
Abstract
Introduction
Several DNA polymorphisms have been associated with high production of fetal hemoglobin (HbF), although the molecular basis is not completely understood. In order to identify and characterize novel HbF-associated elements, we focused on five probands and their four families (from Egypt, Iraq and Iran) with thalassemia major (either β0-IVSII-1 or β0-IVSI-1) and unusual HbF elevation (>98 %), congenital or acquired after rejection of bone marrow transplantation, suggesting an anticipated favorable genetic background to high HbF expression.
Methods
Patient recruitment, genomic DNA sequencing, western blotting, electrophoretic mobility shift assays, surface plasmon resonance (SPR) biospecific interaction analysis, bioinformatics analyses based on docking experiments.
Results
A polymorphism of the Aγ-globin gene is here studied in four families with β0-thalassemia (β0-IVSII-1 and β0-IVSI-1) and expressing unusual high HbF levels, congenital or acquired after rejection of bone marrow transplantation. This (G→A) polymorphism is present at position +25 of the Aγ-globin genes, corresponding to a 5′-UTR region of the Aγ-globin mRNA and, when present, is physically linked in chromosomes 11 of all the familiar members studied to the XmnI polymorphism and to the β0-thalassemia mutations. The region corresponding to the +25(G→A) polymorphism of the Aγ-globin gene belongs to a sequence recognized by DNA-binding protein complexes, including LYAR (Ly-1 antibody reactive clone), a zinc-finger transcription factor previously proposed to be involved in down-regulation of the expression of γ-globin genes in erythroid cells.
Conclusion
We found a novel polymorphism of the Aγ-globin gene in four families with β0-thalassemia and high levels of HbF expression. Additionally, we report evidence suggesting that the Aγ-globin gene +25(G→A) polymorphism decreases the efficiency of the interaction between this sequence and specific DNA binding protein complexes.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
A novel Aγ-globin-gene polymorphism is found associated with XmnI Gγ-globin-gene polymorphism and β0-globin-gene mutations. |
This genetic background might be required, although not sufficient for high expression of γ-globin-genes and production of HbF. |
The Aγ-globin-(G→A) polymorphism decreases the binding efficiency of nuclear factors to the binding sequences present in the Aγ-globin gene. |
1 Introduction
Increased expression of γ-globin gene and high fetal hemoglobin (HbF) production in β-thalassemia patients are associated with an amelioration of clinical severity [1–5]. In several cases, β-thalassemia patients producing high HbF do not need transfusion regimen and, consequently, chelation therapy [6, 7]. This well recognized finding has prompted the search for HbF inducers to treat β-thalassemia patients expressing low HbF levels [8–16]. On the other hand, the search for HbF-associated polymorphisms has recently gained great attention, in order to stratify β-thalassemia patients with respect to expectancy of the first blood transfusion, need for annual intake of blood, response to HbF inducers (the most studied of which is hydroxyurea) [17–19].
The issue of the so called “HbF modifiers” and associated polymorphisms within genes directly or indirectly linked with high production of HbF has been the object of several studies in the field of β-thalassemia, well discussed in a key paper by Thein and Menzel [20], reporting the progress in the understanding of the persistence of HbF in adults. Three major loci (XmnI-HBG2 single nucleotide polymorphism, HBS1L-MYB intergenic region on chromosome 6q, and BCL11A) contribute to high HbF production. It should be pointed out that the identification of these three major loci might help in identifying putative HbF-associated transcription factors (most of which belong to transcription repressors of the γ-globin gene) as well as binding sites for these transcription repressors [21–24].
The fact that several HbF-related polymorphisms probably act in synergy maintains high interest in finding novel HbF-associated genetic biomarkers. This field of investigation, in addition to a clear interest in diagnostics and prognostics, might contribute to the development of novel therapeutic options, in the case the polymorphism(s) is (are) associated with novel therapeutic markers [20].
In order to identify novel HbF-associated elements, we focused on five probands and their four families (from Egypt, Iraq and Iran) with thalassemia major (either β0-IVSII-1 or β0-IVSI-1) and unusual HbF elevation (>98 %), congenital or developed after rejection of allogeneic bone marrow transplantation (BMT) [25–27] suggesting an anticipated favorable genetic background to high HbF expression. In such selected families, analysis of the XmnI HbF-associated polymorphisms and full sequencing of the Aγ- and Gγ-globin genes were performed.
2 Materials and Methods
2.1 Patients and Clinical Examination
Among consecutive patients affected by β-hemoglobin disorders and referred at the International Centre for Transplantation in Thalassemia and Sickle Cell Anaemia, (Mediterranean Institute of Haematology, Policlinic of “Tor Vergata” University, Rome, Italy) to perform allogenic BMT, we observed four families with unusual elevations of the expression of fetal hemoglobin. In these four families we identified five thalassemic patients with very high HbF levels (99 %), congenital or acquired after transplant rejection and associated to transfusion independency. We performed genetic studies of patients and their families. The present study has been conducted according to the principles expressed in the Declaration of Helsinki and in full compliance with the guidelines of the Mediterranean Institute of Hematology, Policlinic of Tor Vergata, Rome, Italy. Ethics Committee’s approval for the research has been obtained. All patients and family members provided appropriate informed consent. Clinical parameters of patients and family members are reported in Table 1. In order to compare the genetic data reported on these families with other thalassemic patients and respective family members, we screened a cohort of 183 subjects from randomly selected β-thalassemia families from East or West Mediterranean areas and 43 healthy subjects.
2.2 Capillary electrophoresis (CE)
High voltage CE was performed by using the Minicap Flex Piercing capillary system (Sebia, Lisses, France). Sample processing required a 1:6 dilution of 50 μl whole blood with hemolysing solution and vortexing for 5 s. After loading the primary sample tubes into the carousel, the instrument performed automated bar code reading, mixing of the samples by inversion, cap piercing, sampling, and dilution. Electrophoresis was performed at alkaline pH (9.4), high voltage (9500 V), and controlled temperature. The hemoglobin bands were detected by absorption photometry, and optical density measurements were converted to an electropherogram. The migration position was measured in arbitrary units between 0 and 300 and was quantified as a percentage. Results were acquired and examined by using the Sebia Phoresis REL 8.6.2 Software [28, 29]. The standard controls were the purified HbA (Sigma-Aldrich) and HbF (Alpha Wassermann, Milano, Italy).
2.3 Extraction of Genomic DNA
The DNA was extracted from 500 μl of whole blood by using the QIAamp DNA Blood Mini Kit (Qiagen, Hilden, Germany), according to the manufacturer’s instructions. The DNA obtained was analyzed by agarose gel electrophoresis and quantified by using the SmartSpec Plus spectrophotometer (Bio-Rad, Hercules, CA, USA).
