
Abstract
Vascular calcification in chronic kidney disease (CKD) patients is associated to increased mortality. Osteoprotegerin (OPG) is a soluble tumor necrosis factor (TNF) superfamily receptor that inhibits the actions of the cytokines receptor activator of nuclear factor kappa-B ligand (RANKL) and TNF-related apoptosis-inducing ligand (TRAIL) by preventing their binding to signaling receptors in the cell membrane. OPG-deficient mice display vascular calcification while OPG prevented calcification of cultured vascular smooth muscle cells and protected kidney cells from TRAIL-induced death. OPG may be a biomarker in patients with kidney disease. Circulating OPG is increased in predialysis, dialysis and transplant CKD patients and may predict vascular calcification progression and patient survival. By contrast, circulating OPG is decreased in nephrotic syndrome. In addition, free and exosome-bound urinary OPG is increased in human kidney disease. Increased urinary OPG has been associated with lupus nephritis activity. Despite the association of high OPG levels with disease, experimental functional information available suggests that OPG might be protective in kidney disease and in vascular injury in the context of uremia. Thus, tissue injury results in increased OPG, while OPG may protect from tissue injury. Recombinant OPG was safe in phase I randomized controlled trials. Further research is needed to fully define the therapeutic and biomarker potential of OPG in patients with kidney disease.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Chronic kidney disease (CKD) is present in around 8–16 % of the worldwide population [1]. CKD may evolve to end-stage renal disease (ESRD) requiring dialysis and/or transplantation. In addition, CKD is a cardiovascular risk factor [2] and both cardiovascular and non-cardiovascular mortality are increased in CKD patients [3]. Understanding the cellular and molecular basis of the adverse outcomes of CKD patients is a research priority. CKD may be associated to disease-specific cardiovascular risk factors [2], such as CKD mineral bone disorders (CKD–MBD) [4]. CKD–MBD includes bone disease, hyperparathyroidism, altered calcium and phosphate metabolism, and cardiovascular calcification. Most recent attention has focused on phosphate metabolism and disposal, and its relationship with vascular calcification [5]. Indeed, one of the few medical interventions that impacts survival in CKD patients is the choice of oral phosphate binders [6, 7]. The survival advantage of non-calcium-based phosphate binders may be related to protection from vascular calcification [6, 7]. In this regard, vascular calcification and hyperphosphatemia are features of the accelerated aging of Klotho deficient mice; and human CKD is also characterized by Klotho deficiency, vascular calcification and hyperphosphatemia [8, 9]. Furthermore, vascular calcification is also found in human progeria [10]. From this point of view, there is increasing interest in understanding the complex mechanisms regulating vascular calcification [11–13].
Osteoprotegerin (OPG) is a soluble protein from the tumor necrosis factor (TNF) receptor superfamily named after its best-characterized effect: bone protection [14, 15]. In addition, OPG protects from vascular calcification [16]. More recent evidence has linked OPG to kidney injury. We here review the biology of OPG and the role and biomarker potential of OPG in kidney disease.
TNF superfamily of ligands and receptors
Members of the TNF receptor superfamily are usually type I transmembrane glycoproteins, with some exceptions that include OPG [17]. TNF receptor superfamily proteins share a common extracellular cysteine-rich domain, which may be repeated up to six times in a receptor monomer chain. Some members of the family present a cytoplasmic ~180 amino acid interaction domain known as the death domain. TNF receptors bind proteins of the TNF superfamily, of which the best-known member is TNF-α [18]. Both ligands and receptors trimerize to trigger intracellular signals [19, 20]. TNF superfamily receptors are usually membrane-bound, whereas ligands can be membrane-bound or soluble [21]. Most TNF superfamily members bind to a unique receptor, but there is crosstalk between certain receptors and different ligands [17]. In addition TNF receptor superfamily members may be soluble either because they are encoded as soluble proteins or because of proteolytic processing yielding a soluble protein.
OPG structure and expression
Mature OPG is a 380-amino acid soluble glycoprotein which lacks transmembrane or cytoplasmic domains. OPG is found as either a 60-kDa monomer or 120-kDa dimer linked by disulfide bonds and by noncovalent interactions mediated by death domains and, to a lesser extent, by a C-terminal heparin-binding region [22]. The dimer is more bioactive than the monomer. Both OPG dimers and monomers circulate in serum [23]. Because of its size, under physiological conditions OPG is unlikely to be filtered at the glomerulus [24]. OPG has several structural regions. Four amino terminal cystein-rich domains provide binding sites for the TNF superfamily cytokines receptor activator of nuclear factor kappa-B ligand (RANKL) and TNF-related apoptosis-inducing ligand (TRAIL). Two death domains are typical of TNF superfamily receptors and a carboxy-terminal basic heparin-binding domain is a common feature of peptide growth factors [25–30] (Fig. 1a). Mouse and human OPG proteins are ~85 % identical (Fig. 1b), indicating a high conservation of the OPG gene throughout evolution, suggestive of an important function [31].

Structure of osteoprotegerin (OPG). a Domain structure of 380-amino acid, mature OPG forming a dimer after shedding the signal peptide. Dimerization of OPG results from non-covalent interactions mediated by the death domains and to a lesser extent by a C-terminal heparin-binding region. A C-terminal intermolecular disulfide bond does not contribute to the formation or stability of OPG dimers [22]. CR cystein-rich domains, DD death domain, HB heparin binding region. b Conservation of the OPG gene throughout evolution, as seen in the similarity between mouse [35] and human [104] OPG proteins, according to http://www.rcsb.org/pdb/. Helix in red, loops in green and sheets in yellow (color figure online)
The human OPG gene is located on chromosome 8q and consists of five exons. OPG is expressed in multiple adult organs and cell types, including the lung, heart, kidney, liver, spleen, stomach, intestine, skin, thymus, lymph nodes, bone marrow, osteoblasts, vascular smooth muscle cells, dendritic cells, B-lymphocytes, articular chondrocytes, brain, spinal cord, and thyroid gland [25–29]. Vascular smooth muscle cells are thought to be the main source of OPG in the vascular wall, but endothelial cells also secrete OPG [29, 32]. The widespread expression of OPG suggests that it may have poorly understood roles beyond bone biology regulation. During murine embryogenesis (day 15) in situ hybridization localized OPG to the cartilaginous parts of developing bone, the aorta, the gastrointestinal tract, and the skin [28].
