
Abstract
Osteoporosis is a highly prevalent condition, characterized by compromised bone strength and fragility fractures and with an important associated socio-economic burden. Bisphosphonates are well established as the first line treatment for osteoporosis. However, while randomized control trials have in general demonstrated reasonable anti-fracture efficacy at the spine, they have shown moderate reduction in fracture incidence for non-vertebral sites. Furthermore, oral bisphosphonates are commonly associated with adverse gastrointestinal effects and both oral and parenteral bisphosphonates have been linked with osteonecrosis of the jaw and atypical femoral fracture, two rare but debilitating side effects. In addition, bisphosphonates are not recommended in patients with GFR <35 ml/min/1.73 m2. Hence, there is a clear requirement for newer agents, which are able to reduce fracture risk further, whilst overcoming the limitations of bisphosphonates. Over the past 20 years, knowledge and a deeper understanding of the various signalling pathways involved in bone remodelling has increased, enabling identification of additional targets for therapy. This review focuses on these newer therapies and includes anti-resorptive agents such as raloxifene and other selective oestrogen receptor modulators, the monoclonal antibody denosumab (which inhibits the RANKL pathway), odanacatib, a cathepsin K inhibitor and the anabolic agents, PTH analogue; PTH (1–34) and anti-sclerostin antibodies (activator of the Wnt pathway). Strontium ranelate will not be reviewed as recent reports highlight concerns surrounding its cardiovascular safety and together with an apparent increased risk of thrombosis, its future use remains uncertain. Some of these agents such as raloxifene, denosumab and teriparatide are already in clinical use whilst others are at varying stages of development. This review will provide an overview of the mechanisms of action of these therapeutic agents on the skeleton and assess their efficacy in osteoporosis and fracture prevention.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Osteoporosis is a skeletal disorder in which bone strength is compromised, predisposing the affected individual to fragility fractures. Over 2 million people suffer from osteoporosis in the United Kingdom (UK) and there are an estimated 300,000 fragility fractures per year, with hip fractures costing the National Health Service (NHS) as much as 1.9 billion in hospital and social care. Fractures are a grave source of morbidity and mortality for the patient and occur as a result of microarchitectural deterioration as well as low bone mass [1].
The process of bone remodelling
Impaired bone remodelling is one important mechanism which leads to osteoporosis. In order to understand the pathophysiology of the disorder, we will first describe the process of normal bone remodelling. Skeletal remodelling is a physiological process which occurs throughout adult life and relies on the coupled and balanced processes of bone formation and resorption within every basic multicellular unit (BMU). The function of the BMU is important in the repair of micro-damage caused by mechanical strain and fatigue which occur throughout the skeleton. It is thought apoptotic osteocytes at areas of micro-damage signal the location and extent of the damage which leads to the targeting of bone remodelling to the site of damage. Remodelling occurs over a time span of 90–130 days and relies on the coupled activity of osteoclasts and osteoblasts. This ensures that bone resorption and formation take place at the same rate and facilitate repair of the skeleton and its microcracks without adversely affecting bone mass (Fig. 1).

The bone remodelling cycle. Bone is resorbed by osteoclasts and deposited by osteoblasts in a coupled process that serves to maintain the integrity of bone. Osteoblasts trapped within the matrix become known as osteocytes and function as mechanical sensors capable of detecting microdamage. Odanacatib inhibits cathepsin K, an enzyme imperative to the resorptive activity of osteoclasts, in order to halt loss of bone mass
Remodelling is initiated by osteoclastic resorption, which occurs over several weeks. Osteoclasts attach to bone via a specialized membrane known as the ruffled border and secrete substances that solubilize bone mineral and degrade the matrix, most notably, chloride ions, protons and cathepsin K, a lysosomal cysteine proteinase. Cathepsin K is crucial to the resorptive function of osteoclasts and in humans who have a mutation in the gene that codes for the enzyme, a dense bone phenotype, known as pycnodysostosis, is seen [2]. Indeed, this is the rationale for the development of cathepsin K inhibitors for the treatment of osteoporosis.
The dissolution of bone liberates factors including transforming growth factor β (TGF-β), insulin-like growth factors (IGFs), and bone morphogenetic proteins (BMPs) from the calcified matrix and attracts lining cells and osteoblastic precursors to the resorptive pit and stimulates their differentiation. Furthermore, there is evidence to suggest that osteoclasts themselves are capable of synthesizing compounds that are pro-osteogenic, e.g. BMP-6, sphingosine-1-phosphate and cardiotrophin-1 [3, 4]. Newly formed bone is deposited by osteoblasts into existing lacunae over a period of 3 months. The osteoid is subsequently mineralized and lining cells cover the area. Osteoblasts that do not undergo programmed cell death are trapped within the matrix and are referred to as osteocytes. Osteoclasts undergo apoptosis once resorption is complete [5, 6].
The bone remodelling compartment
Bone remodelling is carried out in a specialized vascular structure known as the bone remodelling compartment (BRC). The BRC is a closed cavity, separate from the bone marrow, which is lined by a canopy of cells capable of secreting Receptor Activator of Nuclear factor Kappa-B ligand (RANKL) and Osteoprotegerin (OPG) [7]. It is penetrated by capillaries that serve as a conduit for transporting osteoblast and osteoclast precursor cells into the basic multicellular unit (BMU), a term that has become synonymous with Harold Frost after he showed osteoblasts, osteoclasts and osteocytes to be present within the remodelling cavity 40 years ago [8]. Furthermore, capillary endothelial cells secrete angiogenic factors such as Vascular Endothelial Growth Factor (VEGF), angiopoietin and endothelin that have a dual role in regulating bone cell activity [9]. It is not unexpected then that osteoclasts and osteoblasts have been shown to possess VEGF receptors and are capable of VEGF production. VEGF is thought to contribute to the early phases of modelling and remodelling, possibly via its role as a chemoattractant in directing cell migration to sites of active remodelling [10–12].
Since the work of Frost in 1975, our understanding of the signalling pathways involved in bone remodelling has progressed greatly. It is now known that the confined space of the BRC is crucial for maintaining the delicate balance between resorption and formation and that this process is tightly regulated by local and systemic factors. Unregulated access to the remodelling space would result in interference from growth factors that are present in high concentrations within the marrow microenvironment and would offset local regulation. Enhanced bone loss results when the coupling of bone resorption and new bone formation is disrupted. This eventually results in reduction of bone mass, deterioration of bone architecture, loss of trabecular numbers and connectivity, increased cortical porosity, leading to osteoporosis and increased susceptibility to fracture [6].
Signalling pathways in bone cells
Osteoclast differentiation and the RANKL-RANK-OPG pathway
Bone remodelling relies on the complex interplay between numerous cell types. We have already discussed how factors derived from the bone matrix and/or osteoclasts during resorption serve to modulate osteoblastic activity. Similarly, osteoclastic differentiation relies on osteoblasts and its secretion of RANKL and macrophage-colony stimulating factor (M-CSF) [13].
Signalling of M-CSF via its receptor c-fms upregulates RANK expression in mononuclear precursor cells during the initial stages of osteoclastic differentiation. Its ligand, RANKL, is synthesized by osteoblasts and marrow stromal cells in response to hormonal stimulation and together with M-CSF is sufficient for promoting the development of mature osteoclasts. Activation of RANK leads to stimulation of the transcription factors NF-κB, AP-1 and NFATc1 [14], which in turn regulate transcriptional expression of genes essential for normal osteoclastic function, namely cathepsin K, MMP-9, TRAcP, DC-STAMP and β3 integrin. The lifespan of osteoclasts is that of several weeks and survival is dependent on the ongoing signalling of M-CSF and RANKL. Osteoprotegerin (OPG) is a decoy receptor for RANKL secreted by osteoblasts and marrow cells and can interfere with RANK-RANKL interaction [13, 15]. A recently launched antibody directed against RANKL has proved to be an effective anti-catabolic treatment in the management of osteoporosis.
Osteoblast differentiation and Wnt signalling
Wnt is a glycoprotein that is crucial for normal bone formation. Canonical signalling (Wnt/β-catenin) regulates differentiation, function and survival of all osteoblastic-type cells (osteoprogenitors, pre-osteoblasts, osteoblasts, osteocytes and bone lining cells) and plays a role in committing multipotent mesenchymal stem cells to the osteoblastic lineage [16]. Although Wnt may also participate in non-canonical signalling (Wnt/Ca2+; Wnt/planar polarity), it is the canonical pathway that is of particular importance in osteoblasts and will thus be the focus [17].
During canonical signalling, Wnt3a binds to a receptor complex comprised of the Frizzled receptor and low-density lipoprotein receptor-related protein 5 or 6 (LRP 5/6). This leads to downstream intracellular events that converge on the prevention of GSK-3β-directed breakdown of the protein β-catenin. Aided by T-cell factor/lymphoid enhancer factors (Tcf/Lef), β-catenin initiates transcriptional upregulation of osteoblastic marker genes, such as RUNX2 and Osterix. RUNX2 is the first osteoblastic marker expressed during cell differentiation and has been shown to regulate gene expression for VEGF, osteocalcin, RANKL, sclerostin and Dentin-Matrix Protein-1 [18, 19].
Endogenous inhibitors secreted by osteocytes and late osteoblasts antagonize Wnt signalling. Inhibitors fall into two major groups: those that bind directly to Wnt and impair its ability to activate its receptor complex (e.g. secreted frizzled protein (sFRP) and Wnt inhibitory factor-1 (WIF-1), and those that interfere with the LRP constituent (e.g. sclerostin and Dickkopf (DKK1) [20, 21]. Neutralizing antibodies directed against sclerostin have so far proven to be an effective anabolic approach in phase II trials for treating osteoporosis (Fig. 2). The differentiation and maturation of osteoblasts are also regulated by a number of autocrine, paracrine and endocrine factors.

