
Abstract
Drug-induced liver injury (DILI) remains a rare but serious complication in drug therapy that is a primary cause of drug failure during clinical trials. Conventional biomarkers, particularly the serum transaminases and bilirubin, serve as useful indicators of hepatocellular or cholestatic liver injury, respectively, but only after substantial and sometimes irreversible tissue damage. Ideally, more sensitive biomarkers that respond very early before irreversible injury has occurred would offer improved outcomes. Novel biomarkers are initially being developed in animal models exposed to intrinsically hepatotoxic stimuli. However, the eventual translation to human populations, even those with known risk factors that predispose the liver to drug toxicity, would be the fundamental goal. Ultimately, some might even be applicable for the early identification of individuals predisposed to idiosyncratic hepatotoxicity potential. This article reviews recent progress in the discovery and qualification of novel biomarkers for DILI and delineates the path to eventual utilization for risk assessment. Some major categories of plasma or serum biomarkers surveyed include proteins, cytokines, circulating mRNAs, and microRNAs.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
1 Introduction
It is estimated that up to 40 % of potentially hepatotoxic compounds in humans go undetected in preclinical studies that utilize conventional clinical pathology procedures [1]. In a survey of 12 pharmaceutical companies and 150 compounds, liver and hypersensitivity/cutaneous reactions were the two human toxicities with the poorest correlation with animal studies [2]. Postmarketing, drug-induced liver injury (DILI) was reported to be responsible for more than 50 % of the cases of acute liver failure, with the hepatotoxicity caused by overdose of acetaminophen (APAP) responsible for 39 % and idiosyncratic liver injury elicited by other drugs accounting for 13 % [3].
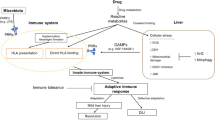
DILI can be either intrinsic to the drug itself or idiosyncratic; that is, of unknown causes and attributable to the individual with one or more susceptibility factors that can include underlying diseases, immune system perturbations, environmental factors, and stresses. Idiosyncratic DILI is a rare phenomenon that cannot be evaluated readily in animals due to the lack of suitable experimental models. Although the incidence of idiosyncratic liver injury has been considered independent of drug exposure, recent studies [4] have suggested that a daily oral dose >10 mg/day and significant hepatic drug metabolism increase the risk of this type of DILI.
Categories of liver injury are distinguished by histopathological phenomena that include liver hypertrophy, acute hepatocellular necrosis (predominant), cholestatic injury, or mixed injuries. DILI also can include fibrosis/cirrhosis, microvesicular steatosis, nonalcoholic steatohepatitis (NASH), phospholipidosis, vascular lesions, and neoplasms [5]. Mechanistic phenomena associated with liver injury include apoptosis/necrosis, inflammation, oxidative stress, and the alteration of metabolic enzymes. In addition to direct drug injury to hepatocytes, Kupffer cells can activate cytokines that may amplify injury, stellate cells may augment injury, and some drugs can injure sinusoidal endothelial cells leading to veno-occlusive disease [6]. One major confounding difference between human versus animal drug effects involves host susceptibility in the former due to pre-existing liver anomalies such as fatty liver, NASH, and chronic hepatitis B or C [7–9]. Another difference can result from susceptibility factors such as class I and II human leucocyte antigen (HLA) genotypes, genetic polymorphisms of drug-metabolizing enzymes, nongenetic host factors such as age or sex, and the concomitant use of potentially hepatotoxic drugs to treat these conditions [7, 10, 11].
As defined by an expert working group at the National Institutes of Health, a biomarker is “A characteristic that is objectively measured and evaluated as an indicator of normal biologic processes, pathogenic processes, or pharmacologic responses to a therapeutic intervention” [12]. Biomarkers are classified by the US Food and Drug Administration (FDA) as exploratory, probable valid, or known valid. A valid biomarker is further defined as: “A biomarker that is measured in an analytical test system with well-established performance characteristics and for which there is an established scientific framework or body of evidence that elucidates the physiologic, toxicologic, pharmacologic, or clinical significance of the test results” [13]. By necessity, biomarker discovery research is typically performed in animal models, but translatable or bridging hepatic biomarkers that would provide both preclinical screening and subsequent clinical monitoring are the ultimate goal.
Conventional biomarkers of liver injury are of two basic types: (1) those that indicate alterations of normal liver function or homeostasis and (2) specific markers of tissue and cell integrity. Normal liver functions include the metabolism of endogenous substances such as bile acids, protein synthesis, and the excretion of metabolic end products such as bilirubin and urea. Examples of conventional biomarkers indicative of altered liver function include drug-induced or disease-induced changes in plasma bile acids, plasma total bilirubin (Tbil), and total plasma protein. Conventional biomarkers of hepatic cell and tissue integrity are normal cellular constituents that are released from damaged or dying cells into the blood where they can be monitored. Examples of this category used in preclinical studies include: alanine aminotransferase (ALT), aspartate aminotransferase (AST), glutamate dehydrogenase (GLDH), gamma-glutamyl transferase (GGT), alkaline phosphatase (ALP), and 5′-nucleotidase [14]. To the clinical pathologist, these are indicators of either hepatocellular injury (ALT, AST, GLDH, total bile acids) or hepatobiliary injury (GGT, ALP, 5′-nucleotidase, Tbil, or total bile acids) [15]. Supplementary tests can include tests for albumin (Alb), total protein, triglycerides, and coagulation measurements [16].
Although a variety of biomarkers are used to diagnose hepatocellular DILI, Hy’s law is regarded as a gold standard [17, 18]. This paradigm is based on detection of greater than threefold elevations of serum ALT in combination with greater than twofold increase of total bilirubin (Tbil) in absence of hepatobiliary injury assessed using ALP. Although the criteria of Hy’s law are widely considered for detection of severe DILI [17], modified criteria for assessment of DILI that address the shortcomings of Hy’s law have been recently developed by a working group of clinicians and scientists [19]. In this refined paradigm, the ALT elevation threshold was raised to greater than fivefold in order to exclude clinically irrelevant drug-related events as well as low-level non-DILI elevations of serum ALT. To address the importance of the damage to the hepatobiliary system, an increase of ALP greater than twofold was also included. Taken together, Hy’s law with the additional criteria described above comprises a refined approach for assessment of DILI in the clinic [19]. This definition recently has been utilized as a diagnostic measure of liver damage in the clinical translation of several emerging biomarkers of liver injury in the clinic, enabling evaluation of the markers in the absence of histopathological evaluation that was not feasible due to ethical and practical reasons [20]. Despite the progress in the development of biomarker-driven paradigms for assessment of DILI in humans, drug development continues to be negatively affected because of unexplained ALT elevations in clinical trials. Thus, the development of alternative biomarkers and approaches is important [21].
Search terms used in the preparation of this review include: drug-induced liver injury, plasma or serum biomarkers, proteomics, metabonomics, cytokines, circulating mRNAs, and microRNAs. The period covered for the selection of articles in the preparation of this review was 1978 through 2013.