2.4 Polymerase Chain Reaction and Sequencing Reaction
Each polymerase chain reaction (PCR) was carried out in a final volume of 50 μl, in the presence of 200 ng of template genomic DNA, 1× Ex Taq Buffer with 2 mM MgCl2 (TaKaRa, Otsu, Shiga, Japan), 0.8 mM dNTPs, 0.5 μM forward and reverse primers, 1.25 U of TaKaRa Ex Taq DNA polymerase (TaKaRa) and ultrapure water. After an initial denaturation step of 2 min at 94 °C, the 35 PCR cycles used were as follows: denaturation, 30 s at 94 °C; annealing, 30 s at 65 °C; elongation, 1 min at 72 °C. In order to amplify sequences of β, Aγ- and Gγ-globin genes, the following primer pairs were employed, respectively: βF1: 5′-GTG CCA GAA GAG CCA AGG ACA GG-3′; βR1: 5′-AGT TCT CAG GAT CCA CGT GCA-3′; βF2: 5′-GCC TGG CTC ACC TGG ACA-3′; βR2: 5′-GTT GCC CAG GAG CTG TGG-3′; βF3: 5′-ACA ATC CAG CTA CCA TTC TGC TTT-3′; βR3: 5′-CAC TGA CCT CCC ACA TTC CCT TTT-3′; AγF: 5′-TTT CCT TAG AAA CCA CTG CTA ACT GAA A-3′; AγR: 5′-TTG TGA TAG TAG CCT TGT CCT CCT CT-3′; GγF: 5′-TTC TTA TTT GGA AAC CAA TGC TTA CTA AAT-3′; GγR: 5′-TTG TGA TAG TAG CCT TGT CCT CCT CT-3′. PCR products were analyzed by agarose gel electrophoresis and then purified from unincorporated primers by using MicroCLEAN (Microzone Limited, Haywards Heath, West Sussex, UK) reagent, according to the manufacturer’s instructions. Finally, purified PCR products were sequenced by using the forward and reverse PCR primers and the ABI PRISM BigDye Terminator Cycle Sequencing Ready Reaction Kit, v1.0 (Applied Biosystems, Life Technologies, Carlsbad, CA, USA). Sequencing reactions were performed in a final volume of 20 μl, containing 60 ng of PCR template, 3.2 pmoles of sequencing primer, 8 μl of Terminator Ready Reaction Mix and ultrapure water. A total of 45 amplification cycles were performed, as follows: denaturation, 96 °C, 10 s; annealing, 65 °C, 5 s; elongation, 65 °C, 3 min. The reaction products were purified from unincorporated dideoxyribonucleotides (ddNTPs) by using a 96-well MultiScreen plate (Merck Millipore, Merck KGaA, Darmstadt, Germany) containing Sephadex G-50 (GE Healthcare, Little Chalfont, Buckinghamshire, UK). Sequencing was finally performed by BMR Genomics (Padua, Italy) and the obtained sequence data were analyzed by the Sequence Scanner, version 1.0 software (Applied Biosystems, Life Technologies).
2.5 Western Blotting
For protein extract preparation, K562 cells [30] were lysed in an ice-cold RIPA buffer (10 Mm Tris-HCl, pH 8.0, 0.5 mM EDTA, 150 mM NaCl, 1 % NP40, 0.1 % SDS, 5 mg/ml DeoxyCholic acid, 1 mM DTT, 2 mM PMSF, 2 mM Na3VO4, 10 mM NaF, 1 μg/ml Leupeptin, 1 μg/ml Aprotinin). Briefly, K562 cells (8 × 106 cells) were collected and washed twice with cold PBS (Phosphate-Buffered Saline, Lonza-Biowhittaker, Basel, Switzerland). Cellular pellets were then suspended with 400 µl of cold RIPA buffer, incubated on ice for 20 min and subjected to five cycles of freeze-thawing. Samples were finally centrifuged at 14,000×g for 3 min at 4 °C and the supernatants were collected and immediately frozen at −80 °C. Protein concentration was determined using Pierce™ BCA Protein Assay Kit (Thermo Fisher Scientific, Rockford, USA). Twelve μg of protein extracts were denatured for 5 min at 98 °C in 1× SDS sample buffer (62.5 mM Tris-HCl pH 6.8, 2 % SDS, 50 mM Dithiotreithol (DTT), 0.01 % bromophenol blue, 10 % glicerol) and loaded on SDS-PAGE gel (10 cm × 8 cm) in Tris-glycine Buffer (25 mM Tris, 192 mM glycine, 0.1 % SDS). The protein standard Precision Plus Protein WesternC Standard (size range of 10–250 kDa) (Bio-Rad Laboratories, MI, Italy) was used as standard to determine molecular weight. Proteins were blotted onto a 20 microns nitrocellulose membrane using the ready to use Trans-Blot Turbo Transfer Packs in combination with the Trans-Blot Turbo transfer system (BioRad) at 2.5 A and ~25 V for 10 min. The membrane were pre-stained in Ponceau S Solution (Sigma Aldrich) to verify the transfer, washed with 25 ml TBS (10 mM Tris-HCl pH 7.4, 150 mM NaCl) for 10 min at room temperature and incubated in 25 ml of blocking buffer for 2 h at room temperature. The membranes were washed three times for 5 min each with 25 ml of TBS/T (TBS, 0.1 % Tween-20) and incubated with LYAR primary rabbit polyclonal antibody (1:500) (Cat. TA315594, OriGene Technologies, Rockville, MD, USA) in 10 ml primary antibody dilution buffer with gentle agitation over night at 4 °C. The day after, the membrane were washed three times for 5 min each with 20 ml of TBS/T and incubated in 15 ml of blocking buffer, in gentle agitation for 2 h at room temperature, with an appropriate HRP-conjugated secondary antibody (1:2000) and a Precision Protein StrpTactin-HRP conjugate (1:10,000) (Bio-Rad) used to detect with great affinity and specificity the primary antibody and the Strep-tag amino acid sequences present in the protein marker, respectively. Finally, after three washes each with 20 ml of TBS/T for 5 min, the membranes were incubated with 10 ml LumiGLO (0.5 ml 20× LumiGLO, 0.5 ml 20× Peroxide and 9.0 ml Milli-Q water) (Cell Signaling) in gentle agitation for 5 min at room temperature and exposed to X-ray film (GE Healthcare, Buckinghamshire, UK). As necessary, after stripping procedure using the Restore Western Blot Stripping Buffer (Pierce) membranes were re-probed with primary and secondary antibodies. X-ray films for chemiluminescent blots were analyzed by Gel Doc 2000 (Bio-Rad) using Quantity One program to elaborate the intensity data of our specific target protein. The primary antibody against p70S6K (cat. 2708, Cell Signaling) was used as normalization control [31, 32].