Cross talk between bone cells, and sites of therapeutic intervention for osteoporosis. Osteocyte/Osteoblast cross talk: Wnt signalling in osteoblasts is antagonized by inhibitory factors sclerostin and DKK1. In addition, the Wnt pathway stimulates the production of osteoprotegerin (OPG), a soluble decoy receptor for the RANKL, preventing osteoclast (OC) differentiation and function. PTH attenuates osteocyte production of such inhibitors, thus favouring bone formation. Teriparatide, a recombinant form of PTH, and antibodies directed against sclerostin, have been shown to exhibit anabolic effects on bone. Osteoblasts stimulate and inhibit osteoclast differentiation via RANKL and OPG expression, respectively. Both denosumab and oestrogen target the RANK-RANKL pathway to disrupt osteoclast maturation
Cross talk
Cross talk between the OPG/RANKL/RANK system and Wnt pathways may play an important part in the pathophysiology of postmenopausal osteoporosis. There is evidence to suggest that β-catenin positively regulates OPG expression and that serum β-catenin levels are significantly reduced in postmenopausal women (PMW) with osteoporosis in relation to PMW without osteoporosis [22, 23]. Recently it was also shown that sclerostin, a key antagonist of Wnt signalling, could stimulate RANKL expression [24]. Based on findings such as these, we now know that Wnt signalling in osteoblasts can decrease osteoclast activation and differentiation whilst simultaneously enhancing bone formation [25].
Systemic regulation of remodelling
There are four main hormones involved in the regulation of bone remodelling, parathyroid hormone (PTH), 1,25(OH)2D3, calcitonin and oestrogen. Secretion of the first three hormones is propelled by a need to keep calcium concentration between 2.2 and 2.6 mM, and deficiency of the latter is of particular significance in postmenopausal osteoporosis. Hormonal regulation is modified by environmental cues such as paracrine cytokines and mechanical strain.
Parathyroid hormone
Role in calcium and phosphate metabolism
PTH is an 84-amino acid peptide produced by the parathyroid glands and has a key role in calcium and phosphate metabolism. It is secreted in response to low blood calcium and restores calcium levels in three main ways: first, it has a direct action on the kidney where it reduces renal excretion of calcium whilst promoting phosphorous excretion; second, PTH stimulates osteoclastic-mediated resorption to liberate calcium and phosphorous from bone and into the circulation; and last, PTH indirectly increases intestinal absorption of calcium and phosphorous through its activation of 1α-cholecalciferol hydroxylase—an important enzyme involved in the synthesis of active vitamin D [26].
Stimulation of the vitamin D receptor (VDR) and retinoid X receptors in osteoblasts by 1,25(OH)2D3 increases M-CSF and RANKL production and indirectly stimulates osteoclastic bone resorption. In contrast, calcitonin can directly inhibit bone resorption by binding to receptors on the osteoclast [15, 27].
Role in skeletal homeostasis
Intermittent vs. continuous PTH exposure
The anabolic effect of PTH on bone is somewhat more complex than its endocrine role in the maintenance of calcium homeostasis. Continuous PTH production in primary hyperparathyroidism is associated with enhanced osteoclastogenesis and bone loss, whereas intermittent exposure to low doses of PTH has anabolic effects and leads to improved bone microarchitecture and increased bone volume. PTH is a potent inducer of osteoblastic activity when administered intermittently. This pattern of pulsatile treatment is employed by teriparatide injections (PTH 1–34 given daily) and stimulates bone formation earlier and to a greater extent than bone resorption, thus generating a temporal “anabolic window” [28, 29].
Anabolic and catabolic action of PTH
The biological effects of PTH are mediated by PTH1R, a high-affinity G-protein coupled receptor that is expressed on the surface of osteoblastic (and renal tubular) cells. Following PTHR1 stimulation two signalling pathways are activated: the protein kinase A (PKA) and the protein kinase C pathway (PKC). The main route of PTH signalling in bone is the cAMP-PKA pathway [30].
PTH reduces osteoblast apoptosis, accelerates the recruitment of newly formed osteoblasts from bone lining cells and increases the number of active osteoblasts. PTHR1 activation in osteoblasts directly promotes canonical Wnt signalling through its rapid phosphorylation of LRP6, which results in subsequent Axin recruitment and β-catenin stabilization [31, 32]. PTH also transcriptionally suppresses SOST gene expression in osteocytes to facilitate canonical signalling and perpetuate the pro-osteoblastogenic signal (Fig. 2) [33].
Although PTH and Wnt represent the two major anabolic pathways in the skeleton and promote bone formation, only Wnt inhibits resorption of bone. PTH induces osteoblastic production of RANKL to indirectly stimulate osteoclast activity [5].
The RANKL:OPG ratio ultimately determines the extent of osteoclastic differentiation and activation and is regulated differentially depending on the mode of PTH administration. In rats, continuous PTH infusion resulted in a sustained increase and decrease in RANKL and OPG mRNA, respectively, thus favouring a resorptive state, whereas intermittent PTH injections resulted in a transient elevation and decline in OPG and RANKL mRNA, respectively, with levels rapidly returning to baseline levels within 3 h of the injection [34].
Oestrogen
Effect on bone cells
Oestrogen is crucial for maintaining a normal ratio of osteoblasts and osteoclasts, and hormone deficiency following menopause is a significant risk factor for the development of osteoporosis in women. Oestrogen activates oestrogen receptor-alpha (ER-α) via 3 mechanisms, classic genomic signalling, oestrogen response element (ERE)-independent and non-genotropic signalling, in order to regulate bone remodelling [35].
Genomic action of oestrogen directly controls osteoclast lifespan and induces apoptosis of osteoclasts and pre-osteoclasts via Fas and Fas ligand (FasL) signalling. Gene transcription of FasL is regulated through the binding of ligand-bound ER-α dimers to EREs present on DNA. Because oestrogen is able to stimulate FasL upregulation in osteoclasts and osteoblasts, it exhibits both autocrine and paracrine behaviour, respectively. Tamoxifen and raloxifene, two selective oestrogen receptor modulators (SERMs), act via an osteoblast-dependent mechanism to attenuate osteoclastic-mediated resorption of trabecular bone [36, 37].
Ligand-activated ER-α can also function independently of ERE, by binding to alternative transcription factors and forestalling interaction with their response elements. The inhibition of interleukin-6 (IL-6) transcription, a cytokine that would otherwise contribute to bone loss in oestrogen deficiency states, occurs as a result of ER-α binding to P50 and P65 subunits of the NF-κB complex [35].
During growth, bone is deposited at the periosteal surface to increase cross sectional area and removed from the endocortical compartment to increase the size of the medullary cavity. Non-genomic signalling of oestrogen in osteoblast progenitors acts to retard endocortical resorption and preserve cortical bone mass [35, 38].
Effect on the immune system
Components involved in the regulation of the immune system can influence the production of osteoclastic factors. These factors have a number of effects and can influence bone resorption. For instance, IL-6, IL-1 and TNF-α have been shown to enhance RANKL and OPG expression [39]. Part of oestrogen’s action on bone is therefore due to its inhibitory effects on RANKL-inducing cytokines. Oestrogen attenuates production of the paracrine signals IL-1β and TNF-α from immune cells such as monocytes [40] and synthesis of IL-6 from marrow and osteoblastic cells [41]. IL-1 and TNF-α have also been shown to promote stromal cell M-CSF expression in ovariectomised mice [42]. Hence, a reduction in circulating oestrogen is linked to cytokine elevation and favours osteoclastic development. However, correlating circulating cytokine levels to osteoporosis in postmenopausal women has proved difficult for it is likely that cytokine production occurs within the local microenvironment of the BRC. Measuring systemic concentrations would reflect production by a whole host of different tissues [43].
Furthermore, immune cells such as B and T lymphocytes can directly produce RANKL and contribute to the bone loss associated with sex steroid deficiency. In one experiment it was found that T-cell deficient mice that had undergone ovariectomy were protected from bone loss, and a study comparing postmenopausal women to premenopausal controls found the former to have significantly increased RANKL production by T-cells [44–46].
ER-α signalling
Accumulating evidence suggests that ER-α signals independently of oestrogen, in order to transduce mechanical strain into pro survival cues in bone forming cells.
In osteoblast progenitor cells, ER-α has been shown to potentiate canonical Wnt signalling and accrual of cortical bone in response to mechanical stimulation in vitro [38]. Through this mechanism, ER-α signalling sensitizes osteoblastic cells to mechanical loading and encourages the transition of osteoblast progenitors (expressing Osterix-1 gene) to mature osteoblasts, whilst suppressing adipocytic differentiation [47]. Additionally, strain and not oestrogen instigates membrane localization of ER-α, where ER-α interacts with caveolin-1 resulting in mitogen-activated protein kinase (MAPK) activation and subsequent inhibition of osteoblast/osteocyte apoptosis [48, 49].
Interestingly, although the response of bone cells to strain and oestrogen both require ER-α, evidence gathered from rat osteosarcoma cells suggests that it is only the latter that regulates receptor cellular concentration. Therefore, down regulation of ER-α in the absence of oestrogen would impair the bone’s anabolic response to strain [48].
Pathophysiology of postmenopausal osteoporosis
For the reasons stated, declining oestrogen levels after menopause enhance the age-related changes in bone remodelling and predispose PMW to primary osteoporosis. Hormonal deficiency prolongs the survival of osteoclastic cells and favours adipocytic differentiation over osteoblastic, thereby shifting skeletal equilibrium in the direction of increased bone resorption. The average age of menopause is 51 years old and affects trabecular bone more than cortical bone, which is unsurprising given that trabecular bone has a far greater surface area [50].
Treatment of postmenopausal osteoporosis
Issues with bisphosphonates
Bisphosphonates are the first line treatment for osteoporosis and are approved for use in postmenopausal, glucocorticoid-induced and male osteoporosis. However, bisphosphonate treatment is not without its issues.
