
Abstract
There is a profound need in oncology to detect cancer earlier, guide individualized therapies, and better monitor progress during treatment. Currently, some of this information can be achieved through solid tissue biopsy and imaging. However, these techniques are limited because of the invasiveness of the procedure and the size of the tumor. A liquid biopsy can overcome these barriers as its non-invasive nature allows samples to be collected over time. Liquid biopsies may also allow earlier detection than traditional imaging. Liquid biopsies include the analysis of circulating tumor cells (CTCs), cell-free nucleic acid (cfNA), or extracellular vesicles obtained from a variety of biofluids, such as peripheral blood. In this review, we discuss different liquid biopsy types and how they fit into the current regulatory landscape.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Description of current liquid biopsy types. |
Advantages and disadvantages of liquid biopsies over solid tumor biopsies. |
Current regulatory landscape for the use of liquid biopsies in clinical assays. |
1 Introduction to Liquid Biopsies
The current standard of care for cancer diagnostics is usually an evaluation of biopsied tissue (i.e., tumor) under a microscope by a highly trained pathologist. In most cases, there are additional protein and nucleic acid (NA) markers that can help aide a pathologist in diagnosis and in determining the best treatment regimen. This is especially true because newer cancer therapies are focused on targeting and interfering with specific mechanisms that block the growth and spread of cancer. Unfortunately biopsies can be challenging to access, expensive, and painful for patients [1]. These factors make serial collection nearly impossible despite the information it might provide, such as resistance to a cancer therapy. Finally, the heterogeneity of solid tumors is a major issue when sampling tissue biopsies as results may vary between sites even though they are within the same tumor tissue [2, 3]. Given the importance of the information a biopsy can supply, there is a critical need for a surrogate that can provide the same information while overcoming the current limitations.
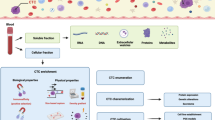
The great hope of liquid biopsies in precision medicine is that they can augment traditional biopsies because they are non-invasive tests that can evaluate tumoral material. This material includes circulating tumor cells (CTCs) and fragments of NAs, either freely circulating or encapsulated in extracellular vesicles (EVs). CTCs were the first liquid biopsy, originally recognized in 1869 [4]. However, their clinical utility was only recognized in 1994 when Immunicon, later known as Veridex, built a platform to isolate and detect them. Sensitivity and specificity issues have meant that other liquid biopsies have lagged behind CTCs, but this is rapidly changing. Similar to CTCs, cell-free NA (cfNA) was first described in 1948, but their clinical importance has only recently been highlighted [5]. cfNA is either shed or released from tumors or CTCs circulating in a variety of biofluids, including peripheral blood, urine [6, 7], amniotic fluid [8], and cerebrospinal fluid [9–11]. Finally, although EVs are a recent arrival on the liquid biopsy scene, their importance is rapidly increasing because of their perceived ability to add yet another important perspective to cancer diagnostics. EVs are membranous vesicles encapsulating NAs and proteins that are also either released or shed into a variety of biofluids. Liquid biopsies therefore have enormous diagnostic and treatment implications in oncology as an integral part of precision medicine (Fig. 1).

Liquid biopsy materials reflect spatial and temporal heterogeneity. The different types of liquid biopsies released from solid tumors were not drawn to actual yields
In this review, we discuss the advantages and disadvantages of liquid biopsies, focusing on oncology applications. We also describe the different types of liquid biopsies and the current companies developing assays. We then discuss the differences between FDA-approved and laboratory-developed tests (LDTs) specifically as they relate to liquid biopsies. We also highlight current regulatory discussions concerning liquid biopsies. Finally, we conclude with a brief discussion on the future of liquid biopsies and what is required for them to become a part of the clinical paradigm.
2 Advantages/Disadvantages of Liquid Biopsies
It has been well documented that tumors develop heterogeneity over time as well as spatially within a single tumor. Spatial heterogeneity exists because of the pressure of the tumor microenvironment, including hypoxia, immune infiltrations, nutrients, and drug exposure. Tumors also express heterogeneity over time and have been shown to evolve after replication errors and selective pressure from treatments [3] (Fig. 1). Therefore, initial solid tumor biopsies performed before treatment may bear little resemblance to the patient’s current solid tumor or tumor status. Conversely, liquid biopsies are believed to better reflect tumor heterogeneity as they contain cells or NAs shed into circulation from a potentially wide range of regions of the primary and metastatic tumor sites. A few clinical correlations of liquid biopsies have already been established, but more are needed [12, 13].