2 The Multistep Process for DILI Biomarker Research
Preclinical biomarker research proceeds in three basic steps: discovery, validation, and qualification. The discovery process typically involves the generation of a candidate database that is populated through genomic or proteomic technologies or through the curation of candidates gleaned from the scientific literature [22]. Biomarker protein discovery typically is performed in analytical laboratories equipped with gas chromatography (GC), high-pressure liquid chromatography (LC), or combined GC and LC mass spectrometric (MS) procedures such as LC–MS, LC–MS–MS, GC–MS, and GC–MS–MS. Multiple reaction monitoring (MRM) is a targeted approach that refers to a tandem MS (MS–MS) scan mode used with triple quadrupole MS instrumentation that provides rapid, sensitive, and specific quantitation of analytes in highly complex sample matrices [23]. When stable isotope-labeled internal standards are included, MRM can provide highly reproducible concentration determinations for the quantitation of low molecular mass analytes (<1,000 Da) in pharmaceutical and clinical applications [23, 24]. For a recent review of MS-based proteomics, see Angel et al. [24]. A typical approach to the identification of RNA and mircoRNA (miRNA) biomarkers is to monitor the gene expression profiles utilizing high-throughput assay platforms such as quantitative polymerase chain reaction (PCR), microarrays, and most recently the next generation sequencing (NGS). For a recent review of the discovery of miRNAs and other noncoding RNAs from deep-sequencing data, see Yang et al. [25]. Candidate biomarkers are also “discovered” by a thorough review of the scientific literature. The Safer And Faster Evidence-based Translation (SAFE-T) consortium’s biomarker candidates were originally selected based on solid scientific evidence and rationale based on the available literature before progressing through a specific validation/qualification strategy [26].
Validation is the formal technical procedure used to prove the suitability of an analytical assay, and toxicity biomarker method validation differs from drug bioanalysis or diagnostic applications. A validated biomarker is: “A biomarker that is measured in an analytical test system with well-established performance characteristics and for which there is an established scientific framework or body of evidence that elucidates the physiologic, toxicologic, pharmacologic, or clinical significance of the test results” [13]. Analytical method validation for biomarkers can be performed according to the FDA Guidance for Bioanalytical Method Validation [27]. A detailed “fit-for-purpose” biomarker assay development and method validation approach has been described by Lee et al. [28] and Lee and Hall [29]. The validation process for serum protein biomarkers based on three primary multianalyte immunoassay platforms recently has been described by Köhler and Seitz [30].
Qualification refers to the process of associating a candidate biomarker with a specific biological phenomenon and clinical outcome. The US FDA has defined that: “Qualification is a conclusion that within the stated context of use, the results of assessment with a drug development tool (biomarker) can be relied upon to have a specific interpretation and application in drug development and regulatory review” [31]. The principles of the biomarker qualification process have been designed at the US FDA [32] and later adopted worldwide [33]. In reality, the biomarker qualification process is a dialog between the sponsor and regulatory agency that results in the understanding and characterization of biomarker performance under the defined context of use [34]. A central component of the process is the Voluntary Biomarker Data Submission (VXDS) that enables discussing initial data on the biomarker and its context of use. If successful, the Biomarker Qualification Review Team recommends proceeding with a full submission that must provide a full characterization of biomarker performance and its context of use. After the review of the full submission is completed, the FDA issues a decision about the qualification [33]. Because the biomarker qualification process is complex and requires considerable resources, the qualification efforts are mainly pursued by consortia such as the Health and Environmental Sciences Institute (HESI), the Predictive Safety Testing Consortium (PSTC), and SAFE-T. Three biomarker qualification submissions have been successfully completed to date. The first two biomarker submissions qualified several emerging biomarkers of acute kidney injury for preclinical use and the third application resulted in nonclinical qualification of circulating cardiac troponins as biomarkers of morphologic cardiac damage [35]. While the three successful submissions to date have focused on preclinical qualification, the consortia are also progressing toward the clinical qualification of these and additional biomarkers spanning multiple organ toxicities. The SAFE-T consortium, started under the EU Innovative Medicines Initiative (IMI), is currently qualifying clinical biomarkers of liver, kidney, and vascular injury. Despite the progress in preclinical qualification of biomarkers, clinical qualification of novel biomarkers has not yet been achieved.
3 Categories of Emerging DILI Biomarkers
For the clinical chemist, major categories of potential biomarker sources can include proteins and the enzymatic activities of proteins, cytokines, mRNAs, microRNA and genomic endpoints, metabolites, and bile acids. These are discussed as follows.
3.1 Proteomic Applications
Proteomics is a discovery technology that refers to the identification by analytical methods of the proteome or entire complement of proteins present in a cell or tissue at a specific time. Estimates for the number of proteins in the proteome range up to 100,000 excluding posttranslational modifications (PTMs) [36]. Improved analytical platforms that are capable of detecting as little as 100 pmol/L (approximately 5 μg/L) has made it possible to detect as many as 1,000–2,000 different proteins in a single laboratory [37]. Most human proteins are regulated by reversible, enzymatic PTMs also referred to as the subproteome, which provide dynamic control of protein function. Although proteomic discovery is technically more difficult than transcriptomics, it is much more meaningful to the toxicologist or clinician. The identification of specific proteins involves a combination of (1) a separation technique (e.g., two-dimensional gel electrophoresis, HPLC) followed by (2) tandem mass spectrometry. Ninety-nine percent of the serum mass is comprised of the 22 most abundant proteins while proteins of interest as biomarkers may only be present in nanomolar to femtomolar quantities [38]. These high abundance proteins are depleted and protein mixtures are usually digested to smaller peptide fragments prior to analysis. Identification of specific peptides is based on high-resolution mass analysis and sequential degradation of the peptides. The relative abundance of protein isoforms present in biological samples can span up to 12 orders of magnitude providing an additional analytical challenge [24]. Conventional hepatotoxicity biomarkers are predominantly proteins measured with enzymatic activity-based assays in automated clinical pathology laboratories and include markers of hepatocellular injury (primarily ALT and AST) and hepatobiliary injury (primarily GGT, ALP, and Tbil). Any new protein biomarkers will either be developed from prospective candidates currently described in the literature, those recently identified from proteomic models, or those suggested by gene expression models. Some of these novel markers including paraoxonase 1 (PON1), purine nucleoside phosphorase (PNP), malate dehydrogenase (MDH), arginase 1 (ARG1), and α-glutathione S-transferase (α-GST) are currently being evaluated through the PSTC and SAFE-T consortia in the US and EU, respectively. While some of these new biomarkers can be measured by enzymatic assays, others will have to rely on other assay technologies and platforms, including immune-based assays [39].
3.1.1 Global proteomics
In the past 2 years, some interesting candidate protein biomarkers for DILI have been reported. These are of two categories: protein panels versus individual biomarkers. Bell et al. [40] have recently used a label-free quantitative proteomic (LFQP) approach based on mass spectrometry to investigate protein expression in sera samples obtained from 74 DILI patients and compared these to 40 control subjects. Proteins revealed by pattern analysis included components of inflammation and immune system activation along with several pathways specific to hepatotoxicity and implicated in the pathogenesis of idiosyncratic DILI. Ninety-two priority-1 proteins (i.e., those most likely to be correctly identified) were differently expressed between DILI and control groups. Apolipoprotein E had the greatest success in differentiating DILI from control patients and along with 65 additional proteins demonstrated significant differential expression that was unique to the DILI analysis.
3.1.2 Targeted Proteomics and Individual Protein Assessments
Recently, emphasis has shifted from global proteomic studies to the precise relative quantification of known proteins in complex mixtures [41]. LC–MRM/MS and immunoassays are the methods of choice. Selected reaction monitoring-based (SRM also known as MRMFootnote 1) proteomics refers to a mass spectrometric procedure used most effectively in LC–MS and particularly well suited for the detection and quantification of specific predetermined analytes having known fragmentation properties in complex backgrounds [42]. The application of SRM mass spectrometry has been successfully applied to quantitatively measure many specific target molecules within a complex mixture [43]. A sample preparation technique referred to as stable isotope standards with capture by antipeptide antibodies (SISCAPA) has been used to enrich targeted low abundance peptides [44]. Although a number of clinically important plasma proteins can be measured with precision comparable to current clinical immunoassays, some important limitations of the MRM technique include the necessity to know and design into the assay any PTMs in order to detect those modified peptides, the fact that some proteins may not produce suitable peptides, and the presence of genetic variants that involve the alteration of a single amino acid peptide that will preclude their detection [45]. The advantage of immunoassays (enzyme linked immunosorbent assays or ELISAs) for protein biomarker assessments is their direct clinical application and potential for diagnostic assay development; however, these are dependent upon the commercial availability of suitable antibodies.