Firstly, bisphosphonates reduce non-vertebral and hip fractures by only 20–40 % whilst reducing vertebral fractures by approximately 60–70 % [51, 52]. Secondly, when administered orally (daily/weekly/monthly) the drugs predispose to esophageal irritation and gastrointestinal side effects and leads to discontinuation of the drug in up to 20 % of subjects. Intravenous (quarterly/yearly) bisphosphonate infusion is associated with an acute phase reaction which leads to transient flu-like symptoms in about 20–30 % of cases, although these symptoms tend to be more pronounced after the first dose. Third, given that drug clearance occurs via the kidney, and that high affinity of bisphosphonates for bone mineral results in prolonged skeletal retention and accumulative drug exposure, intravenous bisphosphonates are not advised in those with renal dysfunction (eGFR <35 ml/min/1.73 m2) [53]. And lastly, while uncommon long term treatment is linked to an increased incidence of atypical femoral fracture (AFF) and osteonecrosis of the jaw (ONJ) [54, 55].
In summary, the sole use of bisphosphonates as treatment for osteoporosis is imperfect and there is an important requirement for the development of newer drugs that reduce fractures with equivalent or greater efficacy to bisphosphonates, whilst at the same time free from their constraints. Treatment methods fall into two main categories, namely anti-resorptive and anabolic.
Anti resorptive versus anabolic therapy
Anti-resorptive drugs inhibit not only bone resorption but also bone formation indirectly since the two processes are tightly coupled. The inevitable decline in remodelling rate associated with these agents benefits bone strength by increasing mineral deposition/unit volume of bone tissue and, therefore, BMD, and by maintaining bone architecture, thus leading to a reduction in fracture risk. Oestrogen, SERMs, denosumab and bisphosphonates are all anti-resorptives.
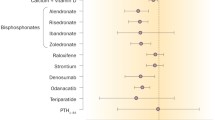
In contrast, anabolic agents enhance bone formation and as coupled to resorption increase the rate of remodelling, but in favour of formation, which increases the amount of bone laid down within each remodelling compartment. The result is an ongoing gain in bony tissue with subsequent enhancement of bone strength, and it is this that differentiates the effects of anabolic therapy (e.g. intermittent PTH) from other high remodelling disease states otherwise detrimental to bone health (e.g. oestrogen deficiency). Teriparatide is an anabolic agent approved for use in postmenopausal osteoporosis, male osteoporosis and glucocorticoid-induced osteoporosis [56]. Although true fracture reduction is the most important outcome, surrogate markers such as changes in turnover markers and BMD, which are more easily demonstrated, are sometimes used when evaluating new treatment options.
Anti-resorptive treatment
Oestrogen
Prior to bisphosphonates, oestrogen was commonly used to treat osteoporosis. Although findings published by the Women’s Health Initiative revealed that oestrogen (conjugated equine oestrogens, 0.625 mg/day, plus medroxyprogesterone acetate, 2.5 mg/day) was efficacious at improving BMD and preventing osteoporotic fractures, its use declined dramatically after the same study demonstrated an increase in the risk of breast cancer and cardiovascular events associated with use [57, 58].
Selective oestrogen receptor modulators (SERMs)
Indications and use
Non-steroidal SERMs have beneficial skeletal effects and are capable of inducing tissue-specific ER activity [59]. Therefore, they do not exhibit the same adverse effects on the breast, endometrium and heart as oestradiol; in fact raloxifene (60 mg orally/day), which is the only FDA approved SERM for treatment of postmenopausal osteoporosis, reduces the risk of breast cancer by 65 %, and its use is associated with a 30–50 % reduction in risk of vertebral fracture, although this may not be comparable to the 50 % fracture risk reduction achieved with bisphosphonates due to differences in population selection and severity of osteoporosis [60, 61].
Pivotal trials
The MORE trial, a multicenter, blinded, randomized, placebo-controlled study, enrolled 7705 PMW with osteoporosis who had spine or hip T scores of <−2.5, with or without existing fractures. Participants were randomized to receive raloxifene orally (60 mg/day or 120 mg/day) or placebo, but all received supplemental cholecalciferol and calcium. The reduction in risk of new vertebral fractures and percentage increase in BMD were evaluated at the end of a 36-month follow-up period. It was found that raloxifene induced a modest increase in BMD at both the spine and hip and significantly decreased the risk of new vertebral fractures (Fig. 3). Unfortunately, raloxifene failed to reduce the incidence of hip and other non-vertebral fractures, demonstrating it to be the least potent of the anti-resorptive agents (Table 1). Moreover, its effect on bone turnover and BMD was modest when compared to that of bisphosphonates and denosumab, which suggests that a greater degree of bone suppression must be achieved to decrease risk of non-vertebral fractures [62, 63]. Indeed, one study found Zoledronic acid to reduce urinary N-telopeptide of type 1 collagen (NTX) to a significantly greater extent than raloxifene (p < 0.001) at all time points (2, 4 and 6 months). Similar findings were obtained with serum bone-specific ALP in PMW with low bone mass [64].

New vertebral fracture reduction amongst osteoporotic PMW (n = 6,828), and percentage change in lumbar spine and femoral neck BMD after 36 months of Raloxifene. a The risk of new vertebral fractures was reduced by 55 % (60 mg/day) and 40 % (120 mg/day) in women with no prevalent baseline fractures and by 31 % (60 mg/day) and 49 % (120 mg/day) in women with prevalent baseline fractures compared to placebo, (p < 0.001 for all comparisons). b Compared to placebo, 60 mg raloxifene increased BMD by 2.6 and 2.1 % and 120 mg raloxifene increased BMD by 2.7 and 2.4 % at the spine and femoral neck, respectively (p < 0.001 for all comparisons). RR relative risk, CI confidence interval. Reproduced with permission from Ettinger et al. [62]
Head to head trials
A meta analysis of seven head to head randomized controlled trials comparing efficacy of raloxifene with ALN concluded that, despite ALN being more effective at increasing BMD, the efficacy of the drugs in preventing fractures; vertebral (p = 0.45) and non vertebral (p = 0.87) did not differ significantly at the end of the 2-year follow-up period [65].
Side effects and precautions
Raloxifene increases the risk of stroke in PMW at risk of coronary heart disease and increases venous thromboembolic events and hot flushes [66]. Other SERMs have been studied and abandoned for various reasons. Rather more recently, the agent lasofoxifene was investigated in a randomized control trial and was found to significantly reduce risk of vertebral and non-vertebral fractures, along with stroke, coronary heart disease and ER-positive breast cancer. But despite these encouraging findings, there was a 37 % rise in mortality when one of the two dosages was evaluated (0.25 mg/day); hence the future for lasofoxifene is unsure [67].
Denosumab
Indications and use
Denosumab is a human monoclonal antibody that opposes RANKL and inhibits osteoclast function and survival, to combat the excessive bone loss of osteoporosis. In May 2010 the European Commission afforded marketing authorization (Prolia, Amgen) for the treatment of postmenopausal women with osteoporosis who have elevated fracture risk, and this was quickly followed by approval from the FDA [68].
Pivotal trials
Although numerous clinical trials have been conducted with denosumab, the 3-year randomized placebo-controlled study, named the FREEDOM trial is the largest study to date. The FREEDOM trial enrolled 7,808 women between 60 and 90 years of age who had T scores ranging from −2.5 to −4.0 (assessed at the spine or total hip) and randomized them to receive either denosumab (60 mg biannually as a subcutaneous injection) or placebo. All participants received a daily minimum of 400 IU vitamin D and 1,000 mg calcium supplementation. When fracture reduction at 36 months was assessed, denosumab was found to significantly reduce the incidence of new vertebral fractures compared to placebo (with a cumulative incidence of 7.2 % placebo vs 2.3 % denosumab, a relative decrease of 68 %, p < 0.0001) and also non-vertebral (1.2 vs 0.7 %, 40 %, p = 0.04) and hip fractures (8.0 vs 6.5 %, 20 % p = 0.01) (Table 1) [69]. Fracture risk reduction occurred by the same order of magnitude as bisphosphonates and was irrespective of baseline fracture risk [53, 70]. Denosumab also increased BMD and decreased bone turnover (Fig. 4).

Percentage change in BMD (n = 441) and biochemical bone turnover markers (n = 160), compared to placebo. a, b After 36 months, participants assigned to denosumab experienced a relative increase of 9.2 % at the lumbar spine, and 6.0 % at the total hip, with respect to placebo. c, d Denosumab decreased serum C-telopeptide by 86 and 72 %, and P1NP by 18 and 76 %, at 1 and 36 months, respectively, compared to placebo. p < 0.001 for all comparisons between groups and at all time points. Reproduced with permission from Cummings et al. [69]
Of note, studies conducted with denosumab in women with breast cancer receiving aromatase inhibitor treatment and in men with prostatic cancer on androgen deprivation therapy, also demonstrated an increase in BMD and reduction in vertebral fractures [71, 72].
Head to head trials
Evidence suggests that denosumab is as efficacious as Zoledronate (ZOL), although this does not take into account the observed 28 % reduction in mortality seen following treatment with zoledronate after a hip fracture [73]. Zoledronate is the bisphosphonate with the greatest potency and is more potent than oral Alendronic acid (ALN), the first line treatment in osteoporosis. This is supported by findings from the DECIDE study, a phase III, double blind trial in which 1,189 PMW (with a T score of ≤−2.0 at the total hip or lumbar spine) were randomized to receive either denosumab (60 mg/6 months) or oral ALN (70 mg/week) over a 1-year period. Percentage change in BMD and bone turnover markers from baseline was assessed. There was a significantly larger increase in BMD at all skeletal sites at 12 months with denosumab compared to ALN, particularly at the total hip (3.5 vs 2.6 %, p < 0.0001). Treatment difference at the femoral neck was 0.6 %; trochanter, 1.0 %; lumbar spine, 1.1 % and 1/3 distal radius, 0.6 % (p ≤ 0.0002 at all sites). The same trial showed a significant reduction in bone turnover markers with denosumab compared to ALN, although bone quality, an important determinant of fracture risk, was not assessed [74].
