
Abstract
Background
Statins are among the most prescribed drugs worldwide to reduce the risk of cardiovascular events. Interindividual variability in drug response is a major clinical problem and is of concern during drug development. Statins, such as atorvastatin, are taken orally and access to their site of action in the liver is greatly facilitated by both intestinal and hepatic transporters.
Objective
To examine the impact of polymorphisms of the multidrug resistance 1(MDR1) and solute carrier organic anion transporter 1B1 (SLCO1B1) genes on the therapeutic response to atorvastatin as well as the presence of gender–gene interaction.
Methods
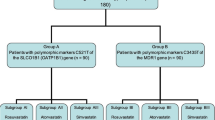
Serum lipid levels were determined at baseline and 4 weeks following 40 mg/day atorvastatin treatment in 50 Egyptian hypercholesterolemic patients (27 males and 23 females). Identification of MDR1 C3435T and SLCO1B1 A388G gene polymorphisms was performed using a polymerase chain reaction–restriction fragment length polymorphism (PCR-RFLP) method.
Results
Treatment with atorvastatin resulted in a mean reduction of total cholesterol (TC), low density lipoprotein cholesterol (LDL-C), and triglyceride (TG) of 8.7 %, 9.2 %, and 4.1 %, respectively, and a mean increase of high density lipoprotein cholesterol (HDL-C) of 1 %. Baseline and post-treatment HDL-C levels were statistically significantly higher in the MDR 1 TT homozygotes when compared with the CC wild type. The percentage change in TC, LDL-C, TG, and HDL-C did not show any statistically significant difference when compared among the different MDR 1 C3435T or SLCO1B1 A388G genotypes. The SLCO1B1 GG homozygotes showed a decrease in TG, whereas there was an increase in TG following atorvastatin treatment in AA and AG carriers in females; however, males did not show any statistically significant difference. There was no statistically significant association between either the coronary artery disease (CAD) risk factors (family history of CAD, hypertension, diabetes mellitus, smoking) or concomitant medications with the percentage change in different lipid parameters.
Conclusion
MDR1 C3435T was associated with baseline and post-treatment HDL-C variation. SLCO1B1 A388G showed gender-related effects on TG change following atorvastatin treatment. None of the comorbidities or the concomitant medications influenced the percentage change of lipid parameters following atorvastatin treatment. The results of this study may lead to an improved understanding of the genetic determinants of lipid response to atorvastatin treatment.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
1 Introduction
Circulatory diseases are the leading cause of death worldwide with lipid metabolism being one of the main determinants of cardiovascular risk [1]. To reduce the risk of cardiovascular events, statins are among the most prescribed drugs worldwide. The statins’ mechanism of action is to inhibit the HMG-CoA reductase enzyme, which catalyzes the biosynthesis of cholesterol in the liver. Once inhibited, it stops the formation of mevalonic acid and reduces intracellular cholesterol synthesis, which causes reduced secretion of apolipoprotein B-containing lipoproteins from the liver and the upregulation of low density lipoprotein (LDL) receptor activity, both of which contribute to lowering LDL cholesterol (LDL-C) in the plasma [2]. Statins differ in absorption, plasma protein binding, solubility, and excretion and exhibit variable dose-related efficacy in reducing LDL-C. Interindividual variability in drug response is a major clinical problem and is of concern during drug development [3]. In addition to known environmental factors, sequence polymorphic variants in genes encoding drug-metabolizing enzymes, drug transporters, or drug targets may have important pharmacokinetic and pharmacodynamic consequences and thus affect drug response [4]. Statins, such as atorvastatin, are taken orally and access to their site of action in the liver is greatly facilitated by both intestinal and hepatic transporters [5]. Similarly, atorvastatin and its metabolites are excreted primarily into the bile by transporters, represented by the ATP binding cassette (ABC) family. ABC transporters are a superfamily of integral transmembrane proteins that uses energy of ATP hydrolysis to translocate a broad spectrum of molecules across the cell membrane. ABCB1 is a 170-kDa transporter protein named P-glycoprotein (P-gp) that has been associated with the transport of cellular lipids and drugs [6]. P-gp is encoded by a polymorphic gene named multidrug resistance 1 (MDR1) located on chromosome 7. The MDR1 gene has more than 20 polymorphisms, some of which have been associated with altered P-gp expression and activity in vivo [7]. The C3435T synonymous polymorphism in exon 26 of the MDR1 gene is at a wobble position but does not change the amino acid from an isoleucine at position 1,145 [8]. Since it has been proposed that atorvastatin alters P-gp activity, it is possible that variations in MDR1 may also affect lipid response to atorvastatin [6].
