. Stem Cell Res. 2013;11:782–92.
Bruns I, Lucas D, Pinho S, Ahmed J, Lambert MP, Kunisaki Y, et al. Megakaryocytes regulate hematopoietic stem cell quiescence through CXCL4 secretion. Nat Med. 2014;20:1315–20. This paper shows that HSC are located in contact with megakaryocytes in the BM and that megakryocytes maintain HSC quiscent by secreting CXCL4.
Zhao M, Perry JM, Marshall H, Venkatraman A, Qian P, He XC, et al. Megakaryocytes maintain homeostatic quiescence and promote post-injury regeneration of hematopoietic stem cells. Nat Med. 2014;20:1321–6. This paper shows that HSC are located in contact with megakaryocytes in the BM and that megakryocytes maintain HSC quiscent by secreting TGF-β1 that activates SMAD signaling in HSC.
Levesque JP, Zannettino AC, Pudney M, Niutta S, Haylock DN, Snapp KR, et al. PSGL-1-mediated adhesion of human hematopoietic progenitors to P-selectin results in suppression of hematopoiesis. Immunity. 1999;11:369–78.
Winkler IG, Snapp KR, Simmons PJ, Levesque J-P. Adhesion to E-selectin promotes growth inhibition and apoptosis of human and murine hematopoietic progenitor cells independent of PSGL-1. Blood. 2004;103:1685–92.
Sipkins DA, Wei X, Wu JW, Runnels JM, Cote D, Means TK, et al. In vivo imaging of specialized bone marrow endothelial microdomains for tumour engraftment. Nature. 2005;435:969–73.
Zarbock A, Ley K, McEver RP, Hidalgo A. Leukocyte ligands for endothelial selectins: specialized glycoconjugates that mediate rolling and signaling under flow. Blood. 2011;118:6743–51.
Goetz DJ, Greif DM, Ding H, Camphausen RT, Howes S, Comess KM, et al. Isolated P-selectin glycoprotein ligand-1 dynamic adhesion to P- and E-selectin. J Cell Biol. 1997;137:509–19.
Dimitroff CJ, Lee JY, Rafii S, Fuhlbrigge RC, Sackstein R. CD44 is a major E-selectin ligand on human hematopoietic progenitor cells. J Cell Biol. 2001;153:1277–86.
Sreeramkumar V, Leiva M, Stadtmann A, Pitaval C, Ortega-Rodríguez I, Wild MK, et al. Coordinated and unique functions of the E-selectin ligand ESL-1 during inflammatory and hematopoietic recruitment in mice. Blood. 2013;122:3993–4001. Using mice knocked-out for either or both the PSGL-1 and ESL-1 genes, these authors show that ESL-1 cooperates with PSGL-1 for E-selectin binding on neutrophils and their recruitment to sites of inflammation. Likewise ESL-1 and PSGL-1 cooperate to promote E-selectin binding to HSPC and their homing within the BM. While neutrophils deficient for both ESL-1 and PSGL-1 bind poorly to E-selectin with defective extravasation into the peritoneum, HSPC deficient for both ESL-1 and PSGL-1 still bind to E-selectin showing that HSPC express additional E-selectin ligands.
Nimrichter L, Burdick MM, Aoki K, Laroy W, Fierro MA, Hudson SA, et al. E-selectin receptors on human leukocytes. Blood. 2008;112:3744–52.
Frenette PS, Mayadas TN, Rayburn H, Hynes RO, Wagner DD. Susceptibility to infection and altered hematopoiesis in mice deficient in both P- and E-selectins. Cell. 1996;84:563–74.
Nabors LK, Wang LD, Wagers AJ, Kansas GS. Overlapping roles for endothelial selectins in murine hematopoietic stem/progenitor cell homing to bone marrow. Exp Hematol. 2013;41:588–96.
Winkler IG, Barbier V, Nowlan B, Jacobsen RN, Forristal CE, Patton JT, et al. Vascular niche E-selectin regulates hematopoietic stem cell dormancy, self renewal and chemoresistance. Nat Med. 2012;18:1651–7. E-selectin plays a unique role amongst seletins by inhibiting HSC proliferation in HSC perivascular niches. Deletion of the gene or administration of a small synthetic E-selectin antagonist increases HSC quiescence, self-renewal potential upon transplantation and resitance to irradiation and cytotoxic chemotherapy.