Many molecules positively or negatively regulate OPG expression. Cytokines and growth factors [interleukin (IL)-1α, IL-6, IL-11, IL-17, IL-18, TNF-α, TNF-β, bone morphogenetic protein (BMP)-2, transforming growth factor (TGF)-β1, angiotensin II, pigment epithelium-derived factor (PEDF), and platelet-derived growth factor (PDGF)], bone mineral metabolism molecules (calcium, vitamin D), hormones (17β-estradiol), and Wnt signaling upregulate OPG expression. Parathyroid hormone (PTH), glucocorticoids, prostaglandin E2, immunosuppressant drugs, peroxisome proliferator-activated receptor-gamma (PPAR-γ) activators, insulin-like growth factor (IGF)-1 and basic fibroblast growth factor (bFGF) downregulate OPG [25, 26, 29, 30]. It is yet unclear which of these multiple regulators is most important in vivo or how OPG contributes to disease modulation when the regulators are overexpressed.
Functions of OPG
Osteoprotegerin is a soluble, non-signaling decoy receptor that binds RANKL and TRAIL with an affinity in the same order of magnitude, although greater for RANKL [25–27, 29, 33]. In addition, OPG binds glycosaminoglycans such as heparin, heparan sulfate, chondroitin sulfate and dermatan sulfate [30]. Binding to these molecules regulates both OPG disposal and the function of the OPG ligand. Thus, binding of OPG to any of these three ligands prevents binding to the others [34] providing a three-way interaction. OPG molecules lacking the glycosaminoglycan-binding domain are still active in inhibiting TRAIL binding to its cell membrane receptors [34] but are devoid of activity related to glycosaminoglycan-binding, such as adhesion of leukocytes to endothelium.
The OPG/RANKL complex is internalized either through lipid rafts by interaction with cell membrane syndecan-1 or by clathrin-coated pit formation. By contrast, binding to glycosaminoglycans competes with OPG/RANKL interaction and prevents OPG internalization [25].
The monomeric and the dimeric cytokine-binding regions of OPG bind RANKL with approximately 500-fold higher affinity than the cell membrane signaling receptor RANK [35]. Thus, OPG prevents RANKL activation of RANK and RANKL-induced activation of osteoclast maturation and function as well as bone loss [29, 36, 37].
The RANKL/RANK axis also regulates the crosstalk between the bone and the immune system. RANKL-expressing T cells can express RANKL and promote osteoclastogenesis, while OPG from B cells may counteract RANKL from T cells during the immune response [38]. OPG is also involved in the regulation of B cell maturation and efficient antibody responses [29, 39].
The affinity of TRAIL for OPG is weaker than that for other transmembrane TRAIL receptors [40]. Thus, it has been suggested that OPG interaction with TRAIL may be less physiologically significant than the interaction with RANKL. However, cell culture studies have clearly demonstrated that OPG protects from TRAIL cytotoxicity [25, 41, 42]. TRAIL is a potentially lethal TNF superfamily protein normally expressed in many human tissues including the kidney [43]. Under physiological conditions normal parenchymal cells have protective mechanisms that prevent TRAIL-induced death. Both breast cancer cells and human kidney tubular cells release OPG that protects them from TRAIL cytotoxicity in culture [25, 41]. By contrast, in an inflammatory environment, parenchymal cells, such as kidney tubular cells, are sensitized to TRAIL-induced apoptosis [42]. However, conclusive demonstration of the functional relevance of OPG neutralization of TRAIL in vivo is lacking.
Binding of endothelial OPG to leukocyte RANKL or TRAIL may prevent endothelial cell apoptosis [44]. In addition, circulating OPG may directly interact with heparan sulfates in endothelium favoring leukocyte adhesion [45]. The pro-adhesive effect of OPG is prevented by heparin. It is unclear whether the pro-adhesive action of OPG is dependent on binding to TRAIL or RANKL in leukocytes, since OPG binding to glycosaminoglycans prevents binding to TRAIL or RANKL [34]. OPG also enhances the proangiogenic properties of endothelial cells and endothelial colony-forming cells, thus promoting vasculogenesis in vivo [46, 47]. OPG increased endothelial colony-forming cell viability and adhesion to activated endothelium under shear stress conditions. The proangiogenic properties of OPG appeared to require binding to syndecan-1, although OPG 1-194, that lacks the heparin-binding domain, still had pro-vasculogenic effects [47]. OPG also promoted pulmonary artery smooth muscle cell proliferation and migration and has survival factor activity for serum-deprived vascular smooth muscle cells, potentially contributing to vascular injury [48, 49].
Genetically modified mice have provided further clues as to the key roles of OPG in vivo [16, 31, 50] (Table 1). Overexpression of OPG is associated to increased bone density (osteopetrosis), while targeted deletion of OPG results in severe premature osteoporosis [51]. Osteoporosis in OPG-deficient mice was explained by unrestricted RANKL promotion of osteoclast activity and bone resorption. Interestingly, OPG deficiency also results in calcification of the aorta and renal arteries. OPG−/−·ApoE−/− mice had more atherosclerotic plaques and calcified vascular lesions than OPG+/+·ApoE−/− controls [48, 49], indicating that OPG protects from atherosclerosis and vascular calcification. The molecular mechanisms of vascular calcification in these mice are unclear [16, 39, 52, 53]. Furthermore, human mutations that inactivate OPG cause juvenile Paget disease [54], an osteopathy characterized by rapidly remodeling woven bone, osteopenia and vascular injury in the form of aneurysms [55–57]. Thus, OPG also regulates vascular wall resistance to stress. As discussed above, vascular calcification is a key feature of CKD–MBD, while aortic aneurysms and CKD are frequent clinical companions in which CKD confers an increased risk of mortality [58, 59]. Thus, unraveling the role of OPG in kidney disease-associated vascular injury is of particular importance. While genetically modified mice lack a kidney phenotype beyond renal artery calcifications, their response to kidney injury has not been addressed.