The solute carrier organic anion transporter 1B1 (SLCO1B1) gene codes for a hepatic influx transporter, organic anion transporter polypeptide 1B1 (OATP1B1), that is thought to play a key role in statin transport into hepatocytes [9]. OATP1B1 is expressed predominantly on the basolateral membrane of human hepatocytes, where it mediates active hepatocyte uptake of many endogenous substrates and a wide range of compounds including statins [10]. A reduced OATP1B1-mediated uptake from the bloodstream into the liver because of genetic variants or drug interactions may lead to reduced statin concentrations in the liver, but an increased systemic exposure. This will result in a reduced lipid-lowering effect, but an increased risk of muscle toxicity [11]. A number of single nucleotide polymorphisms (SNPs) have been found within the SLCO1B1 gene, located on chromosome 12. Tirona et al. [12] identified 14 non-synonymous SNPs, represented by 16 distinct haplotypes. One of these non-synonymous SNPs, which is present in exon 4, is SCLOB1 c.388A > G involving the substitution of asparagine to aspartic acid at amino acid 130 (Asn 130 Asp). This results in a substrate-specific effect [9] and is associated with altered transport activity.
The present study was designed to examine the possible role of polymorphisms in two genes, namely MDR1 and SLCO1B1, coding for an efflux transporter and an influx transporter, respectively, on the response to a very widely prescribed statin, atorvastatin, which is administered to unrelated hypercholesterolemic Egyptian patients and to assess the presence of gender–gene interaction. Moreover, we also aimed to examine whether there is an interaction between the patients’ characteristics, comorbidities, the intake of concomitant medications, and the percentage change of lipids following atorvastatin treatment.
2 Subjects and Methods
2.1 Subjects
Fifty unrelated Egyptian hypercholesterolemic patients (27 males and 23 females; mean age 55.2 ± 9.9 years) were recruited from the Cardiology Department in Kasr El-Ainy University Hospital (Cairo, Egypt) from June 2010 to October 2011. Fasting baseline lipids were measured 1 day before and 4 weeks after 40 mg/day atorvastatin treatment (Ator, manufactured by Egyptian International Pharmaceutical Industries Co. (EIPICO), Egypt). The patients underwent dietary consultation. Inclusion criteria included hypercholesterolemic patients (age >18) requiring statin treatment according to the recommendations of the third report of the National Cholesterol Education Program [13]. Exclusion criteria included pregnancy, clinical diagnosis of familial hypercholesterolemia, acute coronary syndrome, hepatic disease, kidney disease, endocrinological disorders, and malignant disease. Subjects who were receiving concomitant lipid-lowering therapy or those who were under treatment with medications that could affect the lipid profile, e.g., contraceptive pills, were not included in this study. The study protocol was approved by the local scientific ethics committee and all patients provided informed consent to participate in this study.
2.2 Methods
2.2.1 Lipid and Lipoprotein Measurements
Blood samples were collected from the individuals after 12 h fast, before and 4 weeks after atorvastatin administration. Serum total cholesterol (TC), high density lipoprotein cholesterol (HDL-C), and triglyceride (TG) concentrations were measured using standard enzymatic methods with an automated analyzer (Hitachi 917, Hitachi Ltd., Tokyo, Japan) using kits supplied by Boehringer Mannheim (BM, Germany). HDL-C was measured after phosphotungstic acid and magnesium precipitation. LDL-C concentration was calculated using the Friedewald formula [14].
2.2.2 Genomic DNA Analysis
Genomic DNA was extracted from EDTA-anticoagulated blood by means of a Thermo Scientific Gene JET whole blood genomic DNA purification mini kit (Thermo Fisher Scientific Inc., USA). MDR1 (C3435T) and SCLO1B1 (A388G) gene polymorphisms were detected using a polymerase chain reaction–restriction fragment length polymorphism (PCR-RFLP) method.