Somers WS, Tang J, Shaw GD, Camphausen RT. Insights into the molecular basis of leukocyte tethering and rolling revealed by structures of P- and E-selectin bound to sLex and PSGL-1. Cell. 2000;103:467–79.
Maly P, Thall A, Petryniak B, Rogers CE, Smith PL, Marks RM, et al. The alpha(1,3)fucosyltransferase Fuc-TVII controls leukocyte trafficking through an essential role in L-, E-, and P-selectin ligand biosynthesis. Cell. 1996;86:643–53.
Leiva M, Quintana JA, Ligos JM, Hidalgo A. Haematopoietic ESL-1 enables stem cell proliferation in the bone marrow by limiting TGFβ availability. Nat Commun. 2016;7:10222. ESL-1 induces HSC quiscence by promoting secretion of TGF-β. Inhibition of HSC quiscence by ESL-1 is mechanistically independent from inhibition of HSC quiscence by ESL-1 even if E-selectin and ESL-1 interact with each other
Leon-Rico D, Aldea M, Sanchez R, Segovia JC, Weiss LA, Hidalgo A, et al. Brief report: reduced expression of CD18 leads to the in vivo expansion of hematopoietic stem cells in mouse bone marrow. Stem Cells. 2014;32:2794–8. Mice with a hypomorphic mutation in the Itgb2 gene have increased number of HSC and HPC in the BM and HSC have enhanced engraftment potenital upon transplantation.
Katayama Y, Hidalgo A, Peired A, Frenette PS. Integrin α4β7 and its counter receptor MAdCAM-1 contribute to hematopoietic progenitor recruitment into bone marrow following transplantation. Blood. 2004;104:2020–6.
Levesque JP, Leavesley DI, Niutta S, Vadas M, Simmons PJ. Cytokines increase human hemopoietic cell adhesiveness by activation of very late antigen (VLA)-4 and VLA-5 integrins. J Exp Med. 1995;181:1805–15.
Wagers AJ, Allsopp RC, Weissman IL. Changes in integrin expression are associated with altered homing properties of Lin−/loThy1.1loSca-1+c-kit+ hematopoietic stem cells following mobilization by cyclophosphamide/granulocyte colony-stimulating factor. Exp Hematol. 2002;30:176–85.
Notta F, Doulatov S, Laurenti E, Poeppl A, Jurisica I, Dick JE. Isolation of single human hematopoietic stem cells capable of long-term multilineage engraftment. Science. 2011;333:218–21.
Grassinger J, Haylock DN, Storan MJ, Haines GO, Williams B, Whitty GA, et al. Thrombin-cleaved osteopontin regulates hemopoietic stem and progenitor cell functions through interactions with α9β1 and α4β1 integrins. Blood. 2009;114:49–59.
Nilsson SK, Debatis ME, Dooner MS, Madri JA, Quesenberry PJ, Becker PS. Immunofluorescence characterization of key extracellular matrix proteins in murine bone marrow in situ. J Histochem Cytochem. 1998;46:371–7.
Nilsson SK, Johnston HM, Whitty GA, Williams B, Webb RJ, Denhardt DT, et al. Osteopontin, a key component of the hematopoietic stem cell niche and regulator of primitive hematopoietic progenitor cells. Blood. 2005;106:1232–9.
Storan MJ, Heazlewood SY, Heazlewood CK, Haylock DN, Alexander WS, Neaves RJ, et al. Brief report: factors released by megakaryocytes thrombin cleave osteopontin to negatively regulate hematopoietic stem cells. Stem Cells. 2015;33:2351–7.
Jacobsen K, Kravitz J, Kincade PW, Osmond DG. Adhesion receptors on bone marrow stromal cells: in vivo expression of vascular cell adhesion molecule-1 by reticular cells and sinusoidal endothelium in normal and gamma-irradiated mice. Blood. 1996;87:73–82.
Simmons PJ, Masinovsky B, Longenecker BM, Berenson R, Torok-Storb B, Gallatin WM. Vascular cell adhesion molecule-1 expressed by bone marrow stromal cells mediates the binding of hematopoietic progenitor cells. Blood. 1992;80:388–95.
