
Abstract
Objective
Summarize and analyze the characteristics of patients with Multiple Endocrine Neoplasia type 1 (MEN-1) who were diagnosed with malignant tumors that do not belong to MEN-1 components.
Methods
Clinical data from patients with MEN-1 who visited Peking Union Medical College Hospital between April 2012 and April 2022 were collected. We compared the clinical characteristics of patients with malignant tumors outside of their MEN-1 components to those without additional tumors. MEN-1 gene testing was performed on most of these patients using Sanger sequencing, whole-exome sequencing, or MLPA.
Results
A total of 221 MEN-1 patients were diagnosed, of which 23 (10.40%) were found to have malignant tumors that did not belong to MEN-1 components, including papillary thyroid carcinoma (PTC) (4.52%), breast cancer (1.81%), urologic neoplasms (1.35%), primary hepatic carcinoma (PCC) (0.09%), meningeal sarcoma (0.05%), glioblastoma (0.05%), cervical cancer (0.05%), and lung carcinoma (0.05%). MEN-1 gene mutations were identified in 11 patients, including missense mutations, frameshift mutations, and splice mutations. The prevalence of each endocrine neoplasm, particularly gastroenteropancreatic neuroendocrine tumor, was higher in MEN-1 patients with other malignant tumors compared to MEN-1 patients without malignant tumors.
Conclusion
Our retrospective study revealed a higher incidence of non-MEN-1 component malignant tumors in MEN-1 patients, especially breast cancer, PTC, and urologic neoplasms. These patients also exhibit more severe clinical phenotypes of MEN-1.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Multiple endocrine neoplasia (MEN) is a condition characterized by the occurrence of neoplasms involving two or more endocrine glands. The types of MEN include MEN-1, MEN-2, and MEN-4, depending on the specific endocrine glands affected and the involved gene mutations. While most patients diagnosed with MEN inherit the condition as an autosomal dominant trait, sporadic cases can occur even without family history [1,2,3].
MEN-1 is caused by mutations in the MEN1 gene and is characterized by hyperparathyroidism, pituitary neuroendocrine tumors (PitNET), and gastroenteropancreatic neuroendocrine tumors (GEP-NET). Its estimated prevalence in the population is 1 in 30,000 [4]. Other uncommon manifestations in MEN-1 patients include thymic and bronchial neuroendocrine tumors (NET), adrenal adenomas, and cutaneous/mucosal or visceral abnormalities such as lipomas, hypomelanotic macules, collagenomas, and meningiomas [3].
Results from clinical studies and preclinical experiments indicate the possible involvement of MEN-1 gene, which encodes menin, in the development of non-endocrine tumors [3]. There have been several case reports about MEN-1 complicating with other malignancies, such as breast cancer, melanoma, renal cell carcinoma, and prostate cancer [5,6,7]. Further observations are needed to determine whether the relationship between MEN-1 and malignancies is direct or coincidental. Several studies have confirmed an increased risk of breast cancer in MEN-1 patients, with a relative risk of 2.83 categorizes for women [5]. Additionally, a study has discovered a previously unrecognized function of menin in controlling melanoma cell proliferation, migration, metastasis, and apoptosis [6].
Here, we present a summary of cases diagnosed with MEN-1 and concurrent diagnosis of other malignant tumors, evaluated in a tertiary clinical center. To our knowledge, this is the first study to summarize patients with co-occurring malignant tumors and MEN-1.
Materials and methods
The study was conducted at Peking Union Medical College Hospital between April 2012 and April 2022, where patients diagnosed with MEN-1 were included in the data collection. The criteria for patients diagnosed with MEN-1 were any one of these conditions: (1) two or more tumors located within one of the three primary endocrine sites (parathyroid glands, pancreas/duodenum/foregut, pituitary gland), (2) a first-degree relative with clinical diagnosis of MEN-1 and a confirmed diagnosis of a MEN-1 associated tumor, or (3) identified as harboring a MEN-1 germline mutation without the development of MEN-1 associated tumors [3].