A variety of factors currently limit the ability to obtain solid tumor biopsy samples over the course of treatment. Liquid biopsies are less invasive and more cost effective than traditional tumor biopsies, so they are better suited for obtaining frequent biopsies and information over time. This information can help detect resistance to therapies (thereby redirecting the treatment strategy) or disease reoccurrence earlier than traditional biopsies or imaging methods. Finally, liquid biopsies could also be used for early cancer detection, could provide invaluable information on cancer subtyping and prognosis, and could predict treatment responsiveness [1].
Although the advantages of using liquid biopsies are many, limitations continue to hinder their widespread use. One of the main challenges of utilizing CTCs, cfNA, or EVs is the ability to distinguish between tumor-derived biomarkers from those from other tissue sources. Although downstream analysis methods such as next-generation sequencing (NGS) have a low limit of detection, there are limitations when the yield of NA material is extremely low. Selective or semi-selective amplification of DNA and RNA is often performed to enhance analytical sensitivity, which can introduce biases, resulting in false-positive and false-negative findings. Sophisticated software and highly trained personnel are often required to perform data analysis because of the large amounts of data acquired, background noise, and algorithms used for analysis. Additionally, there are no standards for the amount of CTCs, cfNA, or EVs that must be analyzed to adequately represent tumor heterogeneity.
3 Circulating Tumor Cells
CTCs are cells that have been shed from a primary or metastatic tumor and are potentially capable of forming metastases. Metastases are responsible for 90 % of cancer-related deaths [14]. The metastatic process is accomplished when a CTC successfully carries out a series of processes. First, the CTC must detach from the primary tumor and intravasate into the bloodstream. Once in circulation, the CTC must survive attacks from immune cells and shear stress to extravasate into microvessels of a distant tissue [15]. It must then adapt and survive in the new microenvironment of the distant tissue and colonize to form a metastatic lesion [14]. CTCs have been found in the bloodstream of patients with early-stage and metastatic cancer as single cells or cell clusters. CTC enumeration has been shown to be associated with progression-free and overall survival and response to therapy [13, 16] in breast, colon, and prostate cancer [17–19]. However, at this time, no molecular biomarkers extracted from CTCs have been shown to have clinical utility.
CellSearch, characterized in 2004, is the only FDA-cleared method to enumerate CTCs [13], but many other methods have been developed or are in development [20]. Enrichment and isolation can be based on immunophenotypic properties, most commonly on expression of epithelial cell-adhesion molecule (EpCAM). In addition to CellSearch, several other EpCAM-based enrichment methods are commercially available. One example is the AdnaTest (Qiagen), which is based on immunomagnetic enrichment of EpCAM, cancer antigen (CA)-15-3, and human epidermal growth factor receptor (HER)-2 receptors [21]. Enrichment can also be based on other physical properties of CTCs, including density, size, deformability, or electrical properties. For example, RareCell has a commercially available device called the ISET that isolates CTCs based on size using a microfluidic device [22]. Methods to detect CTCs are based on immunophenotypic identification of cytokeratin, enzymatic methods, and reverse transcriptase quantitative polymerase chain reaction (RT-qPCR) [23–25]. Other post-isolation analysis of NA from single CTCs has been performed to determine quantity and sequencing to find genetic alterations [26].
There are several limitations to using CTCs clinically, including their rarity and the need for highly specialized equipment to isolate them. Additionally, heterogeneity of CTC surface protein expression is an obstacle, as several methods isolate CTCs based on the expression of these proteins, such as the epithelial marker EpCAM. Therefore, these methods will not detect any CTC undergoing an epithelial to mesenchymal transition (EMT) [27]. In fact, close to 40 % of patients with metastatic breast cancer have no detectable epithelial CTCs [28, 29]. Many researchers are developing methods to capture CTCs that do not express EpCAM. One such group using a tumor antigen-independent method has reported an ability to sort up to 107 cells using deterministic lateral displacement, inertial focusing, and magnetophoresis on a microfluidic chip, known as the CTC-iChip [30]. Other groups are using EMT-based markers in addition to EpCAM to capture CTCs. One such company, On-Q-ity (Waltham, MA, USA) reports >70 % capture efficiency of an EpCAM-negative cell line with their OnQChip [31]. A more comprehensive review of non-epCAM-based approaches for cell enrichment and isolation was published earlier this year [32].
Overall, CTCs hold promise as a surrogate for traditional biopsies, although many limitations remain. Because not all CTCs express epithelial markers, much effort is being put into developing methods able to detect mesenchymal as well as epithelial CTCs [32]. Many assays for predictive biomarkers have been explored, including RT-PCR, fluorescence in situ hybridization (FISH), and sequencing. While some have demonstrated success in early clinical exploratory studies, more comprehensive validation and standardization will be necessary before they can be used in the clinic. A commercially successful CTC technology will be a cost-effective and reproducible method with proven clinical utility.