Based on previous reports in animal models and clinical studies, Antoine et al. [46] have investigated the serum proteins High Mobility Group Box-1 Protein (HMGB1) and keratin-18 (K18) with the goal of assessing the mechanistic utility of HMGB1 and full-length K18 to establish the balance between apoptosis, necrosis, and immune cell activation throughout the duration of clinical APAP hepatotoxicity. A combination of established ELISAs and novel LC–MS/MS-based assays were used in this targeted study. They concluded that if used in conjunction with established biomarkers, these mechanism-based biomarkers augment the understanding of basic cell death mode dynamics and progression during the early stage of clinical APAP hepatotoxicity.
Bailey et al. [47] have recently completed a performance evaluation of three biomarkers to diagnose drug-induced hepatocellular and biliary injury in 34 acute toxicity studies in the rat. The selected biomarkers were α-GST, ARG1, and 4-hydroxyphenylpyruvate dioxygenase (HPD). α-GST is a cytoplasmic member of a complex family of multifunctional proteins involved in phase II detoxification of xenobiotics. ARG1 performs the critical function of catalyzing the hydrolysis of arginine to ornithine and urea. The enzyme HPD is strongly expressed in hepatocytes where it is involved in tyrosine catabolism. Using fit-for-purpose ELISAs, these biomarkers were compared for specificity and sensitivity relative to ALT. All three biomarkers, alone or in combination with ALT, were found to improve specificity when compared to ALT alone. All three detected hepatocellular necrosis and/or degeneration in the majority of the animals. ARG1 demonstrated the best sensitivity for the detection of biliary injury with or without ALT.
In a very recent study, Schomaker et al. [20] evaluated the utility of four emerging biomarkers, GLDH, PNP, MDH, and PON1 as potential indicators of liver injury in cohorts of human subjects that included healthy individuals and individuals with a variety of liver impairments that were not necessarily the result of DILI. Three of these proteins (PON1, PNP, and MDH) were originally identified by proteomic methods as serum biomarkers associated with rat liver toxicity or hypertrophy [48]. In this extensive clinical study, a group of healthy volunteers (n = 186) consisted of male and female subjects between the ages of 18 and 55 years with absence of clinically relevant abnormalities detected by a detailed medical history, full physical examination, including blood pressure and pulse rate measurement, electrocardiograms, and lack of positive findings in routine clinical laboratory tests. A second group, healthy subjects (n = 364), were defined as healthy based on the normal levels of ALT, AST, ALK, Tbil, glucose, blood urea nitrogen, serum creatinine, and creatine kinase. A third group, hepatic injury subjects (n = 469), had abnormal hepatic enzyme profiles with both AST and ALT levels greater than two times normal healthy levels and with a diagnosed disease resulting in impaired liver function. The results indicated that GLDH and MDH levels were stable across both healthy groups and were not impacted by sex or age. GLDH had the strongest correlation with elevated ALT levels, and exhibited the greatest predictive power for liver injury, as determined by receiver operating characteristic (ROC) analysis. MDH also had a high correlation with ALT levels and a high predictive ability in this study. The evaluation of a small subset (n = 6) of patients with APAP-induced liver injury provided evidence that both GLDH and MDH might have utility as biomarkers of DILI in humans.
3.2 Cytokines
Cytokines comprise a diverse group of soluble peptides characterized by short half-lives and transient activities that provide signaling between cells and elicit a number of biological responses. Due to the role of inflammation and the inflammatory cascade, changes in extrahepatic cytokine levels in the blood following exposure to hepatotoxic drugs have been proposed as a potential source for DILI biomarkers [49]. The infiltration of inflammatory cells into the liver is often observed even in cases of direct DILI. This includes the primary proinflammatory cytokines such as tumor necrosis factor α, interleukin-1β, and interleukin-6 [50] that are expressed sequentially. The quantification of soluble cytokines is typically performed by immunoassays or bioassays. However, cytokines generally lack tissue specificity, and for quantitative purposes, their short half-lives in serum and low to undetectable baseline control levels present a challenge. For a comprehensive review of blood cytokines as potential biomarkers of preclinical safety assessment, see Tarrant [51].
3.3 Nucleic Acid-Based Biomarkers mRNAs and MicroRNAs
Major technological advantages of nucleic acid-based biomarker platforms are their relatively simple and straightforward assay design and the availability of high-throughput assay platforms such as quantitative PCR, microarrays, and most recently the NGS. The nucleic acid chemistry eliminates issues associated with the development of species-specific antibodies used protein-based assays. However, the rapid development of new assay technologies and expansion of genomic sequence information leading to the discovery of new RNAs including miR isomers (isomiRs) make the comparison of datasets across assay platforms challenging [52]. Despite the discrepancies in identified genes and/or miRs in individual studies, when data is analyzed in light of biological pathways (system biology approach), the biological interpretation reported has been largely consistent [53].
The NGS allows for unprecedented speed in sequencing of genomic DNA and all RNA species including microRNAs. Several NGS platforms have become established such as SOLiD, HiSeq, 454 FLX, MiSeq, and IonTorrent. Because NGS enables surveying of the whole genome at a reasonable price that is steadily decreasing, these technologies have the potential to revolutionize medicine and biomarker development [54]. Recently, Wang et al. [55] reported an extensive list of circulating RNA-based biomarker candidates based on their RNA profiling study results from the APAP overdose mouse model on plasma and liver using microarray and NGS platforms, respectively. A recent publication by Su et al. [56] suggests that NGS is more sensitive than traditional microarray technologies for detection of transcriptomic changes caused by toxicants. In addition, the open nature of the NGS technologies enables the identification of novel transcripts, splice variants, microRNA, and other noncoding regulatory elements in one sequencing run. Because several challenges such as the standardization of methods and procedures need to be addressed before NGS can take a prominent role in the biomarker area, an international project called Sequencing Quality Control (SEQC) modeled after successful Microarray Quality Control (MAQC) has been initiated [57].
The presence of extracellular RNA in blood and other body fluids has been well documented [58]. The extracellular RNA is thought to be released into the circulation from intact and viable cells as well as necrotic cells. The RNA molecules are protected against degradation by ubiquitous RNases in the extracellular space by forming complexes with other molecules, mainly proteins, or are components of circulating microvesicles or exosomes [59]. The notion that tissue-specific RNA molecules (mRNA or miR) are released from injured cells in a similar fashion as proteins led to evaluating circulating RNAs as biomarkers of tissue injury. Because exosomes and shedding vesicles are thought to be part of a cell-to-cell communication and multisignaling processes [60], one can even speculate that changes in circulating RNA species can provide additional information that could be useful for assessing the prodromal stage of tissue injury or cellular stresses or possibly provide mechanistic insights into tissue injury.