OPG as a therapeutic agent
The role of OPG in bone mass regulation sparked research on its potential therapeutic role in human osteoporosis. Escherichia coli-expressed Fc-OPG is a recombinant protein with an N-terminal Fc fragment, while CHO-expressed OPG-Fc (AMGN-0007) has a C-terminal Fc fragment and is ten times more potent [60]. In animal models, OPG-Fc and soluble RANK-Fc fusion proteins inhibited RANKL-mediated osteoclastogenesis [39]. OPG-Fc lacks the signal-peptide, heparin-binding domain and death domain-homologous regions of native OPG, has a longer half-life and lower affinity for TRAIL, while affinity for RANKL is preserved [22, 61]. Administration of Fc-OPG reduced the area of calcified lesions without modulating atherosclerosis in atherogenic diet-fed ldlr (−/−) mice [62]. However, there is conflicting evidence on the potential of exogenous OPG to impact on disease. Secchiero et al. [61] in a series of papers have alerted on the potential risks of OPG administration by documenting adverse effects on pancreatic islet cells, endothelium and vascular fibrosis associated with up-regulation of arterial TGF-β1 in ApoE (−/−) mice [63–65]. Of particular interest to CKD, OPG inhibits vascular calcification induced by warfarin or by vitamin D in rats [66]. The use of oral anticoagulants targeting vitamin K is a key risk factor for calciphylaxis, the most severe form of vascular calcification [67].
Phase 1 clinical trials of two molecular forms of OPG (Fc-OPG and OPG-Fc) concluded that these constructs were safe in postmenopausal women or cancer patients and reduced biochemical markers of bone resorption [68, 69]. Despite the promising results, OPG was not further developed as a therapeutic agent since the anti-RANKL monoclonal antibody denosumab was more effective and there were concerns that OPG-mediated blockade of TRAIL might favor tumor development or growth [68–70]. Denosumab is now in clinical use for prevention of skeletal-related events in adults with bone metastases from solid tumors and for sex hormone-deprivation induced osteoporosis [71]. By contrast, no active clinical trials using OPG were found in the clinicaltrials.gov web page in November 2013 [72].
Biomarker potential of circulating OPG in kidney disease
There is increasing interest in biomarkers that allow risk stratification in CKD and eventually guide the use of new therapeutic approaches [73]. In the general population high serum OPG was associated with cardiovascular risk factors and/or predicted cardiovascular mortality [74]. Circulating OPG levels are increased in CKD patients [75–81] (Fig. 2). However there is not enough information on the in vivo role of OPG to consider OPG a uremic toxin [82]. In addition, circulating OPG is decreased in nephrotic syndrome [83]. Circulating OPG levels have been associated with adverse outcomes in CKD patients [74, 81, 84–88].

Serum osteoprotegerin (OPG) levels in chronic kidney disease (CKD) patients. a Non-dialysis CKD patients. Four different ELISA kits were used: Biovendor (Laboratory Medicine Inc., Brno, Czech Republic) in references [75, 77, 78, 81]; Rapid Bio Lab (West Hills, CA, USA) in [76]; ALPCO (Salem, NH, USA) in [79] and Immunodiagnostic (UK) in [80]. b Hemodialysis patients. Data as reported in references [23, 76, 80, 87, 91]. c Peritoneal dialysis patients. Data as reported in [80, 88]. d Renal transplant recipients. Data as reported in [23, 92]. The n represents the number of studies used to calculate the mean. Data presented as mean and SD of several studies (empty columns) or mean or median and SD of individual studies (black columns). Conversion from conventional to International System (SI) units was performed if needed according to instructions in each ELISA datasheet (*Conversion factor 1 pmol/l = 120 pg/ml and **Conversion factor 1 pg/ml = 0.05 pmol/l according to datasheet)
Chronic kidney disease
Diabetes mellitus is the most frequent cause of CKD that progresses to ESRD. In addition, diabetes is associated to macrovascular and microvascular disease, including a high prevalence of vascular calcification [85]. By contrast, there is a negative association between diabetes and abdominal aortic aneurysms [89]. Serum OPG has been associated to the presence of diabetes. There is an independent positive correlation between circulating OPG and basal glycemia [86] or hemoglobin A1c levels [90]. Diabetic patients with CKD have higher OPG levels than diabetics without CKD. In 1,939 non-dialysis adult type 1 diabetics, those with macroalbuminuria and/or renal impairment had higher OPG concentrations than those without overt kidney disease [32].
Circulating OPG levels increase as estimated glomerular filtration rate (eGFR) decreases in diabetic and non-diabetic CKD patients [32, 75–78, 81, 90] (Fig. 2a). However different authors have reported very different values. This may be related to differences in enzyme-linked immunosorbent assay (ELISA) kits, conversion factors, technical issues, or to real differences between patient populations.
Renal replacement therapy
Serum OPG levels are increased in patients undergoing renal replacement therapy (hemodialysis, peritoneal dialysis or kidney transplantation) [23, 80, 87, 88, 91, 92]. In hemodialysis patients, OPG values were similar in three reports, but much lower in a fourth one [87]. The conversion factor to International System (SI) units indicated in the datasheet of the Immunodiagnostik kit used this fourth report (MW 19.9 kD) and it is very different from the molecular weight of OPG and the values used by other assays (MW 120 kD) (Fig. 2b). There was a significant correlation between OPG levels, dialysis vintage and age in hemodialysis patients [23]. It was suggested that serum OPG might help discriminate hemodialysis patients with biopsy-proven low turnover bone disease from those with high turnover renal osteodystrophy when intact PTH (iPTH) is ≤300 pg/ml [87]. Within this PTH range, mean OPG was lower in patients with adynamic bone disease than in those with hyperparathyroidism and mixed osteodystrophy. However, no formal assessment of test performance was reported. Two studies [80, 88] measured serum OPG levels in peritoneal dialysis (Fig. 2c).
Two studies measuring serum OPG levels in renal transplant recipients were very discordant (Fig. 2d) [23, 92]. The reason for the discrepancy is unclear. Renal function may have contributed, but was not presented in one of the reports.