2.2.2.1 Genotyping of MDR1 3435C > T (rs1045642) SNP
A 197-bp fragment of the MDR1 gene was generated by PCR amplification with the primers: (F) 5′-TGT TTT CAG CTG CTT GAT GG-3′, (R) 5′-AAG GCA TGT ATG TTG GCC TC-3′. Amplification was done in a total volume of 25 μl of reaction mixture which included 50 ng genomic DNA, 25 pmol of each primer (BIONEER Corporation, Korea), 12.5 μl Dream Taq™ Green PCR master mix (Thermo Fisher Scientific Inc., USA). PCR was performed in a Hybaid (express) thermal cycler (Promega Corporation, WI, USA) according to the protocol proposed by Cascorbi et al. [15], which consisted of an initial denaturation at 94 °C for 2 min, followed by 35 cycles of 30 s at 94 °C, 30 s at 60 °C, and 30 s at 72 °C, and lastly, a final elongation step of 7 min at 72 °C. The restriction enzyme Sau3AI (Thermo Fisher Scientific Inc., USA) was used to distinguish the MDR1 C3435Tgenotypes. The digested DNA fragments were separated by gel electrophoresis on 3 % ethidium bromide stained agarose gel. The three possible genotypes were the CC homozygote appearing as 158- and 39-bp fragments, the C/T heterozygote with 197-,158-, and 39-bp fragments, and the TT homozygote with a 197-bp fragment (Supplementary Fig. I).
2.2.2.2 Genotyping of SLCO1B1 388A > G (rs2306283) SNP
A 274-bp fragment of the SLCO1B1 gene was generated by PCR amplification with 60 pmol of each primer; (F): 5′-GCA AAT AAA GGG GAA TAT TTC TC-3′ and (R): 5′-AGA GAT GTA ATT AAA TGT ATA C-3′ (BIONEER Corporation, Korea). PCR was performed according to the protocol proposed by Tirona et al. [12], which consisted of an initial denaturation at 94 °C for 5 min, followed by 37 cycles of denaturation at 94 °C for 30 s, annealing at 46 °C for 30 s, and extension at 72 °C for 30 s. A final 5-min extension at 72 °C was adopted. After amplification, the PCR products (274 bp) were digested with the CLaI restriction endonuclease (Thermo Fisher Scientific Inc., USA). The three possible genotypes were the GG homozygote appearing as 155- and 119-bp fragments, the A/G heterozygote with 274-, 155-, and 119-bp fragments, and the AA homozygote with 274-bp fragments (Supplementary Fig. II).
2.3 Statistical Analysis
The Statistical Program for Social Sciences (SPSS) version 12.0 was used for data management (SPSS Inc., Chicago, IL). The observed genotype frequencies were compared with those expected under Hardy–Weinberg equilibrium using a X 2 test. Mean and standard deviation described quantitative data. Change of lipid profile after treatment was calculated as a percentage change from pretreatment values. Parametric and non-parametric t tests compared two independent groups and parametric and non-parametric ANOVA (analysis of variance) for more than two independent groups. Post hoc tests made pairwise comparisons. The P value was considered significant at the 0.05 level.
3 Results
The demographic characteristics of the 50 patients recruited in this study are shown in Supplementary Table I.
3.1 Therapeutic Response to Treatment
Treatment with atorvastatin resulted in a significant reduction in TC, LDL-C, and TG. However, the increase in HDL-C did not reach statistical significance (Table 1).
3.1.1 Gender-Related Therapeutic Response to Treatment
The mean HDL-C level in females was significantly higher than males pre- and post-treatment. After treatment, there was a 13.2 % decrease in TG level in male patients, whereas the female patients showed a 6.7 % increase in TG level, but this difference was not statistically significant as shown in (Table 2).
3.2 Genotype and Allele Frequencies of MDR1 C3435T and SLCO1B1 A388G Gene Polymorphisms
The genotype frequencies of MDR1 C3435T polymorphism were 38, 40, and 22 % for CC, CT, and TT, respectively; the allele frequencies were C = 0.58 and T = 0.42. Regarding SLCO1B1 A388G polymorphism, the genotype frequencies were 18, 56, and 26 % for AA, AG, GG respectively; the allele frequencies were A = 0.46 and G = 0.54.The observed genotype distributions were consistent with the Hardy–Weinberg equilibrium (p = 0.07 and 0.2, respectively).