The presence of liver-specific mRNAs in peripheral blood as potential DILI biomarkers has been proposed by several investigators. Kudo et al. [61] evaluated the utility of circulating mRNA as a molecular marker to detect hepatic injury using rat liver fibrosis models by monitoring plasma-circulating mRNAs of the major liver-derived genes, Alb and haptoglobin, by real-time quantitative reverse transcription PCR (qRT-PCR). The sensitivity and kinetics of plasma-circulating mRNA were compared with those of plasma ALT activity. Their results indicated that circulating plasma Alb mRNA might serve as a molecular marker for hepatic injury using the acute and chronic rat liver fibrosis models. The measurement of plasma-circulating Alb mRNA enabled sensitive and early detection of hepatic injury with a large assay window when compared with that of plasma ALT activity. In a study by Wetmore et al. [62], significant increases of liver-specific mRNAs were observed in plasma of rats treated with model hepatotoxicants, even at doses that had no effect on serum aminotransferases or liver histopathology. Furthermore, liver-specific mRNAs were associated with both necrotic debris and microvesicles, and the transcriptomics analysis of circulating mRNA in plasma of rats treated with d-galactosamine (d-gal) and APAP revealed chemical-specific profiles. Cell-free, liver specific α(1)-microglobulin/bikunin precursor (Ambp) and Alb mRNAs have been detected by RT-PCR 24 h after d-gal or APAP administration to Sprague Dawley rats [63]. Alb mRNA was detected as early as 2 h after d-gal administration, while AST and ALT levels were still unchanged. More recently, liver-specific Ambp, Alb, apolipoprotein h (Apoh), and group-specific component (Gc) mRNAs have been quantified by RT-PCR in the plasma from rats dosed acutely with seven hepatotoxicants [64]. The increased levels of these four mRNAs were comparable and correlated with both ALT values and the scoring of hepatocellular necrosis 24 h after dosing. Plasma levels of Gc and Alb mRNAs were increased substantially earlier than ALT. Although these data are very promising, additional studies are necessary to assess whether transcriptomics analysis of plasma can supplement or even replace a liver tissue sample/biopsy as material needed for toxicogenomic analysis of liver injury.
miRNAs comprise a family of small (20–30 nucleotides), short-lived regulatory molecules that function by modulating protein production. This regulation can occur at important levels of genome function, including chromatin structure, chromosome segregation, transcription, RNA processing, RNA stability, and translation and is generally inhibitory [65]. In recent years, stable miRNAs have been detected in serum, plasma, urine, saliva, and other body fluids. Some publications report miRNA concentrations to be higher in serum than in plasma from the same individuals, presumably due to platelet sources [66]. However, miRNA measurements between serum and plasma have been found to be highly correlated and thus both types of specimens are acceptable for circulating miRNA analysis [67]. There are a large number of known mammalian miRNA genes, and each of these miRNA may regulate hundreds of different protein-coding genes [68]. Over 1,000 human miRNAs have been identified to date [66]. miR-122 is of particular interest as a potential specific and sensitive biomarker of liver-specific injury.
Although the biological function of circulating miRNAs is generally unknown, they offer certain advantages as biomarkers of DILI. They are stable, much less complex than other biological molecules, do not undergo postprocessing modifications and are reliably extracted and assayed in serum or plasma [69]. Wang et al. [69] found miR-122 and miR-192 in particular exhibit dose- and exposure-duration-dependent changes in plasma that parallel liver degeneration and serum ALT levels in animal models. miR-122 is highly expressed in hepatocytes, comprising 70% of the total miRNA pool of the healthy liver where it regulates gene networks that control lipid metabolism, cell differentiation, and other processes [70, 71]. miR-122 is highly conserved in at least 12 different species including humans [72], and, like other miRNAs, circulating miR-122 is very stable in serum. Laterza et al. [73] have assessed miR-122 as a biomarker of liver toxicity in rats treated with CCl4 or CBrCl3, both of which produce hepatocellular degeneration and liver necrosis. They observed consistent plasma miR-122 concentrations in the treated animals that were correlated with the histopathological data. These increases were orders of magnitude higher than concurrent observations for ALT with greater sensitivity noted. Other studies have reported that miR-122 concentrations were increased in the plasma of patients and animal (mouse) models with viral-induced liver diseases as well as chemically induced liver injury [74]. When Starckx et al. [75] exposed rats to three well-established liver toxicants (APAP, allyl alcohol, and α-naphthyl isothiocyanate), the liver-enzyme inducer phenobarbital, or the cardiotoxicant doxorubicin; they found a clear increase in plasma miR-122 following the administration of APAP, allyl alcohol, and α-naphthyl isothiocyanate. This miR-122 response paralleled that of other hepatotoxicity markers and was consistent with liver injury as confirmed by histopathological evaluation. These changes in miR-122 were detected earlier than standard liver injury markers and exhibited a wide dynamic range. In contrast, miR-122 responses to phenobarbital and doxorubicin were low. Recent human studies have confirmed the utility of miR-122 as a biomarker.
In clinical studies, serum miR-122 and miR-192 have been found to be substantially higher in humans with APAP poisoning and non-APAP acute liver injury (ALI), but not in patients with kidney injury or in healthy volunteers [76]. Recently, Antoine et al. [77] studied the potential of a panel of novel biomarkers to identify patients with APAP-induced ALI at first presentation to the hospital when currently used markers are within the normal range. In this study, miR-122, HMGB1, full-length and caspase-cleaved K18 and GLDH were measured in plasma samples from patients (n = 129) at first presentation to the hospital. ROC curve analysis and positive/negative predictive values then were used to compare sensitivity to report liver injury versus ALT and international normalized ratio (INR). In patients presenting with normal ALT or INR, miR-122, HMGB1, and K18 identified the development of liver injury (n = 15) or not (n = 84) with a high degree of accuracy that significantly outperformed ALT, INR, and plasma APAP concentration. These results indicated that elevations in plasma miR-122, HMGB1, and K18 identified subsequent ALI development in patients on admission to hospital, soon after APAP overdose, and in patients with ALTs in the normal range. miR-122 elevations have also been reported in the literature in response to several liver diseases including hepatic carcinoma, hepatitis, and nonalcoholic fatty liver disease (NAFLD) [78–80]. Zhang et al. [74] reported miR-122 to be elevated in patients with hepatitis B compared to healthy patients. These elevations correlated with the histopathology severity score determined from biopsies and were both more sensitive and specific than modification in ALT activity.
3.4 Metabonomics
The liver is the major site for synthesis of endogenous compounds and degradation of xenobiotics. Therefore, the metabolites and/or their profiles have been explored as sources of more sensitive and predictive biomarkers for DILI. Metabolites embody a diverse group of compounds with low molecular weight that includes lipids, amino acids, peptides, nucleic acids, organic acids, vitamins, thiols, and carbohydrates. The study of endogenous metabolites, or the “metabolome,” provides the basis of metabonomics (also known as “metabolomics”). Metabonomics has been defined as “the quantitative measurement of the time-related multiparametric metabolic response of living organism to pathophysiological or genetic modification” [81]. Because of the complexity of the metabolome and the diverse properties of metabolites, no single analytical platform can be applied to detect all metabolites in a biological sample. Currently, nuclear magnetic resonance spectrometry and mass spectrometry technologies provide methods of choice for monitoring of metabolic profiles. The advantages and limitations of these methods are extensively reviewed by Wang et al. [82]. Statistical and bioinformatic analyses of metabolic profiles employ a myriad of tools that identify patterns of metabolites and define the context of their changes in light of the etiology and/or pathogenesis of liver injury. There is substantial interindividual variability caused mainly by metabolic competency of gastrointestinal flora and environmental factors such as the diet that limits the final interpretation of the data. Because metabonomics is an emerging discipline, the data reproducibility across and within the analytical platform is challenging. Therefore, the development of standardized procedures that would allow between-laboratory comparisons and define the minimum reporting requirements is necessary. To address these issues, the Metabonomics Standards Initiative (MSI) has been established [83].
Despite the challenges associated with analytical platforms and inherent interindividual variability in metabolite profiles, the metabonomics approach has identified several profiles that correlated with liver injury in rodents treated with model hepatotoxicants such as APAP [84, 85], galactosamine [86], carbon tetrachloride [87], and alcohol [88]. The most interesting aspect of metabonomics is its potential to identify individuals predisposed to liver injury based on predose metabonomics measurements. This approach has been termed “pharmacometabonomics.” In an initial study, a pattern of endogenous metabolites in urine predicted susceptibility to APAP-induced liver injury in rats [89]. In a study evaluating human responders and nonresponders to a mild liver injury after treatment with therapeutic doses of APAP, the pharmacometabonomic approach relying on the pre-exposure urine metabolite profiles as baseline did not differentiate subjects that are more susceptible to APAP liver injury [90]. On the other hand, serum metabonomic profiles taken shortly after the exposure but before significant increases of ALT as a measure of liver injury identified responders to APAP-induced liver injury. Although the metabonomic approaches for biomarker development are promising, significant challenges regarding interindividual variability, analytical standardization, and interpretation of the data currently limit the application of this approach.