Ulyanova T, Scott LM, Priestley GV, Jiang Y, Nakamoto B, Koni PA, et al. VCAM-1 expression in adult hematopoietic and nonhematopoietic cells is controlled by tissue-inductive signals and reflects their developmental origin. Blood. 2005;106:86–94.
Scott LM, Priestley GV, Papayannopoulou T. Deletion of α4 integrins from adult hematopoietic cells reveals roles in homeostasis, regeneration, and homing. Mol Cell Biol. 2003;23:9349–60.
Fathman JW, Fernhoff NB, Seita J, Chao C, Scarfone VM, Weissman IL, et al. Upregulation of CD11A on hematopoietic stem cells denotes the loss of long-term reconstitution potential. Stem Cell Reports. 2014;3:707–15. This paper shows that integrin αL expression on HSC is bimodal and that HSC without integrin αL ensure long-term reconstitution upon transplantation whereas HSC expressing integrin αL have lost their long-term reconstitution potential.
Inlay MA, Serwold T, Mosley A, Fathman JW, Dimov IK, Seita J, et al. Identification of multipotent progenitors that emerge prior to hematopoietic stem cells in embryonic development. Stem Cell Reports. 2014;2:457–72.
Gekas C, Graf T. CD41 expression marks myeloid-biased adult hematopoietic stem cells and increases with age. Blood. 2013;121:4463–72. First paper demonstrating that integrin αIIb is also expressed on adult mouse HSC with long-term reconstitution potential and that deletion of the Itga2b gene results in increased HSC proliferation and apoptosis.
Ferkowicz MJ, Starr M, Xie X, Li W, Johnson SA, Shelley WC, et al. CD41 expression defines the onset of primitive and definitive hematopoiesis in the murine embryo. Development. 2003;130:4393–403.
Plow EF, Pierschbacher MD, Ruoslahti E, Marguerie GA, Ginsberg MH. The effect of Arg-Gly-asp-containing peptides on fibrinogen and von Willebrand factor binding to platelets. Proc Natl Acad Sci U S A. 1985;82:8057–61.
Ding L, Saunders TL, Enikolopov G, Morrison SJ. Endothelial and perivascular cells maintain haematopoietic stem cells. Nature. 2012;481:457–62. First paper demonstrating that conditional deletion of the Kit ligand gene in endothelial cells or in prevascular stromal cells severely reduces the number of HSC in the BM and impairs their reconstitution potential upon transplantation. Therefore these two cell types are critical to HSC maintenance.
Levesque JP, Haylock DN, Simmons PJ. Cytokine regulation of proliferation and cell adhesion are correlated events in human CD34+ hemopoietic progenitors. Blood. 1996;88:1168–76.
Takamatsu Y, Simmons PJ, Levesque JP. Dual control by divalent cations and mitogenic cytokines of alpha 4 beta 1 and alpha 5 beta 1 integrin avidity expressed by human hemopoietic cells. Cell Adhes Commun. 1998;5:349–66.
Cao B, Zhang Z, Grassinger J, Williams B, Heazlewood CK, Churches QI, et al. Therapeutic targeting and rapid mobilization of endosteal HSC using a small molecule integrin antagonist. Nat Commun. 2016;7:11007. Paper showing that a small synthetic inhibitor of integrins α4β1 and α9β1 mobilizes mouse and human HSC and synergizes with the CXCR4 inhibitor Plerixafor.
Fore H, Morton RA. The manganese in bone. Biochem J. 1952;51:598–600.
Versieck J, Barbier F, Speecke A, Hoste J. Manganese, copper, and zinc concentrations in serum and packed blood cells during acute hepatitis, chronic hepatitis, and posthepatitic cirrhosis. Clin Chem. 1974;20:1141–5.
Dimitroff CJ, Lee JY, Fuhlbrigge RC, Sackstein R. A distinct glycoform of CD44 is an L-selectin ligand on human hematopoietic cells. Proc Natl Acad Sci U S A. 2000;97:13841–6.
Ellis SL, Grassinger J, Jones A, Borg J, Camenisch T, Haylock D, et al. The relationship between bone, hemopoietic stem cells, and vasculature. Blood. 2011;118:1516–24.