3.3 Association of Gene Polymorphisms with Baseline Lipid Levels
Baseline HDL-C was statistically significantly higher in the MDR 1 3435TT homozygotes when compared with the CC wild type. As for the SLCO1B1 A388G genotypes, there was no statistically significant association between baseline lipid parameters and the different genotypes (Table 3).
3.4 Association of Gene Polymorphisms with Response to Treatment
In MDR1 3435 TT homozygotes, post-treatment HDL-C level was significantly higher than in CC and CT genotype carriers. As for the SLCO1B1 A388G genotypes, there was no statistically significant association of post-treatment lipid parameters with the different genotypes. The percentage change in TC, HDL-C, LDL-C, and TG did not show any statistically significant difference when compared among the different MDR1 C3435T genotypes using ANOVA, dominant model (CC versus CT + TT), or recessive model (TT versus CC + CT). The same applies for SLCO1B1 A388G genotypes, where there was no statistically significant difference on comparing the percentage change in TC, TG, LDL-C, and HDL-C among the different genotypes using ANOVA, dominant model (AA versus AG + GG), or recessive model (GG versus AG + AA) as shown in Table 3 and Fig. 1a, b.

Percentage change of TC, TG, HDL-C, and LDL-C according to a C3435T polymorphism in MDR1 gene, b A388G polymorphism in SLCO1B1 gene, c C3435T polymorphism in MDR1 gene in male patients, d A388G polymorphism in SLCO1B1 gene in male patients, e C3435T polymorphism in MDR1 gene in female patients, f A388G polymorphism in SLCO1B1 gene in female patients. TC total cholesterol, TG triglycerides, HDL-C high density lipoprotein cholesterol, LDL-C low density lipoprotein cholesterol, MDR1 multidrug resistance 1, SLCO1B1 solute carrier organic anion transporter 1B1
3.5 Gender-Related Association of Gene Polymorphisms with Therapeutic Response to Treatment
To compare the percentage change of TC, TG, LDL-C, and HDL-C in males and in females with respect to C3435T SNP of the MDR1 gene, there was no statistically significant difference in either males or females as shown in Table 4 and Fig. 1c, e. In females, SLCO1B1 GG homozygotes showed a decrease in TG, whereas AA and AG carriers showed an increase in TG following atorvastatin treatment (P = 0.008). However, there was no statistically significant difference in males as shown in Table 4 and Fig. 1d, f.
3.6 Percentage Change in Lipid Profile after Atorvastatin Treatment According to Patients’ Characteristics and Concomitant Medication
There was no statistically significant association between any of the coronary artery disease (CAD) risk factors (family history of CAD, hypertension, diabetes mellitus, smoking) and the percentage change in lipid profile. None of the concomitant medications showed a statistically significant association with the percentage change in the different lipid parameters as shown in (Supplementary Table II).
4 Discussion
Interindividual variations in drug toxicity and efficacy are well established. Although a number of factors may contribute to interindividual variability, including environmental interactions and drug–drug interactions, a patient’s genotype is increasingly understood to influence drug disposition and activity and thus may provide a method to individualize drug therapy [16]. Polymorphisms in genes that encode drug-metabolizing enzymes, drug targets, and drug transporter proteins are among the most clinically important genotypic variations for many medications. Transport proteins may influence drug disposition by impacting the absorption, distribution, and excretion of many drugs [17]. More than 40 genes that could affect response to statins have been investigated, related to both pharmacokinetics (metabolizing enzymes and transport proteins) and pharmacodynamics (receptors and signal transduction pathways), although with contradictory results [18].
In this study, we examined the possible role of polymorphisms in two genes, namely MDR1 and SLCO1B1, coding for an efflux transporter and an influx transporter, respectively, on the response to atorvastatin. In the present study, the percentage change of lipids after 4 weeks of treatment with atorvastatin revealed a significant reduction in TC, TG, and LDL-C. However, the increase in HDL-C did not reach statistical significance. Conflicting results were reported in previous studies as regards the therapeutic response to atorvastatin. A large patient interindividual variability resulting in an overall broad range of response was noted. Rebecchi et al. [19] concluded that LDL-C varied largely from a reduction of 64 % to an increase of 8.1 % after atorvastatin treatment (10 mg/day for 4 weeks) and no increase in HDL-C levels was observed. Using the same 10 mg dose, Rodrigues et al. [20] observed a statistically significant decrease in HDL-C levels.