4 Cell-Free Nucleic Acids
cfNAs circulating in blood were first described in 1948 [5]. However, it took another 56 years and the detection of NRAS gene fragments in the blood of cancer patients to capture scientists’ attention [33, 34]. Since then, cfNA levels have been found to be higher in diseased than in healthy patients [35]. Additionally, other studies have detected tumor-related genetic and epigenetic alterations, such as in PIK3CA, TP53, and KRAS, that drive cancer development [1, 36, 37]. Progress towards the use of cfNA in liquid biopsies has lagged until recently because of technological limitations in the analytical sensitivity of downstream detection schemes and the ability to differentiate between physiological and pathological states [38–40]. As these hurdles are overcome, the ability to easily access and process cfNA throughout the course of a disease is quickly making it the leading type of liquid biopsy. Further, like other forms of liquid biopsy, cfNA has the potential to examine heterogeneity because it overcomes sample biasing by serving as a pool of material from several tumor sites [41]. In this review, we mainly discuss cfNA in terms of DNA, as this currently provides more examples [34, 42].
The release of cfNA into biofluid involves multiple potential mechanisms that are not yet fully understood. The main mechanism proposed is passive release during apoptotic or necrotic events within the tumor microenvironment or from CTCs, with macrophages or other scavenger cells being unable to clean up all the cell debris [43]. Additionally, these same scavenger cells can release the digested NA back into its external environment. The second hypothesized mechanism is active secretion of individual cfNA fragments or incorporation into protein or lipid complexes [42, 44]. The average amount of DNA cfNA in healthy donors and cancer patients is approximately 30 ng [35] and 180 ng [43, 45–47] per ml of blood, respectively. The reported DNA cfNA half-life varies from 15 minutes to several hours [35], with yields influenced by clearance, degradation, and other physiological filtering events [48]. cfNA release has also been found to dramatically decrease after tumor removal and to increase again if metastasis occurs [49].
As with all liquid biopsies, standardization of assays remains a huge hurdle because blood processing (time, blood tube, plasma vs. serum, etc.) and cfNA extraction technologies are not agreed upon. While extraction of cfNA directly from biofluid would help provide standardization, there is currently no commercially available option [50]. Therefore, several groups have tested various commercial spin columns [51]. Results have revealed the extraction methods were highly dependent on fragment size. More traditional methods such as phenol-chloroform extraction, salting out, and PAGE (polyacrylamide gel electrophoresis) isolation have the advantage of extracting a broader range of fragment sizes. More recently, others have introduced cfNA extraction kits that isolate a broader range of cfNA fragments and have larger starting volumes: NucleoSpin® (Macherey-Nagel), EpiQuik (EpiGentek), and QIAamp® (Qiagen). Others have reported data to suggest that the QIAamp® is superior in terms of cfNA yield and reproducibility [52]. Finally, several groups have been utilizing dielectrophoretic devices to rapidly isolate and detect DNA fragment sizes directly from whole blood [53] with the goal of eliminating an extraction step.
Several companies have developed assays utilizing cfNA in oncology and prenatal applications. The majority of these post-enrichment cfNA detection and analysis strategies rely on newer and more sensitive techniques, such as digital PCR and NGS; however, at the time of writing, none of these applications have been FDA approved. Leading companies include Foundation Medicine, Qiagen, Genomic Health, Guardant Health, GRAIL, Myriad Genetics, Personal Genome Diagnostics, Sequenom, and Trovagene. The only FDA-approved cfNA test is the BRACAnalysis CDx® developed by Myriad Genetics as a companion diagnostic for use with Lynparza™ (olaparib), a poly ADP ribose polymerase (PARP) inhibitor. Qiagen’s Therascreen EGFR Plasma RGQ and Roche’s cobas® EGFR Mutation Test v2 have also received the European CE mark for in vitro diagnostics (IVD) [12, 54]. While technology is rapidly enabling cfNA for use as a liquid biopsy, large-scale studies to prove clinical utility are still needed.
5 Extracellular Vesicles
EVs are membranous lipid structures released to the external environment by both healthy and non-healthy cells. EVs encapsulate proteins and NAs specific to their cellular origin and function in intercellular communication, cellular remodeling, immune regulation, and microenvironment modulation [55]. As EVs can migrate from anatomical compartments to biofluids (i.e., blood, urine, cerebral spinal fluid), there is a growing interest in analyzing them as a type of liquid biopsy for disease diagnosis and treatment monitoring [56]. Additionally, NAs entrapped in EVs appear to remain more stable because they are protected from ubiquitous RNases [57]. Finally, EVs are also being explored as delivery vehicles for targeted therapies [58].