The STAND trial explored the effect of denosumab on BMD in PMW transitioning from ALN treatment, thus simulating a potential clinical scenario. Total hip BMD increased by a further 1.90 % at 12 months when switched to denosumab compared to 1.05 % in those who remained on ALN, a statistically significant difference (p < 0.0001); lumbar spine BMD increased by 3.03 and 1.85 % (p < 0.0001), respectively. There was also a greater decrease in serum CTX in transitioning patients [75].
Denosumab was compared to Zoledronic acid in a study assessing skeletal related events (SRE) in patients with bony metastases from advanced cancer. A combined analysis of three pivotal phase III trials showed denosumab to be superior to ZOL in delaying the duration of the first SRE (median delay of 8 months). Incidence of ONJ was similar between groups (p = 0.13) [76].
Side effects and precautions
Denosumab can induce hypocalcaemia in susceptible individuals who have severe renal impairment or are receiving dialysis [77]. In addition, the FREEDOM trial found the incidence of eczema and cellulitis including erysipelas, to be significantly greater in denosumab-treated women compared to placebo (3 vs. 1.7 % and 0.3 vs. <0.1 %, respectively), although the risk of serious side effects such as cancer, infection and cardiovascular events remained consistent between groups [69]. The small increase in recurrent neoplasms observed with denosumab was deemed statistically insignificant; however, because of shared signalling between the immune and skeletal systems further scrutiny of the potential risks of therapy is warranted [78].
One case of ONJ was reported during the aftermath of the FREEDOM trial, and several cases have been noted since [79]. The mechanism is likely to be due to its potent anti-resorptive effect as ONJ has also been described as a rare complication of bisphosphonate therapy, occurring with an incidence of 1:100,000 to 1:10,000 in osteoporotic individuals [55]. To reduce the risk of ONJ, patients taking denosumab with additional risk factors (e.g. chemotherapy, glucocorticoids, dental diseases, radiation) should be aware of the importance of good dental hygiene. Of note, atypical femoral fracture is associated with denosumab use for similar reasons to ONJ.
Denosumab vs. bisphosphonates in clinical practice
Denosumab does not accumulate in the skeleton in the same way as bisphosphonates do since it targets RANKL. It is thus a potent suppressor of bone turnover and has a rapid onset of action. A dose-dependent inhibition of serum CTX can be observed as early as 3 days after administration. Furthermore, drug discontinuation swiftly restores CTX levels to values above baseline and in one study even exceeded those of the placebo group but normalized shortly after. It is not yet known if this rebound effect leads to an increased risk of fractures [80]. The reversible nature of denosumab action necessitates that a reminder system be put in place for patients approaching their next injection and that there be a follow up regime with an alternative anti-osteoporotic agent in the event of discontinuation.
Additional differences exist between denosumab and bisphosphonate therapy. Denosumab has a longer dosing interval compared to oral bisphosphonates and is administered biannually and subcutaneously. This coupled with the fact that it is relatively free from gastrointestinal side effects translates into improved drug adherence long term. Denosumab has a shorter skeletal retention time compared to bisphosphonates and is not limited to those patients with good renal function, since drug excretion does not occur via the kidneys [81].
Anabolic treatment
Teriparatide
Indications and use
Teriparatide is a recombinant form of human PTH (1–34 N-terminal fragment) and is FDA approved for use in PMW with osteoporosis, hypogonadal or primary osteoporosis in men and in both men and women suffering from glucocorticoid-induced osteoporosis (GIOP). Because of cost and inconvenience of daily injections, use is reserved for those with severe disease in UK or Europe. Selected patients should be at high risk of fracture, for example those with multiple risk factors, those with a previous fragility fracture and those who have experienced drug failure/intolerance. In patients with a T score of <−3 who have other risk factors for fracture, teriparatide can be used as first line treatment [82].
Teriparatide is given subcutaneously as a once daily 20 μg injection into the abdomen or thigh. Drug use was initially approved for 18 months but has since been extended to 2 years, as there is some concern about prolonged drug use and osteosarcoma risk.
Clinical trials with teriparatide
Teriparatide induces the largest increase in BMD to date when compared to any other osteoporosis therapy and reduces the risk of vertebral and non-vertebral fracture (excluding hip fracture) in PMW with previous vertebral fracture. A phase III trial showed a 65 % reduction in the risk of new radiographic vertebral fractures (9.7 % increase in lumbar spine BMD vs. 1.1 % placebo) and a 53 % reduction in non-vertebral fractures in patients treated with 20 μg teriparatide for 12 months compared to placebo. The same study also demonstrated that dosing with 40 μg/day was not superior to 20 μg/day in its effects (Table 1) [83].
PTH-treated patients exhibit the largest increase in BMD (10–14 %) at the lumbar spine (comprised of trabecular bone), a less marked increase (<5 %) at the femoral neck (mixed trabecular/cortical bone), with the measured BMD even falling somewhat (by 1–2 %) at the distal radius (cortical bone); however, the significance of the last finding is unknown [56].
Studies assessing the effect of teriparatide on glucocorticoid-induced osteoporosis (GIO) are in keeping with the trend whereby the greatest increase in BMD occurs in the lumbar region, followed by a less marked increase in the region of the hip. Furthermore, they show that teriparatide is able to induce an early rise in markers of bone formation, along with a slower increase in resorptive markers, reinforcing the concept of the ‘anabolic window’, in which there is an initial uncoupling of bone turnover in favour of bone formation. For instance, in the first 3 months of PTH (1–34) treatment, formation markers increased by 150 % and resorption markers by only 100 % [84, 85].
Head to head trials
A head to head trial comparing the effects of teriparatide (40 μg/day, greater than the approved dose) with that of ALN (10 mg/day) showed a significantly greater increase in BMD and markers of bone formation from baseline with teriparatide than with ALN after 14 months of treatment. Although this study also demonstrated the frequency of non-vertebral fractures to be significantly lower in those given teriparatide compared to the ALN-treated group (4 versus 14 %), some of these incidents may have been due to high-impact trauma [86].
Similar outcomes were obtained when teriparatide was compared to calcitonin and strontium ranelate, and in the latter study P1NP levels rose significantly with teriparatide at 1 month and continued to rise until 6 months [87, 88]. Another study found a positive significant correlation between bone turnover status at baseline and BMD changes, with P1NP being the most accurate predictor of lumbar BMD response at 18 months [89].
Pretreatment with anti-resorptive therapy
Pretreatment with antiresorptive drugs can impair the anabolic action of PTH and its ability to stimulate osteoblastic activity, due to a global reduction in bone remodelling. The scenario of sequential therapy arises when a patient continues to fracture and/or lose BMD despite being on antiresorptive therapy or else exhibits drug intolerance and is therefore switched to an anabolic agent.
There is accumulating evidence to suggest that long-term therapy with a bisphosphonate prior to initiating teriparatide blunts the effectiveness of PTH [90–92]. In one study it was found that the increase in lumbar spine BMD after 18 months of teriparatide (20 μg/day) was only 4.1 % in PMW who had been previously treated with ALN for 18–36 months. In contrast, patients pretreated with raloxifene, also an antiresorptive agent, resulted in an incremental increase of 10.2 % (ALN vs. raloxifene, p < 0.05), suggesting that this observation does not hold true for SERMs [92]. However, it is possible that the differences in incremental increase in BMD may be related, in part, to variations in disease severity which may have affected the selection of first line treatment agents.
Combination therapy with antiresorptive agents
Two randomized controlled trials have looked at the effects of combining teriparatide with other drugs for osteoporosis, with one study conducted in PMW treated with intact PTH (100 μg/day) [93] and the other in men treated with teriparatide (40 μg/day) [94]. Neither study found a synergy between teriparatide and ALN. Furthermore, there were no additive effects on BMD gains when intact PTH and ALN were combined, and ALN was even found to reduce BMD gains when compared to teriparatide alone. However, the effects of PTH alone failed to surpass combination therapy at the total hip (p = 0.08). These findings have important ramifications when planning optimal PTH treatment for severely osteoporotic patients, given they are likely to be receiving bisphosphonates before starting teriparatide. In contrast, combined treatment with teriparatide and denosumab led to a greater increase in spinal, femoral neck and hip BMD (9.1, 4.2, 4.9 %, respectively) compared to teriparatide (6.2 %, p = 0.014, 0.8 %, p < 0.007, 0.7 %, p < 0.001) or denosumab alone (5.5 %, p = 0.0005, 2.1 %, p = 0.0238, 2.5 %, p = 0.001) [95].
Interestingly, neither oestrogen nor SERMs (such as raloxifene) appear to blunt the effects of PTH, despite being anti resorptive in nature. When teriparatide was administered to PMW on oestrogen replacement, the response that occurred was consistent with that of previous trials (13 % increase spinal BMD, 2.7–4.4 % hip) [96, 97].
Antiresorptive therapy after PTH
Because BMD has a tendency to fall after the discontinuation of teriparatide, it has been suggested that anti-resorptives be used in the follow up period, to maintain the newly accrued bone and attenuate loss. In a follow-up study of the fracture prevention trial (FPT), participants who were treated with bisphosphonates for at least 24 months during the 30-month post-teriparatide treatment phase demonstrated additional increases in BMD [4.3 % increase in total hip BMD from FPT baseline in the former 20 μg group (p = 0.005 versus placebo) and a 6.8 % increase in the former 40 μg group (p < 0.001 versus placebo)]. In contrast, those who did not receive any anti-resorptive therapy during this time period experienced a reduction in BMD that was similar to placebo (p < 0.05) [98]. A comparable improvement in BMD occurred when teriparatide was followed by raloxifene [99, 100].
Side effects and precautions
The side effects of teriparatide are usually mild but can include weakness, muscle pain, nausea, headache and dizziness. Orthostatic hypotension may also occur, usually within the first 4 h of teriparatide injection. However, this can be avoided by advising patients to remain seated during initial administration, since orthostasis is typically limited to the first few doses [82].