Gender differences in response to atorvastatin treatment showed that the mean baseline and post-treatment HDL-C levels in females were significantly higher than those in males. In accordance with our data, Pedro-Botet et al. [21] concluded that women had higher HDL-C than men at both baseline and 12 months after atorvastatin treatment. Estrogen appears to increase HDL-C. The estrogen receptor (ER)-mediated pathway may play a role in HDL-C response to statin treatment. Kajinami et al. [22] concluded that ERα, PvuII(−) XbaI(+) haplotype was significantly and independently associated with a greater HDL-C increase in hypercholesterolemic women, but not in men, treated by atorvastatin. Our study has also shown that the TG reduction in women was less than that of men, but this difference did not reach statistical significance. This is in contrast to the study by Sakabe et al. [23], who concluded that atorvastatin decreased the TG in women but not in men, after 3 months of atorvastatin (10 mg/day) therapy. Gender-related pharmacokinetic differences of atorvastatin have been reported. Lennernäs [24] reported higher equivalent maximum concentration, lower mean area under the concentration–time curve, and shorter half-life of atorvastatin in women than in men. Women appear to have a slightly lower plasma exposure to atorvastatin for a given dose [25]. However, it remains unclear whether these differences are clinically relevant.
In this study, the contributions of MDR1 C3435T and SLCO1B1 A388G polymorphisms to the plasma lipid therapeutic response after 4 weeks of atorvastatin therapy revealed that the MDR1 TT carriers had significantly higher baseline and post-treatment HDL-C than CC carriers. The increase of baseline HDL-C in TT genotype subjects when compared to CC and CT genotypes may be explained by the results of Jeannesson et al. [26] who reported that levels of ApoA1, the major apolipoprotein in the HDL, were statistically significantly higher in 3435TT carriers when compared to CC and CT carriers in healthy individuals. Several facts support a role for ABCB1 in cholesterol homeostasis. ABCB1 could modulate the different steps of cholesterol trafficking such as cholesterol endogenous biosynthesis, cholesterol esterification, exogenic cholesterol import from LDL and export to HDL [27]. Since ABCB1 allows the efflux of cholesterol from the cells to the HDL, and ABCB1 polymorphisms modify the expression and the activity of the transporter, then it is conceivable that ABCB1 polymorphisms could be associated with variations in ApoA1 plasma levels [26]. The mechanisms involved in the HDL response to statin therapy are, however, not fully understood. It was suggested that P-gp plays a role in the alterations in HDL levels caused by statin treatment [28]. Atorvastatin has been shown to be a substrate of P-gp and polymorphisms in MDR1 gene have been associated with a variable response to atorvastatin [28]. Hoffmeyer et al. [29] reported that the TT genotype of the MDR1 C3435T is associated with more than twofold lower duodenal P-gp protein expression levels compared with CC genotype; therefore, individuals with the TT genotype would be expected to have a twofold lower drug efflux which would result in higher oral bioavailability and higher tissue concentrations. Moreover, the 3435C > T polymorphism in the MDR1 gene was associated with lower mRNA levels in the liver [30]. Atorvastatin is also capable of activating human pregnane X receptor which regulates ABCB1 expression and disrupts ABCB1 ubiquitination, which results in a decrease of the function of the transporter. Atorvastatin treatment also inhibits ABCB1 synthesis in peripheral blood mononuclear cells and hepatocytes. Therefore, inhibition of ABCB1 synthesis may increase the atorvastatin efficacy, leading to a more pronounced reduction of plasma cholesterol. A negative correlation between TC reductions and ABCB1 mRNA levels after atorvastatin treatment was reported [31]. This may explain why TT genotype subjects had higher post-treatment HDL-C when compared to CC and CT genotypes. However, we found no effect of these two polymorphisms on the other baseline or post-treatment lipid parameters. Previous studies reported no association between C3435T polymorphism and baseline lipid parameters [19, 25]. In discordance with our findings, Rodrigues et al. [6] observed that hyperlipemic carriers of the MDR1 haplotype 2677T–3435T had higher baseline serum TC and LDL-C.