2.6 Preparation of K562 Nuclear Extracts
Nuclear extracts from K562 cells were prepared as described elsewhere [33]. Briefly, K562 cells were collected, washed twice with ice-cold phosphate-buffered saline, and suspended in 0.4 ml/107 cells of hypotonic lysis buffer (10 mM Hepes/KOH, pH 7.9, 10 mM KCl, 1.5 mM MgCl2, 0.5 mM DTT, and 0.2 mM phenylmethanesulfonyl fluoride). After incubation on ice for 10 min, the mixture was vortexed for 10 s, and nuclei were pelleted by centrifugation at 12,000g for 10 s, then nuclear proteins were extracted by incubation of the nuclei for 20 min on ice with intermittent gentle vortexing in 20 mM Hepes/KOH, pH 7.9, 25 % glycerol, 420 mM NaCl, 1.5 mM MgCl2, 0.2 mM EDTA, 0.5 mM DTT, 0.2 mM phenylmethanesulfonyl fluoride, 1 μg ml−1 aprotinin, 1 μg ml−1 leupeptin, 2 mM Na3VO4, and 10 mM NaF (Sigma, St Louis, MO, USA); cell debris was removed by centrifugation at 12,000 g for 5 min at 4 °C. The Pierce BCA Protein Assay Kit (Thermo Fisher Scientific, Waltham, MA, USA) was used to measure the protein concentration in the extract, which was then stored in aliquots at −80 °C.
2.7 Electrophoretic Mobility Shift Assays (EMSA)
The sequence of the double-stranded oligonucleotides (ODNs) mimicking the +25 Aγ-globin gene sequences and used in EMSA were 5′-AAC GTC TGA GGT TAT CAA TAA GCT-3′ (+25 Aγ-globin gene sequence) and its complementary stretch. Three pmol of the +25 Aγ-globin ODNs were 32P-labeled using OptiKinase (GE Healthcare, Chalfont St Giles, UK), annealed to an excess of complementary ODN and purified from [γ-32P]ATP (Perkin Elmer, Wellesley, MA, USA). Binding reactions were performed by incubating 2–4 µg of K562 nuclear extract and 16 fmol of 32P-labeled double-stranded of +25 Aγ-globin ODN in a final volume of 20 µL of binding buffer (20 mM Tris-HCl, pH 7.5, 50 mM KCl, 1 mM MgCl2, 0.2 mM EDTA, 5 % glycerol, 1 mM DTT, 0.01 % TritonX100, 0.05 µg µL−1 of poly dI–dC, 0.05 µg µL−1 of a single-stranded ODN). After 20 min incubation at room temperature, samples were immediately loaded onto a 6 % non-denaturing polyacrylamide gel containing 0.25 × Tris/borate/EDTA (22.5 mM Tris, 22.5 mM boric acid, 0.5 mM EDTA, pH 8) buffer. Electrophoresis was carried out at 200 V. Gels were vacuum heat-dried and subjected to autoradiography [32, 34–36]. Assays in the presence of anti-LYAR monoclonal antibody were performed as described previously [31, 36, 37] by using rabbit polyclonal anti-LYAR antibody (AB89469, ABCAM, Cambridge, MA, USA).
2.8 SPR-BIA Technology
Interactions of K562 nuclear factors with double-stranded DNA sequences were studied by Surface Plasmon Resonance (SPR) analysis on Biacore X100 (Biacore, GE Healthcare) [38, 39]. SA sensor chips (Biacore, GE Healthcare) were employed pre-coated with streptavidin. SA sensor chips containing single-stranded normal or mutated +25 and MUT(+25) Aγ-globin ODNs (+25 Aγ-globin gene sequence: 5′-AAC GTC TGA GGT TAT CAA TAA GCT-3′; MUT +25 Aγ-globin gene sequence, G→A mutation underlined: 5′-AAC GTC TGA GAT TAT CAA TAA GCT-3′) were obtained following injection of 1 μM biotinylated ODNs in HBS-EP+ buffer (10 mM HEPES, 150 mM NaCl, 3 mM EDTA, 0.05 % v/v Surfactant P20, pH 7.4) (Biacore, GE Healthcare) on the sensor chip surface, in order to reach an immobilization level of about 700–800 resonance units (RU). Double-stranded target ODNs immobilized on the sensor chips were obtained by a 4 min injection of 2.5 μM complementary ODN diluted in HBS-EP + buffer on the flow cell containing the biotinylated single-stranded of +25 and MUT(+25) Aγ-globin ODN. HBS-EP + buffer was employed as running buffer. In order to study protein interactions to the immobilized double-stranded ODNs, 3.5 μg of K562 nuclear extracts were pre-incubated with 0.6 μg of poly(dI:dC) for 5 min and then injected in the sensor chip flow cells at the concentration of 50 μg/ml for 6 min. The injection of K562 nuclear extracts was performed in binding buffer (buffer TF: 50 mM KCl, 20 mM Tris-HCl pH 7.5, 1 mM MgCl2, 0.2 mM EDTA, 0.01 % Triton X-100). For determining the stability of protein/DNA complexes, injection of TF buffer was performed. The chips were regenerated with a 30-s injection of 1 M NaOH. All procedures were performed at 25 °C and 5 μl/min flow rate.
2.9 Bioinformatic Analyses
All the computational studies were carried out on a 4 CPU (Intel Core2 Quad CPU Q9550, 2.83 GHz) ACPI x64 Linux workstation with Ubuntu 12.04 operating system [40]. The DNA sequence (GAGGTTATCA) containing the consensus sequence GGTTAT was built through the 3D-DART modeling server [41]. The solution structure of the N-terminal zinc finger domain of LYAR (NMR spectroscopy; PDB ID: 1WJV) was downloaded from the Protein Data Bank. The protein was derived from Mus musculus, but the resolved sequence showed high similarity with the one of Homo sapiens (Homo sapiens sequence: MVFFTCNAC GESVKKIQVE KRVSVCRNCE CLSCIDCGKD FWGDDYKNHV KCISEDQKYG GKGYEGK; Mus musculus sequence: MVFFTCNAC GESVKKIQVE KHVSVCRNCE CLSCIDCGKD FWGDDYKSHV KCISEDQKYG GKGYEAK (differences in the sequences are underlined). The 1WJV pdb file contained 20 different structures all characterized by an initial GSSGSSG and terminal SGPSSG sequences that were removed. Before docking simulations, all the molecules were independently contained in a box, hydrated and neutralized adding chlorine and sodium ions and fully minimized with Gromacs 4.6.7 software [42], using the CHARMM27 force field. The HADDOCK web server was used for docking simulations [43, 44]. The amino acid 1, 2, 14, 15, 16, 17, 18, 19, 20, 48, 49 and 50 were set as the protein binding site. There were 200 different binding poses generated and clustered by the server, finally furnishing the most plausible complex. The selected binding pose was finally submitted to energy minimization using Gromacs software [42]. The binding mode was analyzed with PyMOL software [45]. The protein electrostatic potential surface was computed with the APBS tools of PyMOL, starting from a “.pqr” file generated with the pdb2pqr web server, using the CHARMM force field [46].