As the use of EVs is still in its infancy, much debate surrounds their origin and nomenclature because of unanswered questions regarding their biogenesis [55, 59]. Current consensus classifies EVs into three categories based on their size, cell origin, and secretion mechanisms. In brief, exosomes (<100 nm) are the most commonly studied and originate from intracellular vesicles that fuse with and are secreted from the plasma membrane. Microvesicles (100–2000 nm) directly bud from the cell plasma membrane. Apoptotic bodies (50–5000 nm) are released from cells undergoing apoptosis and vary greatly in size [9, 60]. Finally, large oncosomes (1–10 µm) derived from tumor cells are thought to potentially constitute a fourth category [61]. In the future, EV-isolation methods that better separate individual types will bring more specificity to the definition of EVs.
The gold standard for EV isolation is differential ultracentrifugation to concentrate and partially purify EVs. The addition of density gradients helps to improve EV purification by better removing protein aggregates, lipoproteins, and other contaminants. Because of the increasing interest in EVs, commercially available kits that are easy to use and do not require specialized equipment (i.e., ultracentrifuge) are being developed and marketed. These kits are based on the physical properties of EVs, including affinity, size exclusion, and precipitation [9]. Affinity methods to isolate EVs are based on specific membrane markers or physiochemical characteristics (Immunobeads and ELISA-plates, Hansa BioMed; Dynabeads, ThermoFisher Scientific; exoEasy, Qiagen) [62]. Isolated EV yields by affinity methods are often lower than other methods but result in a highly purified EV population with few contaminants [63]. Size exclusion separates EVs based on their size and passage through a physical barrier such as a filter or chromatography resin (qEV, Izon Sciences; ExoMir, Biooscientific). Finally, several kits precipitate EVs (ex. ExoQuick™, System Bioscience [64], Total Exosome Isolation Kit, ThermoFisher Scientific, Exo-Prep, Hansa BioMed; mirCURY, Exiqon). Exo-spin by cell guidance systems isolates EVs with a combination of precipitation and size exclusion.
Characterization of isolated EVs is based on optical and non-optical methods to assess size, concentration, and surface marker composition [65]. Electron microscopy, atomic force microscopy, nanoparticle tracking analysis (NTA), resistive pulse sensing (RPS), and dynamic light scattering (DLS) are used to determine size distribution [9]. NTA, RPS, and DLS are also used to determine concentration. NTA uses an optical-based method to measure EV mean velocity that can be inserted into the Stokes–Einstein equation to determine EV size [66, 67]. RPS relies on separate EVs causing a change in an ionic current passed through a nanopore embedded in a membrane [68–70]. Flow cytometry, western blots, and ELISAs are used to detect surface proteins [71, 72]. Markers used for EV identification include tetraspanins (CD9, CD63, CD81, CD82), major histocompatibility complex (MHC) molecules, milk-fat globule-EGF-factor VIII (MFGE8 or lactadherin), transport protein Rab-5b, cytosolic proteins (Tsg101, Alix), and cytoskeletal proteins (actin, tubulin) [9].
Most clinical assays currently in development use mass spectrophotometry to measure protein expression, but NA-based endpoints are rapidly increasing as their cost decreases and sensitivity improves (i.e., digital PCR and NGS). Exosome Sciences, in collaboration with their majority shareholder, Aetholon Medical, is focused on diagnosing chronic traumatic encephalopathy (CTE) by detection of the exosomal tau protein. A clinical trial, the DETECT study, is ongoing at the Boston University CTE Center. Exosome Diagnostics is the first to offer tests to patients utilizing EV material and has established collaboration and distribution agreements with Eli Lily and Qiagen. Exosome Diagnostics isolates EVs using an affinity membrane and uses RNA for biomarker identification. In early 2016, they launched ExoDx™ Lung (anaplastic lymphoma kinase [ALK]), an LDT, to detect EML-4-ALK fusions to guide ALK-inhibitory therapy, and they plan to launch their second LDT in 2016 (ExoIntelliScore™ Prostate uses urine samples to predict the aggressiveness of prostate cancer to assist in determining the best treatment course). Their final LDT release in 2016 will be a solid tumor mutation-detection panel, which will be available as a clinical development tool for pharmaceutical companies. While several isolation kits for EVs have been developed, none are standardized, and large multicenter clinical trial data to support their use are lacking.
