
Abstract
There is increasing interest in metformin’s effects on the development, treatment, and/or progression of breast cancer. This emerges from observational studies that diabetic women treated with metformin in comparison to other antidiabetic compounds had lower breast cancer incidence and/or mortality rates. The mechanism of action is considered to be activation of hepatic AMPK resulting in reduced gluconeogenesis. Calorie restriction, which consistently reduces mammary tumorigenesis in rodents, is also thought to act through this pathway leading to the hypothesis that metformin’s anticancer effects are mediated in a similar fashion. Here, we review the literature evaluating metformin’s anticancer effects in relation to breast/mammary tumorigenesis. We include clinical observations, as well as studies utilizing rodent models and mammary cell lines. In addition to the anticancer effect of metformin mediated through the AMPK pathway, additional mechanisms of action that directly target tissues have been identified including effects on stem cells, apoptosis, STAT3, and HER2.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Identifying compounds with chemopreventive and adjuvant actions to protect against breast cancer development and recurrence is an active area of research. However, only a few compounds such as tamoxifen, a selective estrogen receptor modulator, and the aromatase inhibitors have been taken into the clinical arena. One limitation of inhibitors that target the functioning of the estrogen receptor has been lack of enduring efficacy in the adjuvant setting, illustrated by recent demonstration of the ATLAS and aTTom trials that 10 years of tamoxifen is better than 5 years in terms of distant disease-free survival and overall survival, and the benefit of 10 compared to 5 years of tamoxifen is realized in years 10 to 15. These trial results suggest that tamoxifen alone may be insufficient adjuvant therapy, particularly in pre-menopausal patients. Not only do we need to start thinking about a longer time horizon for breast cancer chemoprevention, we also need to be thinking about more effective prevention and adjuvant strategies. This unmet need has led to interest in repurposing the diabetes drug metformin for potential roles in breast cancer treatment, adjuvant therapy and long-term prevention. This interest has stemmed from epidemiological studies that support an anticancer role for metformin in breast cancer and other solid tumor malignancies [1–3] and the observation that metformin exhibits low toxicity and can be given to non-diabetic patients without inducing clinical hypoglycemia [4]. Metformin has been in the forefront of approved drugs that could be repurposed for breast cancer therapeutics as a result of reports that metformin use in type 2 diabetic patients is associated with reduced overall cancer incidence and/or death rates in comparison to other treatments [1, 2]. Several recent original research as well as meta-analyses/review articles present additional support and discussion of the overall anticancer effects of metformin in diabetic subjects [5–8] although not all data are consistent with an anticancer effect [9]. Here, we will focus on reviewing the potential for metformin to specifically be used to prevent breast cancer in humans and in experimental rodent studies.
Human Studies
Epidemiological evidence of an association between metformin use and reduced cancer mortality including breast cancer mortality was first published almost 10 years ago [1, 2]. It was also reported that diabetic women who were new metformin users had a significant decrease in cancer diagnosis when followed for up to 10 years, including a 40–50 % reduction in breast cancer diagnosis [3]. An additional study reported that diabetic women treated with metformin who were diagnosed with breast cancer had a better pathologic complete response rate (pCR) to neoadjuvant therapy than did those using other diabetic treatments [10].
Several other epidemiological studies have supported a protective effect of metformin in diabetic women with breast cancer. For example, Taiwanese women who were followed after being diagnosed with type 2 diabetes had a reduced incidence of breast cancer incidence if they were metformin users and there appeared to be a relationship between dose of metformin and effect [11]. In another study from Turkey, newly diagnosed breast cancer patients (average age of 57) taking metformin and matched to women not taking metformin [12] had a significantly lower incidence of stage 3 tumors and triple negative tumors and higher incidence of ER+/PR+ tumors. Of note, these observational studies have all been done in diabetic women, leaving open the question of whether metformin only has an effect in the presence of diabetes. In contrast, other studies have reported that the choice of glucose control agent has no influence on cancer development [13, 14].
Several short-term intervention studies of the effects of metformin on breast tumor cell proliferation have recently been published. Niraula et al. [15] treated newly diagnosed non-diabetic breast cancer patients with 500 mg of metformin three times daily between diagnostic biopsies and breast surgery (median ∼18 days). Tumor Ki67 labeling index, the primary endpoint, was significantly decreased from 36.5 to 33.5 % following metformin treatment. In contrast, Bonanni et al. [16] found that treatment with 850 mg metformin twice daily for 4 weeks between biopsy and surgery in newly diagnosed nondiabetic women did not significantly affect tumor Ki67 in comparison to a placebo group. There was a non-significant mean proportional decrease in Ki-67 of 10.5 % in women with a HOMA (homeostasis assessment model) of >2.8 and a non-significant increase in women with a HOMA of <2.8, suggesting that particular attention must be paid to the study population when investigating metformin effects in a window of opportunity study. A third study by Hadad et al. [17] which had a control arm randomized in a blinded fashion to metformin 1 g twice daily vs. no drug showed significant reductions of the Ki67 LI in two cohorts of patients, a pilot cohort (P = 0.041) and the metformin arm (0.027), whereas there was no reduction in the Ki67 LI in the control group. Perhaps some of the other ongoing intervention trials will clarify these discrepancies (http://clinicaltrials.gov/ct2/show/NCT00897884?term=breast+cancer+AND+metformin).
Implementation of interventions to prevent breast cancer is feasible due to the fact that women at risk can be identified in a number of different ways. This includes calculating the risk of developing breast cancer using the Gail Model (and the factors included in it such as age, family history, previous breast biopsies) [18]. Additional factors such as higher breast density [19] and overweight/obesity [20] have been reported to increase breast cancer risk. Given the number of overweight/obese women in the USA and worldwide, this potentially provides a large number of at risk women who could be identified and targeted in future prevention studies particularly if other risk factors are also identified.
In Vitro Studies
There are numerous publications presenting data on metformin’s effects on the growth of different human breast cancer cell lines which focused on cell proliferation as well as AMPK-associated proteins and apoptosis. A summary of these findings is presented in Table 1.