The study protocol adhered to the ethical guidelines specified by the 1975 Declaration of Helsinki and received approval from the Ethics Committee of PUMCH (reference number JS-1663). Patient data, including clinical information such as age of onset, sex, characteristics of tumors, treatment received, and family history, as well as information on malignant tumors including age of onset, pathological findings, treatment, and prognosis were obtained from both electronic medical records and clinic charts.
Genomic DNA was extracted from peripheral blood lymphocytes using the QIAamp Blood DNA Kit (Qiagen; Hilden, Germany). All coding exons and the exon–intron boundaries of MEN1 were specifically amplified using polymerase chain reaction (PCR) and subsequently analyzed by Sanger sequencing. Samples from cases that tested negative for MEN1 mutation by PCR-sequencing and whole-exome sequencing were further analyzed by for large deletions using the MLPA assay.
Statistical analysis was performed using IBM SPSS Statistics 21 (IBM Corp., Armonk, NY). Continuous variables were expressed as mean ± standard deviation (SD), and categorical variables were expressed as frequencies. Independent samples t-test was used to compare the two groups, while χ2 test was used to assess count data.
Results
Two hundred and twenty-one patients were diagnosed with MEN-1. Of these, 23 (10.40%) patients were found to have malignant tumors that did not belong to the MEN-1 components.
Clinical features of patients diagnosed with MEN-1 and other malignant tumors
In total, 23 cases were identified with co-occurrence of MEN-1 and other malignant tumors, comprising of 6 males and 17 females. The first component tumors of MEN-1 were diagnosed by an average age of 44.35 ± 13.11 years old. Of these diagnoses, PHPT accounted for 39.13% (nine cases), GEP-NET accounted for 43.48% (ten cases), and PitNET accounted for 17.39% (four cases). Eleven patients had a family history of MEN-1. Of these patients with MEN-1, 21 cases had hyperparathyroidism, with an incidence rate of 91.30%. Moreover, 19 cases (82.61%) had PitNETs, including ten cases (52.63%, six microadenomas and four macroadenomas) of pituitary nonfunctioning adenomas, four cases (21.05%) of prolactinomas, four cases (21.05%, two were microadenomas and two were macroadenomas) of GH-secreting adenomas, and one case (5.26%) of TSH/GH-secreting adenoma. GEP-NETs occurred in 19 patients (82.61%), including ten cases (52.63%) of nonfunctioning GEP-NETs, five cases (26.32%) of gastrinomas, two cases (10.53%) of insulinomas, one case (5.26%) with both gastrinoma and insulinoma, and one case (5.26%) of vasoactive intestinal polypeptide tumor (VIPoma). Eleven cases (47.83%) were diagnosed with adrenal adenomas or adrenal hyperplasia, three cases (13.04%) were diagnosed with thymic NETs, and one case with thymoma.
The average age of diagnosis for non-MEN-1 component malignant tumors in these patients was 52.43 ± 11.14 years. Among them, ten cases (4.52%) were diagnosed with papillary thyroid carcinoma (PTC), three cases (1.36%) were diagnosed with urologic neoplasms, including renal papillary cell carcinoma, renal clear cell carcinoma (RCC), and ureteropelvic urothelial carcinoma. Breast cancer was found in four cases (1.81%), while two cases (0.90%) had PCC with Hepatitis B infection. One patient was diagnosed with meningeal sarcoma 16 years after undergoing resection and radiotherapy for pituitary nonfunctioning adenomas. Moreover, one patient was diagnosed with glioblastoma, one with cervical cancer, and another with squamous cell lung carcinoma. Among the 23 patients, six patients had a family history of malignancy (See Table 1 for details).
Nineteen patients underwent genetic testing, which revealed that 11 patients had MEN1 gene mutations. Among these mutations, seven cases had missense mutations, occurring in four cases of thyroid cancer, one case of breast cancer, one case of RCC, and one case of squamous cell lung carcinoma. Additionally, three cases had frameshift mutations, with diagnoses of PCC, meningeal sarcoma, and breast cancer. Lastly, one case had a splice mutation and was diagnosed with breast cancer.