6 Regulatory of Liquid Biopsies
As defined by the FDA, “A laboratory developed test (LDT) is a type of in vitro diagnostic (IVD) test that is designed, manufactured, and used within a single laboratory”. LDTs can be used to measure or detect a variety of different analytes (proteins, NAs, and chemical substances) and vary in complexity [73]. A Clinical Laboratory Improvement Amendment (CLIA) license, governed by the Center for Medicare and Medicaid Services (CMS), is required to perform laboratory testing, including LDTs [74]. CLIA is also overseen by two other federal agencies, the FDA and the Center for Disease Control (CDC), each with different roles, to ensure the quality of laboratory testing [75]. Currently, to perform an LDT, CLIA requires performance specifications for accuracy, precision, analytical sensitivity, analytical specificity, reportable range of test results, reference intervals, and any other performance characteristics deemed important to the test. Quality systems such as proficiency testing must also be implemented to ensure accurate, reliable, and timely results [76]. However, under CLIA, accreditors do not evaluate LDT validation prior to marketing or assess clinical validity. This is in contrast to the FDA, which assesses clinical validity prior to patient testing. Additionally, the FDA requires controls to ensure appropriate design, manufacture, and safety/effectiveness of the device [77]. These agency regulations are important to the development of liquid biopsies, because they will govern how quickly the tools will be available for clinical use.
In 1976, the FDA was given the power to regulate medical devices through the Medical Device Amendment [78]. However, at the time, they chose not to enforce regulatory requirements for LDTs as they were relatively simple tests, confined to single local labs, and often used for rare diseases. However, as technology advances and business models change, LDTs have increased in prevalence and some see them as more complex than ever intended. Therefore, the FDA has become concerned that patient safety is in jeopardy as appropriate controls and evidence for clinical utility is lacking. This has led to draft legislation released in August 2014 that allows the FDA to have more oversight for both pre-market and post-market review of LDTs. This legislation seeks to further define LDTs as a type of IVD device and thus require them to be classified based on risk. Each classification will require different regulations to be met and at different times upon legislature approval [73]. These classifications include low, moderate, and high risk and require different regulations that will be phased in over 9 years. It is important to note that, under this draft legislation, LDTs used in forensics, histocompatibility, stem cell, or tissue transplantation would be exempt from notification and other requirements. Additionally, LDTs tend to pose little risk for rare disease or unmet needs and would also be exempt [79].
While the release of the FDA draft LDT legislation has met with strong objections, it has started an important conversation about improving current LDT regulations, with several influential groups releasing their own legislative proposals to congress. Concerns include whether LDTs are medical devices and that LDT risk categories are not currently well defined. Additionally, several groups are concerned with the financial burden to laboratories to achieve pre-market approval, which can rely on current literature but might also require clinical trials to prove clinical utility. This would set up a model that would provide no incentive for laboratories to develop LDTs, which could stifle innovation and thus limit patient care. The College of American Pathologists (CAP), Association for Molecular Pathology (AMP), and Diagnostic Test Working Group (DTWG) [80] have all issued proposals with suggestions for new legislation regarding LDT regulation. All groups agree that patient safety should always be the main concern. However, if regulation is too stringent, the burden of cost and time could hamper the very innovation and advances that could improve patient outcomes.
7 Future and Conclusions
Liquid biopsies hold great promise as a surrogate for or alongside traditional solid tumor biopsies to provide patients with an improved level of precision care. This is because liquid biopsies are cost effective, are non-invasive, and allow patient monitoring throughout treatment. Additionally, liquid biopsies act as a pool of material, allowing us to better assess heterogeneity across primary and metastatic tumors and giving clinicians a more complete picture of molecular changes over time. Finally, liquid biopsies provide invaluable information for cancer subtyping, prognosis, determining the optimal treatment strategy, detecting recurrence, and predicting responsiveness to drugs [1].
The issue of FDA oversight is important because current draft legislation would likely classify liquid biopsies as high-risk LDTs based on their claims and intended use. While new legislation is clearly needed to ensure patient safety, a balance must be struck so as not to limit the great promise offered by liquid biopsies. Burdensome extra regulations could slow down innovation and adoption, and increased regulatory costs would decrease financial incentives to develop liquid biopsy LDTs. This could be an enormous hurdle in the advancement of patient care, as liquid biopsies are seen as the next leap forward in precision medicine. At this point, the FDA has continued to exercise its authority over LDTs, including LDTs for liquid biopsies. The outcome remains to be seen, as the debate continues and is critical to the development of liquid biopsies in patient testing.
References
Crowley E, Di Nicolantonio F, Loupakis F, Bardelli A. Liquid biopsy: monitoring cancer-genetics in the blood. Nat Rev Clin Oncol. 2013;10(8):472–84.
Swanton C. Intratumor heterogeneity: evolution through space and time. Cancer Res. 2012;72(19):4875–82.
Gerlinger M, Rowan AJ, Horswell S, Larkin J, et al. Intratumor heterogeneity and branched evolution revealed by multiregion sequencing. N Engl J Med. 2012;366(10):883–92.
Ashworth TR. A case of cancer in which cells similar to those in the tumours were seen in the blood after death. Aust Med J. 1896;14:146–7.