3.5 Biomarkers of Excretory Function
Appearance of ALT, AST, and other “leakage” biomarkers in serum is a consequence of their release from damaged or dying hepatocytes. The functional impairment of liver, namely the loss of its excretory function, is detected as an increase of total bilirubin (Tbil), conjugated bilirubin, or bile acids. Because of the substantial liver bilirubin and bile acid-excreting capacity, their serum level increases are usually associated with extensive liver injury. Because Woolbright and Jaeschke [91] reviewed biomarkers of cholestatic liver injury in great detail, we will focus on recently published data regarding individual bile acid profiles as biomarkers of DILI. Bile acids represent a class of structurally similar compounds that play an essential role in cholesterol homeostasis, lipid absorption, and intestinal signaling. They are synthesized in the liver and excreted into the small intestine via the bile duct mainly as glycine or taurine conjugates, and then undergo enterohepatic circulation with further metabolism by bacterial and hepatic enzymes [92]. Because liver and gastrointestinal diseases often affect bile acid synthesis and clearance, serum bile acid levels and/or profiles have been used as biomarkers of liver injury elicited by carbon tetrachloride [93], thioacetamide [94], and human diseases [95, 96]. Yamazaki et al. [97] have shown that alterations of bile acid profiles were mostly evident after treatment with agents causing hepatocellular necrosis and/or cholestasis. More subtle effects were associated with steatosis or treatment with idiosyncratic hepatotoxicants. A recent study by Ellinger-Ziegelbauer et al. [98] provided an interesting observation suggesting the potential of a bile acid profile to differentiate hepatocellular and hepatobiliary injury. In this study, the authors treated rats with a set of agents that produced primarily hepatocellular damage, primarily hepatobiliary injury, or both types of injury. The data indicates that increased unconjugated bile acids in urine and serum might be potential markers of hepatocellular injury, whereas increased levels of conjugated bile acids correspond to potential hepatobiliary injury. If confirmed, bile acid profiles might become a useful tool for interrogating pathogenesis of liver injury.
4 Challenges for Clinical Applications
Table 1 summarizes the current status of traditional and emerging circulating biomarkers of DILI covered by this review. If they are to be employed in the clinical laboratory, analytic techniques used to measure novel biomarkers should be adaptable for routine clinical use. Although proteomic profiling has identified a number of potentially useful biomarkers to date, the initial methods used for the discovery of potential biomarkers are not necessarily well suited for clinical laboratory applications. Existing clinical laboratory applications require the use of internal standards for mass spectrometry, identifying measured components, developing standards for calibration and QC, identifying peak sets in spectra, and the application of established standards for method evaluation such as measures of reproducibility, detection limits, linearity, and recovery; evaluation of calibration curves, potential interferences, reference intervals, and peak characteristics; and separations for profiling methods [99]. The rigorous validation, standardization, and quality assurance demanded for clinical assays can present a formidable challenge for any novel biomarker candidate. A current limitation for the clinical verification of novel biomarker candidates is the requirement to test them in a meaningfully large and well-defined patient population [100]. The cost and complexity of a clinical study needed for the qualification of each biomarker is immense, and each biomarker qualification will require multiple supporting clinical studies. Ultimately, DILI biomarker candidates that complete the discovery and development process chain and gain approval by regulatory agencies will result in a commercial product such as an immunoassay. However, it has been estimated that developing a clinically deployable immunoassay could cost as much as US $100,000 to $250,000 per biomarker candidate for a research grade assay or US $2–4 million for an FDA-approvable assay and could require 1–1.5 years for completion [101]. The magnitude and cost of a clinical qualification effort limits the ability of individual entities to undertake the process on their own. The SAFE-T consortium is a public–private partnership comprising 20 partners from the pharmaceutical industry, small to medium enterprises, academic institutions, and clinical units of excellence with external advisors from the European Medicines Agency [26] focused on the clinical translation of safety biomarkers. Through initiatives like the SAFE-T consortium, the clinical application of safety biomarkers will become possible despite the numerous challenges.
5 Conclusions
Because ALT is a very sensitive biomarker of liver injury, the differentiation of ALT increases that spontaneously resolve from ones that signal serious liver injury (especially in early stages) is very difficult. On the other hand, increases of TBil levels that are considered a hallmark of severe liver injury occur only after significant damage has already occurred. Therefore, the development of new biomarkers capable of assessing the risk of clinically relevant liver injury in early stages is essential for better diagnosis and treatment of DILI. Biomarkers capable of identifying subjects that are susceptible to liver injury would be essential for limiting the incidence of DILI.
The discovery of new biomarkers requires resources. For instance, in a published survey of clinical assays for proteins in plasma and serum that was based on an analysis of FDA approvals through 2008, Anderson [102] concluded that there were serious deficiencies in the protein biomarker pipeline because no proteomics-discovered analytes had yet emerged at that time. The situation in 2013 essentially remains unchanged. Nevertheless, recently published work suggests that some miRNAs may offer promising biomarkers for the detection of DILI.
Because prospective clinical studies evaluating performance of biomarkers of DILI are ethically not feasible, biomarker research of DILI requires new approaches. For instance, the utilization of samples from subjects with a variety of liver damage etiologies via retrospective study design significantly facilitates clinical evaluation of candidate biomarkers [20]. Furthermore, employing samples from cases of accidental poisonings and/or DILI cases from clinical trials will build confidence in identified biomarkers.
Prior to the widespread acceptance of any novel DILI biomarker, it must undergo a rigorous assay validation process that includes the documentation of critical assay parameters such as specificity, sensitivity, accuracy, and precision, as well as demonstrate species relevance and an association with clinical endpoints. Given that this process is quite complex and costly, the support of international collaborative projects spearheaded by HESI, PSTC, and SAFE-T that include academic researchers, clinicians, industry, and regulatory agencies is essential.
Notes
The terms SRM and MRM are cited based on the specific reference.
References
Zhang M, Chen M, Tong W. Is toxicogenomics a more reliable and sensitive biomarker than conventional indicators from rats to predict drug-induced liver injury in humans? Chem Res Toxicol. 2012;25(1):122–9.
Olson H, Betton G, Robinson D, Thomas K, Monro A, Kolaja G, Lilly P, Sanders J, Sipes G, Bracken W, Dorato M, Van Deun K, Smith P, Berger B, Heller A. Concordance of the toxicity of pharmaceuticals in humans and in animals. Regul Toxicol Pharmacol. 2000;32(1):56–67.
Ostapowicz G, Fontana RJ, Schiodt FV, et al. Results of a prospective study of acute liver failure at 17 tertiary care centers in the United States. Ann Intern Med. 2002;137:947–54.
Lammert C, Einarsson S, Saha C, Niklasson A, Bjornsson E, Chalasani N. Relationship between daily dose of oral medications and idiosyncratic drug-induced liver injury: search for signals. Hepatology. 2008;47(6):2003–9.
Verma S, Kaplowitz N. Diagnosis, management and prevention of drug-induced liver injury. Gut. 2009;58(11):1555–64.
Lee WM. Drug-induced hepatotoxicity. N Engl J Med. 2003;349(5):474–85.
Chalasani N, Björnsson E. Risk factors for idiosyncratic drug-induced liver injury. Gastroenterology. 2010;138(7):2246–59.