Differences in clinical characteristics between patients diagnosed with MEN-1 with malignant tumors and those without malignant tumors
The clinical characteristics of MEN-1 patients with and without malignant tumors were compared in this study. There were no significant differences observed in the age at diagnosis of the initial component of MEN-1, incidence of obesity, and family history of malignant tumors between the two groups. Although not reaching statistical significance, there was a higher proportion of diabetes and female patients in the MEN-1 group with malignant tumors. In addition, the incidence of MEN-1 components, including PitNET (82.61% vs. 68.69%, p = 0.167), GEP-NET (86.96% vs. 65.15%, p = 0.035), adrenal adenoma (30.43% vs. 23.23%, p = 0.558), and thymic NET (13.04% vs. 6.56%, p =0. 256), was higher in MEN-1 patients with malignancies than in those without other malignancies. Of these, the incidence of GEP-NET was statistically significantly higher as indicated in Table 2.
Clinical management of MEN-1 in patients with other malignant tumors
In terms of the management of MEN-1-related tumors, 16 cases of hyperparathyroidism patients underwent parathyroid surgery, with at least two or more parathyroid glands removed in ten cases. One patient experienced recurrence after surgery. Six patients underwent pituitary tumor surgery, and one patient continued to receive gamma knife and bromocriptine treatment due to incomplete relief after surgery. One patient with pituitary macroadenoma developed postoperative intracranial and pulmonary infections, ultimately resulting in mortality. Three patients with prolactinomas and one patient with GH-secreting tumor received bromocriptine treatment, while one patient with TSH-GH mixed tumor received octreotide treatment. Other patients with pituitary nonfunctioning microadenoma were under observation. Ten patients with GEP-NET underwent surgical treatment, three patients received octreotide treatment, one patient received radioactive isotope therapy, and one patient of gastrinoma received omeprazole treatment alone. The remaining nonfunctioning GEP-NETs were placed under long-term observation. Three cases of thymic NET underwent surgical treatment. One case of adrenal nodule underwent surgical treatment.
In the management of malignant tumors, all cases of PTC underwent radical surgery. One patient with breast cancer and lymph node metastasis underwent breast-conserving surgery, followed by 30 sessions of radiotherapy and six cycles of chemotherapy (docetaxel plus cyclophosphamide) along with endocrine therapy (goserelin and tamoxifen). Another patient with invasive breast cancer underwent radical surgery, followed by four cycles of chemotherapy (Paclitaxel and Epirubicin) and endocrine therapy (goserelin and anastrozole). One patient only underwent radical surgery for breast cancer, and no recurrence was observed in all cases. One patient with breast cancer refused further treatment. Three patients with urologic neoplasms underwent radical surgery, and no recurrence was observed after surgery. Among two cases of primary liver cancer, one patient underwent surgery, while the other refused further treatment. One patient with meningeal sarcoma underwent surgery and postoperative radiotherapy, but the condition did not improve.
Discussion
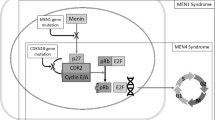
The MEN1 gene, located on the 11q13 chromosome, is responsible for the development of multiple endocrine neoplasia type 1 (MEN-1), an inherited tumor syndrome [3]. It codes for the menin protein, which aids in regulating transcription, chromatin structure, genome stability, and cellular proliferation by either directly associating with interacting protein partners or modulating key cellular signaling pathways [4]. The development of parathyroid, pancreatic, pituitary, and adrenal tumors in MEN1 knockout mice mimics the human MEN1 syndrome, demonstrating that MEN1 acts as a tumor suppressor gene in endocrine tumors [8]. Recent reports have demonstrated that the menin protein is also associated with the development of non-endocrine tumors. For example, menin has been shown to associate with trxG family proteins in a histone methyltransferase complex, it is required for the maintenance of Hox family gene expression, and is involved in the initiation of MLL-mediated leukemogenesis and myeloid transformation [9, 10].