Mandel P, Metais P. C R Seances Soc Biol Fil. 1948;142(3–4):241–3.
Raj DA, et al. A multiplex quantitative proteomics strategy for protein biomarker studies in urinary exosomes. Kidney Int. 2012;81(12):1263–72.
Wiggins RC, et al. Procoagulant activity in normal human urine associated with subcellular particles. Kidney Int. 1986;29(2):591–7.
Asea A, et al. Heat shock protein-containing exosomes in mid-trimester amniotic fluids. J Reprod Immunol. 2008;79(1):12–7.
Witwer KW, Buzás EI, Bemis LT, et al. Standardization of sample collection, isolation and analysis methods in extracellular vesicle research. J Extracell Vesicles. 2013;2. doi:10.3402/jev.v2i0.20360.
Caby MP, et al. Exosomal-like vesicles are present in human blood plasma. Int Immunol. 2005;17(7):879–87.
Street JM, et al. Identification and proteomic profiling of exosomes in human cerebrospinal fluid. J Transl Med. 2012;10:5.
Douillard JY, et al. First-line gefitinib in Caucasian EGFR mutation-positive NSCLC patients: a phase-IV, open-label, single-arm study. Br J Cancer. 2014;110(1):55–62.
Cristofanilli M, et al. Circulating tumor cells, disease progression, and survival in metastatic breast cancer. N Engl J Med. 2004;351(8):781–91.
Fidler IJ. The pathogenesis of cancer metastasis: the ‘seed and soil’ hypothesis revisited. Nat Rev Cancer. 2003;3(6):453–8.
Dunn GP, Old LJ, Schreiber RD. The immunobiology of cancer immunosurveillance and immunoediting. Immunity. 2004;21(2):137–48.
Nalejska E, Maczynska E, Lewandowska MA. Prognostic and predictive biomarkers: tools in personalized oncology. Mol Diagn Ther. 2014;18(3):273–84.
Hayes DF, et al. Circulating tumor cells at each follow-up time point during therapy of metastatic breast cancer patients predict progression-free and overall survival. Clin Cancer Res. 2006;12(14 Pt 1):4218–24.
Danila DC, et al. Circulating tumor cell number and prognosis in progressive castration-resistant prostate cancer. Clin Cancer Res. 2007;13(23):7053–8.
Cohen SJ, et al. Relationship of circulating tumor cells to tumor response, progression-free survival, and overall survival in patients with metastatic colorectal cancer. J Clin Oncol. 2008;26(19):3213–21.
Millner LM, Linder MW, Valdes R Jr. Circulating tumor cells: a review of present methods and the need to identify heterogeneous phenotypes. Ann Clin Lab Sci. 2013;43(3):295–304.
Gorges TM, et al. Circulating tumour cells escape from EpCAM-based detection due to epithelial-to-mesenchymal transition. BMC Cancer. 2012;12:178.
Vona G, et al. Isolation by size of epithelial tumor cells: a new method for the immunomorphological and molecular characterization of circulating tumor cells. Am J Pathol. 2000;156(1):57–63.
Ferreira MM, Ramani VC, Jeffrey SS. Circulating tumor cell technologies. Mol Oncol. 2016;10(3):374–94.
Fizazi K, et al. High detection rate of circulating tumor cells in blood of patients with prostate cancer using telomerase activity. Ann Oncol. 2007;18(3):518–21.
Stahlberg A, Bengtsson M. Single-cell gene expression profiling using reverse transcription quantitative real-time PCR. Methods. 2010;50(4):282–8.
Hodgkinson CL, et al. Tumorigenicity and genetic profiling of circulating tumor cells in small-cell lung cancer. Nat Med. 2014;20(8):897–903.
Kalluri R, Weinberg RA. The basics of epithelial-mesenchymal transition. J Clin Invest. 2009;119(6):1420–8.
Riethdorf S, et al. Detection of circulating tumor cells in peripheral blood of patients with metastatic breast cancer: a validation study of the Cell Search system. Clin Cancer Res. 2007;13(3):920–8.
Schneck H, et al. EpCAM-independent enrichment of circulating tumor cells in metastatic breast cancer. PLoS One. 2015;10(12):e0144535.
Karabacak NM, et al. Microfluidic, marker-free isolation of circulating tumor cells from blood samples. Nat Protoc. 2014;9(3):694–710.
Maimonis P, et al. Poster no. 4898. American Association of Cancer Research 102nd Annual Meeting, Orlando, Florida, April 2–6 2011.
Gabriel MT, et al. Circulating tumor cells: a review of non-EpCAM-based approaches for cell enrichment and isolation. Clin Chem. 2016;62(4):571–81.
Vasioukhin V, et al. Point mutations of the N-ras gene in the blood plasma DNA of patients with myelodysplastic syndrome or acute myelogenous leukaemia. Br J Haematol. 1994;86(4):774–9.