Corsini A, Ganey P, Ju C, Kaplowitz N, Pessayre D, Roth R, Watkins PB, Albassam M, Liu B, Stancic S, Suter L, Bortolini M. Current challenges and controversies in drug-induced liver injury. Drug Saf. 2012;35(12):1099–117.
Tarantino G, Conca P, Basile V, Gentile A, Capone D, Polichetti G, Leo E. A prospective study of acute drug-induced liver injury in patients suffering from non-alcoholic fatty liver disease. Hepatol Res. 2007;37(6):410–5.
Farmer AD, Brind A. Drug-induced liver injury. Medicine. 2011;39(3):536–40.
Lucena MI, Molokhia M, Shen Y, Spanish DILI Registry, EUDRAGENE, DILIN, DILIGEN, International SAEC, et al. Susceptibility to amoxicillin-clavulanate-induced liver injury is influenced by multiple HLA class I and II alleles. Gastroenterology. 2011;141(1):338–47.
Biomarkers Definitions Working Group. Biomarkers and surrogate endpoints: preferred definitions and conceptual framework. Clin Pharmacol Ther. 2001;69:89–95.
FDA. Guidance for Industry. pharmacogenomic data submission (2005). http://www.fda.gov/downloads/Drugs/GuidanceComplianceRegulatoryInformation/Guidances/UCM079849.pdf.
Boone L, Meyer D, Cusick P, Ennulat D, Bolliger AP, Everds N, Meador V, Elliott G, Honor D, Bounous D, Jordan H. Selection and interpretation of clinical pathology indicators of hepatic injury in preclinical studies. Vet Clin Pathol. 2005;34(3):182–8.
Weingand K, Brown G, Hall R, Davies D, et al. Harmonization of animal clinical pathology testing in toxicity and safety studies. The Joint Scientific Committee for International Harmonization of Clinical Pathology Testing. Fundam Appl Toxicol. 1996;29(2):198–201.
Evans GO. Assessment of hepatotoxicty. In: Animal clinical chemistry. 2nd ed. Boca Raton: CRC Press; 2009. p. 37–66.
FDA. Guidance for Industry. Drug-induced liver injury: premarketing clinical evaluation. 2007. http://www.fda.gov/downloads/Drugs/GuidanceComplianceRegulatoryInformation/Guidances/UCM174090.pdf.
Temple R. Hy’s law: predicting serious hepatotoxicity. Pharmacoepidemiol Drug Saf. 2006;15(4):241–3.
Aithal GP, Watkins PB, Andrade RJ, Larrey D, Molokhia M, Takikawa H, Hunt CM, Wilke RA, Avigan M, Kaplowitz N, Bjornsson E, Daly AK. Case definition and phenotype standardization in drug-induced liver injury. Clin Pharmacol Ther. 2011;89(6):806–15.
Schomaker S, Warner R, Bock J, Johnson K, Potter D, Van Winkle J, Aubrecht J. Assessment of emerging biomarkers of liver injury in human subjects. Toxicol Sci. 2013;132(2):276–83.
Lee WM, Senior JR. Recognizing drug-induced liver injury: current problems, possible solutions. Toxicol Pathol. 2005;33(1):155–64.
Paulovich AG, Whiteaker JR, Hoofnagle AN, Wang P. The interface between biomarker discovery and clinical validation: the tar pit of the protein biomarker pipeline. Proteomics Clin Appl. 2008;2(10–11):1386–402.
Kondrat RW, McClusky GA, Cooks RG. Multiple reaction monitoring in mass spectrometry/mass spectrometry for direct analysis of complex mixtures. Anal Chem. 1978;50(14):2017–21.
Angel TE, Aryal UK, Hengel SM, Baker ES, Kelly RT, Robinson EW, Smith RD. Mass spectrometry-based proteomics: existing capabilities and future directions. Chem Soc Rev. 2012;41(10):3912–28.
Yang J, Qu L. deepBase: annotation and discovery of microRNAs and other noncoding RNAs from deep-sequencing data, vol 822. 1st ed. New York: Springer Science Business Media; 2012. p. 233–48.
Matheis K, Laurie D, Andriamandroso C, Arber N, Badimon L, Benain X, Bendjama K, Clavier I, Colman P, Firat H, Goepfert J, Hall S, Joos T, Kraus S, Kretschmer A, Merz M, Padro T, Planatscher H, Rossi A, Schneiderhan-Marra N, Schuppe-Koistinen I, Thomann P, Vidal JM, Molac B. A generic operational strategy to qualify translational safety biomarkers. Drug Discov Today. 2011;13–14:600–8.
FDA Guidance for Industry Bioanalytical Method Validation. 2001. http://www.fda.gov/downloads/Drugs/GuidanceComplianceRegulatoryInformation/Guidances/UCM070107.pdf.
Lee JW, Devanarayan V, Barrett YC, Weiner R, Allinson J, Fountain S, Keller S, Weinryb I, Green M, Duan L, Rogers JA, Millham R, O’Brien PJ, Sailstad J, Khan M, Ray C, Wagner JA. Fit-for-purpose method development and validation for successful biomarker measurement. Pharm Res. 2006;23(2):312–28.
Lee JW, Hall M. Method validation of protein biomarkers in support of drug development or clinical diagnosis/prognosis. J Chromatogr B Analyt Technol Biomed Life Sci. 2009;877(13):1259–71.
Köhler K, Seitz H. Validation processes of protein biomarkers in serum—a cross platform comparison. Sensors (Basel). 2012;12(9):12710–28.
FDA. Guidance for industry. Qualification process for drug development tools. 2010. http://www.fda.gov/downloads/Drugs/GuidanceComplianceRegulatoryInformation/Guidances/UCM230597.pdf.
Goodsaid F, Frueh F. Biomarker qualification pilot process at the US Food and Drug Administration. AAPS J. 2007;9(1):E105–8.
Goodsaid F, Papaluca M. Evolution of biomarker qualification at the health authorities. Nat Biotechnol. 2010;28(5):441–3.
Goodsaid FM, Frueh FW, Mattes W. Strategic paths for biomarker qualification. Toxicology. 2008;245(3):219–23.
FDA. Biomarker Qualification Program. 2012. http://www.fda.gov/Drugs/DevelopmentApprovalProcess/DrugDevelopmentToolsQualificationProgram/ucm284076.htm. Accessed 12 April 2013.
Collings FB, Vaidya VS. Novel technologies for the discovery and quantitation of biomarkers of toxicity. Toxicology. 2008;245:167–74.
Anderson NL. Counting the proteins in plasma. Clin Chem. 2010;56(11):1775–6.
Rifai N, Gillette MA, Carr SA. Protein biomarker discovery and validation: the long and uncertain path to clinical utility. Nat Biotechnol. 2006;24(8):971–83.
Ozer J, Reagan W, Schomaker S, Palandra J, Baratta M, Ramaiah S. Biomarkers in medicine, drug discovery, and environmental health. 1st ed. Hoboken: Wiley; 2010. p. 203–36.
Bell LN, Vuppalanchi R, Watkins PB, Bonkovsky HL, Serrano J, Fontana RJ, Wang M, Rochon J, Chalasani N, US Drug-Induced Liver Injury Network (DILIN) Research Group. Serum proteomic profiling in patients with drug-induced liver injury. Aliment Pharmacol Ther. 2012;35:600–12.
Hoofnagle AN, Becker JO, Oda MN, Cavigiolio G, Mayer P, Vaisar T. Multiple-reaction monitoring-mass spectrometric assays can accurately measure the relative protein abundance in complex mixtures. Clin Chem. 2012;58(4):777–81.
Picotti P, Aebersold R. Selected reaction monitoring-based proteomics: workflows, potential, pitfalls and future directions. Nat Methods. 2012;9(6):555–66.