With respect to proliferation, the ER+ MCF-7 cell line has been consistently reported to respond to the addition of metformin with reduced proliferation as well as increased apoptosis [21, 23, 25–27, 29]. Other breast cancer cell lines with varying hormonal receptor and HER2 status, i.e., MCF-7-HER2, SKBR3, BT20, T47D, MDA-MB-453, BT549, BT-474, and MB-468 were also reported to exhibit reduced proliferation in response to metformin treatment [23–27, 29, 32, 33••, 34]. In contrast, studies using the triple-negative MDA-MB-231 cells have not reported consistent findings, with one study reporting no effect of metformin on cell number by direct cell counting [23], while in three other studies, reduced proliferation was reported when dye assessment methods were used [24, 27, 29]. One of these publications reported effects on triple negative breast cancer cell lines (MDA-MB-468, MDA-MB-231, BT20, and BT549) with no effect reported on the other cell lines tested (MCF-7, BT474, SKBR3, and MDA-MB-453) [24].
A number of these above citations also presented data on effects of metformin treatment on pAMPK activity as well as other proteins in this pathway. In most cases, pAMPK activity was increased while mTOR-associated factors were decreased [21–23, 25, 26, 33••, 34]. Effects of metformin on apoptosis in breast cancer cell lines have also been reported whereby in most cases, enhanced cell death has been found [23, 24, 32].
A recently published study indicated that media glucose concentrations enhanced metformin’s effects on cell death in HeLa, MCF-7, and MDA-MB-231 cell lines [35]. This suggests that cell culture conditions may be an important factor to consider when evaluating metformin’s in vitro actions. In summary, although there appears to be discrepancies with respect to the responses of specific human breast cancer cell lines, in general, metformin appears to have an impact on human breast cancer cell proliferation. However, in most cases, high concentrations of metformin were used so it is difficult to assess the application of these findings to human therapeutics. Additional aspects of in vitro studies are also presented in the “Mechanisms of Action” section.
Rodent Studies
To obtain a better idea of the potential effects metformin might have on either tumor development and/or progression numerous preclinical animal model studies have been conducted. This has included xenograft experiments primarily using human breast cancer cell lines examining the effects of metformin treatment on tumor progression, as well as studies determining the effects on mammary tumor development in carcinogen-induced and transgenic models. Summaries of these studies are presented in Tables 2 and 3, respectively. In general, metformin appears to have demonstrated effects in ER- xenograft experiments and HER2 positive transgenic mammary carcinoma models as will now be described.
Several xenograft studies have assessed the effect of metformin treatment on growth of the triple negative MDA-MB-231 human breast cancer cell line in immunocompromised mice. For example, Cheong et al. [39] implanted MDA-MB-231 cells into the mammary gland fat pad of 4-week-old female CD-1 nu/nu mice and initiated metformin treatment 14 days later (tumor size 50 mm3) at a dose of 250 mg/kg body weight by daily intraperitoneal injection with additional study groups included: control, 2-deoxyglucose (500 mg/kg) or metformin plus 2-deoxyglucose. Following 36 days of treatment, there was no effect of either metformin or 2-deoxyglucose alone, but the combined treatment reduced tumor growth by half. In another study, 5-week-old nude mice were injected subcutaneously with MDA-MB-231 cells, and metformin treatment (2000 μg/ml in drinking water equal to 200 mg/kg body weight) was initiated 8 days later. Metformin treatment significantly reduced tumor growth and Ki67 staining [24]. In the second part of this experiment, metformin treatment was initiated 7 days prior to cell inoculation resulting in an extension of the time until tumor palpability from 10 days in control mice to 20 days in metformin treated mice. Tumor incidence was also significantly decreased from 100 % in control mice to 50 % in metformin-treated mice.
Several studies have been published using additional breast cancer cell lines. Iliopoulos et al. [38•] injected MDA-MB-231, ER-Src (estrogen receptor regulated Src), or BT-474 breast cancer cell lines into the right flank of female nu/nu mice and treated the mice with metformin (200 ug/mL in water) beginning 10 days later. Additional groups received doxorubicin (4 mg/kg), combined doxorubicin and metformin or vehicle. Metformin treatment alone reduced tumor growth and an even greater effect was found with combined doxorubicin and metformin treatment for all cell lines. Notably, the ER-Src model is not a direct model of estrogen receptor driven breast cancer, but rather an estradiol inducible MCF10A estrogen-receptor v-Src model. In another study from this research group, metformin treatment alone did not affect tumor growth, but when combined with doxorubicin, the suppression of tumor growth was far greater than with doxorubicin alone [37]. Metformin treatment was administered every 3 days injected near the tumor after the mass reached ∼50 mm3 (Personal communication K.Struhl). In another aspect of this study, cancer stem cells obtained from this cell line were pretreated with metformin 20 days prior to inoculation into mice, and this prevented tumor formation. How to translate these findings to humans is unclear, but these data implicate metformin-targeted pathways in tumor engraftment and/or mammary carcinoma cell viability, possibly through inhibition of stem cell-specific mechanisms.
A higher dose of metformin, 750 mg/kg/day (5000 ug/ml in drinking water) for 5 weeks, did not impart a protective effect against tumor growth from implanted MDA-MB-435 cells [36]. While there is some concern related to the relevance of this cell line for breast cancer [46, 47], recent data suggest that the MDA-MB-435 line may indeed share some aspects of gene regulation with triple negative breast cancer.
A very recent study presented a direct comparison of metformin (2 mg/mL in drinking water) to phenformin (1.65 mg/mL in drinking water) treatments on local and metastatic growth of a mouse cell line which overexpresses HER2 (MMTV-Erbb2) [41••]. Cells were implanted in both immunodeficient and immunocompetent mice and both treatments effectively and significantly reduced tumor growth and lung metastases with a greater response observed with phenformin treatment. Similar results were obtained performing the study with the MDA-MB-436 triple negative human breast cancer cell line.