Sorenson GD, et al. Soluble normal and mutated DNA sequences from single-copy genes in human blood. Cancer Epidemiol Biomark Prev. 1994;3(1):67–71.
Fleischhacker M, Schmidt B. Circulating nucleic acids (CNAs) and cancer: a survey. Biochim Biophys Acta. 2007;1775(1):181–232.
Medicine Kaiser J. Keeping tabs on tumor DNA. Science. 2010;327(5969):1074.
Higgins MJ, et al. Detection of tumor PIK3CA status in metastatic breast cancer using peripheral blood. Clin Cancer Res. 2012;18(12):3462–9.
Castells A, et al. K-ras mutations in DNA extracted from the plasma of patients with pancreatic carcinoma: diagnostic utility and prognostic significance. J Clin Oncol. 1999;17(2):578–84.
Shinozaki M, et al. Utility of circulating B-RAF DNA mutation in serum for monitoring melanoma patients receiving biochemotherapy. Clin Cancer Res. 2007;13(7):2068–74.
Bettegowda C, et al. Detection of circulating tumor DNA in early- and late-stage human malignancies. Sci Transl Med. 2014;6(224):224ra24.
Esposito A, et al. Monitoring tumor-derived cell-free DNA in patients with solid tumors: clinical perspectives and research opportunities. Cancer Treat Rev. 2014;40(5):648–55.
Gahan PB, Swaminathan R. Circulating nucleic acids in plasma and serum. Recent developments. Ann N Y Acad Sci. 2008;1137:1–6.
Schwarzenbach H, Hoon DS, Pantel K. Cell-free nucleic acids as biomarkers in cancer patients. Nat Rev Cancer. 2011;11(6):426–37.
Anker P, et al. Circulating nucleic acids in plasma or serum. Clin Chim Acta. 2001;313(1–2):143–6.
Allen D, et al. Role of cell-free plasma DNA as a diagnostic marker for prostate cancer. Ann N Y Acad Sci. 2004;1022:76–80.
Chun FK, et al. Circulating tumour-associated plasma DNA represents an independent and informative predictor of prostate cancer. BJU Int. 2006;98(3):544–8.
Sunami E, et al. Quantification of LINE1 in circulating DNA as a molecular biomarker of breast cancer. Ann N Y Acad Sci. 2008;1137:171–4.
Schwarzenbach H, et al. Detection and monitoring of cell-free DNA in blood of patients with colorectal cancer. Ann N Y Acad Sci. 2008;1137:190–6.
Diehl F, et al. Circulating mutant DNA to assess tumor dynamics. Nat Med. 2008;14(9):985–90.
Umetani N, et al. Prediction of breast tumor progression by integrity of free circulating DNA in serum. J Clin Oncol. 2006;24(26):4270–6.
Fleischhacker M, et al. Methods for isolation of cell-free plasma DNA strongly affect DNA yield. Clin Chim Acta. 2011;412(23–24):2085–8.
Slatko BE, Hiraizumi Y. Mutation induction in the male recombination strains of Drosophila melanogaster. Genetics. 1973;75(4):643–9.
Sonnenberg A, et al. Dielectrophoretic isolation and detection of cancer-related circulating cell-free DNA biomarkers from blood and plasma. Electrophoresis. 2014;35(12–13):1828–36.
Lam DC, et al. Plasma EGFR mutation detection associated with survival outcomes in advanced-stage lung cancer. Clin Lung Cancer. 2015;16(6):507–13.
Raposo G, Stoorvogel W. Extracellular vesicles: exosomes, microvesicles, and friends. J Cell Biol. 2013;200(4):373–83.
Santiago-Dieppa DR, et al. Extracellular vesicles as a platform for ‘liquid biopsy’ in glioblastoma patients. Expert Rev Mol Diagn. 2014;14(7):819–25.
Yanez-Mo M, et al. Biological properties of extracellular vesicles and their physiological functions. J Extracell Vesicles. 2015;4:27066.
Andaloussi SEL, Mäger I, Breakefield XO, Wood MJ. Extracellular vesicles: biology and emerging therapeutic opportunities. Nat Rev Drug Discov. 2013;12(5):347–57.
van der Pol E, et al. Classification, functions, and clinical relevance of extracellular vesicles. Pharmacol Rev. 2012;64(3):676–705.
Akers JC, et al. Biogenesis of extracellular vesicles (EV): exosomes, microvesicles, retrovirus-like vesicles, and apoptotic bodies. J Neurooncol. 2013;113(1):1–11.
Minciacchi VR, Freeman MR, Di Vizio D. Extracellular vesicles in cancer: exosomes, microvesicles and the emerging role of large oncosomes. Semin Cell Dev Biol. 2015;40:41–51.