We found that 1.81% of MEN-1 cases were associated with breast cancer, indicating a significantly higher incidence compared to the general population. The 10-year prevalence in different regions in China ranges from 134 to 367 per 100,000 individuals [11]. Studies have shown that the silencing of the MEN1 gene can result in changes in proliferative gene expression among primary mammary luminal progenitor cells in human breast tissue [12]. For instance, Romain T et al. analyzed 354 human breast cancers and identified a lower expression of menin that correlated with ER-negative breast cancer (p = 0.041) [13]. Rachel S.et al. found that MEN-1 carriers were at an increased risk for breast cancer, which was not related to other known risk factors or familial cancer history, and was associated with an earlier age of onset (45 vs 57.5 years old) [14]. However, in our study, the age of onset for breast cancer ranged from 42 to 71 years (average 53 years), which is not earlier than the mean age at diagnosis of breast cancer in Chinese populations (49 years) [11]. There is robust evidence suggesting a strong connection between MEN-1 and breast cancer, which highlights the significance of breast cancer screening for female MEN-1 patients starting at age 40. In our study, we identified one patient who was diagnosed with both breast cancer and prolactinoma. In animal experiments, prolactin was shown to act as a cancer initiator and promoter [15]. Nevertheless, a recent study indicated that the relative risk of breast cancer in 1342 patients with treated hyperprolactinemia was 1.07, which does not suggest an increased risk of breast cancer [16].
Our study found that the incidence of MEN-1 complicated with thyroid papillary carcinoma was 4.52%, which is higher than the general population incidence of PTC in China (0.5–6.9/100,000) [17]. Although the current view is that the association of thyroid abnormalities in patients with MEN-1 may be incidental [3], genetic analysis of papillary cancer tissue did not show loss of heterozygosity (LOH) in MEN-1 patients. Furthermore, there is no evidence from in vitro, animal, or human studies linking germline mutations of MEN1 with the development of PTC [7]. While there is insufficient evidence to support an association between MEN-1 and the development of thyroid cancer, potential factors contributing to the higher incidence of PTC in MEN-1 patients were examined. In our study, two cases of PTC with pituitary growth hormone adenoma were identified, which is a risk factor for the occurrence of PTC [18]. Thyroid ultrasound before surgery of hyperparathyroidism was needed in our center, which may increase the detection rate of thyroid cancer which is asymptomatic, low-malignant, slow-growing tumor, and often overlooked. Frequent radioactive examinations such as CT and nuclear medical examination, which are required in MEN-1 patients to evaluate tumors, can also increase thyroid exposure to excessive radiation and may be a risk factor for the increased incidence of thyroid cancer.
Three cases of MEN-1 were presented with urologic neoplasms, which comprised of RCC, renal papillary cell carcinoma, and upper tract urothelial carcinoma (UTUC). In a study by Perakakis, a German family with MEN-1 was diagnosed with atypical tumors including renal cell carcinoma, papillary thyroid cancer, and prostate cancer [7]. However, no studies have confirmed that the mutation of MEN1 gene is directly associated with the development of urinary tumors. It is hypothesized that the high incidence of urologic neoplasms observed in MEN-1 patients may be linked to urolithiasis caused by early hyperparathyroidism. A prospective study conducted by Jeroen A revealed that an early diagnosis of kidney stones (≤ 40 years) was linked to a heightened risk of RCC (HR: 3.08, 95% CI 1.55–6.11) and UTUC (HR: 1.66, 95% CI 1.03–2.68) [19]. Nevertheless, the pathophysiological mechanisms underlying the development of renal cell carcinoma and urothelial cancer in patients with urolithiasis remain undiscovered.
In this study, there were few relevant reports regarding other atypical tumor entities among MEN-1 patients, such as brain glioma, cervical cancer, and lung cancer. However, it has been found that menin represses PTN transcription via polycomb gene-mediated trimethylation of H3K27, which has been associated with lung adenocarcinoma. Interestingly, menin has been shown to be expressed not only in endocrine tumors, but also in many non-endocrine human cancer cells, including breast, cervical, brain glioma, and gastric cancer cell lines [20]. This suggests potential correlations between MEN-1 and these non-endocrine tumors. Further investigations are required to clarify both the general role of menin in carcinogenesis, as well as the importance of specific mutations.