2.10 Statistical Analysis
All the data were normally distributed and presented as mean ± SD. Statistical differences between groups were compared using one-way ANOVA (ANalyses Of VAriance between groups) software. The p values were obtained using the paired t test of the GraphPad Prism Software. Statistical differences were considered significant when p < 0.05 (*), highly significant when p < 0.01 (**) [36].
3 Results
3.1 HbF-Associated XmnI Polymorphism
The levels of HbF in the members of the four studied families are reported in Figs. 1 and 2. Sequence analysis of the β-globin gene of patients and parents revealed that they carry mutated β0-IVSI-1 or β0-IVSII-1 globin genes. Notably, three β-thalassemia patients (A3, B3 and D3), who required regular blood transfusion pre-transplant, developed a high-HbF phenotype (>97 % HbF) after rejection of allogeneic bone marrow transplantation (BMT). Supplementary Figure 1 shows that these increased levels of HbF are highly significant when compared to HbF starting pre-transplant levels (see also Table 1). Indeed, 40–182 days after transplant and after the autologous reconstitution, the patients developed full and stable production of HbF (about 99 %), maintaining steady hemoglobin levels over 10.5 g/dl without transfusion support. The post-rejection HbF levels were significantly increased (see Table 1; Supplementary Figure 1). In detail, HbF was increased from 1.4–6.6 to 10.0–12.9 g/dl in the three studied patients. The pattern of hemoglobin production is shown in Supplementary Figure 2 and compared to patient B5, who produced high HbF levels (>98 %) since birth and was transfusion free despite the genetic diagnosis of β0-thalassemia. Patients A4, A5, B4 and C3 have been successfully transplanted and produce HbA in agreement with the genetic background and HbF levels of the respective donors (see Table 1 for clinical parameters). In accordance with the clinical parameters, patient B5 did not require BMT, while the patient C3 was enrolled for allogeneic BMT considering the severe secondary erythroid hyperplasia and severe anemia despite of the long hydroxyurea treatment performed in order to maintain high HbF levels. Without exception, these families exhibited a segregation of the XmnI(+) polymorphism with the β0-IVSI-1 and β0-IVSII-1 globin genes. In fact, all the homozygous β0-IVSII-1/β0-IVSII-1 and β0-IVSI-1/β0-IVSI-1 subjects were XmnI(+/+), while all the heterozygous parents (see for instance subjects A1, A2, B2 and C2) transmitted to the heterozygous or homozygous β0-IVSII-1 and β0-IVSI-1 progeny both β0-IVSII-1 (or β0-IVSI-1) and XmnI(+) polymorphism. In consideration of this strong genetic linkage, the full sequencing of both the Gγ-globin and Aγ-globin genes was performed in order to identify other possibly linked polymorphisms.

Genetic tree of the first β-thalassemia family studied. The fetal hemoglobin (HbF) levels (% of total hemoglobins analyzed by capillary electrophoresis) are indicated in red characters. Notably, patient A3, who exhibited intermediated pre-transplantation HbF levels (75.8 %), developed a high-HbF phenotype after BMT rejection (see Table 1 for clinical details). Patients A4 and A5 have been successfully transplanted and produce HbF in agreement with the genetic background and HbF levels of the respective donors. The status of the Gγ-globin-158 XmnI and Aγ-globin +25(G→A) polymorphisms are indicated in green and blue characters, respectively. The β0-thalassemia mutation of the family is indicated on the top. The probands are indicated by black arrows

Genetic trees of the other three β-thalassemia families studied. As in Fig. 1, the fetal hemoglobin (HbF) levels are indicated in red characters. The status of the Gγ-globin-158 XmnI and Aγ-globin +25(G→A) polymorphisms are indicated in green and blue characters, respectively. The β0-thalassemia mutations of all the families are indicated on the top. The probands are indicated by black arrows. Patients B3 and D3, who exhibited low pre-transplantation HbF levels (76.3 and 77.4 %, respectively), developed a high-HbF phenotype after BMT rejection (see Table 1 for clinical details). On the other hand, patient B5 produces high HbF levels (>98 %) since birth and is transfusion free despite the genetic diagnosis of β0-thalassemia. Patients B4 and C3 have been successfully transplanted and produce HbF in agreement with the genetic background and HbF levels of the respective donors. BMT bone marrow transplantation, N.A. not available
3.2 Identification of the +25(G→A) Polymorphism within the Aγ-Globin Gene
Supplementary Figure 3A shows the sequence of the Aγ-globin gene as it can be found in the majority of the β-thalassemia population of the north/north-west Mediterranean area. As it is evident, from nucleotide +22 to nucleotide +31 of the Aγ-globin gene the 5′-GAG GTT ATC A-3′ sequence is present. Interestingly, we found that in all the patients with XmnI(+/+) polymorphism the sequence is mutated in both alleles to 5′-GAG ATT ATC A-3′ (mutated nucleotide underlined). The representative sequences of these patients are shown in Supplementary Figure 3, panels b–g. In particular, sequences of patient A3 (Supplementary Figure 3, d and g) indicate an homozygous state of Gγ-XmnI(+/+) and Aγ-(G→A)(A/A) polymorphisms. Interestingly, the genotypes Gγ-XmnI(+/+) and Aγ-(G→A)(A/A) were found in all the patients producing high HbF levels, including those who developed high HbF levels following BMT rejection (A3, B3 and D3 see Figs 1, 2). It should be noted that this single nucleotide variation (schematically represented in Fig. 3a) is present in the NCBI-dbSNP (rs368698783) although not associated with β-thalassemia and any biological function. In this respect, this Aγ-globin-gene polymorphism corresponds to the 5′-UTR sequence of the Aγ-globin mRNA and belongs to the binding site of LYAR (5′-GGTTAT-3′), a recently described repressor of γ-globin gene in K562 cells [37].