Eissler CL, Bremmer SC, Martinez JS, Parker LL, Charbonneau H, Hall MC. A general strategy for studying multisite protein phosphorylation using label-free selected reaction monitoring mass spectrometry. Anal Biochem. 2011;418(2):267–75.
Anderson NL, Anderson NG, Haines LR, Hardie DB, Olafson RW, Pearson TW. Mass spectrometric quantitation of peptides and proteins using Stable Isotope Standards and Capture by Anti-Peptide Antibodies (SISCAPA). J Proteome Res. 2004;3(2):235–44.
Anderson L, Hunter CL. Quantitative mass spectrometric multiple reaction monitoring assays for major plasma proteins. Mol Cell Proteomics. 2006;5(4):573–88.
Antoine DJ, Jenkins RE, Dear JW, Williams DP, McGill MR, Sharpe MR, Craig DG, Simpson KJ, Jaeschke H, Park BK. Molecular forms of HMGB1 and keratin-18 as mechanistic biomarkers for mode of cell death and prognosis during clinical acetaminophen hepatotoxicity. J Hepatol. 2012;56(5):1070–9.
Bailey WJ, Holder D, Patel H, Devlin P, Gonzalez RJ, Hamilton V, Muniappa N, Hamlin DM, Thomas CE, Sistare FD, Glaab WE. A performance evaluation of three drug-induced liver injury biomarkers in the rat: alpha-glutathione S-transferase, arginase 1, and 4-hydroxyphenyl-pyruvate dioxygenase. Toxicol Sci. 2012;130(2):229–44.
Amacher DE, Adler R, Herath A, Townsend RR. Use of proteomic methods to identify serum biomarkers associated with rat liver toxicity or hypertrophy. Clin Chem. 2005;51(10):1796–803.
Laverty HG, Antoine DJ, Benson C, Chaponda M, Williams D, Kevin Park B. The potential of cytokines as safety biomarkers for drug-induced liver injury. Eur J Clin Pharmacol. 2010;66(10):961–76.
Lacour S, Gautier JC, Pallardy M, Roberts R. Cytokines as potential biomarkers of liver toxicity. Cancer Biomark. 2005;1(1):29–39.
Tarrant JM. Blood cytokines as biomarkers of in vivo toxicity in preclinical safety assessment: considerations for their use. Toxicol Sci. 2010;117(1):4–16.
Etheridge A, Lee I, Hood L, Galas D, Wang K. Extracellular microRNA: a new source of biomarkers. Mutat Res. 2011;717(1–2):85–90.
Su Z, Li Z, Chen T, Li QZ, Fang H, Ding D, Ge W, Ning B, Hong H, Perkins RG, Tong W, Shi L. Comparing next-generation sequencing and microarray technologies in a toxicological study of the effects of aristolochic acid on rat kidneys. Chem Res Toxicol. 2011;24(9):1486–93.
Gullapalli RR, Desai KV, Santana-Santos L, Kant JA, Becich MJ. Next generation sequencing in clinical medicine: challenges and lessons for pathology and biomedical informatics. J Pathol Inform. 2012;3:40.
Wang K, Yuan Y, Li H, Cho JH, Huang D, Gray L, Qin S, Galas DJ. The spectrum of circulating RNA: a window into systems toxicology. Toxicol Sci. 2013;132(2):478–92.
Su YW, Chen X, Jiang ZZ, Wang T, Wang C, Zhang Y, Wen J, Xue M, Zhu D, Zhang Y, Su YJ, Xing TY, Zhang CY, Zhang LY. A panel of serum microRNAs as specific biomarkers for diagnosis of compound- and herb-induced liver injury in rats. PLoS One. 2012;7(5):e37395.
FDA. MicroArray Quality Control (MAQC). 2011. http://www.fda.gov/ScienceResearch/BioinformaticsTools/MicroarrayQualityControlProject/default.htm.
Swaminathan R, Butt AN. Circulating nucleic acids in plasma and serum: recent development. Ann New York Acad Sci. 2006;1075:1–9.
Fleischacker M. Biology of circulating mRNA: still questions than answers? Ann. New York Acad Sci. 2006;1075:40–9.
Cocucci E, Racchetti G, Meldolesi J. Shedding microvesicles: artifacts no more. Trends Cell Biol. 2009;19:43–51.
Kudo Y, Ochi T, Shimada H, Ogawa S, Shinjo K. Utility of plasma circulating mRNA as a marker to detect hepatic injury. J Vet Med Sci. 2008;70(9):993–5.
Wetmore BA, Brees DJ, Singh R, Watkins PB, Andersen ME, Loy J, Thomas RS. Quantitative analyses and transcriptomic profiling of circulating messenger RNAs as biomarkers of rat liver injury. Hepatology. 2010;51(6):2127–39.
Miyamoto M, Yanai M, Ookubo S, Awasaki N, Takami K, Imai R. Detection of cell-free, liver-specific mRNAs in peripheral blood from rats with hepatotoxicity: a potential toxicological biomarker for safety evaluation. Toxicol Sci. 2008;106(2):538–45.
Okubo S, Miyamoto M, Takami K, Kanki M, Ono A, Nakatsu N, Yamada H, Ohno Y, Urushidani T. Identification of novel liver-specific mRNAs in plasma for biomarkers of drug-induced liver injury and quantitative evaluation in rats treated with various hepatotoxic compounds. Toxicol Sci. 2013;132(1):21–31.
Carthew RW, Sontheimer EJ. Origins and mechanisms of miRNAs and siRNAs. Cell. 2009;136(4):642–55.
Wang K, Yuan Y, Cho JH, McClarty S, Baxter D, Galas DJ. Comparing the microRNA spectrum between serum and plasma. PLoS One. 2012;7(7):e41561.
Kroh E, Parkin R, Mitchell P, Tewari M. Analysis of circulating microRNA biomarkers in plasma and serum using quantitative reverse transcription-PCR (qRT-PCR). Methods. 2010;50:298–301.
Williams AE. Functional aspects of animal microRNAs. Cell Mol Life Sci. 2008;65(4):545–62.
Wang K, Zhang S, Marzolf B, Troisch P, Brightman A, Hu Z, Hood LE, Galas DJ. Circulating microRNAs, potential biomarkers for drug-induced liver injury. Proc Natl Acad Sci USA. 2009;106(11):4402–7.
Elfimova N, Schlattjan M, Sowa JP, Dienes HP, Canbay A, Odenthal M. Circulating microRNAs: promising candidates serving as novel biomarkers of acute hepatitis. Front Physiol. 2012;3:476.
Tsai WC, Hsu SD, Hsu CS, Lai TC, Chen SJ, Shen R, Huang Y, Chen HC, Lee CH, Tsai TF, Hsu MT, Wu JC, Huang HD, Shiao MS, Hsiao M, Tsou AP. MicroRNA-122 plays a critical role in liver homeostasis and hepatocarcinogenesis. J Clin Invest. 2012;122(8):2884–97.
Jopling C. Liver-specific microRNA-122: biogenesis and function. RNA Biol. 2012;9(2):137–42.
Laterza OF, Lim L, Garrett-Engele PW, Vlasakova K, Muniappa N, Tanaka WK, Johnson JM, Sina JF, Fare TL, Sistare FD, Glaab WE. Plasma microRNAs as sensitive and specific biomarkers of tissue injury. Clin Chem. 2009;55(11):1977–83.
Zhang Y, Jia Y, Zheng R, Guo Y, Wang Y, Guo H, Fei M, Sun S. Plasma microRNA-122 as a biomarker for viral-, alcohol-, and chemical-related hepatic diseases. Clin Chem. 2010;56(12):1830–8.