Two patients in this study were diagnosed with a co-occurrence of PCC and MEN-1. However, both patients were suffering from Hepatitis B, which is a major risk factor for PCC. Interestingly, heterozygous ablation of MEN-1 in female mice has been shown to reduce chemical carcinogen-induced liver carcinogenesis, and repress activation of the inflammation pathway [21].
In our study, we compared the clinical characteristics of MEN-1 patients with and without other malignancies. A higher prevalence of PitNET, GEP-NET, thymic NET, and adrenal adenoma in MEN-1 patients with malignant tumors compared to those without malignancies was found, and the prevalence of GEP-NET in MEN-1 with malignant tumors group was statistically higher. Previous studies had found that GEP-NETs occurred more frequently in presence of a positive family history for non-neuroendocrine GEP cancer, obesity, and in presence of diabetes [22]. We also evaluated risk factors such as obesity, diabetes, family history of other malignancies, and smoking in both groups of patients, but no significant statistical differences were observed. Although we did not find more severe pattern of mutations in MEN-1 with other malignant tumors. We hypothesized that loss of function from menin protein may be more severe in MEN-1 patients with malignant tumors based on the more serious clinical features observed.
In this study, the diagnosis of all MEN-1 patients was based on one of the three diagnostic criteria proposed in the 2012 MEN-1 guidelines [3]. However, it is worth noting that some scholars have raised concerns about the potential confusion in clinical and familial diagnosis of MEN-1 due to the occurrence of phenotypic variations in both patients and their relatives [23]. Another research has also indicated that mutation-positive and mutation-negative MEN1 patients have a different phenotype and clinical course. Mutation-negative patients develop MEN1 manifestations at higher age and have a life expectancy, did not develop a third main MEN1 manifestation during follow-up [24]. However, in our study, 6 out of the 12 patients who lacked a genetic diagnosis exhibited the presence of all three tumors associated with MEN-1 which is inconsistent with previous research. So, further studies involving larger case cohorts and longer follow-up periods are needed to determine whether the diagnostic criteria for MEN-1 should be updated and whether there are clinical and prognostic differences between patients with and without MEN-1 gene mutations. Additionally, in patients who do not show MEN-1 mutations through routine whole exome sequencing or Sanger sequencing, the possibility of large genomic deletions or intronic splice site mutations should be considered. A small subset of patients may have other genetically related syndromes, such as MEN-4. In our study, four patients (cases 4, 5, 8, 10) underwent whole exome sequencing, but no mutations were identified in CDKNB1 or other pathogenic genes.
This study summarizes the largest number of patients diagnosed concurrently with MEN-1 and malignant tumors not related to MEN in a single center. Our study found a high prevalence of breast cancer, PTC, and urologic neoplasms in MEN-1 patients. Additionally, occurrences of malignant tumors such as brain glioma, squamous cell lung carcinoma, PCC, cervical cancer were found, which had rarely been reported in the past. 10.4% patients were found to have malignant tumors that did not belong to the MEN-1 components, which suggested that a small proportion of patients with MEN-1 may be at risk for additional, unpredictable cancers outside of the expected disease manifestations. Further investigation is needed to determine the etiology and pathogenesis of these tumors. Nevertheless, the more severe clinical features observed in MEN-1 patients with other malignant tumors compared to those without malignant tumors may suggest a more serious loss of function from menin protein. A limitation of our study was that it is retrospective in nature, we are unable to obtain cancer tissue from patients for evaluation the LOH to analyze the possible underlying mechanisms of malignancies in MEN-1 patients.
In conclusion, the data reported in the current study have showed in our retrospective analysis a potentially relevant difference in the occurrence of concurrent neoplasms in MEN-1 patients. Further larger and prospective studies are needed to confirm the achieved results, also in accordance with more accurate genetic testing and survival analyses. The specific mechanism underlying this relationship is still unclear, but the clinical manifestations of MEN-1 patients with malignant tumors are more severe than those without malignant tumors.
Data availability
Some or all data generated or analyzed during this study are included in this published article or in the data repositories listed in “References.”