Findings on lipid response to atorvastatin with respect to MDR1 and SLCO1B1 polymorphisms described in our work are in agreement with three previous studies that could not prove any significant contribution of C3435T MDR1 polymorphisms to the lipid response to atorvastatin at a dosage of 10 mg/day for 4 weeks and 80 mg/day for 24 months. respectively, in hypercholesterolemic patients [6, 25, 32]. Moreover, other studies concluded that SLCO1B1 388A > G polymorphism did not exert a significant effect on the lipid therapeutic responses to either of simvastatin, rosuvastatin, or pravastatin examined in their studies [22, 33]. In contrast to our results, Donnelly et al. [34] concluded that the 388G allele was associated with a slightly lower mean baseline TC and carriers of the 388G variant were associated with a greater LDL-C response to statins. Moreover, Rodrigues et al. [20] concluded that a significantly high reduction of LDL-C in response to atorvastatin treatment was found in individuals homozygous for SLCO1B1 c.388G allele when compared to c.388A allele carriers, regardless of gender.
A large study by The Heart Protection Study Collaborative Group [35] comprising 16,664 genotyped participants concluded that rs2306283 variants were not associated with significant differences in pretreatment LDL-C levels and LDL-C reductions with simvastatin (40 mg/day) was 0.62 % per SLCO1B1 rs2306283 G (388G) allele. Moreover, The SEARCH Collaborative Group [36] reported that in haplotype with rs4149056, the SLCO1B1 rs2306283 G (388G) allele was associated with a significantly lower risk of myopathy.
Our findings prove that there was no gender gene–interaction with respect to C3435T SNP of the MDR1 gene. In disagreement with our results, Kajinami et al. [28] reported that post-treatment HDL-C levels in TT homozygotes were significantly lower than in CC homozygotes in men. However, female CC homozygotes experienced significantly smaller reductions in LDL-C, but larger increases in HDL-C, relative to variant allele carriers following atorvastatin (10 mg/day for 52 weeks) therapy. Those gender-specific differences were, however, not reproduced in a subsequent study [37]. Our results show that women displayed a significant gene–drug interaction with respect to the different SLCO1B1 A388G genotypes, whereas in men the SLCO1B1 locus did not account for any significant fraction of the variability in response. This may be related to the fact that estrone-3-sulfate and 17α-ethinylestradiol sulfate are OATP1B1 substrates, which show biphasic kinetics suggesting the possibility of multiple substrate binding sites in OATP1B1 [38].
In this work, there was no statistically significant association between any of the CAD risk factors (family history of CAD, hypertension, diabetes mellitus, smoking) and the percentage change in lipid profile after atorvastatin treatment. Kokaze et al. [39] showed that daily cigarette consumption was significantly and positively correlated with TC, LDL-C, and TG levels and negatively with HDL-C levels. Moreover, Voora et al. [18] concluded that nonsmokers have an enhanced LDL-C-lowering response to statins. Hoenig and Sellke [40] stated that insulin-resistant patients showed statistically significant higher percentage reductions in LDL-C compared to insulin-sensitive patients, after a 6-week course of atorvastatin 80 mg.
Minimal numbers of studies were conducted to examine the interaction between intake of concomitant medication and the lipid-lowering response to atorvastatin. In an attempt to study whether the intake of concomitant medication may influence the percentage change of lipids following 4 weeks of atorvastatin treatment, it was observed that none of the mentioned drugs influenced the lipid response to atorvastatin treatment. Concomitant clopidogrel treatment resulted in an increase in percentage reduction in TC which did not reach statistical significance (P = 0.077). In agreement, Mitsios et al. [41] stated that atorvastatin does not significantly influence the clopidogrel-induced inhibition of platelet activation, nor does clopidogrel influence the therapeutic efficacy of atorvastatin. Our results show that intake of β-blockers was associated with a higher percentage increase in HDL-C which did not reach statistical significance (P = 0.09). It is noteworthy that Ozbilen et al. [42] reported that there were no statistically significant changes in the lipid parameters in either of the two studied groups that were treated with carvedilol and metoprolol.