The impact of metformin treatment on prevention of mammary tumorigenesis in the clinically relevant (ER-/HER2/neu) transgenic mouse MMTV-neu line 202 has been investigated. In the first published study, metformin treatment (100 mg/kg/day) was initiated at 8 weeks of age and the mice followed until 52 weeks of age. At study termination, fewer metformin-treated mice had high tumor multiplicity, and these mice had extended tumor latency and increased life expectancy compared to the non-treated mice [42•]. A more recent study from this research group used the same experimental protocol except that the mice were followed for their lifespan [43]. The results obtained confirmed the delay in mammary tumor development in MMTV-neu mice although tumor number and metastases rates were not affected. This group has also reported that at a similar dose of metformin in the SHR mouse strain increased life span, but metformin did not influence the development of spontaneous malignant tumors [48].
A very recently published paper used MMTV-neu mice treated with a dose of 250 mg/kg daily of metformin administered by IP injections from 8–18 weeks of age. Since this was a short-term study, mammary tumor incidence was not the endpoint, rather mammary morphogenesis was evaluated [33••]. Mammary gland whole mounts from the metformin-treated mice exhibited decreased lateral branching and alveolar structure in comparison to glands from the control mice. Additional findings indicated that sphere formation was reduced from MECs obtained from mammary tissue of metformin-treated mice and ErbB2 and EGFR expression was down regulated while AMPK activation was enhanced.
Effects of metformin treatment in female rats administered the carcinogen N-methyl-N-nitrosourea (MNU) to induce mammary tumors have also been reported. When female Sprague Dawley rats were treated with either 5 or 50 mg/kg/day of metformin in their drinking water, neither dose affected tumor incidence although the higher dose extended tumor latency slightly [44]. In another publication, results were presented from three different experiments on the effects of metformin in a rapidly emerging mammary tumor model in which MNU is administer at 20 rather than ∼50 days of age [29]. In experiment 1, metformin was included in the diet at either 0.5 or 1 % beginning at 28 days of age for 5 days, and then, the doses were lowered to 0.05 and 0.25 %, respectively, for an additional 28 days for a total of 33 days of treatment. There was no effect of either metformin dose on mammary tumor incidence, although at the higher dose, latency was significantly extended and mammary tumor multiplicity and weight were reduced. It was also reported that tumors from the metformin-treated rats exhibited activation of the AMPK pathway. Further, at the higher dose of metformin, serum insulin and leptin concentrations were reduced, but there were no effects observed on IGF-I, adiponectin or glucose levels. In experiment 2, metformin was included in the diet at 0.3 % from 4 until 13 weeks of age, but there was no effect of metformin treatment on mammary tumorigenesis. In experiment 3, 0.25 % metformin was included in the diet and combined with dietary energy restriction of 40 %. There was no additional benefit to the protective effect of calorie restriction with the higher dose of metformin. In another chemical carcinogenesis study, Wistar rats were given MNU at ∼52 days of age and from 10 weeks of age fed a high fat diet (45 % fat by calories) to increase body weight gain. At 18 weeks of age, some of the rats were ovariectomized, and metformin (2 mg/ml in drinking water) treatment was initiated. After 3 weeks of metformin treatment, mammary tumor burden was reduced fivefold. Notably, progesterone receptor expression of the mammary tumors was reduced with no effects on either estrogen receptor alpha or HER2 in the metformin-treated obese rats compared to tumors obtained from non-treated obese rats [45].
In an attempt to more accurately reflect a human intervention trial, we are conducting a long-term metformin treatment study using MMTV-TGF-α mice that develop mammary tumors in the second year of life. The mice are fed a moderately high fat diet from 10 weeks of age, and metformin treatment is initiated at 30 weeks of age and maintained to 90 weeks of age. In addition to an ad libitum control group, we have included a group with a 25 % reduction in caloric intake to make a direct comparison to metformin treatment. Interestingly, although metformin is frequently referred to as a calorie restriction mimetic, few attempts have been made to make a direct comparison of these two interventions. We anticipate results of this study to be available in 2015.
Results of a study in another transgenic mouse model of triple negative breast cancer, MMTV-PyMT, were recently reported with metformin treatment compared to phenformin [41••]. In comparison to findings above evaluating these two compounds on tumor progression where phenformin appeared to be more effective than metformin, in this transgenic mouse model, the compounds had a similar effect on reducing mammary tumor weight, tumors per mouse, and metastases to lungs. The anticancer effects of phenformin had been investigated in several earlier studies. In one study, phenformin was administered at a dose of 5 mg/day for 2.5 months or at 10 mg/day for 5 months in female rats treated with DMBA to induce mammary tumors [49]. Both doses significantly reduced mammary tumor incidence (100 vs 43 % and 88.7 vs 55 % control vs phenformin). In a second rodent model, phenformin was given to 3.5 month old C3H/Sn mice (80 mg/kg/day, 5 days/week = ∼2.4 mg/mouse) [50]. This treatment regimen reduced spontaneous mammary tumor incidence 3.8-fold (20 versus 80 %) and extended lifespan by over 20 %. Due to concerns of causing serious lactic acidosis, phenformin is no longer used clinically for diabetes treatment and thus has not been investigated clinically as an anticancer drug.
Although limited in number, these published reports indicate potential cancer preventive effects for metformin treatment particularly to extend tumor latency. However, metformin has been administered in a number of different ways, i.e., in water or food or by ip injection and in one case locally so it is difficult to make direct comparisons of outcomes. Further, most studies have been undertaken in rodents fed low fat diets while humans usually consume diets with higher fat levels. How this might impact drug availability and effects on animal physiology remain to be determined. Clearly, studies using mice fed a high fat diet would be more a reflection of the human situation. Overall though metformin’s effect on cancer latency and/or progression appears to be more robust than its effect on prevention.