Nilsson SK, Haylock DN, Johnston HM, Occhiodoro T, Brown TJ, Simmons PJ. Hyaluronan is synthesized by primitive hemopoietic cells, participates in their lodgment at the endosteum following transplantation, and is involved in the regulation of their proliferation and differentiation in vitro. Blood. 2003;101:856–62.
Legras S, Levesque JP, Charrad R, Morimoto K, Le Bousse C, Clay D, et al. CD44-mediated adhesiveness of human hematopoietic progenitors to hyaluronan is modulated by cytokines. Blood. 1997;89:1905–14.
Charrad RS, Li Y, Delpech B, Balitrand N, Clay D, Jasmin C, et al. Ligation of the CD44 adhesion molecule reverses blockage of differentiation in human acute myeloid leukemia. Nat Med. 1999;5:669–76.
Jin L, Hope KJ, Zhai Q, Smadja-Joffe F, Dick JE. Targeting of CD44 eradicates human acute myeloid leukemic stem cells. Nat Med. 2006;12:1167–74.
Finlayson M. Modulation of CD44 activity by A6-peptide. Front Immunol. 2015;6:135.
Piotrowicz RS, Damaj BB, Hachicha M, Incardona F, Howell SB, Finlayson M. A6 peptide activates CD44 adhesive activity, induces FAK and MEK phosphorylation, and inhibits the migration and metastasis of CD44-expressing cells. Mol Cancer Ther. 2011;10:2072–82.
Lai H, Zhang S, Wu C, Chen L, Liu G, Wu R, et al. Selective cytotoxicity of A6 peptide against ZAP-70 expressing CLL B-cells. Cancer Res. 2014;74:950 (abstract).
Coulthard MG, Morgan M, Woodruff TM, Arumugam TV, Taylor SM, Carpenter TC, et al. Eph/ephrin signaling in injury and inflammation. Am J Pathol. 2012;181:1493–503.
Boyd AW, Bartlett PF, Lackmann M. Therapeutic targeting of EPH receptors and their ligands. Nat Rev Drug Discov. 2014;13:39–62. Comprehensive review on the role of Eph receptors and ephrins and possible therapeutic targeting.
Lazarova P, Wu Q, Kvalheim G, Suo Z, Haakenstad KW, Metodiev K, et al. Growth factor receptors in hematopoietic stem cells: EPH family expression in CD34+ and CD133+ cell populations from mobilized peripheral blood. Int J Immunopathol Pharmacol. 2006;19:49–56.
Inada T, Iwama A, Sakano S, Ohno M, Sawada K, Suda T. Selective expression of the receptor tyrosine kinase, HTK, on human erythroid progenitor cells. Blood. 1997;89:2757–65.
Nguyen TM, Arthur A, Gronthos S. The role of Eph/ephrin molecules in stromal-hematopoietic interactions. Int J Hematol. 2016;103:145–54.
Ting MJ, Day BW, Spanevello MD, Boyd AW. Activation of ephrin A proteins influences hematopoietic stem cell adhesion and trafficking patterns. Exp Hematol. 2010;38:1087–98.
Charmsaz S, Beckett K, Smith FM, Bruedigam C, Moore AS, Al-Ejeh F, et al. EphA2 is a therapy target in EphA2-positive leukemias but is not essential for normal hematopoiesis or leukemia. PLoS One. 2015;10:e0130692.
Wrobel T, Pogrzeba J, Stefanko E, Wojtowicz M, Jazwiec B, Dzietczenia J, et al. Expression of Eph A4, Eph B2 and Eph B4 receptors in AML. Pathology oncology research: POR. 2014;20:901–7.
Alonso CL, Trinidad EM, de Garcillan B, Ballesteros M, Castellanos M, Cotillo I, et al. Expression profile of Eph receptors and ephrin ligands in healthy human B lymphocytes and chronic lymphocytic leukemia B-cells. Leuk Res. 2009;33:395–406.
Wimmer-Kleikamp SH, Nievergall E, Gegenbauer K, Adikari S, Mansour M, Yeadon T, et al. Elevated protein tyrosine phosphatase activity provokes Eph/ephrin-facilitated adhesion of pre-B leukemia cells. Blood. 2008;112:721–32.