Osteosarcoma
Two of the major trials [83, 86] with teriparatide were terminated early after it was found by a carcinogenicity study in rats that the drug could induce osteosarcoma [101]. However, to date there is no substantive clinical evidence to suggest that osteosarcoma is induced in states of high and/or prolonged PTH secretion, e.g. renal osteodystrophy. Moreover, no osteosarcomas were found during the pivotal teriparatide trial, questioning the relevance of the rat carcinogenicity findings [83]. Since the launch of teriparatide in 2002, Eli Lilly has identified one potential osteosarcoma case but the cause–effect relationship remains unproven [102]. Despite this, teriparatide is best avoided in patients with an elevated risk of osteosarcoma and history of cancer. This includes adolescents in whom the epiphyses have not yet closed, those with Paget’s disease or prior skeletal radiation, and patients with unexplained increases in ALP [82]. A number of countries recommend the use of teriparatide only after the menopause, although there is no reliable evidence to support this.
Novel therapies
Cathepsin K inhibitors
Given that the protease cathepsin K is central to enzymatic bone degradation, inhibitors of cathepsin K represent a novel therapeutic approach in the treatment of osteoporosis. Currently, the only agent under clinical evaluation is odanacatib, since it alone displays adequate affinity and specificity for cathepsin K (rather than cathepsins S, L and B). This is significant given that trials with less specific cathepsin K inhibitors have been halted following the discovery of skin reactions such as rashes and scleroderma-like thickening [103, 104].
A phase II trial conducted with odanacatib (50 mg/week) in 399 PMW with low BMD (T scores of between −2 and −3.5) found that after 24 months of oral therapy, BMD was increased by 5.7 % at the lumbar spine, 4.1 % at the total hip and 4.7 % at the femoral neck compared to placebo. Further to this, a dose-dependent decrease of resorption markers was noted, along with a transient and moderate decline in formation markers without suppression of bone formation rate. Cutaneous lesions resembling scleroderma were not observed and adverse outcomes were similar to that of placebo [105]. A large phase III trial (NCT00529373) has now been completed.
A second cathepsin K inhibitor called ONO-5334 was evaluated at varying doses as part of the phase II OCEAN study. Lumbar spine BMD (LSBMD) at 12 months was compared to baseline BMD in 265 PMW with low BMD. LSBMD increased significantly with ONO-5334 [3.7 ± 0.5 % (50 mg twice daily), 3.1 ± 0.48 % (100 mg once daily) and 5.1 ± 0.49 % (300 mg once daily)] compared to placebo (0.6 ± 0.48 %). Total hip and femoral neck BMD exhibited significant increases of 3.0 ± 0.36 % and 2.6 ± 0.44 %, respectively, with 300 mg ONO-5334. Further clinical trials are required to evaluate drug efficacy and safety long term [106].
The OCEAN study also showed a reduction in resorptive markers with ONO-5334 comparable to placebo, but no accompanying suppression of bone formation rate, hence providing a clue to the mechanism of action of cathepsin K inhibitors [106]. Since these agents interfere with the process of resorption rather than impairing osteoclast viability, signalling between osteoclasts and osteoblasts is preserved and bone formation unaffected. This uncoupling action of the drugs contrasts that of denosumab and bisphosphonates, two antiresorptives that function by reducing osteoclast differentiation and promoting apoptosis, respectively [81].
Inhibitors of Wnt antagonists
Sclerostin
The observation that SOST gene inactivation occurs in sclerosteosis and van Buchem, two rare diseases characterized by high bone mass, has provided the rationale for targeting sclerostin in osteoporosis. An antibody against sclerostin tested in a rat model of postmenopausal osteoporosis, increased BMD at all sites and prevented oestrogen deficiency-associated bone loss [107].
A human monoclonal sclerostin antibody, called AMG 785, which inhibits the binding of sclerostin to LRP5/6, has been developed and evaluated as part of a randomized, double-blind placebo controlled phase I trial. The study recruited 72 postmenopausal women and men and demonstrated a 5.3 % increase in BMD at the lumbar spine and 2.8 % at the total hip after 85 days in participants given a solitary subcutaneous dose of 10 mg/kg compared to placebo. Furthermore, bone formation markers (P1NP, osteocalcin, bone-specific ALP) increased whilst resorption markers (serum CTX) decreased, indicating an uncoupling action and large anabolic window, possibly due to cross talk with RANKL/OPG with a subsequent increase in OPG [108]. A phase II study [NCT00896532] is currently underway, in which the efficacy of the sclerostin antibody will be compared with ALN and teriparatide. Zoledronic acid, a potent bisphosphonate, leads to increases in sclerostin when given for post-menopausal osteoporosis [109]. This explains, in part, the suppressive effects of zoledronate on bone formation. It is, therefore, interesting to speculate whether the use of a sclerostin antibody with zoledronate may attenuate the negative effect of zoledronate on bone formation and, therefore, enhance its efficacy in fracture prevention.
Enhanced Wnt signalling has been linked to malignancies such as hepatocellular and colorectal cancer [110]. Indeed, 75 % of osteosarcomas were found to have a deficiency of Wnt inhibitory factor 1 (WIF), leading to increased Wnt signalling [111]. As a result phase III trials gauging the long-term safety of the antibody are awaited.
Dkk-1
Dkk-1 is also an inhibitor of Wnt signalling. The assessment of Dkk1 inhibitors has thus far been restricted to preclinical trials and has yet to be considered in the context of osteoporosis, although it has been shown to be effective at preventing bone loss in rheumatoid arthritis [112] and multiple myeloma [113]. However, the wide tissue expression of DKK1 may limit the use of DKK1 antibodies.
Conclusion
Recent discoveries in the field of bone cell biology have led to the development of novel therapeutic compounds for osteoporosis and fracture prevention which is the goal of all treatment. These new drugs coupled with existing therapies are increasing the range of non-bisphosphonate treatment options that will become available for patients. Although established antiresorptive drugs such as denosumab suppress bone remodelling via a coupling effect, newer drugs in development such as odanacatib exhibit an uncoupling effect, enabling greater bone formation. Sclerostin antibodies offer an exciting new anabolic treatment which until now is limited to teriparatide only. Teriparatide induces the greatest increase in BMD at the spine compared to anti-resorptive agents. It is anticipated that sclerostin antibodies will have a similar if not greater t anabolic profile as unlike teriparatide it does not increase bone resorption. As these drugs transition from preclinical evaluation to use in the clinical setting, patients will be able to receive increasingly individualized therapy, targeted to their specific clinical requirements.
Abbreviations
- BRC:
-
Bone remodelling compartment
- RANKL:
-
Receptor activator of nuclear factor kappa-B ligand
- OPG:
-
Osteoprotegerin
- BMU:
-
Basic multicellular unit
- VEGF:
-
Vascular endothelial growth factor
- M-CSF:
-
Macrophage colony stimulating factor
- LRP 5/6:
-
Low-density lipoprotein receptor-related protein 5 or 6
- GSK:
-
3beta-glycogen synthase kinase
- Tcf/Lef:
-
T cell factor/lymphoid enhancer factors
- RUNX2:
-
Runt-related transcription factor 2
- PMW:
-
Postmenopausal women
- PTHR1:
-
Parathyroid hormone receptor
- PKA:
-
Protein kinase A
- PKC:
-
Protein kinase C
- SOST:
-
Sclerostin gene
- ER-alpha:
-
Oestrogen receptor-alpha
- FasL:
-
Fas-ligand
- MAPK:
-
Mitogen-activated protein kinase
- AFF:
-
Atypical femoral fracture
- ONJ:
-
Osteonecrosis of the jaw
- SRE:
-
Skeletal-related events
- GIOP:
-
Glucocorticoid-induced osteoporosis
References
National Osteoporosis Society. Key facts and figures. http://www.nos.org.uk/page.aspx?pid=328. Updated 2013. Accessed 3 Dec 2013
Gelb BD, Shi GP, Chapman HA, Desnick RJ (1996) Pycnodysostosis, a lysosomal disease caused by cathepsin K deficiency. Science 273:1236–1238
Pederson L, Ruan M, Westendorf JJ, Khosla S, Oursler MJ (2008) Regulation of bone formation by osteoclasts involves Wnt/BMP signaling and the chemokine sphingosine-1-phosphate. Proc Natl Acad Sci USA 105:20764–20769