Mechanisms of Action
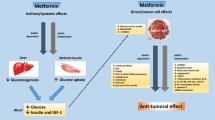
A downstream target for metformin was unknown until the discovery of the AMP protein kinase. AMPK is an AMP-activated protein kinase that is an essential factor in maintaining energy homeostasis following multiple types of cellular stress including heat shock, metabolic poisoning, glucose starvation, oxygen deprivation, and disruption of blood supply [51]. Through acute phosphorylation of multiple downstream targets, as well as long-term effects on gene and protein expression, activated AMPK switches off ATP consumption pathways and switches on ATP production pathways [52]. Based on metformin’s effects on diabetes, its action has been attributed to activation of AMPK in the liver with eventual lowering of gluconeogenesis and then the reduction of serum glucose and subsequently the need for less insulin. Since insulin and IGF-I are potent growth-promoting proteins, reducing their levels could explain a whole body approach to metformin’s anticancer effect. Recently, AMPK has been discovered to be a negative regulator of the dysregulated aerobic glycolysis in cancer cells (the Warburg effect) and a direct suppressor of tumor growth, based on studies in which genetic ablation of the AMPK alpha 1 subunit promoted Myc-induced tumor progression [53•]
In addition, direct effects of metformin on cell proliferation and apoptosis have been documented in multiple human breast cancer cell lines as was described above in the in vitro section. In many cases, metformin concentrations far exceeded clinical in vivo levels, although these in vitro findings have indeed indicated that metformin can directly affect cells if it can access organs and tissues and tissue levels can exceed plasma levels several fold [54]. A possible explanation for some of the varying results is that there may be a relationship between metformin sensitivity and glucose exposure in vivo that is difficult to reproduce with cultured cells. Another possibility is that tissue concentrations of metformin may be much higher than previously thought [54]. While many different pathways have been shown to be impacted by metformin in breast cancer cell lines, the general consensus is that metformin can suppress mTOR-mediated protein translation and cell growth. This inhibition of mTOR may be mediated through AMPK activation [55]. However, inhibition of mTOR is also known to cause feedback activation of Akt, whose over activation can lead to the development of cancer [56]. Thus, the mechanism underlying metformin’s effect on cancer cells is still elusive.
AMPK can be activated through reversible phosphorylation at the Thr172 site within its α-subunit by upstream kinases [52]. LKB1 is an upstream kinase of AMPK that phosphorylates and activates AMPK in response to a decrease in energy storage. However, previous studies have shown that AMPK can also be activated without direct activation of LKB1, indicating the existence of other upstream AMPK kinases [57, 58]. Among other potential AMPK kinase candidates, ATM can phosphorylate LKB1, the upstream kinase of AMPK, in response to DNA damage [59, 60]. In addition, ATM can also activate AMPK in an LKB1-independent manner [61•]. Recently, a genome-wide association study (GWAS) identified ATM as a gene whose variation affects glucose response of diabetic patients to metformin treatment [62••]. In this study, metformin-mediated AMPK activation in hepatic H4IIE cells was strongly inhibited by the ATM specific inhibitor KU-55933. These findings provide novel insights as to how metformin acts as a potential pharmaceutical agent for cancer prevention and treatment.
ATM is a protein kinase that is deficient in ataxia-telangiectasia (A-T), an autosomal recessive childhood disorder characterized by cerebellar ataxia and oculocutaneous telangiectasias [63•]. The gene mutated in this disease, ATM (A-T, mutated), encodes a 370-kDa Ser/Thr protein kinase. While ATM has been reported to function in controlling cell cycle progression by phosphorylating p53 after DNA damage, it is also known that ATM plays an important role in regulating cellular glucose homeostasis [63•]. It has recently become clear that p53, a downstream target of ATM, also regulates multiple steps of glucose metabolism pathways. p53 inhibits glycolysis by suppressing the expression of multiple enzymes involved in the glycolytic process [64]. Recent reports also indicate that p53 inhibits glycolysis in cancer cells by stimulating the activity of multiple enzymes that participate in the TCA cycle and the oxidative phosphorylation process [65]. This theory is supported by multiple lines of recent evidence showing that p53 is activated along with ATM and AMPK following the addition of metformin to various cancer cell lines [66–68]. These results suggest that p53 may play a key role in inhibiting aberrant glucose metabolism following metformin treatment. It remains to be determined if this response is important in the prevention and/or progression of breast cancer.
Interestingly, a recent finding indicated that metformin can activate Chk2 kinase, a key component of the DNA damage-like response (DDR) pathway, through activation of ATM [69]. Though actual DNA damage is not observed upon metformin treatment, the activation of Chk2 may likely protect cells from DNA damage caused by oxidative stress-induced during the lipid oxidation process. While the conventional role for p53 is to act as a tumor suppressor by sensing DNA damage and inducing cell cycle arrest and cell apoptosis, the possibility should not be ruled out that metformin may induce ATM/chk2’s and ATM/p53’s ability to promote the DDR process, thus preventing malignant transformation [70].
However, results of in vitro experiments suggest that metformin may also be activated through AMPK-independent mechanisms. For example, in a glioblastoma model, metformin treatment exerted antiproliferative effects through an AMPK independent mechanism directly inhibiting mTOR by enhancing PRAS40’s association with RAPTOR [71]. In another study, using prostate cancer cells when the AMPK pathway was inhibited, metformin was still able to exert antiproliferative effects [72]. Additional work by the same group found that REDD, which is a negative regulator of mTOR, was required for the reduction in cell proliferation [73]. Other studies have suggested that metformin can affect stem cells or self-renewal of some breast cancer cell lines [40, 74, 75]. Further, in a MCF-7 mammosphere model, the addition of metformin at an 11-mM concentration reduced their size and number [76]. In addition, when mammosphere formation was enhanced with addition of estrogen, metformin reduced the expression of OCT4, which is considered a cancer stem cell marker.