Nephrotic syndrome
Nephrotic syndrome represents the most severe form of proteinuric kidney disease. As a result of protein loss through proteinuria and a compensatory increase in protein production mainly by the liver, nephrotic syndrome is characterized by a wide spectrum of changes in serum protein concentrations. In childhood nephrotic syndrome, serum OPG was lower, even in newly diagnosed patients (prior to steroid therapy) than in normal controls (0.84 ± 0.18 vs. 1.1 ± 0.22 pg/ml) [83]. OPG was even lower in steroid-dependent frequent relapsers (0.24 ± 0.14 pg/ml). There was a negative correlation between OPG and markers of disease severity such as serum cholesterol and urinary protein excretion. Low OPG was hypothesized to result from urinary losses and to potentially contribute to bone resorption. Steroids, which are known to increase RANKL and to reduce OPG expression, were thought to further decrease OPG values [83].
Vascular calcification
Higher circulating OPG levels have been associated with vascular calcification in CKD patients [16, 77, 78]. High circulating OPG levels were associated with the presence of coronary artery calcifications (CAC) in non-dialyzed CKD patients, peritoneal dialysis, and renal transplant patients [77, 78, 80, 88, 89, 92–95]. Circulating OPG levels also correlated with factors associated with vascular calcification. In hemodialysis patients OPG levels correlated with dialysis vintage and age [23]. In adults with type 1 diabetes, high circulating OPG was associated to peripheral vascular events such as revascularization procedures or amputation of lower extremities [32]. OPG levels increased rapidly in association with early vascular calcification stages and then reached a plateau. It was hypothesized that OPG may be a marker of atherosclerosis/vascular calcification onset rather than its severity or progression [78].
Aortic pulse wave velocity is an indirect, noninvasive method to assess aortic stiffness and increases in the presence of vascular calcification. CKD patients with the highest serum OPG levels had a 10 % higher aortic pulse wave velocity compared to those with the lowest levels [79].
Figure 3 integrates data from three studies that related serum OPG levels to coronary artery calcium (CAC) score in CKD patients. In non-dialysis CKD patients, serum OPG levels in the higher tertile (>8.8 pmol/l, conversion 1 pmol/l = 120 pg/ml) were associated with the presence of CAC [77]. In 195 non-dialysis CKD patients, OPG ≥ 10.71 pmol/l was the only variable significantly associated with moderate CAC (100–400) [odds ratio (OR) 2.73 [1.03;7.26]; p = 0.04] [78]. In renal transplant recipients, receiver operating characteristic (ROC) curve analysis yielded an optimal plasma OPG cutoff value for predicting a CAC score of 8.3 pmol/l. The percentage of calcified (CAC > 100) subjects with OPG > 8.3 pmol/l was 75 % (OR 5.72 [1.94;16.8], p = 0.002) [92]. Baseline CAC, but not baseline or 1-year OPG, was associated with CAC progression. In a different set of 107 transplanted patients, vascular calcification progression was again observed almost exclusively in patients with a prior vascular calcification, but in a multivariate analysis, serum calcium, OPG, and eGFR were independently associated with progression at 1 year [96].

Relation between coronary artery calcification (CAC) score and serum osteoprotegerin (OPG) levels. Data are summarized from [77, 78, 92] and correspond to non-dialysis chronic kidney disease (CKD) and transplantation CKD patients. Patients with score 100–400 and ≥400 from Ref. [78] were grouped together (score ≥ 100)
Mortality
Cardiovascular mortality is the leading cause of death in patients with CKD even after successful transplantation [92, 95]. For pre-dialysis CKD patients, the risk for cardiovascular death may be higher than the risk for ESRD requiring renal replacement therapy [75]. In patients with vascular calcification the risk of cardiovascular death is particularly high [91]. Serum OPG has been associated with cardiovascular mortality in pre-dialysis CKD patients [76].
In renal transplant recipients, baseline OPG levels were associated with long-term (7–9 years) cardiovascular or all-cause mortality [93]. Circulating OPG was higher in non-survivors and in patients who died from cardiovascular disease than in survivors and patients who died from other causes [93].
In a prospective cohort study of 1,157 elderly women, multivariable-adjusted linear regression models showed that elevated OPG levels at baseline were associated with faster decline in eGFR at 5- and 10-year follow-up and a higher risk of hospitalizations or death [97]. However, baseline OPG levels were higher in patients with lower baseline eGFR.
In summary, circulating OPG is a potential biomarker for vascular calcifications and mortality in CKD. However, a causal role of OPG in these associations remains uncertain. In mice, OPG deficiency is associated to diffuse medial calcification of the aorta and renal arteries. By contrast, in humans with CKD, increased OPG is associated with vascular calcification. Several hypotheses may explain this potential discrepancy [79, 95]. The high OPG levels in patients with vascular disease may be triggered by inflammation or by the presence of osteoblast-like cells in calcified vessels. OPG might thus be a marker of injury while playing a protective role in vascular injury by inhibiting vascular wall calcification. This concept is supported by observations in OPG-deficient mice and by the demonstration that OPG dose-dependently within a pathophysiologically relevant range (100–10,000 pg/ml) reduces vascular smooth muscle calcification in culture [16, 98]. Alternatively, medial calcification in OPG-deficient mice might be secondary to severe osteoporosis and not a direct result of OPG deficiency in the vasculature. In this regard, information is missing on the temporal sequence of events. Thus, no prospective study has determined whether serum OPG levels rise in response to the development of vascular calcification or whether elevated levels of serum OPG precede the development of vascular calcification [79, 95].
Kidney and urinary OPG
There is little information on OPG expression and function during kidney injury. A transcriptomics analysis identified TRAIL and OPG as the apoptosis-related genes most highly expressed in human diabetic nephropathy of the tubulointerstitium [41]. Diabetic nephropathy is the most frequent form of CKD [99]. The findings were interpreted as disclosing local renal synthesis of OPG mRNA during tissue injury. Human kidney OPG mRNA levels directly correlated with serum creatinine, proteinuria and histological injury scores, indicating a relationship between OPG expression and severity of kidney injury. While the increased expression of TRAIL was confirmed at the protein level and localized to podocytes and tubular cells by immunohistochemistry, OPG protein was not studied [41]. In more recent studies the Nephromine database of published kidney transcriptome datasets disclosed that OPG mRNA was increased in human kidneys with IgA nephropathy and that renal cortex OPG mRNA levels correlated with chronicity index quartiles in aging humans [100]. OPG was increased in tubular epithelial cells in CKD biopsies and in epithelial cells lining kidney cysts in another form of kidney injury, autosomal dominant polycystic kidney disease [100]. Furthermore, while in normal kidney OPG was present at the basolateral side of tubular cells, a diffuse staining pattern was found in injured kidney suggesting that directional secretion of OPG might be affected by kidney disease. Specifically, immunostaining suggested that during kidney disease tubular cells might secrete OPG to the tubular lumen, thus favoring the presence of OPG in urine [100].