References
Thakker RV (1998) Multiple endocrine neoplasia—syndromes of the twentieth century. J Clin Endocrinol Metab 83:2617–2620
Wells SA Jr, Asa SL, Dralle H et al (2015) Revised American Thyroid Association guidelines for the management of medullary thyroid carcinoma. Thyroid 25:567–610
Thakker RV, Newey PJ, Walls GV et al (2012) Clinical practice guidelines for multiple endocrine neoplasia type 1 (MEN1). J Clin Endocrinol Metab 97:2990–3011
Melmed S, Richard J, Allison B et al (2019) Williams textbook of endocrinology, 14th edn. Elsevier
Dreijerink KM, Goudet P, Burgess JR, Valk GD, International Breast Cancer in MEN1 Study Group (2014) Breast-cancer predisposition in multiple endocrine neoplasia type 1. N Engl J Med 371(6):583–584
Gao SB, Feng ZJ, Xu B, Chen Y, Zheng HH, Yin P, Hua X, Jin GH (2011) Menin represses malignant phenotypes of melanoma through regulating multiple pathways. J Cell Mol Med 15(11):2353–2363. https://doi.org/10.1111/j.1582-4934.2010.01222.x
Perakakis N, Flohr F, Kayser G et al (2016) Multiple endocrine neoplasia type 1 associated with a new germline Men1 mutation in a family with atypical tumor phenotype. Hormones (Athens) 15(1):113–117. https://doi.org/10.14310/horm.2002.1626
Crabtree JS, Scacheri PC, Ward JM et al (2001) A mouse model of multiple endocrine neoplasia, type 1, develops multiple endocrine tumors. Proc Natl Acad Sci U S A 98:1118–1123
Hughes CM, Rozenblatt-Rosen O, Milne TA et al (2004) Menin associates with a trithorax family histone methyltransferase complex and with the hoxc8 locus. Mol Cell 13:587–597
Jin S, Zhao H, Yi Y et al (2010) c-Myb binds MLL through menin in human leukemia cells and is an important driver of MLL-associated leukemogenesis. J Clin Invest 120:593–606
Li T, Mello-Thoms C, Brennan PC (2016) Descriptive epidemiology of breast cancer in China: incidence, mortality, survival, and prevalence. Breast Cancer Res Treat 159(3):395–406. https://doi.org/10.1007/s10549-016-3947-0
van Leeuwaarde RS, Dreijerink KM, Groner AC, Vos ES et al (2017) Indication for breast cancer screening in MEN1? J Clin Endocrinol Metab 102(6):2083–2090
Teinturier R, Abou Ziki R, Kassem L et al (2021) Reduced menin expression leads to decreased ERα expression and is correlated with the occurrence of human luminal B-like and ER-negative breast cancer subtypes. Breast Cancer Res Treat 190(3):389–401. https://doi.org/10.1007/s10549-021-06339-9
van Leeuwaarde RS, Dreijerink KM, Ausems MG, Beijers HJ, Dekkers OM, de Herder WW, van der Horst-Schrivers AN, Drent ML, Bisschop PH, Havekes B, Peeters PHM, Pijnappel RM, Vriens MR, Valk GD (2017) MEN1-dependent breast cancer: indication for early screening? Results from the Dutch MEN1 Study Group. J Clin Endocrinol Metab 102(6):2083–2090. https://doi.org/10.1210/jc.2016-3690
Rose-Hellekant TA, Arendt LM, Schroeder MD, Gilchrist K, Sandgren EP, Schuler LA (2003) Prolactin induces ERa-positive and ERa-negative mammary cancer in transgenic mice. Oncogene 22:4664–4674
Dekkers O, Romijn J, de Boer A, Vandenbroucke J (2010) The risk for breast cancer is not evidently increased in women with hyperprolactinemia. Pituitary 13:195–198. https://doi.org/10.1007/s11102-009-0214-y
Bray F, Ferlay J, Soerjomataram I, Siegel RL, Torre LA, Jemal A (2018) Global cancer statistics 2018: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J Clin 68(6):394–424. https://doi.org/10.3322/caac.21492
Gullu BE, Celik O, Gazioglu N, Kadioglu P (2010) Thyroid cancer is the most common cancer associated with acromegaly. Pituitary 13(3):242–248. https://doi.org/10.1007/s11102-010-0224-9
van de Pol JAA, van den Brandt PA, Schouten LJ (2019) Kidney stones and the risk of renal cell carcinoma and upper tract urothelial carcinoma: the Netherlands Cohort Study. Br J Cancer 120(3):368–374. https://doi.org/10.1038/s41416-018-0356-7