3.3 The +25(G→A) Polymorphism within the Aγ-Globin Gene is Associated with the XmnI(+) Polymorphism
Interestingly, the (G→A) Aγ-globin-gene polymorphism was found to be linked to the XmnI (+) polymorphism in all of the family members studied. Moreover, when the family trees are comparatively analyzed, it appears clear that in these families the β0-IVSII-1 and the β0-IVSI-1 genotypes are associated with the Gγ-globin-XmnI(+) and Aγ-globin-(G→A) polymorphisms (see the scheme of Fig. 3a underlying the chromosome 11 pattern found in the majority of the subjects analyzed), strongly suggesting that this sequences association was selected in these populations. Of great interest is to note that in these families we have no evidence for a Gγ-globin-XmnI(+) polymorphism in the absence of an Aγ-globin-(G→A) polymorphisms and, conversely, for an Aγ-globin-(G→A) polymorphisms without a Gγ-globin-XmnI(+) polymorphism. The association between Gγ-globin-XmnI(+) and Aγ-globin-(G→A) polymorphisms is not restricted to the patients presented in this study, but this was confirmed in a cohort of 89 subjects from randomly selected β-thalassemia families of the eastern Mediterranean area, which includes Egypt, Kurdistan, Iran and Iraq (supplementary Table 1). Moreover, it was firmly demonstrated in a cohort of 94 subjects from β-thalassemia families of the western Mediterranean area, including 31 β039/β039, 9 β039/β+IVSI-110, 33 β+IVSI-110/β+IVSI-110, one β0IVSI-1/β+IVSI-6 and one β039/β+IVSI-6 patients (Breveglieri et al., manuscript in preparation and Supplementary Table 2) and in a cohort of 43 healthy Caucasian subjects randomly selected in the western Mediterranean area (supplementary Table 3). In the cohorts of subjects reported in the present paper we were unable to find a lack of association between the Aγ-globin-(G→A) and the XmnI(+) polymorphisms. Overall, subjects showing a lack of association between Gγ-globin-XmnI(+) and Aγ-globin-(G→A) polymorphisms are not frequent (<5 %, see Supplementary Tables).

Genetic linkage and biological effects of the Aγ-globin +25(G→A) polymorphism. a Genetic linkage of the β-globin IVSII-1 (or IVSI-1) gene, Aγ-globin +25(G→A) polymorphism and Gγ-globin-158 XmnI polymorphism. b Western blotting showing the presence of LYAR in K562 cell extracts. c Electrophoretic mobility shift assay (EMSA) studies showing the effects of an anti-LYAR monoclonal antibody on the interactions between K562 nuclear factors and +25 Aγ-globin-gene sequences. In this experiment K562 nuclear extracts were incubated for 15 min with increasing amounts on anti-LYAR antibody or control antibody, as indicated (see Sect. 2) and then added to 32P-double stranded +25 Aγ-globin ODN. After a further 15-min incubation, the complexes (arrowed) were analyzed by polyacrylamide-gel electrophoresis. d, e SPR-BIA study performed using K562 nuclear extracts and sensor chips containing immobilized +25 (solid lines) and MUT(+25) (dotted lines) Aγ-globin oligonucleotides (ODNs). After double-stranded ODN generation (see Supplementary Fig. S4) K562 nuclear extracts were injected in TF buffer (TFB). After this association step, dissociation was studied by injecting TFB on the flow cells. RUfin (final RUs, i.e. the RU levels obtained after the association phase) and RUres (residual RUs, i.e. the RU levels obtained after the dissociation phase) are indicated in the representative experiment shown in panel d. Panel e shows a summary of values of RUfin and RUres obtained after injection of K562 nuclear extracts to immobilized +25 (black boxes) and MUT(+25) (grey boxes) Aγ-globin double-stranded ODNs. Results represent the average ± SD of four independent experiments
3.4 Effects of the Aγ-Globin-(G→A) Gene Polymorphism
A possible hypothesis explaining how the Aγ-globin-(G→A) gene polymorphism contributes in elevated HbF levels is the altered recognition by DNA-binding proteins involved in γ-globin-gene regulation. Interestingly, the Aγ-globin (G→A) polymorphism belongs to the 5′-GGTTAT-3′ binding site of LYAR (Ly-1 antibody reactive clone), a zinc-finger transcription factor recently proposed as a repressor of γ-globin gene in K562 cells [37]. To verify the possibility that the Aγ-globin-(G→A) gene polymorphism affects the binding efficiency of nuclear factors to the LYAR 5′-GGTTAT-3′ binding site, SPR-BIA [38, 39] and molecular docking simulations [40] were performed. When direct binding was performed in EMSA [34–36] using K562 nuclear extracts, interaction to the +25 Aγ-globin gene LYAR sequences was detected. In these nuclear extracts LYAR is present (Fig. 3b) and, more importantly, plays a critical role in binding to the +25 Aγ-globin-gene sequences. In fact, the generation of protein/DNA complexes is highly reduced when the binding reaction between K562 nuclear extracts and DNA is performed in the presence of an antibody recognizing LYAR (Fig. 3c, left side of the panel). The strong inhibition of protein/DNA complex formation when a LYAR antibody is used was not observed when a control antibody was employed (Fig. 3c, right side of the panel). Interestingly, when SPR-BIA was performed using sensor chips containing immobilized +25 and MUT(+25) Aγ-globin ODNs (see Supplementary Fig. 4 for probe immobilization and double-strand ODN generation), a significant difference was found in the binding of K562 nuclear extracts to +25 (solid lines) and MUT(+25) (dotted lines) Aγ-globin sequences (Fig. 3d), being the binding less efficient to the MUT(+25) sequence. These differences were evident also during the dissociation phase, indicating that the DNA-protein complexes generated are less stable (see Fig. 3e for a summary of four independent experiments). To determine possible reasons for the differential recognition by LYAR of +25 and MUT(+25) Aγ-globin sequences, molecular docking simulations were performed. Docking simulations showed a marked electrostatic and shape complementarities between LYAR and the DNA consensus sequence 5′-GGTTAT-3′ (Fig. 4a). Besides the expected interactions between the phosphate backbone of the nucleic acid and the largely positive surface of the protein, however, the docking simulations suggested three less obvious key interactions (Fig. 4, panels b, c): (a) the ammonium ion of the LYS20 side chain interacted with the N7 of the GGTTAT (Lysproton/guanineN7 distance = 2.4 Å), with the carbonyl of the GGTTAT (Lysproton/guanineoxygen distance = 2.6 Å) and with the carbonyl of the GGTTAT (Lysproton/guanineoxygen distance = 2.1 Å); (b) the methyl functions of the GGTTAT sequence were accommodated in a cleft generated by the side chains of LYS14, ILE16 and GLN17; (c) the methyl of the thymine in the complement sequence of the GGTTAT was accommodated in a cleft generated by the side chains of MET1, LYS15 and ILE16. As sketched in Fig. 4c, the docking simulation suggested the importance of almost all residues in the GGTTAT sequence (or of its coupled sequence). Interestingly the G→A polymorphism does not allow the generation of one of the hydrogen bonds, suggesting that this polymorphism might be associated with decrease of LYAR-DNA recognition efficiency.