Evidence further supporting local synthesis of OPG by tubular epithelium was obtained in cultured cells. Neutralization of OPG sensitized cultured human tubular cells to TRAIL-induced death, suggesting that OPG is an autocrine cytoprotective factor [41]. OPG was found in tubular cell-secreted exosome-like particles [101]. Exosomes and other microvesicles actively secreted by cells may play a variety of roles, including disposal of unwanted material, intercellular communication and regulation of cell death and of vascular calcification. Thus OPG in exosomes might contribute to any of these functions. OPG was also found in urinary exosomes [100]. In an exploratory study OPG was increased in urinary exosome-like vesicles in CKD patients, including those with diabetic nephropathy, IgA nephropathy and polycystic kidney disease, all of them diseases characterized by increased kidney OPG mRNA or protein [100].
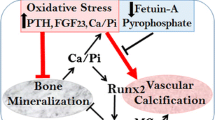
Urinary OPG may also be a potential biomarker of lupus nephritis activity. In 87 lupus nephritis patients urinary OPG levels were associated with evidence of active nephritis such as hematuria, urine protein/creatinine ratio, and the presence of circulating anti-dsDNA antibodies [102]. The authors hypothesized that microvascular endothelial cells from inflamed kidneys were the source of urinary OPG. However, an alternative hypothesis is that urinary OPG originated in stressed tubular cells (Fig. 4). Indeed, as commented above, tubular cell OPG might more readily access the urinary space than endothelial cell-derived OPG, and immunohistochemistry data and the presence of OPG in exosomes suggest a tubular origin of urinary OPG.

Osteoprotegerin (OPG) origin and functions in the kidney and potential systemic functions. Vascular smooth muscle cells are thought to be the main source of OPG in the vascular wall, but endothelial cells also secrete OPG. In the vascular wall the best characterized effect of OPG is prevention of vascular calcification. Tubular cells are a source of OPG in the kidneys. OPG from tubular cells may appear in urine, at least in part associated to exosomes. Tubular cell OPG may be an autocrine response that protects from TRAIL-induced apoptosis
Conclusions and required research
In conclusion, functional animal studies, cell culture studies and human genetic defects suggest that OPG has a key role in downregulating vascular calcification, preventing arterial aneurysms and protecting kidney cells from inflammation-induced death (Fig. 4). In addition increased circulating, urinary and kidney OPG levels are observed in different forms of human CKD. Increased circulating OPG was associated with vascular calcification and mortality in CKD patients. An integration of experimental and clinical data suggests that the increase in OPG observed in vivo may be compensatory and protective, but that it fails to fully prevent injury. OPG may contribute to slower progression of tissue injury as suggested by the more severe vascular injury observed in atherosclerosis-prone OPG-deficient mice. Alternatively, the forms of OPG accumulated in CKD may be dysfunctional and not able to provide tissue protection. There is plenty of experience with this notion. As a clear example, the high PTH levels in CKD patients are in part due to accumulation of non-functional peptides. Still, some authors support the notion that despite preventing calcification, OPG may contribute to injury.
Specific areas require further research in order for OPG to be incorporated into daily clinical practice. Recent studies concluded that the discriminatory capacity of current assays for OPG is not enough for clinical use at least to screen for fractures or vascular calcification in CKD [84, 103]. Thus, OPG may have a role as a biomarker of risk. However, better standardization of assays and definition of cutoff points is required to validate its potential use as a biomarker.
There are great gaps in our understanding of the role of OPG in kidney diseases and the role and meaning of urinary OPG. Despite the promising findings, further studies are needed to establish the role of urinary OPG, either free or associated with exosomes, in monitoring or staging of human kidney injury.
Finally, recombinant OPG has been successfully tested and verified to prevent calcification in mice and shown to be safe in phase I human clinical trials. Although research into OPG as a therapeutic agent in osteoporosis has been abandoned, an improved understanding of its pathophysiology may identify new potential uses in cardiovascular and kidney protection.
References
Jha V, Garcia-Garcia G, Iseki K et al (2013) Chronic kidney disease: global dimension and perspectives. Lancet 382(9888):260
Gansevoort RT, Correa-Rotter R, Hemmelgarn BR et al (2013) Chronic kidney disease and cardiovascular risk: epidemiology, mechanisms, and prevention. Lancet 382(9889):339
de Jager DJ, Grootendorst DC, Jager KJ et al (2009) Cardiovascular and noncardiovascular mortality among patients starting dialysis. JAMA 302(16):1782
Kidney Disease: Improving Global Outcomes (KDIGO) CKD-MBD Work Group (2009) KDIGO clinical practice guideline for the diagnosis, evaluation, prevention, and treatment of chronic kidney disease–mineral and bone disorder (CKD–MBD). Kidney Int Suppl 113:S1
Gonzalez-Parra E, Tuñón J, Egido J, Ortiz A (2012) Phosphate: a stealthier killer than previously thought? Cardiovasc Pathol 21(5):372