Walker EC, McGregor NE, Poulton IJ, Pompolo S, Allen EH, Quinn JM, Gillespie MT, Martin TJ, Sims NA (2008) Cardiotrophin-1 is an osteoclast-derived stimulus of bone formation required for normal bone remodeling. J Bone Miner Res 23:2025–2032
Baron R, Hesse E (2012) Update on bone anabolics in osteoporosis treatment: rationale, current status, and perspective. J Clin Endocrinol Metab 97:311–325
Eriksen EF (2010) Cellular mechanisms of bone remodeling. Rev Endocr Metab Disord 11:219–227
Eriksen EF, Qvesel D, Hauge EM, Melsen F (2005) Further evidence that vascular remodeling spaces are lined by cells of osteogenic origin: characterization of a possible coupling structure. J Bone Miner Res 15:S371
Frost HM (1969) Tetracycline-based histological analysis of bone remodeling. Calcif Tissue Res 3:211–237
Brandi ML, Collin-Osdoby P (2006) Vascular biology and the skeleton. J Bone Miner Res 21:183–192
Tombran-Tink J, Barnstable CJ (2004) Osteoblasts and osteoclasts express PEDF, VEGF-A isoforms, and VEGF receptors: possible mediators of angiogenesis and matrix remodeling in the bone. Biochem Biophys Res Commun 316:573–579
Mayr-Wohlfart U, Waltenberger J, Hausser H, Kessler S, Gunther KP, Dehio C, Puhl W, Brenner RE (2002) Vascular endothelial growth factor stimulates chemotactic migration of primary human osteoblasts. Bone 30:472–747
Street J, Lenehan B (2009) Vascular endothelial growth factor regulates osteoblast survival—evidence for an autocrine feedback mechanism. J Orthop Surg Res 4:1–13
Khosla S. The OPG/RANKL/RANK system. Endocrinol 2001;142:5050–5055
Humphrey MB, Lanier LL, Nakamura MC (2005) Role of ITAM-containing adapter proteins and their receptors in the immune system and bone. Immunol Rev 208:50–65
Crockett JC, Rogers MJ, Coxon FP, Hocking LJ, Helfrich MH (2011) Bone remodelling at a glance. J Cell Sci 124:991–998
Gambardella A, Nagaraju CK, O’Shea PJ, Mohanty ST, Kottam L, Pilling J, Sullivan M, Djerbi M, Koopmann W, Croucher PI, Bellantuono I (2011) Glycogen synthase kinase-3α/β inhibition promotes in vivo amplification of endogenous mesenchymal progenitors with osteogenic and adipogenic potential and their differentiation to the osteogenic lineage. J Bone Miner Res 26:811–821
Khosla S, Westendorf JJ, Oursler MJ (2008) Building bone to reverse osteoporosis and repair fractures. J Clin Invest 118:421–428
Otto F, Lubbert M, Stock M (2003) Upstream and downstream targets of RUNX proteins. J Cell Biochem 89:9–18
Lian JB, Stein GS, Javed A, van Wijnen AJ, Stein JL, Montecino M, Hassan MQ, Gaur T, Lengner CJ, Young DW (2006) Networks and hubs for the transcriptional control of osteoblastogenesis. Rev Endocr Metab Disord 7:1–16
Kawano Y, Kypta R (2003) Secreted antagonists of the Wnt signalling pathway. J Cell Sci 116:2627–2634
Li X, Zhang Y, Kang H, Liu W, Liu P, Zhang J, Harris SE, Wu D (2005) Sclerostin binds to LRP5/6 and antagonizes canonical Wnt signaling. J Biol Chem 280:19883–19887
Xiao-Juan X, Lin S, Yan-Ping Y, Rui Z, Bo S, Cheng-Gang L, Man-Xiang Wu (2013) Serum β-catenin levels associated with the ratio of RANKL/OPG in patients with postmenopausal osteoporosis. Int J Endocrinol 2013:534352
De Toni EN, Thieme SE, Herbst A, Behrens A, Stieber P, Jung A, Blum H, Göke B, Kolligs FT (2008) OPG is regulated by beta-catenin and mediates resistance to TRAIL-induced apoptosis in colon cancer. Clin Cancer Res 14:4713–4718
Wijenayaka AR, Kogawa M, Lim HP, Bonewald LF, Findlay DM, Atkins GJ (2011) Sclerostin stimulates osteocyte support of osteoclast activity by a RANKL-dependent pathway. PLoS One 6:e25900
Baron R, Rawadi G (2007) Targeting the Wnt/beta-catenin pathway to regulate bone formation in the adult skeleton. Endocrinology 148:2635–2643
Pleiner-Duxneuner J, Zwettler E, Paschalis E (2009) Treatment of osteoporosis with parathyroid hormone and teriparatide. Calcif Tissue Int 84:159–170
Zaidi M, Inzerillo AM, Moonga BS, Bevis PJ, Huang CL (2002) Forty years of calcitonin-where are we now? A tribute to the work of Iain Macintyre, FRS. Bone 30:655–663
Vescini F, Grimaldi F (2012) PTH 1-84: bone rebuilding as a target for the therapy of severe osteoporosis. Clin Cases Miner Bone Metab 9:31–36
Roschger P, Dempster DW, Zhou H, Paschalis EP, Silverberg SJ, Shane E, Bilezikian JP, Klaushofer K (2007) New observations on bone quality in mild primary hyperparathyroidism as determined by quantitative backscattered electron imaging. J Bone Miner Res 22:717–723
Qin L, Raggatt LJ, Partridge NC (2004) Parathyroid hormone: a double-edged sword for bone metabolism. Trends Endocrinol Metab 15:60–65
Jilka RL, O’Brien CA, Bartell SM, Weinstein RS, Manolagas SC (2010) Continuous elevation of PTH increases the number of osteoblasts via both osteoclast-dependent and independent mechanisms. J Bone Miner Res 25:2427–2437
Wan M, Yang C, Li J, Wu X, Yuan H, Ma H, He X, Nie S, Chang C, Cao X (2008) Parathyroid hormone signaling through low-density lipoprotein-related protein 6. Genes Dev 22:2968–2979
Keller H, Kneissel M (2005) SOST is a target gene for PTH in bone. Bone 37:148–158
Ma YL, Cain RL, Halladay DL, Yang X, Zeng Q, Miles RR, Chandrasekhar S, Martin TJ, Onyia JE (2001) Catabolic effects of continuous human PTH (1-38) in vivo is associated with sustained stimulation of RANKL and inhibition of osteoprotegerin and gene-associated bone formation. Endocrinology 142:4047–4054
Manolagas SC, O’Brien CA, Almeida M (2013) The role of estrogen and androgen receptors in bone health and disease. Nat Rev Endocrinol 9:699–712
Krum SA, Miranda-Carboni GA, Hauschka PV, Carroll JS, Lane TF, Freedman LP, Brown M (2008) Estrogen protects bone by inducing Fas ligand in osteoblasts to regulate osteoclast survival. EMBO J 27:535–545
Nakamura T, Imai Y, Matsumoto T, Sato S, Takeuchi K, Igarashi K, Harada Y, Azuma Y, Krust A, Yamamoto Y, Nishina H, Takeda S, Takayanagi H, Metzger D, Kanno J, Takaoka K, Martin TJ, Chambon P, Kato S (2007) Estrogen prevents bone loss via estrogen receptor alpha and induction of Fas ligand in osteoclasts. Cell 130:811–823
Almeida M, Iyer S, Martin-Millan M, Bartell SM, Han L, Ambrogini E, Onal M, Xiong J, Weinstein RS, Jilka RL, O’Brien CA, Manolagas SC (2013) Estrogen receptor-α signaling in osteoblast progenitors stimulates cortical bone accrual. J Clin Invest 123:394–404
Kwan Tat S, Padrines M, Theoleyre S, Heymann D, Fortun Y (2004) IL-6, RANKL, TNF-alpha/IL-1: interrelations in bone resorption pathophysiology. Cytokine Growth Factor Rev 15:49–60
Pacifici R (1999) Aging and cytokine production. Calcif Tissue Int 65:345–351
Girasole G, Jilka RL, Passeri G, Boswell S, Boder G, Williams DC, Manolagas SC (1992) 17 beta-estradiol inhibits interleukin-6 production by bone marrow-derived stromal cells and osteoblasts in vitro: a potential mechanism for the anti-osteoporotic effect of estrogens. J Clin Invest 89:883–891
Kimble RB, Srivastava S, Ross FP, Matayoshi A, Pacifici R (1996) Estrogen deficiency increases the ability of stromal cells to support murine osteoclastogenesis via an interleukin-1 and tumor necrosis factor-mediated stimulation of macrophage colony-stimulating factor production. J Biol Chem 271:28890–28897
Lerner UH (2006) Bone remodeling in post-menopausal osteoporosis. J Dent Res 85:584–595
Pacifici R (2012) Role of T cells in ovariectomy induced bone loss-revisited. J Bone Miner Res 27:231–239
Horowitz MC, Fretz JA, Lorenzo JA (2010) How B cells influence bone biology in health and disease. Bone 47:472–479
D’Amelio P, Grimaldi A, Di Bella S, Brianza SZ, Cristofaro MA, Tamone C, Giribaldi G, Ulliers D, Pescarmona GP, Isaia G (2008) Estrogen deficiency increases osteoclastogenesis up-regulating T cells activity: a key mechanism in osteoporosis. Bone 43:92–100
Rodda SJ, McMahon AP (2006) Distinct roles for Hedgehog and canonical Wnt signaling in specification, differentiation and maintenance of osteoblast progenitors. Development 133:3231–3244
Zaman G, Jessop HL, Muzylak M, De Souza RL, Pitsillides AA, Price JS, Lanyon LL (2006) Osteocytes use estrogen receptor alpha to respond to strain but their ERalpha content is regulated by estrogen. J Bone Miner Res 21:1297–1306
Aguirre JI, Plotkin LI, Gortazar AR, Millan MM, O’Brien CA, Manolagas SC, Bellido T (2007) A novel ligand-independent function of the estrogen receptor is essential for osteocyte and osteoblast mechanotransduction. J Biol Chem 282:25501–25508
Lindsay R, Cosman F (2012) Osteoporosis. In: Longo DL, Fauci AS, Kasper DL, Hauser SL, Jameson JL, Loscalzo J (eds) Harrison’s principles of internal medicine, vol 2, 18th edn. McGraw-Hill, New York, pp 3121–3124