Starckx S, Batheja A, Verheyen GR, Jonghe SD, Steemans K, Dijck BV, Singer M, Bogdan N, Snoeys J, Vinken P, Sasaki JC, Gompel JV, Guzzie-Peck P, Lampo A, Lammens L. Evaluation of miR-122 and other biomarkers in distinct acute liver injury in rats. Toxicol Pathol. 2013;41(5):795–804.
Starkey Lewis PJ, Dear J, Platt V, Simpson KJ, Craig DG, Antoine DJ, French NS, Dhaun N, Webb DJ, Costello EM, Neoptolemos JP, Moggs J, Goldring CE, Park BK. Circulating microRNAs as potential markers of human drug-induced liver injury. Hepatology. 2011;54(5):1767–76.
Antoine DJ, Dear JW, Starkey-Lewis P, Platt V, Coyle J, Masson M, Thanacoody RH, Gray AJ, Webb DJ, Moggs JG, Bateman DN, Goldring CE, Park BK. Mechanistic biomarkers provide early and sensitive detection of acetaminophen-induced acute liver injury at first presentation to hospital. Hepatology. 2013. doi:10.1002/hep.26294.
Qi P, Cheng SQ, Wang H, Li N, Chen YF, Gao CF. Serum microRNAs as biomarkers for hepatocellular carcinoma in Chinese patients with chronic hepatitis B virus infection. PLoS One. 2011;6(12):e28486.
Cermelli S, Ruggieri A, Marrero JA, Ioannou GN, Beretta L. Circulating microRNAs in patients with chronic hepatitis C and non-alcoholic fatty liver disease. PLoS One. 2011;6(8):e23937.
Xu J, Wu C, Che X, Wang L, Yu D, Zhang T, Huang L, Li H, Tan W, Wang C, Lin D. Circulating microRNAs, miR-21, miR-122, and miR-223, in patients with hepatocellular carcinoma or chronic hepatitis. Mol Carcinog. 2011;50(2):136–42.
Nicholson JK, Connelly J, Lindon JC, Holmes E. Metabonomics: a platform for studying drug toxicity and gene function. Nat Rev Drug Discov. 2002;1(2):153–61.
Wang JH, Byun J, Pennathur S. Analytical approaches to metabolomics and applications to systems biology. Semin Nephrol. 2010;30(5):500–11.
MSI: Metabonomics Standards Initiative. http://msi-workgroups.sourceforge.net/. Accessed 12 April 2013.
Kumar BS, Chung BC, Kwon OS, Jung BH. Discovery of common urinary biomarkers for hepatotoxicity induced by carbon tetrachloride, acetaminophen and methotrexate by mass spectrometry-based metabolomics. J Appl Toxicol. 2012;32(7):505–20.
Sun J, Schnackenberg LK, Beger RD. Studies of acetaminophen and metabolites in urine and their correlations with toxicity using metabolomics. Drug Metab Lett. 2009;3(3):130–6.
Coen M, Goldfain-Blanc F, Rolland-Valognes G, Walther B, Robertson DG, Holmes E, Lindon JC, Nicholson JK. Pharmacometabonomic investigation of dynamic metabolic phenotypes associated with variability in response to galactosamine hepatotoxicity. J Proteome Res. 2012;11(4):2427–40.
Zira A, Kostidis S, Theocharis S, Sigala F, Engelsen SB, Andreadou I, Mikros E. 1H NMR-based metabonomics approach in a rat model of acute liver injury and regeneration induced by CCl4 administration. Toxicology. 2013;303:115–24.
Gika HG, Ji C, Theodoridis GA, Michopoulos F, Kaplowitz N, Wilson ID. Investigation of chronic alcohol consumption in rodents via ultra-high-performance liquid chromatography-mass spectrometry based metabolite profiling. J Chromatogr A. 2012;1259:128–37.
Clayton TA, Lindon JC, Cloarec O, Antti H, Charuel C, Hanton G, Provost JP, Le Net JL, Baker D, Walley RJ, Everett JR, Nicholson JK. Pharmaco-metabonomic phenotyping and personalized drug treatment. Nature. 2006;440(7087):1073–7.
Winnike JH, Li Z, Wright FA, Macdonald JM, O’Connell TM, Watkins PB. Use of pharmaco-metabonomics for early prediction of acetaminophen-induced hepatotoxicity in humans. Clin Pharmacol Ther. 2010;88(1):45–51.
Woolbright BL, Jaeschke H. Novel insight into mechanisms of cholestatic liver injury. World J Gastroenterol. 2012;18(36):4985–93.
Darkoh C, Lichtenberger LM, Ajami N, Dial EJ, Jiang ZD, DuPont HL. Bile acids improve the antimicrobial effect of rifaximin. Antimicrob Agents Chemother. 2010;54(9):3618–24.
Shimada T, Nakanishi T, Toyama A, Yamauchi S, Kanzaki A, Fujiwake H, Sato TA, Ikegawa M. Potential implications for monitoring serum bile acid profiles in circulation with serum proteome for carbon tetrachloride-induced liver injury/regeneration model in mice. J Proteome Res. 2010;9(9):4490–500.
Kawai H, Kudo N, Kawashima Y, Mitsumoto A. Efficacy of urine bile acid as a non-invasive indicator of liver damage in rats. J Toxicol Sci. 2009;34(1):27–38.
Trottier J, Białek A, Caron P, Straka RJ, Milkiewicz P, Barbier O. Profiling circulating and urinary bile acids in patients with biliary obstruction before and after biliary stenting. PLoS One. 2011;6(7):e22094.
Lake AD, Novak P, Shipkova P, Aranibar N, Robertson D, Reily MD, Lu Z, Lehman-McKeeman LD, Cherrington NJ. Decreased hepatotoxic bile acid composition and altered synthesis in progressive human nonalcoholic fatty liver disease. Toxicol Appl Pharmacol. 2013;268(2):132–40.
Yamazaki M, Miyake M, Sato H, Masutomi N, Tsutsui N, Adam KP, Alexander DC, Lawton KA, Milburn MV, Ryals JA, Wulff JE, Guo L. Perturbation of bile acid homeostasis is an early pathogenesis event of drug induced liver injury in rats. Toxicol Appl Pharmacol. 2013;268(1):79–89.
Ellinger-Ziegelbauer H, Adler M, Amberg A, Brandenburg A, Callanan JJ, Connor S, Fountoulakis M, Gmuender H, Gruhler A, Hewitt P, Hodson M, Matheis KA, McCarthy D, Raschke M, Riefke B, Schmitt CS, Sieber M, Sposny A, Suter L, Sweatman B, Mally A. The enhanced value of combining conventional and “omics” analyses in early assessment of drug-induced hepatobiliary injury. Toxicol Appl Pharmacol. 2011;252(2):97–111.
Hortin GL. Can mass spectrometric protein profiling meet desired standards of clinical laboratory practice? Clin Chem. 2005;51(1):3–5.
Anderson NL, Ptolemy AS, Rifai N. The riddle of protein diagnostics: future bleak or bright? Clin Chem. 2013;59(1):194–7.
Carr SA, Anderson L. Protein quantitation through targeted mass spectrometry: the way out of biomarker purgatory? Clin Chem. 2008;11:1749–1752 (editorial).
Anderson NL. The clinical plasma proteome: a survey of clinical assays for proteins in plasma and serum. Clin Chem. 2010;56(2):177–85.
Acknowledgements
The preparation of this article was not supported by any external funding. The authors have no conflicts of interest that are directly relevant to the content of this article.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Amacher, D.E., Schomaker, S.J. & Aubrecht, J. Development of Blood Biomarkers for Drug-Induced Liver Injury: An Evaluation of Their Potential for Risk Assessment and Diagnostics. Mol Diagn Ther 17, 343–354 (2013). https://doi.org/10.1007/s40291-013-0049-0
Published:
Issue Date:
DOI: https://doi.org/10.1007/s40291-013-0049-0