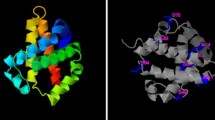
Molecular docking simulations. a H-bonds between LYS20 and the neighboring guanines in GGTTAT sequence. LYAR is depicted as green cartoon; DNA is depicted as cyan cartoon. Details of the protein clefts interacting with the thymine of the GGTTAT sequence and of its complementary CCAATA sequence are shown. LYAR is depicted as electrostatic surface; DNA is depicted as cyan lines. b Schematic representation of the proposed binding mode between LYAR and DNA consensus sequence. All images were done with PyMol. c The binding mode proposed for the LYAR/DNA complex involves the formation of hydrogen bridges between the adjacent LYS27 and the two guanines of the sequence GGTTAT. The thymine of the consensus sequence, with the adjacent thymine paired to adenine are found to establish hydrophobic interactions with the only two lipophilic cavity on the surface of LYAR, formed, respectively, by LYS22, ILE23, MET8 and LYS22, ILE23 and ARG24. These interactions were not detected in the complex LYAR/mutated DNA
4 Conclusions
Clinical observations have shown that increased levels of fetal hemoglobin (HbF) can ameliorate the severity of β-hemoglobin disorders, including β-thalassemia [1–5]. The high HbF level is associated with several and different genetic factors on different chromosomes, including polymorphisms found in the MYB [21] and BCL11A [27, 47] loci and the XmnI polymorphism of the Gγ-globin gene [48]. Our sequencing study on four selected thalassemic families with high HbF levels provided the unique opportunity to define that the XmnI polymorphism is always associated with a novel polymorphism found within the 5′-UTR sequence of the Aγ-globin-gene (the +25 G→A polymorphism). The Gγ-globin-XmnI(+) and Aγ-globin-(G→A) polymorphisms were associated with high HbF production in all the patients studied. Notably, these polymorphisms were identified as well in three patients who underwent allogeneic BMT (A3, B3 and D3) and developed stable high HbF production after BMT rejection, suggesting that this genetic background may be favorable and participates in the high HbF production even in adult age. In addition, our data show that in β-thalassemia the Gγ-globin-XmnI(+)/Aγ-globin-(G→A) genotype is under genetic linkage with the β0-thalassemia mutations. As far as functional roles of the regions involved in the Gγ-globin-XmnI(+) and Aγ-globin-(G→A) polymorphisms, a possible cross-talk between transcription factors binding to these two regions is a very exciting hypothesis. One of the possible transcription factors involved in binding to the region including the Aγ-globin-(G→A) sequence has been recently proposed by Ju et al [37], who identified LYAR as a transcription factor binding to the 5′-GGTTAT-3′ 5′-UTR region of the Aγ-globin gene. Interestingly, (a) LYAR is a repressor of Aγ-globin-gene expression in erythroid cells (K562 cells and erythroid precursor cells) [37] and (b) nucleotide variations (including the G→A polymorphism described here) lead to changes in Aγ-globin gene expression [37]. Based on our SPR-based Biacore experiments and docking analysis, we can conclude that the Aγ-globin-(G→A) polymorphism, frequent in family members producing high HbF levels, modifies the binding efficiency of nuclear factors (including LYAR, a putative repressor of γ-globin-gene expression). Therefore this point mutation may participate in the high HbF production exhibited by these studied patients. However, we should underline that, when informative siblings of these families are comparatively analyzed, this Gγ-globin-XmnI(+) and Aγ-globin-(G→A) polymorphisms association does not justify by itself the high HbF production; rather it can be considered important but not sufficient. For example, individual B5 was transfusion-independent whereas the B3 and B4 siblings with same β-globin genotype and γ-globin-gene polymorphisms were transfusion-dependent. Individuals with the +25 polymorphism in homozygous state but heterozygous for β-thalassemia alleles did not show significant elevation of HbF level (for instance C1 and D1), with the exception of B1, who exhibits a quite relevant level of HbF (17.6 %) and consequently B4 who, after transplant, exhibits the same high HbF levels of B1, her bone marrow donor. Therefore, it is apparent that other factors should also be involved in high HbF production. Further experiments will be required to clarify this point, including transcriptomic and proteomic analyses, as well as studies on other loci known to modify HbF level (BCL11A, HBS1L-MYB and KLF1) [21, 22, 24, 47]. A lack of relationship between MYB-KLF1-BCL11A polymorphisms and high HbF was proposed for one of the family here described [27]. In this particular field of investigation, the analysis of the HbF modifiers loci should be extensive, since preliminary experiments performed on informative samples presented in this paper did not show genotype differences for some BCL11A and HBS1L-MYB loci. For instance, identical sequences for polymorphic BCL11A (rs1427407, rs10189857 and rs11886868) and HBS1L-MYB (rs9399137) were found in the informative B3 and B5 patients exhibiting acquired (B3) or congenital (B5) high HbF levels, respectively. This analysis will be the major issue of a forthcoming study.
In conclusion, (a) we have described a novel polymorphism (+25, G→A) of the Aγ-globin gene; (b) this Aγ-globin-(G→A) polymorphism is always linked to the Gγ-globin-XmnI(+) polymorphism and segregates with the β-globin gene mutations; (c) this polymorphism leads to a decreased binding efficiency of nuclear factors to LYAR-binding sites. Interestingly LYAR has been recently reported to be a repressor of the γ-globin genes [37]. Thus, the Aγ-globin-(G→A) might be proposed as a novel gene HbF modifier functionally associated with the known Gγ-globin-XmnI polymorphism and involved in the high expression of HbF even in adult age. In this respect, our paper suggests that in future functional studies focusing on the control of γ-globin-gene expression, the associated Gγ-globin-XmnI(+) and Aγ-globin-(G→A) polymorphisms should be considered together.
References
Cao A, Galanello R. Beta-thalassemia. Genet Med. 2010;12:61–76. doi:10.1097/GIM.0b013e3181cd68ed.
Giardine B, Borg J, Viennas E, Pavlidis C, Moradkhani K, Joly P, et al. Updates of the HbVar database of human hemoglobin variants and thalassemia mutations. Nucleic Acids Res. 2014;42 (Database issue):D1063–D1069. doi: 10.1093/nar/gkt911.
Old JM. Screening and genetic diagnosis of haemoglobin disorders. Blood Rev. 2003;17:43–53.
Weatherall DJ. Pathophysiology of thalassaemia. Bailliere’s Clin Haematol. 1998;11:127–46.
Weatherall DJ. Phenotype-genotype relationships in monogenic disease: lessons from the thalassaemias. Nat Rev Genet. 2001;2:245–55.