Black DM, Thompson DE, Bauer DC, Ensrud K, Musliner T, Hochberg MC, Nevitt MC, Suryawanshi S, Cummings SR, Fracture Intervention Trial (2000) Fracture risk reduction with alendronate in women with osteoporosis: the Fracture Intervention Trial. FIT research group. J Clin Endocrinol Metab 85:4118–4124
Black DM, Delmas PD, Eastell R, Reid IR, Boonen S, Cauley JA, Cosman F, Lakatos P, Leung PC, Man Z, Mautalen C, Mesenbrink P, Hu H, Caminis J, Tong K, Rosario-Jansen T, Krasnow J, Hue TF, Sellmeyer D, Eriksen EF, Cummings SR (2007) Once-yearly Zoledronic acid for treatment of postmenopausal osteoporosis. N Engl J Med 356:1809–1822
Lippuner K (2012) The future of osteoporosis treatment—a research update. Swiss Med Wkly 142:w13624
Shane E, Burr D, Ebeling PR, Abrahamsen B, Adler RA, Brown TD, Cheung AM, Cosman F, Curtis JR, Dell R, Dempster D, Einhorn TA, Genant HK, Geusens P, Klaushofer K, Koval K, Lane JM, McKiernan F, McKinney R, Ng A, Nieves J, O’Keefe R, Papapoulos S, Sen HT, van der Meulen MC, Weinstein RS, Whyte M, American Society for Bone and Mineral Research (2010) Atypical subtrochanteric and diaphyseal femoral fractures: report of a task force of the American Society for Bone and Mineral Research. J Bone Miner Res 25:2267–2294
Khosla S, Burr D, Cauley J, Dempster DW, Ebeling PR, Felsenberg D, Gagel RF, Gilsanz V, Guise T, Koka S, McCauley LK, McGowan J, McKee MD, Mohla S, Pendrys DG, Raisz LG, Ruggiero SL, Shafer DM, Shum L, Silverman SL, Van Poznak CH, Watts N, Woo SB, Shane E, American Society of Bone and Mineral Research (2007) Bisphosphonate-associated osteonecrosis of the jaw: report of a task force of the American Society for Bone and Mineral Research. J Bone Miner Res 22:1479–1491
Hodsman AB, Bauer DC, Dempster DW, Dian L, Hanley DA, Harris ST, Kendler DL, McClung MR, Miller PD, Olszynski WP, Orwoll E, Yuen CK (2005) Parathyroid hormone and teriparatide for the treatment of osteoporosis: a review of the evidence and suggested guidelines for its use. Endocr Rev 26:688–703
Rossouw JE, Anderson GL, Prentice RL, LaCroix AZ, Kooperberg C, Stefanick ML, Jackson RD, Beresford SA, Howard BV, Johnson KC, Kotchen JM, Ockene J, Writing Group for the Women’s Health Initiative Investigators (2002) Risks and benefits of estrogen plus progestin in healthy postmenopausal women: principal results from the Women’s Health Initiative randomized controlled trial. JAMA 288:321–333
Cauley JA, Robbins J, Chen Z, Cummings SR, Jackson RD, LaCroix AZ, LeBoff M, Lewis CE, McGowan J, Neuner J, Pettinger M, Stefanick ML, Wactawski-Wende J, Watts NB, Women’s Health Initiative Investigators (2003) Effects of estrogen plus progestin on risk of fracture and bone mineral density: the women’s health initiative randomized trial. JAMA 290:1729–1738
McDonnell DP (2003) Mining the complexities of the estrogen signaling pathways for novel therapeutics. Endocrinology 144:4237–4240
Dickler MN, Norton L (2001) The MORE trial: multiple outcomes for raloxifene evaluation. Breast cancer as a secondary end point: implications for prevention. Ann N Y Acad Sci 949:134–142
Seeman E, Crans GG, Diez-Perez A, Pinette KV, Delmas PD (2006) Anti vertebral fracture efficacy of raloxifene: a meta-analysis. Osteoporos Int 17:313–316
Ettinger B, Black DM, Mitlak BH, Knickerbocker RK, Nickelsen T, Genant HK, Christiansen C, Delmas PD, Zanchetta JR, Stakkestad J, Gluer CC, Krueger K, Cohen FJ, Eckert S, Ensrud KE, Avioli LV, Lips P, Cummings SR, for the Multiple Outcomes of Raloxifene Evaluation (MORE) Investigators (1999) Reduction of vertebral fracture risk in postmenopausal women with osteoporosis treated with raloxifene. Results from a 3-year randomized clinical trial. JAMA 282:637–645
Agnusdei D, Iori N (2001) Raloxifene: results from the MORE study. J Musculoskelet Neuron Interact 1:127–132
Bachmann G, Kriegman A, Goncalves J, Kianifard F, Warren M, Simon JA (2011) Effect of Zoledronic acid compared with raloxifene on bone turnover markers in postmenopausal women with low bone density. Menopause 18:851–856
Lin T, Yan SG, Cai XZ, Ying ZM, Yuan FZ, Zuo X (2014) Alendronate versus raloxifene for postmenopausal women: a meta-analysis of seven head to head randomized controlled trials. Int J Endocrinol 2014:1–16. Article ID 796510. doi:10.1155/2014/796510
Barrett-Connor E, Mosca L, Collins P, Geiger MJ, Grady D, Kornitzer M, McNabb MA, Wenger NK, Raloxifene Use for The Heart (RUTH) Trial Investigators (2006) Effects of raloxifene on cardiovascular events and breast cancer in postmenopausal women. N Engl J Med 355:125–137
Cummings SR, Ensrud K, Delmas PD, LaCroix AZ, Vukicevic S, Reid DM, Goldstein S, Sriram U, Lee A, Thompson J, Armstrong RA, Thompson DD, Powles T, Zanchetta J, Kendler D, Neven P, Eastell R, PEARL Study Investigators (2010) Lasofoxifene in postmenopausal women with osteoporosis. N Engl J Med 362:686–696
Rizzoli R, Yasothan U, Kirkpatrick P (2010) Denosumab. Nat Rev Drug Discov 9:591–592
Cummings SR, San Martin J, McClung MR, Siris ES, Eastell R, Reid IR, Delmas P, Zoog HB, Austin M, Wang A, Kutilek S, Adami S, Zanchetta J, Libanati C, Siddhanti S, Christiansen C, FREEDOM Trial (2009) Denosumab for prevention of fractures in postmenopausal women with osteoporosis. N Engl J Med 361:756–765
Boonen S, Adachi JD, Man Z, Cummings SR, Lippuner K, Torring O, Gallagher JC, Farrerons J, Wang A, Franchimont N, San Martin J, Grauer A, McClung M (2011) Treatment with denosumab reduces the incidence of new vertebral and hip fractures in postmenopausal women at high risk. J Clin Endocrinol Metab 96:1727–1736
Ellis GK, Bone HG, Chlebowski R, Paul D, Spadafora S, Smith J, Fan M, Jun S (2008) Randomized trial of denosumab in patients receiving adjuvant aromatase inhibitors for nonmetastatic breast cancer. J Clin Oncol 26:4875–4882
Smith MR, Egerdie B, Hernández Toriz N, Feldman R, Tammela TL, Saad F, Heracek J, Szwedowski M, Ke C, Kupic A, Leder BZ, Goessl C, Denosumab HALT Prostate Cancer Study Group (2009) Denosumab in men receiving androgen-deprivation therapy for prostate cancer. N Engl J Med 361:745–755
Lyles KW, Colon-Emeric CS, Magaziner IS et al (2007) Zoledronic acid and clinical fractures and mortality after hip fracture. N Engl J Med 18:1799–1809
Brown JP, Prince RL, Deal C, Recker RR, Kiel DP, de Gregorio LH, Hadji P, Hofbauer LC, Alvaro-Gracia JM, Wang H, Austin M, Wagman RB, Newmark R, Libanati C, San Martin J, Bone HG (2009) Comparison of the effect of denosumab and alendronate on BMD and biochemical markers of bone turnover in postmenopausal women with low bone mass: a randomized, blinded, phase 3 trial. J Bone Miner Res 24:153–161
Kendler DL, Roux C, Benhamou CL, Brown JP, Lillestol M, Siddhanti S, Man HS, San Martin J, Bone HG (2010) Effects of denosumab on bone mineral density and bone turnover in postmenopausal women transitioning from alendronate therapy. J Bone Miner Res 25:72–81
Lipton A, Fizazi K, Stopeck AT, Henry DH, Brown JE, Yardley DA, Richardson GE, Siena S, Maroto P, Clemens M, Bilynskyy B, Charu V, Beuzeboc P, Rader M, Viniegra M, Saad F, Ke C, Braun A, Jun S (2012) Superiority of denosumab to Zoledronic acid for prevention of skeletal-related events: a combined analysis of 3 pivotal, randomized, phase 3 trials. Eur J Cancer 48:3082–3092
Prescribing information of XGEVA (denosumab) approved by the FDA. Last revision 06/2013. Available under http://www.accessdata.fda.gov/drugsatfda_docs/label/2013/125320s094lbl.pdf
McClung MR, Lewiecki EM, Cohen SB, Bolognese MA, Woodson GC, Moffett AH, Peacock M, Miller PD, Lederman SN, Chesnut CH, Lain D, Kivitz AJ, Holloway DL, Zhang C, Peterson MC, Bekker PJ, AMG 162 Bone Loss Study Group (2006) Denosumab in postmenopausal women with low bone mineral density. N Engl J Med 354:821–831
Aghaloo TL, Felsenfeld AL, Tetradis S (2010) Osteonecrosis of the jaw in a patient on Denosumab. J Oral Maxillofac Surg 68:959–963
Miller PD, Bolognese MA, Lewiecki EM, McClung MR, Ding B, Austin M, Liu Y, San Martin J, Amg Bone Loss Study Group (2008) Effect of denosumab on bone density and turnover in postmenopausal women with low bone mass after long-term continued, discontinued, and restarting of therapy: a randomized blinded phase 2 clinical trial. Bone 43:222–229
Rachner TD, Khosla S, Hofbauer LC (2011) New horizons in osteoporosis. Lancet 377:1276–1287
Sikon A, Batur P (2010) Profile of teriparatide in the management of postmenopausal osteoporosis. Int J Womens Health 2:37–44
Neer RM, Arnaud CD, Zanchetta JR, Prince R, Gaich GA, Reginster JY, Hodsman AB, Eriksen EF, Ish-Shalom S, Genant HK, Wang O, Mellstrom D, Oefjord ES, Marcinowska-Suchowierska E, Salmi J, Mulder H, Halse J, Sawicki AZ, Mitlak BH (2001) Effect of parathyroid hormone (1-34) on fractures and bone mineral density in postmenopausal women with osteoporosis. N Engl J Med 344:1434–1441