Ren F, Xu HW, Hu Y et al (2012) Expression and subcellular localization of menin in human cancer cells. Exp Ther Med 3:1087–1091
Xu B, Li SH, Zheng R, Gao SB, Ding LH, Yin ZY, Lin X, Feng ZJ, Zhang S, Wang XM, Jin GH (2013) Menin promotes hepatocellular carcinogenesis and epigenetically up-regulates Yap1 transcription. Proc Natl Acad Sci U S A 110(43):17480–17485. https://doi.org/10.1073/pnas.1312022110
Feola T, Puliani G, Sesti F, Modica R, Centello R, Minotta R, Cannavale G, Di Meglio S, Di Vito V, Lauretta R, Appetecchia M, Colao A, Lenzi A, Isidori AM, Faggiano A, Giannetta E (2022) Risk factors for gastroenteropancreatic neuroendocrine neoplasms (GEP-NENs): a three-centric case-control study. J Endocrinol Invest 45(4):849–857. https://doi.org/10.1007/s40618-021-01715-0
Turner JJ, Christie PT, Pearce SH, Turnpenny PD, Thakker RV (2010) Diagnostic challenges due to phenocopies: lessons from multiple endocrine neoplasia type1 (MEN1). Hum Mutat 31(1):E1089–E1101. https://doi.org/10.1002/humu.21170
de Laat JM, van der Luijt RB, Pieterman CR, Oostveen MP, Hermus AR, Dekkers OM, de Herder WW, van der Horst-Schrivers AN, Drent ML, Bisschop PH, Havekes B, Vriens MR, Valk GD (2016) MEN1 redefined, a clinical comparison of mutation-positive and mutation-negative patients. BMC Med 14(1):182. https://doi.org/10.1186/s12916-016-0708-1
Funding
Article processing charges (APC)was funded by Chinese Academy of Medical Sciences Innovation Fund for Medical Sciences, grant number is 2021-I2M-1-023; National Key Clinical Specialty Capacity Improvement Project.
Author information
Authors and Affiliations
Contributions
YZ contributed to the study design, acquisition of data, analysis of data, interpretation of data, drafting the article. AS and OW contributed to the acquisition of data, analysis of data. E.I.E. FG contributed to study design, interpretation of data, drafting the article, revising it for intellectual content and final approval of the version to be published. LW contributed to study design, interpretation of data, drafting the article, revising it for intellectual content. LD contributed to study design, interpretation of data, drafting the article, revising it for intellectual content. HY contributed to study design, interpretation of data, drafting the article, revising it for intellectual content. HP contributed to study design, interpretation of data, drafting the article, revising it for intellectual content. HZ contributed to the interpretation of data, drafting the article, revising it for intellectual content, and final approval of the version to be published.
Corresponding author
Ethics declarations
Conflict of interest
The authors have no competing interests to declare that are relevant to the content of this article.
Ethics approval
Study protocol conforms to the ethical guidelines of the 1975 Declaration of Helsinki and was approved by the Ethics Committee of PUMCH (reference number was JS-1663).
Consent to participate
Informed consent was obtained from all individual participants included in the study.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Springer Nature or its licensor (e.g. a society or other partner) holds exclusive rights to this article under a publishing agreement with the author(s) or other rightsholder(s); author self-archiving of the accepted manuscript version of this article is solely governed by the terms of such publishing agreement and applicable law.
About this article

Cite this article
Zhao, Y.x., Wang, O., Song, A. et al. The risk of concurrent malignancies in patients with multiple endocrine neoplasia type 1: insights into clinical characteristics of those with multiple endocrine neoplasia type 1. J Endocrinol Invest 47, 1931–1939 (2024). https://doi.org/10.1007/s40618-023-02288-w
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s40618-023-02288-w