Among studies of other targets of metformin in breast cancer, STAT3 has recently emerged, because metformin inhibits STAT3 phosphorylation in triple negative and HER2 positive breast cancers [33••, 77]. Also, in a Src-induced transformation model, metformin inactivates STAT3 [40]. Metformin reduces the phosphorylation of both Tyr705 and Ser727 residues on STAT3 [77]. Phosphorylation of Tyr705 causes rapid translocation of STAT3 to the nucleus and activates the expression of proliferation and survival genes. Phosphorylation of Ser727 (pSTAT3 S727) has been reported to cause STAT3 to localize to the mitochondria (mitoSTAT3) where it modulates complexes I and II and promotes breast cancer growth [78]. Another group also linked mitoSTAT3 (pSTAT3 S727) with modulation of mitochondrial function, in part, through binding of mitochondrial DNA and regulating transcription of key proteins [79]. This newly discovered role of STAT3 in mitochondrial function regulation is of particular interest since metformin has been reported to inhibit complex I [80], which is believed to be a direct target of metformin [81]. The mechanism of metformin induced suppression of STAT3 phosphorylation is unclear. Several possible upstream pathways have been proposed to be responsible for STAT3 inactivation by metformin. Metformin effects on RTKs, mTOR, and Src have all been implicated as possible mechanisms [77].
Conclusions
Strong in vitro evidence with fairly consistent findings indicate antiproliferative actions for metformin as well as induction of apoptosis in a number of different human breast cancer cell lines. However, the metformin concentrations are frequently much higher than what would be achieved in vivo with currently established therapeutic doses. In human studies, the majority of the results are observational in diabetic women, thus much work remains to be done to determine if non-diabetic women at high risk for breast cancer might benefit from chemopreventive use of metformin. In animal models, the impact of metformin treatment is strongest in xenograft models representing cancer progression. With respect to prevention of mammary tumors, the major effect has been on tumor latency. Novel mechanisms of metformin involving mitochondrial bioenergetics are emerging and may involve both AMPK-dependent and -independent pathways. New regulators, including the ATM protein kinase as part of the AMPK pathway and the modulation of the STAT3 pathway by metformin independent of AMPK, are being investigated. Although considered a calorie restriction mimetic this still has not been directly compared in the breast cancer field. Presently, the concept of secondary prevention is being studied in the phase III MA.32 clinical trial in which breast cancer patients are randomized to metformin or placebo to study the impact of metformin on invasive disease-free survival in early stage breast cancer. This trial is likely to provide new insights to possible roles of metformin in breast cancer adjuvant therapy.
References
Papers of particular interest, published recently, have been highlighted as: • Of importance •• Of major importance
Evans JMM, Donnelly LA, Emslie-Smith AM, Alessi DR, Morris AD. Meformin and reduced risk of cancer in diabetic patients. Brit Med J. 2005;330:1304–5.
Bowker SL, Veugelers PJ, Majumdar SR, Johnson JA. Increased cancer-related mortality for patients with type 2 diabetes who use sulfonylureas or insulin. Diabetes Care. 2006;29:254–8.
Libby G, Donnelly LA, Donnan PT, Alessi DR, Morris AD, Evans JMM. New users of metformin are at low risk of incident cancer. A cohort study among people with type 2 diabetes. Diabetes Care. 2009;32:1620–5.
Goodwin PJ, Pritchard KI, Ennis M, Clemons M, Graham M, Fantus IG. Insulin-lowering effects of metformin in women with early breast cancer. Clin Breast Cancer. 2008;8:501–5.
Noto H, Goto A, Tsujimoto T, Noda M. Cancer risk in diabetic patients treated with metformin: a systematic review and meta-analysis. PLoS One. 2012;7:e33411.
Franciosi M, Lucisano G, Laprice E, Strippoli GFM, Pellegrini F, Nicolucci A. Metformin therapy and risk of cancer inpatients with type 2 diabetes: systematic review. PLoS One. 2013;8:e71583.
Bo S, Ciccone G, Rosato R, Villois P, Appendino G, Ghigo E, et al. Cancer mortality reduction and metformin: a retrospective cohort study in type 2 diabetic patients. Diabetes Obes Metab. 2012;14:23–9.
Zhang P, Li H, Tan X, Chen L, Wang S. Association of metformin use with cancer incidence and mortality: a meta-analysis. Cancer Epidemiol. 2013;37:207–18.
Malek M, Aghili R, Emani Z, Khamseh ME. Risk of cancer in diabetes: the effect of metformin. ISRN Endocrinol. 2013;article ID 636927.
Jiralerspong S, Palla SL, Girdano SH, Meric-Bernstam F, Liedtke C, Barnett CM, et al. Metformin and pathologic complete responses to neoadjuvant chemotherapy in diabetic patients with breast cancer. J Clin Oncol. 2009;27:3297–302.
Tseng C-H. Metformin may reduce breast cancer risk in Taiwanese women with type 2 diabetes. Breast Cancer Res Treat. 2014;145:785–90.
Aksoy S, Sendur MAN, Altundag K. Demographic and clinico-pathological characteristics in patients with invasive breast cancer receiving metformin. Med Oncol. 2013;30:560.
Qiu H, Rhoads GG, Berlin JA, Marcella SW, Demissie K. Initial metformin or sulphonylurea exposure and cancer occurrence among patients with type 2 diabetes mellitus. Diabetes Obes Metab. 2013;15:349–57.
Simó R, Plana-Ripoli O, Puente D, Morros R, Mundet X, Vilca LM, et al. Impact of glucose-lowering agents on the risk of cancer in type 2 diabetic patients. The Barcelona Case–control Study. PLoS One. 2013;8:e79968.
Niraula S, Dowling RJO, Ennis M, Chang MC, Done SJ, Hood N, et al. Metformin in early breast cancer: a prospective window of opportunity neoadjuvant study. Breast Cancer Res Treat. 2012;135:821–30.
Bonanni B, Puntoni M, Cazzaniga M, Pruneri G, Serrano D, Guerrieri-Gongaga A, et al. Dual effect of metformin on breast cancer proliferation in a randomized presurgical trial. J Clin Oncol. 2012;30:2593–600.