Jamal SA, Vandermeer B, Raggi P et al (2013) Effect of calcium-based versus non-calcium-based phosphate binders on mortality in patients with chronic kidney disease: an updated systematic review and meta-analysis. Lancet 382(9900):1268
Ortiz A, Sanchez-Niño MD (2013) The demise of calcium-based phosphate binders. Lancet 382(9900):1232
Izquierdo MC, Perez-Gomez MV, Sanchez-Niño MD et al (2012) Klotho, phosphate and inflammation/ageing in chronic kidney disease. Nephrol Dial Transplant 27(Suppl 4):iv6
Hu MC, Shi M, Zhang J et al (2011) Klotho deficiency causes vascular calcification in chronic kidney disease. J Am Soc Nephrol 22(1):124
Salamat M, Dhar PK, Neagu DL, Lyon JB (2010) Aortic calcification in a patient with Hutchinson–Gilford progeria syndrome. Pediatr Cardiol 31(6):925
Massy ZA, Drüeke TB (2013) Vascular calcification. Curr Opin Nephrol Hypertens 22(4):405
Shroff R, Long DA, Shanahan C (2013) Mechanistic insights into vascular calcification in CKD. J Am Soc Nephrol 24(2):179
de Francisco ALM, Rodríguez M (2013) Magnesium—its role in CKD. Nefrologia 33(3):389
Martin TJ (2013) Historically significant events in the discovery of RANK/RANKL/OPG. World J Orthop 4(4):186
Gallagher JC, Tella SH (2013) Prevention and treatment of postmenopausal osteoporosis. J Steroid Biochem Mol Biol. doi:10.1016/j.jsbmb.2013.09.008
Bucay N, Sarosi I, Dunstan CR et al (1998) Osteoprotegerin-deficient mice develop early onset osteoporosis and arterial calcification. Genes Dev 12(9):1260
Aggarwal BB (2003) Signalling pathways of the TNF superfamily: a double-edged sword. Nat Rev Immunol 3(9):745
Pennica D, Nedwin GE, Hayflick JS et al (1984) Human tumour necrosis factor: precursor structure, expression and homology to lymphotoxin. Nature 312(5996):724
Armitage RJ (1994) Tumor necrosis factor receptor superfamily members and their ligands. Curr Opin Immunol 6(3):407
Cosman D (1994) A family of ligands for the TNF receptor superfamily. Stem Cells 12(5):440
Lotz M, Setareh M, von Kempis J, Schwarz H (1996) The nerve growth factor/tumor necrosis factor receptor family. J Leukoc Biol 60(1):1
Schneeweis LA, Willard D, Milla ME (2005) Functional dissection of osteoprotegerin and its interaction with receptor activator of NF-kappaB ligand. J Biol Chem 280(50):41155
Rashtchizadeh N, Ghorbanihaghjo A, Argani H et al (2012) Serum receptor activator of nuclear factor-κ B ligand, osteoprotegrin, and intact parathyroid hormone in hemodialysis and renal transplant patients. Ther Apher Dial 16(6):600
Sato T, Tominaga Y, Iwasaki Y et al (2001) Osteoprotegerin levels before and after renal transplantation. Am J Kidney Dis 38(4 Suppl 1):S175
Silva I, Branco JC (2011) Rank/Rankl/opg: literature review. Acta Reumatol Port 36(3):209
Tat SK, Pelletier JP, Velasco CR, Padrines M, Martel-Pelletier J (2009) New perspective in osteoarthritis: the OPG and RANKL system as a potential therapeutic target? Keio J Med 58(1):29
Papadopouli AE, Klonaris CN, Theocharis SE (2008) Role of OPG/RANKL/RANK axis on the vasculature. Histol Histopathol 23(4):497
Hofbauer LC, Khosla S, Dunstan CR, Lacey DL, Boyle WJ, Riggs BL (2000) The roles of osteoprotegerin and osteoprotegerin ligand in the paracrine regulation of bone resorption. J Bone Miner Res 15(1):2
Klejna K, Naumnik B, Gasowska K, Myśliwiec M (2009) OPG/RANK/RANKL signaling system and its significance in nephrology. Folia Histochem Cytobiol 47(2):199
Wright HL, McCarthy HS, Middleton J, Marshall MJ (2009) RANK, RANKL and osteoprotegerin in bone biology and disease. Curr Rev Musculoskelet Med 2(1):56
Simonet WS, Lacey DL, Dunstan CR et al (1997) Osteoprotegerin: a novel secreted protein involved in the regulation of bone density. Cell 89(2):309
Gordin D, Soro-Paavonen A, Thomas MC et al (2013) Osteoprotegerin is an independent predictor of vascular events in Finnish adults with type 1 diabetes. Diabetes Care 36(7):1827
Vitovski S, Phillips JS, Sayers J, Croucher PI (2007) Investigating the interaction between osteoprotegerin and receptor activator of NF-kappaB or tumor necrosis factor-related apoptosis-inducing ligand: evidence for a pivotal role for osteoprotegerin in regulating two distinct pathways. J Biol Chem 282(43):31601
Lamoureux F, Picarda G, Garrigue-Antar L et al (2009) Glycosaminoglycans as potential regulators of osteoprotegerin therapeutic activity in osteosarcoma. Cancer Res 69(2):526
Nelson CA, Warren JT, Wang MW, Teitelbaum SL, Fremont DH (2012) RANKL employs distinct binding modes to engage RANK and the osteoprotegerin decoy receptor. Structure 20(11):1971
Boyce BF, Xing L (2007) Biology of RANK, RANKL, and osteoprotegerin. Arthritis Res Ther 9(Suppl 1):S1
Boyce BF, Xing L (2008) Functions of RANKL/RANK/OPG in bone modeling and remodeling. Arch Biochem Biophys 473(2):139
Li Y, Toraldo G, Li A et al (2007) B cells and T cells are critical for the preservation of bone homeostasis and attainment of peak bone mass in vivo. Blood 109(9):3839
Romas E (2009) Clinical applications of RANK-ligand inhibition. Intern Med J 39(2):110
Emery JG, McDonnell P, Burke MB et al (1998) Osteoprotegerin is a receptor for the cytotoxic ligand TRAIL. J Biol Chem 273(23):14363
Lorz C, Benito-Martín A, Boucherot A et al (2008) The death ligand TRAIL in diabetic nephropathy. J Am Soc Nephrol 19(5):904
Sanchez-Niño MD, Benito-Martin A, Gonçalves S et al (2010) TNF superfamily: a growing saga of kidney injury modulators. Mediat Inflamm 2010. doi:10.1155/2010/182958
Lorz C, Benito A, Ucero AC, Santamaría B, Ortiz A (2009) Trail and kidney disease. Front Biosci (Landmark Ed) 14:3740