Maddigan A, Truitt L, Arsenault R, Freywald T, Allonby O, Dean J, et al. EphB receptors trigger Akt activation and suppress Fas receptor-induced apoptosis in malignant T lymphocytes. J Immunol. 2011;187:5983–94.
Pojda Z, Molineux G, Dexter TM. Hemopoietic effects of short-term in vivo treatment of mice with various doses of rhG-CSF. Exp Hematol. 1990;18:27–31.
Matsunaga T, Sakamaki S, Kohgo Y, Ohi S, Hirayama Y, Niitsu Y. Recombinant human granulocyte colony-stimulating factor can mobilize sufficient amounts of peripheral blood stem cells in healthy volunteers for allogeneic transplantation. Bone Marrow Transplant. 1993;11:103–8.
To LB, Haylock DN, Simmons PJ, Juttner CA. The biology and clinical uses of blood stem cells. Blood. 1997;89:2233–58.
Levesque JP, Helwani FM, Winkler IG. The endosteal ‘osteoblastic’ niche and its role in hematopoietic stem cell homing and mobilization. Leukemia. 2010;24:1979–92.
Levesque JP, Takamatsu Y, Nilsson SK, Haylock DN, Simmons PJ. Vascular cell adhesion molecule-1 (CD106) is cleaved by neutrophil proteases in the bone marrow following hematopoietic progenitor cell mobilization by granulocyte colony-stimulating factor. Blood. 2001;98:1289–97.
Levesque JP, Hendy J, Takamatsu Y, Simmons PJ, Bendall LJ. Disruption of the CXCR4/CXCL12 chemotactic interaction during hematopoietic stem cell mobilization induced by GCSF or cyclophosphamide. J Clin Invest. 2003;111:187–96.
Winkler IG, Levesque JP. Mechanisms of hematopoietic stem cell mobilization: when innate immunity assails the cells that make blood and bone. Exp Hematol. 2006;34:996–1009.
Semerad CL, Christopher MJ, Liu F, Short B, Simmons PJ, Winkler I, et al. G-CSF potently inhibits osteoblast activity and CXCL12 mRNA expression in the bone marrow. Blood. 2005;106:3020–7.
Winkler IG, Sims NA, Pettit AR, Barbier V, Nowlan B, Helwani F, et al. Bone marrow macrophages maintain hematopoietic stem cell (HSC) niches and their depletion mobilizes HSCs. Blood. 2010;116:4815–28.
Chow A, Lucas D, Hidalgo A, Méndez-Ferrer S, Hashimoto D, Scheiermann C, et al. Bone marrow CD169+ macrophages promote the retention of hematopoietic stem and progenitor cells in the mesenchymal stem cell niche. J Exp Med. 2011;208:261–71.
Christopher MJ, Rao M, Liu F, Woloszynek JR, Link DC. Expression of the G-CSF receptor in monocytic cells is sufficient to mediate hematopoietic progenitor mobilization by G-CSF in mice. J Exp Med. 2011;208:251–60.
To LB, Levesque J-P, Herbert KE. How I treat patients who mobilize hematopoietic stem cells poorly. Blood. 2011;118:4530–40.
Broxmeyer HE, Orschell CM, Clapp DW, Hangoc G, Cooper S, Plett PA, et al. Rapid mobilization of murine and human hematopoietic stem and progenitor cells with AMD3100, a CXCR4 antagonist. J Exp Med. 2005;201:1307–18.
DiPersio JF, Micallef IN, Stiff PJ, Bolwell BJ, Maziarz RT, Jacobsen E, et al. Phase III prospective randomized double-blind placebo-controlled trial of plerixafor plus granulocyte colony-stimulating factor compared with placebo plus granulocyte colony-stimulating factor for autologous stem-cell mobilization and transplantation for patients with non-Hodgkin’s lymphoma. J Clin Oncol. 2009;27:4767–73.
DiPersio JF, Stadtmauer EA, Nademanee A, Micallef IN, Stiff PJ, Kaufman JL, et al. Plerixafor and G-CSF versus placebo and G-CSF to mobilize hematopoietic stem cells for autologous stem cell transplantation in patients with multiple myeloma. Blood. 2009;113:5720–6.