Hadad S, Iwamoto T, Jordan L, Purdie C, Bray S, Baker L, et al. Evidence for biological effects of metformin in operable breast cancer: a pre-operative, window-of-opportunity, randomized trial. Breast Cancer Res Treat. 2011;128:783–94.
Gail MH, Brinton LA, Byar DP, Green SB, Shairir C, Mulvhihill JJ. Projecting individualized probabilities of developing breast cancer for white females who are being examined annually. J Natl Cancer Inst. 1989;81:1879–86.
Martin LJ, Boyd NF. Potential mechanisms of breast cancer risk associated with mammaographic density: hypothesis based on epidemiological evidence. Breast Cancer Res. 2008;10:201.
Santen RJ, Boyd NF, Chlebowski RT, Cummings S, Cuzik J, Dowsett M, et al. Critical assessment of new risk factors for breast cancer: considerations for development of an improved risk prediction model. Endocrine-Relat Cancer. 2007;14:169–87.
Zakikhani M, Dowling R, Fantus IG, Sonenberg N, Pollak M. Metformin is an AMP kinase-dependent growth inhibitor for breast cancer cells. Cancer Res. 2006;66:10269–73.
Dowling RJO, Zakikhani M, Fantus IG, Pollack M, Sonenberg N. Metformin inhibits mammalian target of rapamycin-dependent translation initiation in breast cancer cells. Cancer Res. 2007;67:10804–12.
Zhuang Y, Miskimins WK. Cell cycle arrest in metformin treated breast cancer cells involves activation of AMPK, downregulation of cyclin D1, and requires p27Kipl or p21Cipl. J Mol Signal. 2008;3:1–11.
Liu B, Fan Z, Edgerton SM, Deng X-S, Alimova IN, Lind SE, et al. Metformin induces unique biological and molecular responses in triple negative breast cancer cells. Cell Cycle. 2009;8:2031–40.
Alimova IN, Liu B, Fan Z, Edgerton SM, Dillon T, Lind SE, et al. Metformin inhibits breast cancer cell growth, colony formation and induces cell cycle arrest in vitro. Cell Cycle. 2009;8:909–15.
Vazquez-Martin A, Oliveras-Ferraros C, Menendez JA. The antidiabetic drug metformin supprresses HER2 (erbB-2) oncoprotein overexpression via inhibition of the mTOR effector p70S6K1 in human breast carcinoma cells. Cell Cycle. 2009;8:88–96.
Zakikhani M, Blouin M-J, Piura E, Pollack MN. Metformin and rapamycin have distinct effects on the AKT pathway and proliferation in breast cancer cells. Breast Cancer Res Treat. 2010;213:271–9.
Jung J-W, Park S-B, Lee S-J, Seo M-S, Trosko JE, Kang K-S. Metformin represses self-renewal of the human breast carcinoma stem cells via inhibition of estrogen receptor-mediated OCT4 expression. PLos One 2011;e28068.
Zhu Z, Jiang W, Thompson MD, McGinley JN, Thompson HJ. Metformin as an energy restriction mimetic agent for breast cancer prevention. J Carcinog. 2011;10:17.
Liu B, Fan Z, Edgerton SM, Yang X, Lind SE, Thor AD. Potent anti-proliferative effects of metformin on trastuzamab-resistant breast cancer cells via inhibition of ErbB2/IGF-1 receptor interactions. Cell Cycle. 2011;10:2959–66.
Cufi S, Corominas-Faja B, Vazquez-Martin A, Oliveras-Ferraros C, Dorca J, Bosch-Barrera J, et al. Metformin-induced preferential killing of breast cancer initiating CD44+CD24-/low cells is sufficient to overcome primary resistance to trastuzumab in HER2+ human breast cancer xenografts. Oncotarget. 2012;3:395–8.
Williams CC, Singleton BA, Llopis SD, Skiripnikova EV. Metformin induces a senescence-associated gene signature in breast cancer cells. J Health Care Poor Underserved. 2013;24:93–103.
Zhu P, Davis M, Blackweider AJ, Backman N, Liu B, Edgerton S, et al. Metformin selectively targets tumor-initiating cells in ErbB2-overexpressing breast cancer models. Cancer Prev Res. 2014;7:199–210. This paper presents details of the effects of metformin both in vitro and in vivo on HER2/neu/ErbB2 overexpressing breast cancer.
Du Y, Zheng H, Wang J, Ren Y, Li M, Gong C, et al. Metformin inhibits histone H2B monoubiquitination and downstream gene transcription in human breast cancer cells. Oncol Lett. 2014;8:809–12.
Choi YW, Lim IK. Sensitization of metformin-cytotoxicity by dichloroacetate via reprogramming glucose metabolism in cancer cells. Cancer Lett. 2014;346:300–8.
Phoenix K, Vumbaca F, Claffey KP. Therapeutic metformin/AMPK activation promotes the angiogenic phenotype in the ERα negative MDA-MB-435 breast cancer model. Breast Cancer Res Treat. 2009;113:101–11.
Hirsch HA, Iliopoulos D, Tsichlis PN, Struhl K. Metformin selectively targets cancer stem cells, and acts together with chemotherapy to block tumor growth and prolong remission. Cancer Res. 2009;69:7507–11.
Iliopoulos D, Hirsch HA, Struhl K. Metformin decreases the dose of chemotherapy for prolonging tumor remission in mouse xenografts involving multiple cancer cell types. Cancer Res. 2011;71:3196–201. This manuscript presents the effects of metformin treatment in xenograft experiments as well as work with potential effects of metformin on breast cancer stem cells.
Cheong J-H, Park ES, Liang J, Dennison JB, Tsavachiidou D, Nguyen-Charles C, et al. Dual inhibition of tumor energy pathway by 2-deoxyglucose and metformin is effective against a broad spectrum of preclinial cancer models. Mol Cancer Ther. 2011;10:2350–62.
Hirsch HA, Iliopoulos D, Struhl K. Metformin inhibits the inflammatory response associated with cellular transformation and cancer stem cell growth. Proc Natl Acad Sci U S A. 2013;110:972–7.