Pritzker LB, Scatena M, Giachelli CM (2004) The role of osteoprotegerin and tumor necrosis factor-related apoptosis-inducing ligand in human microvascular endothelial cell survival. Mol Biol Cell 15(6):2834
Zauli G, Corallini F, Bossi F et al (2007) Osteoprotegerin increases leukocyte adhesion to endothelial cells both in vitro and in vivo. Blood 110(2):536
Kobayashi-Sakamoto M, Isogai E, Holen I (2010) Osteoprotegerin induces cytoskeletal reorganization and activates FAK, Src, and ERK signaling in endothelial cells. Eur J Haematol 85(1):26
Benslimane-Ahmim Z, Poirier F, Delomenie C et al (2013) Mechanistic study of the proangiogenic effect of osteoprotegerin. Angiogenesis 16(3):575
Lawrie A, Waterman E, Southwood M et al (2008) Evidence of a role for osteoprotegerin in the pathogenesis of pulmonary arterial hypertension. Am J Pathol 172(1):256
Bennett BJ, Scatena M, Kirk EA et al (2006) Osteoprotegerin inactivation accelerates advanced atherosclerotic lesion progression and calcification in older ApoE−/− mice. Arterioscler Thromb Vasc Biol 26(9):2117
Fuller K, Wong B, Fox S, Choi Y, Chambers TJ (1998) TRANCE is necessary and sufficient for osteoblast-mediated activation of bone resorption in osteoclasts. J Exp Med 188(5):997
Min H, Morony S, Sarosi I et al (2000) Osteoprotegerin reverses osteoporosis by inhibiting endosteal osteoclasts and prevents vascular calcification by blocking a process resembling osteoclastogenesis. J Exp Med 192(4):463
Collin-Osdoby P (2004) Regulation of vascular calcification by osteoclast regulatory factors RANKL and osteoprotegerin. Circ Res 95(11):1046
Wu Y, Liu J, Guo H et al (2013) Establishment of OPG transgenic mice and the effect of OPG on bone microarchitecture. Int J Endocrinol 2013:125932
Malan E (1975) 23rd Congress of the European Society of Cardiovascular Surgery. J Cardiovasc Surg (Torino) 16(3):215
http://omim.org/entry/602643. Accessed 1 Jan 2014
Rehman T, Ali R, Taylor C, Yonas H (2010) Bilateral giant cavernous carotid artery aneurysms in a child with juvenile Paget’s disease. World Neurosurg 73(6):691
Allen CA, Hart BL, Taylor CL, Clericuzio CL (2008) Bilateral cavernous internal carotid aneurysms in a child with juvenile paget disease and osteoprotegerin deficiency. Am J Neuroradiol 29(1):7
Saratzis A, Sarafidis P, Melas N, Saratzis N, Kitas G (2013) Impaired renal function is associated with mortality and morbidity after endovascular abdominal aortic aneurysm repair. J Vasc Surg 58(4):879
Patel VI, Lancaster RT, Mukhopadhyay S et al (2012) Impact of chronic kidney disease on outcomes after abdominal aortic aneurysm repair. J Vasc Surg 56(5):1206
Lacey DL, Boyle WJ, Simonet WS et al (2012) Bench to bedside: elucidation of the OPG–RANK–RANKL pathway and the development of denosumab. Nat Rev Drug Discov 11(5):401
Secchiero P, Zauli G (2008) Letter by Secchiero and Zauli regarding article, “osteoprotegerin inhibits vascular calcification without affecting atherosclerosis in ldlr(−/−) mice”. Circulation 118(2):e18 (author reply e19)
Morony S, Tintut Y, Zhang Z et al (2008) Osteoprotegerin inhibits vascular calcification without affecting atherosclerosis in ldlr(−/−) mice. Circulation 117(3):411
Toffoli B, Bernardi S, Candido R et al (2011) Osteoprotegerin induces morphological and functional alterations in mouse pancreatic islets. Mol Cell Endocrinol 331(1):136
Corallini F, Rimondi E, Secchiero P (2008) TRAIL and osteoprotegerin: a role in endothelial physiopathology? Front Biosci 13:135
Toffoli B, Pickering RJ, Tsorotes D et al (2011) Osteoprotegerin promotes vascular fibrosis via a TGF-β1 autocrine loop. Atherosclerosis 218(1):61
Price PA, June HH, Buckley JR, Williamson MK (2001) Osteoprotegerin inhibits artery calcification induced by warfarin and by vitamin D. Arterioscler Thromb Vasc Biol 21(10):1610
Hayashi M, Takamatsu I, Kanno Y et al (2012) A case-control study of calciphylaxis in Japanese end-stage renal disease patients. Nephrol Dial Transplant 27(4):1580
Body JJ, Greipp P, Coleman RE et al (2003) A phase I study of AMGN-0007, a recombinant osteoprotegerin construct, in patients with multiple myeloma or breast carcinoma related bone metastases. Cancer 97(3 Suppl):887
Schwarz EM, Ritchlin CT (2007) Clinical development of anti-RANKL therapy. Arthritis Res Ther 9(Suppl 1):S7
Kohli SS, Kohli VS (2011) Role of RANKL-RANK/osteoprotegerin molecular complex in bone remodeling and its immunopathologic implications. Indian J Endocrinol Metab 15(3):175
http://www.ema.europa.eu/. Accessed 1 Jan 2014
http://clinicaltrials.gov/. Accessed 1 Jan 2014
Ortiz A, Massy ZA, Fliser D et al (2012) Clinical usefulness of novel prognostic biomarkers in patients on hemodialysis. Nat Rev Nephrol 8(3):141
Kiechl S, Schett G, Wenning G et al (2004) Osteoprotegerin is a risk factor for progressive atherosclerosis and cardiovascular disease. Circulation 109(18):2175
Mikami S, Hamano T, Fujii N et al (2008) Serum osteoprotegerin as a screening tool for coronary artery calcification score in diabetic pre-dialysis patients. Hypertens Res 31(6):1163
Jiang JQ, Lin S, Xu PC, Zheng ZF, Jia JY (2011) Serum osteoprotegerin measurement for early diagnosis of chronic kidney disease–mineral and bone disorder. Nephrology (Carlton) 16(6):588
Morena M, Dupuy AM, Jaussent I et al (2009) A cut-off value of plasma osteoprotegerin level may predict the presence of coronary artery calcifications in chronic kidney disease patients. Nephrol Dial Transplant 24(11):3389
Morena M, Jaussent I, Halkovich A et al (2012) Bone biomarkers help grading severity of coronary calcifications in non dialysis chronic kidney disease patients. PLoS One 7(5):e36175