Karpova D, Dauber K, Spohn G, Chudziak D, Wiercinska E, Schulz M, et al. The novel CXCR4 antagonist POL5551 mobilizes hematopoietic stem and progenitor cells with greater efficiency than plerixafor. Leukemia. 2013;27:2322–31.
Bonig H, Wundes A, Chang K-H, Lucas S, Papayannopoulou T. Increased numbers of circulating hematopoietic stem/progenitor cells are chronically maintained in patients treated with the CD49d blocking antibody natalizumab. Blood. 2008;111:3439–41.
Jing D, Oelschlaegel U, Ordemann R, Holig K, Ehninger G, Reichmann H, et al. CD49d blockade by natalizumab in patients with multiple sclerosis affects steady-state hematopoiesis and mobilizes progenitors with a distinct phenotype and function. Bone Marrow Transplant. 2010;45:1489–96.
Ramirez P, Rettig MP, Uy GL, Deych E, Holt MS, Ritchey JK, et al. BIO5192, a small molecule inhibitor of VLA-4, mobilizes hematopoietic stem and progenitor cells. Blood. 2009;114:1340–3.
Damiano JS, Cress AE, Hazlehurst LA, Shtil AA, Dalton WS. Cell adhesion mediated drug resistance (CAM-DR): role of integrins and resistance to apoptosis in human myeloma cell lines. Blood. 1999;93:1658–67.
Taylor ST, Hickman JA, Dive C. Epigenetic determinants of resistance to etoposide regulation of Bcl-X(L) and Bax by tumor microenvironmental factors. J Natl Cancer Inst. 2000;92:18–23.
Mudry RE, Fortney JE, York T, Hall BM, Gibson LF. Stromal cells regulate survival of B-lineage leukemic cells during chemotherapy. Blood. 2000;96:1926–32.
de la Fuente MT, Casanova B, Cantero E, Hernandez del Cerro M, Garcia-Marco J, Silva A, et al. Involvement of p53 in alpha4beta1 integrin-mediated resistance of B-CLL cells to fludarabine. Biochem Biophys Res Commun. 2003;311:708–12.
Recher C, Ysebaert L, Beyne-Rauzy O, Mansat-De Mas V, Ruidavets JB, Cariven P, et al. Expression of focal adhesion kinase in acute myeloid leukemia is associated with enhanced blast migration, increased cellularity, and poor prognosis. Cancer Res. 2004;64:3191–7.
Dillmann F, Veldwijk MR, Laufs S, Sperandio M, Calandra G, Wenz F, et al. Plerixafor inhibits chemotaxis toward SDF-1 and CXCR4-mediated stroma contact in a dose-dependent manner resulting in increased susceptibility of BCR-ABL+ cell to imatinib and Nilotinib. Leuk Lymphoma. 2009;50:1676–86.
Noborio-Hatano K, Kikuchi J, Takatoku M, Shimizu R, Wada T, Ueda M, et al. Bortezomib overcomes cell-adhesion-mediated drug resistance through downregulation of VLA-4 expression in multiple myeloma. Oncogene. 2009;28:231–42.
Buchner M, Baer C, Prinz G, Dierks C, Burger M, Zenz T, et al. Spleen tyrosine kinase inhibition prevents chemokine- and integrin-mediated stromal protective effects in chronic lymphocytic leukemia. Blood. 2010;115:4497–506.
Neri P, Ren L, Azab AK, Brentnall M, Gratton K, Klimowicz AC, et al. Integrin β7-mediated regulation of multiple myeloma cell adhesion, migration, and invasion. Blood. 2011;117:6202–13.
Malfuson JV, Boutin L, Clay D, Thepenier C, Desterke C, Torossian F, et al. SP/drug efflux functionality of hematopoietic progenitors is controlled by mesenchymal niche through VLA-4/CD44 axis. Leukemia. 2014;28:853–64.
Hsieh YT, Gang EJ, Shishido SN, Kim HN, Pham J, Khazal S, et al. Effects of the small-molecule inhibitor of integrin α4, TBC3486, on pre-B-ALL cells. Leukemia. 2014;28:2101–4.