Lane NE, Sanchez S, Modin GW, Genant HK, Pierini E, Arnaud CD (1998) Parathyroid hormone treatment can reverse corticosteroid-induced osteoporosis. Results of a randomized controlled clinical trial. J Clin Invest 102:1627–1633
Rehman Q, Lang TF, Arnaud CD, Modin GW, Lane NE (2003) Daily treatment with parathyroid hormone is associated with an increase in vertebral cross-sectional area in postmenopausal women with glucocorticoid-induced osteoporosis. Osteoporos Int 14:77–81
Body JJ, Gaich GA, Scheele WH, Kulkarni PM, Miller PD, Peretz A, Dore RK, Correa-Rotter R, Papaioannou A, Cumming DC, Hodsman AB (2002) A randomized double-blind trial to compare the efficacy of teriparatide [recombinant human parathyroid hormone (1-34)] with alendronate in postmenopausal women with osteoporosis. J Clin Endocrinol Metab 87:4528–4535
Hwang JS, Tu ST, Yang TS, Chen JF, Wang CJ, Tsai KS (2006) Teriparatide vs. calcitonin in the treatment of Asian postmenopausal women with established osteoporosis. Osteoporos Int 17:373–378
Recker RR, Marin F, Ish-Shalom S, Moricke R, Hawkins F, Kapetanos G, de la Peña MP, Kekow J, Farrerons J, Sanz B, Oertel H, Stepan J (2009) Comparative effects of teriparatide and strontium ranelate on bone biopsies and biochemical markers of bone turnover in postmenopausal women with osteoporosis. J Bone Miner Res 24:1358–1368
Chen P, Satterwhite JH, Licata AA, Lewiecki EM, Sipos AA, Misurski DM, Wagman RB (2005) Early changes in biochemical markers of bone formation predict BMD response to teriparatide in postmenopausal women with osteoporosis. J Bone Miner Res 20:962–970
Boonen S, Marin F, Obermayer-Pietsch B, Simões ME, Barker C, Glass EV, Hadji P, Lyritis G, Oertel H, Nickelsen T, McCloskey EV, EUROFORS Investigators (2008) Effects of previous antiresorptive therapy on the bone mineral density response to two years of teriparatide treatment in 30 postmenopausal women with osteoporosis. J Clin Endocrinol Metab 93:852–860
Ma YL, Bryant HU, Zeng Q, Schmidt A, Hoover J, Cole HW, Yao W, Jee WS, Sato M (2003) New bone formation with teriparatide [human PTH (1-34)] is not retarded by long term pretreatment with alendronate, estrogen or raloxifene in ovariectomized rats. Endocrinology 144:2008–2015
Ettinger B, San Martin J, Crans G, Pavo I (2004) Differential effects of teriparatide on BMD after treatment with raloxifene or alendronate. J Bone Miner Res 19:745–751
Black DM, Greenspan SL, Ensrud KE, Palermo L, McGowan JA, Lang TF, Garnero P, Bouxsein ML, Bilezikian JP, Rosen CJ, PaTH Study Investigators (2003) The effects of parathyroid hormone, and alendronate alone or in combination in postmenopausal osteoporosis. N Engl J Med 349:1207–1215
Finkelstein JS, Hayes A, Hunzelman JL, Wyland JJ, Lee H, Neer RM (2003) The effects of parathyroid hormone, alendronate or both in men with osteoporosis. N Engl J Med 349:1216–1226
Tsai JN, Uihlein AV, Lee H, Kumbhani R, Siwila-Sackman R, Mc Kay EA, Burnett-Bowie SA, Neer RM, Leder BZ (2013) Teriparatide and denosumab, alone or combined, in women with postmenopausal osteoporosis: the DATA study randomized trial. Lancet 382(9886):50–56
Lindsay R, Nieves J, Formica C, Henneman E, Woelfert L, Shen V, Dempster D, Cosman F (1997) Randomised controlled study of effect of parathyroid hormone on vertebral-bone mass and fracture incidence among postmenopausal women on oestrogen with osteoporosis. Lancet 350:550–555
Cosman F, Nieves J, Woelfert L, Formica C, Gordon S, Shen V, Lindsay R (2001) Parathyroid hormone added to established hormone therapy: effects on vertebral fracture and maintenance of bone mass after parathyroid hormone withdrawal. J Bone Miner Res 16:925–931
Prince R, Sipos A, Hossain A, Syversen U, Ish-Shalom S, Marcinowska E, Halse J, Lindsay R, Dalsky GP, Mitlak BH (2005) Sustained nonvertebral fragility fracture risk reduction after discontinuation of teriparatide treatment. J Bone Miner Res 20:1507–1513
Adami S, San Martin J, Muñoz-Torres M, Econs MJ, Xie L, Dalsky GP, McClung M, Felsenberg D, Brown JP, Brandi ML, A Sipos (2008) Effect of raloxifene after recombinant teriparatide [hPTH (1-34)] treatment in post-menopausal women with osteoporosis. Osteoporos Int 19:87–94
Minne H, Audran M, Simões ME, Obermayer-Pietsch B, Sigurðsson G, Marín F, Dalsky GP, Nickelsen T, EUROFORS Study Group (2008) Bone density after teriparatide in patients with or without prior antiresorptive treatment: one-year results from EUROFORS study. Curr Med Res Opin 24:3117–3128
Vahle JL, Sato M, Long GG, Young JK, Francis PC, Engelhardt JA, Westmore MS, Linda Y, Nold JB (2002) Skeletal changes in rats given daily subcutaneous injections of recombinant human parathyroid hormone(1-34) for two years and relevance to human safety. Toxicol Pathol 30:312–321
Harper KD, Krege JH, Marcus R, Mitlak BH (2007) Osteosarcoma and teriparatide? J Bone Miner Res 22:334
Gauthier JY, Chauret N, Cromlish W, Desmarais S, le Duong T, Falgueyret JP, Kimmel DB, Lamontagne S, Léger S, LeRiche T, Li CS, Massé F, McKay DJ, Nicoll-Griffith DA, Oballa RM, Palmer JT, Percival MD, Riendeau D, Robichaud J, Rodan GA, Rodan SB, Seto C, Thérien M, Truong VL, Venuti MC, Wesolowski G, Young RN, Zamboni R, Black WC (2008) The discovery of odanacatib (MK-0822), a selective inhibitor of cathepsin K. Bioorg Med Chem Lett 18:923–928
Peroni A, Zini A, Braga V, Colato C, Adami S, Girolomoni G (2008) Drug-induced morphea: report of a case induced by balicatib and review of the literature. J Am Acad Dermatol 59:125–129
Bone HG, McClung MR, Roux C, Recker RR, Eisman JA, Verbruggen N, Hustad CM, DaSilva C, Santora AC, Ince BA (2010) Odanacatib, a cathepsin K inhibitor for osteoporosis: a two year study in postmenopausal women with low bone density. J Bone Miner Res 25:937–947
Eastell R, Nagase S, Ohyama M, Small M, Sawyer J, Boonen S, Spector T, Kuwayama T, Deacon S (2011) Safety and efficacy of cathepsin K inhibitor ONO-5334 in postmenopausal osteoporosis: the OCEAN study. J Bone Miner Res 26:1303–1312
Li X, Ominsky MS, Warmington KS, Morony S, Gong J, Cao J, Gao Y, Shalhoub V, Tipton B, Haldankar R, Chen Q, Winters A, Boone T, Geng Z, Niu QT, Ke HZ, Kostenuik PJ, Simonet WS, Lacey DL, Paszty C (2009) Sclerostin antibody treatment increases bone formation, bone mass, and bone strength in a rat model of postmenopausal osteoporosis. J Bone Miner Res 24:578–588
Padhi D, Jang G, Stouch B, Fang L, Posvar E (2011) Single dose placebo controlled randomized study of AMG 785, a sclerostin monoclonal antibody. J Bone Miner Res 26:19–26
Anastasilakis AD, Polyzos SA, Gkiomisi A, Bisbinas I, Gerou S, Makras P (2013) Comparative effect of zoledronic acid versus denosumab on serum sclerostin and dickkopf-1 levels of naïve postmenopausal women with low bone mass :a randomized, head-to-head clinical trial. J Clin Endocrinol Metab 98(8):3206–3212
Giles RH, van Es JH, Clevers H (2003) Caught up in a Wnt storm: Wnt signaling in cancer. Biochim Biophys Acta 1653:1–24
Kansara M, Tsang M, Kodjabachian L, Sims NA, Trivett MK, Ehrich M, Dobrovic A, Slavin J, Choong PF, Simmons PJ, Dawid IB, Thomas DM (2009) Wnt inhibitory factor 1 is epigenetically silenced in human osteosarcoma, and targeted disruption accelerates osteosarcomagenesis in mice. J Clin Invest 119:837–851
Diarra D, Stolina M, Polzer K, Zwerina J, Ominsky MS, Dwyer D, Korb A, Smolen J, Hoffmann M, Scheinecker C, van der Heide D, Landewe R, Lacey D, Richards WG, Schett G (2007) Dickkopf-1 is a master regulator of joint remodeling. Nat Med 13:156–163
Heath DJ, Chantry AD, Buckle CH, Coulton L, Shaughnessy JD Jr, Evans HR, Snowden JA, Stover DR, Vanderkerken K, Croucher PI (2009) Inhibiting Dickkopf-1 (Dkk1) removes suppression of bone formation and prevents the development of osteolytic bone disease in multiple myeloma. J Bone Miner Res 24:425–436
Seaman E, Vellas B, Benhamou C, Aquino JP et al (2006) Strontium ranelate reduces the risk of vertebral and nonvertebral fractures in women eighty years of age and older. J Bone Miner Res 21:1113–1120
Hampson G, Fogelman I (2012) Clinical role of bisphosphonates. Int J Women Health 4:455–469
Conflict of interest
The authors declare no conflict of interests.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article

Cite this article
Ishtiaq, S., Fogelman, I. & Hampson, G. Treatment of post-menopausal osteoporosis: beyond bisphosphonates. J Endocrinol Invest 38, 13–29 (2015). https://doi.org/10.1007/s40618-014-0152-z
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s40618-014-0152-z