Orecchioi S, Reggiani F, Talarico G, Mancusco P, Calleri A, Gregato G, et al. The biguanides metformin and phenformin inhibit angiogenesis, local and metastatic growth of breast cancer by targeting both neoplastic and microenvironment cells. Int J Cancer. 2015;136:534–44. Results presented comparing metformin to phenformin and show that metformin can have direct activity against breast cancer cells and also has anti-angiogenic activity against white adipose tissue progenitor cells.
Anisimov VN, Berstein LM, Egormin PA, Piskunova TS, Popovich IG, Zabezhinksi MA, et al. Effect of metformin on life span and on the development of spontaneous mammary tumors in HER-2/neu transgenic mice. Exp Gerontol. 2005;40:685–93. Long-term treatment study indicating prevention of mammary tumor development in HER-2/neu transgenic mice.
Anisimov VN, Egormin PA, Piskunova TS, Popovich IG, Tyndyk ML, Yurova MV, et al. Metformin extends life span of HER-2/neu transgenic mice and in combination with melatonin inhibits growth of transplantable tumors in vivo. Cell Cycle. 2010;9:188–97.
Bojková B, Orendas P, Kassayova M, Kutna V, Ahlersová E, Ahlers I. Metformin in chemically-induced mammary carcinogenesis in rats. Neoplasma. 2009;56:269–74.
Giles ED, Wellberg EA, Astling DP, Anderson SM, Thor AD, Jindal S, et al. Obesity and overfeeding affecting both tumor and systemic metabolism activates the progesterone receptor to contribute to postmenopausal breast cancer. Cancer Res. 2012;72:6490–501.
Hadad SM, Appleyard V, Thompson AM. Therapeutic metformin/AMPK activation promotes the angiogenic phenotype in the ERα negative MDA-MB-435 breast cancer model. Breast Cancer Res Treat. 2009;114:391.
Ross DT, Scherf U, Eisen MB, Perou CM, Rees C, Spellman P, et al. Systematic variation in gene expression patterns of human cancer cell lines. Nat Genet. 2000;24:227–35.
Anisimov VN, Berstein LM, Egormin PA, Piskunova TS, Popovich IG, Zabezhinksi MA, et al. Metformin slows down aging and extends life span of female SHR mice. Cell Cycle. 2008;7:2769–73.
Dilman VM, Berstein LM, Zabezhinksi MA, Alexandrov VA, Bobrov YF, Pliss GB. Inhibition of DMBA-induced carcinogenesis by phenformin in the mammary gland of rats. Arch Geschwulstforsc. 1978;48:1–8.
Dilman VM, Anisimov VN. Effect of treatment with phenformin, diphenylhydantoin or L-dopa on life span and tumour incidence in C3H/Sn mice. Gerontology. 1980;26:241–6.
Hardie DG, Hawley SA. AMP-activated protein kinase: the energy charge hypothesis revisited. BioEssays. 2001;23:1112–9.
Carling D. The AMP-activated protein kinase cascade—a unifying system for energy control. Trends Biochem Sci. 2004;29:18–24.
Faubert B, Boily G, Izreig S, Griss T, Samborska B, Dong Z, et al. AMPK is a negative regulator of the Warburg effect and suppresses tumor growth in vivo. Cell Metab. 2013;17:113–24. This paper provides data supporting a link between AMPK and HIF-1α activities.
Choi YH, Lee MG. Pharmacokinetic and pharmacodynamic interaction between nifedine and metformin in rats: competitive inhibition for metabolism of nifedine and metformin by each other via CYP isozymes. Xenobiotica. 2012;42:483–95.
Hahn-Windgassen A, Nogueira V, Chen C-C, Skeen JE, Sonenberg N, Hay N. AKT activates the mammalian target of rapamycin by regulating cellular ATP level and AMPK activity. J Biol Chem. 2005;280:32081–9.
Sun S-Y, Rosenberg LM, Wang X, Zhou Z, Yue P, Fu H, et al. Activation of Akt and eIF4E survival pathways by rapamycin-mediated mammalian target of rapamycin inhibition. Cancer Res. 2005;65:7052–8.
Ratten R, Giri S, Singh AK, Singh I. 5-Aminoimidazole-4-carboxamide-1-β-D-ribofuranoside inhibits cancer cell proliferation in vitro and in vivo via AMP-activated protein kinase. J Biol Chem. 2005;280:39582–93.
Sakamoto K, Gõransson O, Hardie DG, Alessi DR. Activity of LKB1 and AMPK-related kinases in skeletal muscle: effects of contraction, phenformin, and AICAR. Am J Physiol. 2004;287:E310–7.
Fernandes N, Sun Y, Chen S, Paul P, Shaw RJ, Cantley LC, et al. DNA damage-induced association of ATM with its target proteins requires a protein interaction domain in the N terminas of ATM. J Biol Chem. 2005;280:15158–64.
Sapkota GP, Boudeau J, Deak M, Kieloch A, Morrice N, Alessi DR. Identification and characterization of four novel phosphorylation sites (Ser31, Ser325, Thr336 and Thr366) on LKB1/STK11, the protein kinase mutated in Peutz-Jeghers cancer syndrome. Biochem J. 2002;362:481–90.
Sun Y, Connors KE, Yang D-Q. AICAR induces phosphorylation of AMPK in an ATM-dependent LKB1-independent manner. Mol Cell Biochem. 2007;306:239–45. This is the first report documenting that AMPK can be phosphorylated/activated in an ATM-dependent manner.
UKPDS, GoDarts Diabetes Pharmacogenetic Study Group. The Welcome Trust Case Control Consortium 2. Common variants near ATM are associated with glycemic response to metformin in type 2 diabetes. Nat Genet. 2011;43:117–20. This is the first GWAS study that links genetic variances of ATM with type 2 diabetes patients’ response to metformin treatment.
Yang D-Q, Halaby M-J, Li Y, Hibma JC, Burn P. Cytoplasmic ATM protein kinase: an emerging therapeutic target for diabetes, cancer and neuronal degeneration. Drug Dis Today. 2011;16:332–8. This is a comprehensive review that details ATM’s connection with insulin resistance and type 2 diabetes.