Scialla JJ, Leonard MB, Townsend RR et al (2011) Correlates of osteoprotegerin and association with aortic pulse wave velocity in patients with chronic kidney disease. Clin J Am Soc Nephrol 6(11):2612
Sigrist MK, Levin A, Er L, McIntyre CW (2009) Elevated osteoprotegerin is associated with all-cause mortality in CKD stage 4 and 5 patients in addition to vascular calcification. Nephrol Dial Transplant 24(10):3157
Ford ML, Smith ER, Tomlinson LA, Chatterjee PK, Rajkumar C, Holt SG (2012) FGF-23 and osteoprotegerin are independently associated with myocardial damage in chronic kidney disease stages 3 and 4. Another link between chronic kidney disease–mineral bone disorder and the heart. Nephrol Dial Transplant 27(2):727
Duranton F, Cohen G, De Smet R et al (2012) Normal and pathologic concentrations of uremic toxins. J Am Soc Nephrol 23(7):1258
Mohamed GB, Abdel-Latif EA (2011) Serum osteoprotegerin (OPG) in children with primary nephrotic syndrome. Saudi J Kidney Dis Transplant 22(5):955
Liabeuf S, Okazaki H, Desjardins L et al (2013) Vascular calcification in chronic kidney disease: are biomarkers useful for probing the pathobiology and the health risks of this process in the clinical scenario? Nephrol Dial Transplant [Epub ahead of print]
Blázquez-Medela AM, García-Ortiz L, Gómez-Marcos MA et al (2012) Osteoprotegerin is associated with cardiovascular risk in hypertension and/or diabetes. Eur J Clin Invest 42(5):548
Shin JY, Shin YG, Chung CH (2006) Elevated serum osteoprotegerin levels are associated with vascular endothelial dysfunction in type 2 diabetes. Diabetes Care 29(7):1664
Coen G, Ballanti P, Balducci A et al (2002) Serum osteoprotegerin and renal osteodystrophy. Nephrol Dial Transplant 17(2):233
Janda K, Krzanowski M, Chowaniec E et al (2013) Osteoprotegerin as a marker of cardiovascular risk in patients on peritoneal dialysis. Pol Arch Med Wewn 123(4):149
Lederle FA (2012) The strange relationship between diabetes and abdominal aortic aneurysm. Eur J Vasc Endovasc Surg 43(3):254
Rasmussen LM, Tarnow L, Hansen TK, Parving HH, Flyvbjerg A (2006) Plasma osteoprotegerin levels are associated with glycaemic status, systolic blood pressure, kidney function and cardiovascular morbidity in type 1 diabetic patients. Eur J Endocrinol 154(1):75
Pencak P, Czerwieńska B, Ficek R et al (2013) Calcification of coronary arteries and abdominal aorta in relation to traditional and novel risk factors of atherosclerosis in hemodialysis patients. BMC Nephrol 14:10
Bargnoux AS, Dupuy AM, Garrigue V et al (2009) Evolution of coronary artery calcifications following kidney transplantation: relationship with osteoprotegerin levels. Am J Transplant 9(11):2571
Hjelmesaeth J, Ueland T, Flyvbjerg A et al (2006) Early posttransplant serum osteoprotegerin levels predict long-term (8-year) patient survival and cardiovascular death in renal transplant patients. J Am Soc Nephrol 17(6):1746
Matsubara K, Stenvinkel P, Qureshi AR et al (2009) Inflammation modifies the association of osteoprotegerin with mortality in chronic kidney disease. J Nephrol 22(6):774
Svensson M, Dahle DO, Mjøen G et al (2012) Osteoprotegerin as a predictor of renal and cardiovascular outcomes in renal transplant recipients: follow-up data from the ALERT study. Nephrol Dial Transplant 27(6):2571
Meneghini M, Regalia A, Alfieri C et al (2013) Calcium and osteoprotegerin levels predict the progression of the abdominal aortic calcifications after kidney transplantation. Transplantation 96(1):42
Lewis JR, Lim WH, Zhu K et al (2014) Elevated osteoprotegerin predicts declining renal function in elderly women: a 10-year prospective cohort study. Am J Nephrol 39(1):66
Zhou S, Fang X, Xin H, Li W, Qiu H, Guan S (2013) Osteoprotegerin inhibits calcification of vascular smooth muscle cell via down regulation of the Notch1-RBP-Jκ/Msx2 signaling pathway. PLoS One 8(7):e68987
Fernández Fernández B, Elewa U, Sánchez-Niño MD et al (2012) 2012 Update on diabetic kidney disease: the expanding spectrum, novel pathogenic insights and recent clinical trials. Minerva Med 103(4):219
Szteyn S (1988) Types of neurons in nucleus olivaris inferior of the European bison. J Hirnforsch 29(3):353
Benito-Martin A, Ucero AC, Zubiri I et al (2013) Osteoprotegerin in exosome-like vesicles from human cultured tubular cells and urine. PLoS One 8(8):e72387
Kiani AN, Johnson K, Chen C et al (2009) Urine osteoprotegerin and monocyte chemoattractant protein-1 in lupus nephritis. J Rheumatol 36(10):2224
West SL, Lok CE, Jamal SA (2013) Osteoprotegerin and fractures in men and women with chronic kidney disease. J Bone Miner Metab. doi:10.1007/s00774-013-0506-1
Luan X, Lu Q, Jiang Y et al (2012) Crystal structure of human RANKL complexed with its decoy receptor osteoprotegerin. J Immunol 189(1):245
Acknowledgments
FIS PS09/00447, PI13/00047, ISCIII-RETIC RedinRen RD12/0021 FEDER funds, Comunidad de Madrid S2010/BMD-2378. Programa Intensificación Actividad Investigadora (ISCIII) to AO and Sara Borrel to MDSN, ERA/EDTA fellowship to LGE, CYTED IBERERC.
Conflict of interest
None.
Author information
Authors and Affiliations
Corresponding authors
Additional information
A. Montañez-Barragán, I. Gómez-Barrera, L. González-Espinoza and A. Ortiz have contributed equally to this work.
Rights and permissions
About this article

Cite this article
Montañez-Barragán, A., Gómez-Barrera, I., Sanchez-Niño, M.D. et al. Osteoprotegerin and kidney disease. J Nephrol 27, 607–617 (2014). https://doi.org/10.1007/s40620-014-0092-x
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s40620-014-0092-x