El-Beshlawy A, Hamdy M, El Ghamrawy M. Fetal globin induction in beta-thalassemia. Hemoglobin. 2009;33:S197–203. doi:10.3109/03630260903351882.
Thein SL. The emerging role of fetal hemoglobin induction in non-transfusion-dependent thalassemia. Blood Rev. 2012;26:S35–9. doi:10.1016/S0268-960X(12)70011-5.
Atweh G, Fathallah HV. Pharmacologic induction of fetal haemoglobin production. Hematol Oncol Clin N. 2010;24:1131–44. doi:10.1016/j.hoc.2010.08.001.
Fibach E, Bianchi N, Borgatti M, Zuccato C, Finotti A, Lampronti I, et al. Effects of rapamycin on accumulation of alpha-, beta- and gamma-globin mRNAs in erythroid precursor cells from beta-thalassaemia patients. Eur J Haematol. 2006;77:437–41.
Fibach E, Prus E, Bianchi N, Zuccato C, Breveglieri G, Salvatori F, et al. Resveratrol: antioxidant activity and induction of fetal hemoglobin in erythroid cells from normal donors and β-thalassemia patients. Int J Mol Med. 2012;29:974–82. doi:10.3892/ijmm.2012.928.
Finotti A, Gambari R. Recent trends for novel options in experimental biological therapy of β-thalassemia. Expert Opin Biol Ther. 2014;14:1443–54. doi:10.1517/14712598.2014.927434.
Gambari R. Alternative options for DNA-based experimental therapy of β-thalassemia. Expert Opin Biol Ther. 2012;12:443–62. doi:10.1517/14712598.2012.665047.
Gambari R, Fibach E. Medicinal chemistry of fetal hemoglobin inducers for treatment of beta-thalassemia. Curr Med Chem. 2007;14:199–212.
Sankaran VG. Targeted therapeutic strategies for fetal hemoglobin induction. Hematol Am Soc Hematol Educ Program. 2011;459–65. doi:10.1182/asheducation-2011.1.459.
Perrine SP, Pace BS, Faller DV. Targeted fetal hemoglobin induction for treatment of beta hemoglobinopathies. Hematol Oncol Clin N. 2014;28:233–48. doi:10.1016/j.hoc.2013.11.009.
Quek L, Thein SL. Molecular therapies in beta-thalassaemia. Br J Haematol. 2007;136:353–65.
Badens C, Joly P, Agouti I, Thuret I, Gonnet K, Fattoum S, et al. Variants in genetic modifiers of β-thalassemia can help to predict the major or intermedia type of the disease. Haematologica. 2011;96:1712–4. doi:10.3324/haematol.2011.046748.
Banan M, Bayat H, Azarkeivan A, Mohammadparast S, Kamali K, Farashi S. The XmnI and BCL11A single nucleotide polymorphisms may help predict hydroxyurea response in Iranian β-thalassemia patients. Hemoglobin. 2012;36:371–80. doi:10.3109/03630269.2012.691147.
Danjou F, Francavilla M, Anni F, Satta S, Demartis FR, Perseu L, et al. A genetic score for the prediction of beta-thalassemia severity. Haematologica. 2015;100:452–7. doi:10.3324/haematol.2014.113886.
Thein SL, Menzel S. Discovering the genetics underlying foetal haemoglobin production in adults. Br J Haematol. 2009;145:455–67. doi:10.1111/j.1365-2141.2009.07650.x.
Jiang J, Best S, Menzel S, Silver N, Lai MI, Surdulescu GL, et al. cMYB is involved in the regulation of fetal hemoglobin production in adults. Blood. 2006;108:1077–83.
Sankaran VG, Menne TF, Xu J, Akie TE, Lettre G, Van Handel B, et al. Human fetal hemoglobin expression is regulated by the developmental stage-specific repressor BCL11A. Science. 2008;322:1839–42. doi:10.1126/science.1165409.
Thein SL, Menzel S, Lathrop M, Garner C. Control of fetal hemoglobin: new insights emerging from genomics and clinical implications. Hum Mol Genet. 2009;18(R2):R216–23. doi:10.1093/hmg/ddp401.
Trakarnsanga K, Wilson MC, Lau W, Singleton BK, Parsons SF, Sakuntanaga P, et al. Induction of adult levels of β-globin in human erythroid cells that intrinsically express embryonic or fetal globin by transduction with KLF1 and BCL11A-XL. Haematologica. 2014;99:1677–85. doi:10.3324/haematol.2014.110155.
Paciaroni K, Gallucci C, De Angelis G, Alfieri C, Roveda A, Lucarelli G. Sustained and full fetal hemoglobin production after failure of bone marrow transplant in a patient homozygous for beta 0-thalassemia: a clinical remission despite genetic disease and transplant rejection. Am J Hematol. 2009;84:372–3. doi:10.1002/ajh.21392.
Paciaroni K, Lucarelli G. Hemopoietic stem cell transplantation failure followed by switch to stable production of fetal hemoglobin. Blood. 2012;119:1091–2. doi:10.1182/blood-2011-10-388678.
Paciaroni K, Lucarelli G, Martelli F, Migliaccio AR, von Lindern M, Borg J, Gillemans N, van Dijk TB, Philipsen S. Transfusion-independent β(0)-thalassemia after bone marrow transplantation failure: proposed involvement of high parental HbF and an epigenetic mechanism. Am J Blood Res. 2014;4:27–32.
Fibach E, Bianchi N, Borgatti M, Prus E, Gambari R. Mithramycin induces fetal hemoglobin production in normal and thalassemic human erythroid precursor cells. Blood. 2003;102:1276–81.
Lampronti I, Bianchi N, Zuccato C, Dall’acqua F, Vedaldi D, Viola G, et al. Increase in gamma-globin mRNA content in human erythroid cells treated with angelicin analogs. Int J Hematol. 2009;90:318–27. doi:10.1007/s12185-009-0422-2.
Lozzio CB, Lozzio BB, Machado EA, Fuhr JE, Lair SV, Bamberger EG. Effects of sodium butyrate on human chronic myelogenous leukaemia cell line K562. Nature. 1979;281:709–10.
Bianchi N, Finotti A, Ferracin M, Zuccato C, Breveglieri G, Brognara E, et al. Increase of microRNA-210, decrease of raptor gene expression and alteration of mammalian target of rapamycin regulated proteins following mithramycin treatment of human erythroid cells. PLoS One. 2015;10:1–33. doi:10.1371/journal.pone.0121567.
Finotti A, Treves S, Zorzato F, Gambari R, Feriotto G. Upstream stimulatory factors are involved in the P1 promoter directed transcription of the A β H-J-J locus. BMC Mol Biol. 2008;9:110–25. doi:10.1186/1471-2199-9-110.