Jacamo R, Chen Y, Wang Z, Ma W, Zhang M, Spaeth EL, et al. Reciprocal leukemia-stroma VCAM-1/VLA-4-dependent activation of NF-κB mediates chemoresistance. Blood. 2014;123:2691–702. Paper showing that interaction between VCAM-1 and integrin α4β1 activates NF-κB pathway in BM stromal cells and leukemia cells and that inhibition of NF-κB pathway reduces CAM-DR.
Bjorklund CC, Baladandayuthapani V, Lin HY, Jones RJ, Kuiatse I, Wang H, et al. Evidence of a role for CD44 and cell adhesion in mediating resistance to lenalidomide in multiple myeloma: therapeutic implications. Leukemia. 2014;28:373–83.
Burgess M, Gill D, Singhania R, Cheung C, Chambers L, Renyolds BA, et al. CD62L as a therapeutic target in chronic lymphocytic leukemia. Clin Cancer Res. 2013;19:5675–85.
Lafouresse F, Bellard E, Laurent C, Moussion C, Fournie JJ, Ysebaert L, et al. L-selectin controls trafficking of chronic lymphocytic leukemia cells in lymph node high endothelial venules in vivo. Blood. 2015;126:1336–45. Paper demonstrating that primary human B-CLL cells home to human and mouse lymph nodes via high endothelial venules and that this homing is mediated by L-selectin expressed by CLL cells.
Winkler IG, Barbier V, Pattabiraman DR, Gonda TJ, Magnani JL, Levesque J-P. Vascular niche E-selectin protects acute myeloid Leukaemia stem cells from chemotherapy. Blood. 2014;124:620.
Aggoune D, Magnani JL, Van Etten RA, Krause DS. The vascular niche is involved in regulating leukemic stem cells in murine chronic myelogenous leukemia. Blood. 2014;124:516.
Landowski TH, Olashaw NE, Agrawal D, Dalton WS. Cell adhesion-mediated drug resistance (CAM-DR) is associated with activation of NF-kappa B (RelB/p50) in myeloma cells. Oncogene. 2003;22:2417–21.
Zhang B, Li M, McDonald T, Holyoake TL, Moon RT, Campana D, et al. Microenvironmental protection of CML stem and progenitor cells from tyrosine kinase inhibitors through N-cadherin and Wnt–β-catenin signaling. Blood. 2013;121:1824–38.
Nervi B, Ramirez P, Rettig MP, Uy GL, Holt MS, Ritchey JK, et al. Chemosensitization of acute myeloid leukemia (AML) following mobilization by the CXCR4 antagonist AMD3100. Blood. 2009;113:6206–14.
Zeng Z, Xi Shi Y, Samudio IJ, Wang R-Y, Ling X, Frolova O, et al. Targeting the leukemia microenvironment by CXCR4 inhibition overcomes resistance to kinase inhibitors and chemotherapy in AML. Blood. 2009;113:6215–24.
Uy GL, Rettig MP, Motabi IH, McFarland K, Trinkaus KM, Hladnik LM, et al. A phase 1/2 study of chemosensitization with the CXCR4 antagonist plerixafor in relapsed or refractory acute myeloid leukemia. Blood. 2012;119:3917–24.
Acknowledgments
Jean-Pierre Levesque and Ingrid G. Winkler are supported by the Research Fellowships from the National Health and Medical Research Council (NHMRC) of Australia (#1044091 and 1108352, respectively).
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of Interest
Jean-Pierre Levesque and Ingrid G. Winkler declare that GlycoMimetics Inc. has supported a portion of their research involving their E-selectin antagonist.
Human and Animal Rights and Informed Consent
This article does not contain any studies with human or animal subjects performed by any of the authors.
Additional information
This article is part of the Topical Collection on Role of Classical Signaling Pathways in Stem Cell Maintenance
Rights and permissions
About this article

Cite this article
Levesque, JP., Winkler, I.G. Cell Adhesion Molecules in Normal and Malignant Hematopoiesis: from Bench to Bedside. Curr Stem Cell Rep 2, 356–367 (2016). https://doi.org/10.1007/s40778-016-0066-0
Published:
Issue Date:
DOI: https://doi.org/10.1007/s40778-016-0066-0