DeBerardinis RJ, Lum JJ, Hatzivassilou G, Thompson CB. The biology of cancer: metabolic reprogramming fuels cell growth and proliferation. Cell Metab. 2008;7:11–20.
Chen JQ, Russo J. Dysregulation of glucose transport, glycolysis, TCA cycle and glutaminolysis by oncogenes and tumor suppressors in cancer cells. Biochim Biophys Acta. 1826;2012:370–84.
Storozhuk Y, Hopmans SN, Sanli T, Barron C, Tsiani E, Cutz J-C, et al. Metformin inhibits growth and enhances radiation response of non-small cell lung cancer (NSCLC) through ATM and AMPK. Brit J Cancer. 2013;108:2021–32.
Yi G, He Z, Zhou Y, Xian L, Yuan T, Jia X, et al. Low concentration of metformin induces a p53-dependent senescence in hepatoma cells via activation of the AMPK pathway. Int J Oncol. 2013;43:1503–10.
Jones RG, Plas DR, Kubek S, Buzzai MJ, Mu J, Xu Y, et al. AMP-activated protein kinase induces a p53-dependent metabolic checkpoint. Mol Cell. 1994;18:283–93.
Vazquez-Martin A, Oliveras-Ferraros C, Martin-Castillo B, Menendez JA. Metformin activates an ataxia telengiectasia mutated (ATM)/Chk2-regulated DNA damage-like response. Cell Cycle. 2011;10:1499–501.
Del Barco S, Vazquez-Martin A, Cufi S, Oliveras-Ferraros C, Bosch-Barrera B, Joven J, et al. Metformin: multi-faceted protection against cancer. Oncotarget. 2011;2:896–917.
Liu X, Chhipa RR, Pooya S, Wortman M, Yachyshin S, Chow LML, et al. Discrete mechanisms of mTOR and cell cycle regulation by AMPK agonists independent of AMPK. Proc Natl Acad Sci USA 2014.
Ben Sahra I, Laurent K, Loubat A, Giorgetti-Peraldi S, Colosetti P, Auberger P, et al. The antidiabetic drug metformin exerts an antitumoral effect in vitro and in vivo through a decrease of cyclin D1 level. Oncogene. 2008;27:3576–86.
Ben Sahra I, Regazzetti C, Robert G, Laurent K, Le Marchand-Brustel Y, Auberger P, et al. Metformin, independent of AMPK, induces mTOR inhibition and cell-cycle arrest through REDD1. Cancer Res. 2011;71:4366–72.
Vazquez-Martin A, Oliveras-Ferraros C, Del Barco S, Martin-Castillo B, Menendez JA. The anti-diabetic drug metformin suppresses self-renewal and proliferation of trastuzumab-resistant tumor-initiating breast cancer stem cells. Breast Cancer Res Treat. 2011;126:355–64.
Vazquez-Martin A, Oliveras-Ferraros C, Del Barco S, Martin-Castillo B, Menendez JA. Metformin regulates breast cancer stem cell ontogeny by transcriptional regulation of the epithelial-mesenchymal transition (EMT) status. Cell Cycle. 2010;18:3807–14.
Pollak M. The insulin receptor/insulin-like growth factor receptor family as a therapeutic target in oncology. Clin Cancer Res. 2012;18:40–50.
Deng X-S, Wang S, Deng A, Liu B, Edgerton SM, Lind SE, et al. Metformin targets Stat3 to inhibit cell growth and induce apoptosis in triple-negative breast cancers. Cell Cycle. 2012;11:367–76.
Zhang Q, Raje V, Yakovlev VA, Yacoub A, Szczepanek K, Meier J, et al. Mitochondrial localized Stat3 promotes breast cancer growth via phosphorylation of serin 727. J Biol Chem. 2013;288:31280–8.
Macias E, Rao D, Carbajal S, Kiguchi K, DiGiovanni J. Stat3 binds to mtDNA and regulates mitochondrial gene expression in keratinocytes. J Invest Derm. 2014;134:1971–80.
Wheaton WW, Weinberg SE, Hamanaka RB, Soberanes S, Sullivan LB, Anso E, et al. Metformin inhibits mitochondrial complex 1 of cancer cells to reduce tumorigenesis. eLife. 2014;3:e02242.
El-Mir M-Y, Nogueria V, Fontaine E, Avéret N, Rigoulet M, Leserve X. Dimethylbiguanide inhibits cell respiration via an indirect effect targeted on the respiratory chain complex I. J Biol Chem. 2000;275:223–8.
Acknowledgments
Support for this work comes from The Hormel Foundation (MPC), NIH-NCI- R01CA157012 (MPC) and Paint the Town Pink-Grant (MPC), R01CA113570 (DAP), University of Minnesota Office of Discovery and Translation and Clinical Translational Science Institute (DAP), the Randy Shaver Cancer Foundation (DAP) and Community Fund (DAP).
Compliance with Ethics Guidelines
ᅟ
Conflict of Interest
Michael E. Grossmann, Da-Qing Yang, Zhijun Guo, David A. Potter, and Margot P. Cleary declare that they have no conflict of interest.
Human and Animal Rights and Informed Consent
This article does not contain any studies with human or animal subjects performed by any of the authors.
Author information
Authors and Affiliations
Corresponding author
Additional information
This article is part of the Topical Collection on Cancer Chemoprevention
Michael E. Grossmann and Da-Qing Yang contributed equally to this work.
Rights and permissions
About this article

Cite this article
Grossmann, M.E., Yang, DQ., Guo, Z. et al. Metformin Treatment for the Prevention and/or Treatment of Breast/Mammary Tumorigenesis. Curr Pharmacol Rep 1, 312–323 (2015). https://doi.org/10.1007/s40495-015-0032-z
Published:
Issue Date:
DOI: https://doi.org/10.1007/s40495-015-0032-z