Enderle D, et al. Characterization of RNA from exosomes and other extracellular vesicles isolated by a novel spin column-based method. PLoS One. 2015;10(8):e0136133.
Tauro BJ, et al. Comparison of ultracentrifugation, density gradient separation, and immunoaffinity capture methods for isolating human colon cancer cell line LIM1863-derived exosomes. Methods. 2012;56(2):293–304.
Schageman J, et al. The complete exosome workflow solution: from isolation to characterization of RNA cargo. Biomed Res Int. 2013;2013:253957.
van der Pol E, et al. Optical and non-optical methods for detection and characterization of microparticles and exosomes. J Thromb Haemost. 2010;8(12):2596–607.
Dragovic RA, et al. Sizing and phenotyping of cellular vesicles using nanoparticle tracking analysis. Nanomedicine. 2011;7(6):780–8.
Filipe V, Hawe A, Jiskoot W. Critical evaluation of nanoparticle tracking analysis (NTA) by NanoSight for the measurement of nanoparticles and protein aggregates. Pharm Res. 2010;27(5):796–810.
Momen-Heravi F, et al. Alternative methods for characterization of extracellular vesicles. Front Physiol. 2012;3:354.
Garza-Licudine E, et al. Portable nanoparticle quantization using a resizable nanopore instrument—the IZON qNano. Conf Proc IEEE Eng Med Biol Soc. 2010;2010:5736–9.
de Vrij J, et al. Quantification of nanosized extracellular membrane vesicles with scanning ion occlusion sensing. Nanomedicine (Lond). 2013;8(9):1443–58.
van der Vlist EJ, et al. Fluorescent labeling of nano-sized vesicles released by cells and subsequent quantitative and qualitative analysis by high-resolution flow cytometry. Nat Protoc. 2012;7(7):1311–26.
Ueda K, et al. Antibody-coupled monolithic silica microtips for highthroughput molecular profiling of circulating exosomes. Sci Rep. 2014;4:6232.
Framework for Regulatory Oversight of Laboratory Developed Tests (LDTs) DRAFT GUIDANCE, F.a.D. Adminstration, Editor. 2014.
Clinical Laboratory Improvement Amendments of 1988. 1988, US Government Publishing Office.
Clinical Laboratory Improvement Amendments (CLIA). 4-16-14. Available from: http://www.fda.gov/MedicalDevices/DeviceRegulationandGuidance/IVDRegulatoryAssistance/ucm124105.htm. Accessed 3 Mar 2016.
Survey Procedures and Interpretive Guidelines for Laboratories and Laboratory Services. Available from: https://www.cms.gov/Regulations-and-Guidance/Legislation/CLIA/Interpretive_Guidelines_for_Laboratories.html. Accessed 7 Mar 2016.
Centers for Medicare & Medicaid Services. Clinical laboratory improvement amendments (CLIA). Available from: https://www.cms.gov/Regulations-and-Guidance/Legislation/CLIA/index.html. Accessed 7 Mar 2016.
HR 11124 An Act to amend the Federal Food, Drug, and Cosmetic Act to provide for the safety and effectiveness of medical devices intended for human use, and for other purposes. Library of Congress Thomas; 1976.
Draft Guidance for Industry, Food and Drug Administration Staff, and Clinical Laboratories 2014. Available from: http://www.fda.gov/downloads/medicaldevices/deviceregulationandguidance/guidancedocuments/ucm416685.pdf. Accessed 8 Mar 2016.
Ray T. Amid Competing LDT Regulatory Proposals, Common Ground but Key Disagreements for Congress to Consider. 2015. Available from: https://www.genomeweb.com/molecular-diagnostics/amid-competing-ldt-regulatory-proposals-common-ground-key-disagreements. Accessed 8 Mar 2016.
Acknowledgments
LS, LM and ML contributed to the writing of the manuscript. LS created the figure. ML and RV served as editors for the manuscript.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
LS, LM, ML, and RV declare no conflicts of interest.
Funding
This work was funded by the National Institute of Health Grant # HHSN261201500035C and HHSN261201300034C.
Ethical approval
All procedures performed in studies involving human participants were in accordance with the ethical standards of the institutional and/or national research committee and with the 1964 Helsinki declaration and its later amendments or comparable ethical standards.
Rights and permissions
About this article

Cite this article
Strotman, L.N., Millner, L.M., Valdes, R. et al. Liquid Biopsies in Oncology and the Current Regulatory Landscape. Mol Diagn Ther 20, 429–436 (2016). https://doi.org/10.1007/s40291-016-0220-5
Published:
Issue Date:
DOI: https://doi.org/10.1007/s40291-016-0220-5