Andrews NC, Faller DV. A rapid micropreparation technique for extraction of DNA-binding proteins from limiting numbers of mammalian cells. Nucleic Acids Res. 1991;19:2499. doi:10.1093/nar/19.9.2499.
Borgatti M, Lampronti I, Romanelli A, Pedone C, Saviano M, Bianchi N, et al. Transcription factor decoy molecules based on a peptide nucleic acid (PNA)-DNA chimera mimicking Sp1 binding sites. J Biol Chem. 2002;278:7500–9.
Romanelli A, Pedone C, Saviano M, Bianchi N, Borgatti M, Mischiati C, et al. Molecular interactions with nuclear factor kappaB (NF-kappaB) transcription factors of a PNA-DNA chimera mimicking NF-kappaB binding sites. Eur J Biochem. 2001;268:6066–75.
Finotti A, Bianchi N, Fabbri E, Borgatti M, Breveglieri G, Gasparello J, et al. Erythroid induction of K562 cells treated with mithramycin is associated with inhibition of raptor gene transcription and mammalian target of rapamycin complex 1 (mTORC1) functions. Pharmacol Res. 2015;91:57–68. doi:10.1016/j.phrs.2014.11.005.
Ju J, Wang Y, Liu R, Zhang Y, Xu Z, Wang Y, et al. Human fetal globin gene expression is regulated by LYAR. Nucleic Acids Res. 2014;42:9740–52.
Stockley PG, Persson B. Surface plasmon resonance assays of DNA-protein interactions. Method Mol Cell Biol. 2009;543:653–69. doi:10.1007/978-1-60327-015-1_38.
Li ZL, Abe H, Ueki K, Kumagai K, Araki R, Otsuki Y, et al. Identification of c-Jun as bcl-2 transcription factor in human uterine endometrium. J Histochem Cytochem. 2003;51:1601–9.
Marzaro G, Guiotto A, Borgatti M, Finotti A, Gambari R, Breveglieri G, et al. Psoralen derivatives as inhibitors of NF-κB/DNA interaction: synthesis, molecular modeling, 3D-QSAR, and biological evaluation. J Med Chem. 2013;56:1830–42. doi:10.1021/jm3009647.
van Dijk M, Bonvin AM. 3D-DART: a DNA structure modeling server. Nucleic Acids Res. 2009;37(Web Server Issue):W235–W239.
Hess B, Kutzner C, van der Spoel D, Lindahl E. GROMACS 4: algorithms for highly efficient, load-balanced, and scalable molecular simulation. J Chem Theory Comput. 2008;4:435–47. doi:10.1021/ct700301q.
Dominguez C, Boelens R, Bonvin AM. HADDOCK: a protein-protein docking approach based on biochemical or biophysical information. J Am Chem Soc. 2003;125:1731–7.
de Vries SJ, van Dijk M, Bonvin AM. The HADDOCK web server for data-driven biomolecular docking. Nat Protocol. 2010;5:883–97. doi:10.1038/nprot.2010.32.
DeLano WL. The PyMOL molecular graphics system. San Carlos: DeLano Scientific; 2002.
Dolinsky TJ, Nielsen JE, McCammon JA, Baker NA. PDB2PQR: an automated pipeline for the setup of Poisson-Boltzmann electrostatics calculations. Nucleic Acids Res. 2004;32:W665–7.
Sankaran VG, Xu J, Orkin SH. Transcriptional silencing of fetal hemoglobin by BCL11A. Ann NY Acad Sci. 2010;1202:64–8. doi:10.1111/j.1749-6632.2010.05574.x.
Nguyen TK, Joly P, Bardel C, Moulsma M, Bonello-Palot N, Francina A. The XmnI (G)gamma polymorphism influences hemoglobin F synthesis contrary to BCL11A and HBS1L-MYB SNPs in a cohort of 57 beta-thalassemia intermedia patients. Blood Cel Mol Dis. 2010;45:124–7.
Acknowledgments
The Ferrara Association for the Fight against Thalassemia (ALT) is deeply acknowledged for help in patient recruitment. We thank Dr. Marco Andreani and Manuela Testi (Policlinic of “Tor Vergata” University, Rome, Italy), Dr. Maria Rita Gamberini (Ferrara Hospital, Ferrara, Italy) and Dr. Francesco Chiavilli (Rovigo Hospital, Italy) for providing DNA and cellular samples. The funders had no role in the study design, data collection and analysis, decision to publish, or preparation of the manuscript.
Author contribution
RG, KP, GL performed the research, designed the research study, wrote the paper and contributed essential reagents or tools; NB performed the research, designed the research study and wrote the paper; LCC, EF, GDA, CG, CA, MR, AI, MM, PS performed the research and analyzed the data; JG and AM designed the research study and analyzed the data; CZ, IL, AF, GB, CG, MB, EF, GM and AC performed the research.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
NB, LCC, EF, IL, AF, GB, CZ, GM, AC, GDA, CG, CA, MR, AI, MM, JG, AM, PS,GL, RG and KP declare no conflicts of interest.
Funding
Roberto Gambari is funded by Fondazione Cariparo (Cassa di Risparmio di Padova e Rovigo), CIB (Consorzio Interuniversitario per le Biotecnologie), UE THALAMOSS Project (Thalassemia Modular Stratification System for Personalized Therapy of Β-Thalassemia; n. 306201-FP7-HEALTH-2012-INNOVATION-1), Telethon (contract GGP10124) and by COFIN-2010. This research activity has been also supported by Associazione Veneta per la Lotta alla Talassemia (AVLT), Rovigo. Monica Borgatti was funded by Ministero della Salute, Italy (contract 098/GR-2009-1596647). The funders had no role in the study design, data collection and analysis, decision to publish, or preparation of the manuscript.
Ethical approval and informed consent
The present study has been conducted according to the principles expressed in the Declaration of Helsinki and in full compliance with the guidelines of the Mediterranean Institute of Hematology, Rome, Italy. Ethics Committee’s approval for the research has been obtained. All patients and family members provided appropriate informed consent. The study was also approved by the Ethical Committee of Rovigo Hospital and Ferrara Hospital.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
About this article

Cite this article
Bianchi, N., Cosenza, L.C., Lampronti, I. et al. Structural and Functional Insights on an Uncharacterized Aγ-Globin-Gene Polymorphism Present in Four β0-Thalassemia Families with High Fetal Hemoglobin Levels. Mol Diagn Ther 20, 161–173 (2016). https://doi.org/10.1007/s40291-016-0187-2
Published:
Issue Date:
DOI: https://doi.org/10.1007/s40291-016-0187-2