The discrepancy in results concluded from different studies may be due to different study designs. In this work, we recruited Egyptian hypercholesterolemic subjects (mean age 55.2 ± 9.9 years) who received atorvastatin at a daily dosage of 40 mg for 4 weeks. This is in contrast to other studies who recruited healthy normocholesterolemic subjects [43]; elderly patients [44]; or who received different dosage of atorvastatin (10 mg/day) [25]; for a different duration (52 weeks) [28]; or who received another type of statin such as pravastatin [44]. Our study included comparable numbers of men (54 %) and women (46 %) to be able to examine possible gender–gene interactions in the response to atorvastatin treatment. This is in contrast to other studies that exclusively examined men [45] or included a small [25, 28] or a big number [46] of women. To our knowledge, this is the first study examining the lipid responses to atorvastatin in Egyptians; thus, the influence of ethnicity on pharmacogenetic responses must be taken into account by performing and comparing clinical trials in various ethnic groups.
In conclusion, treatment with atorvastatin resulted in a significant reduction of TC, TG, and LDL-C; however, the HDL-C increase did not reach statistical significance. Baseline and post-treatment HDL-C levels were statistically significantly higher in the MDR1 3435TT homozygotes when compared with the CC wild type. Regarding the SLCO1B1 A388G genotypes, there was gender–gene interaction. In females, GG homozygotes showed a decrease in TG, whereas there was an increase in TG following atorvastatin treatment in AA and AG carriers. None of the comorbidities or the concomitant medications influenced the percentage change of lipid parameters following atorvastatin treatment. However, this study is limited by its sample size which may have caused the statistical power of the test performed to be below the desired level. Therefore, our findings require replication in a larger cohort. Pharmacogenetic testing has an increasing impact in the individualization of drug treatment and could contribute significantly to enhanced drug safety and efficacy. Therefore, it would be interesting to complement the present study by investigating whether the MDR1 and SLCO1B1 SNPs may influence the response to other doses of atorvastatin, as well as to other statins available in our country.
Abbreviations
- ABC:
-
ATP binding cassette
- ANOVA:
-
Analysis of variance
- EIPICO:
-
Egyptian International Pharmaceutical Industries Co.
- ER:
-
Estrogen receptor
- HDL-C:
-
High density lipoprotein cholesterol
- LDL-C:
-
Low density lipoprotein cholesterol
- MDR1 :
-
Multidrug resistance 1
- OATP1B1:
-
Organic anion transporter polypeptide 1B1
- PCR-RFLP:
-
Polymerase chain reaction–restriction fragment length polymorphism
- P-gp:
-
P-glycoprotein
- SLCO1B1:
-
Solute carrier organic anion transporter 1B1
- SNP:
-
Single nucleotide polymorphism
- TC:
-
Total cholesterol
- TG:
-
Triglycerides
References
Yusuf S, Hawken S, Ounpuu S, et al. Effect of potentially modifiable risk factors associated with myocardial infarction in 52 countries (the INTERHEART study): case–control study. Lancet. 2004;364(9438):937–52.
Vaughan CJ, Gotto AM Jr, Basson CT. The evolving role of statins in the management of atherosclerosis. J Am Coll Cardiol. 2000;35(1):1–10.
Bercovich Dani, Friedlander Yechiel, Korema Sigal, et al. The association of common SNPs and haplotypes in the CETP and MDR1 genes with lipids response to fluvastatin in familial hypercholesterolemia. Atherosclerosis. 2006;185:97–107.
Evans WE, Johnson JA. Pharmacogenomics: the inherited basis for interindividual differences in drug response. Annu Rev Genomics Hum Genet. 2001;2:9–39.
Mangravite LM, Krauss RM. Pharmacogenomics of statin response. Curr Opin Lipidol. 2007;18:409–14.
Rodrigues AC, Rebecchi IMM, Bertolami MC, et al. High baseline serum total and LDL cholesterol levels are associated with MDR1 haplotypes in Brazilian hypercholesterolemic individuals of European descent. Braz J Med Biol Res. 2005;38:1389–97.
Marzolini C, Paus E, Buclin T, et al. Polymorphisms in human MDR1 (P-glycoprotein): recent advances and clinical relevance. Clin Pharmacol Ther. 2004;75:13–33.
Hamidovic A, Hahn K, Kolesar J. Clinical significance of ABCB1 genotyping in oncology. J Oncol Pharm Practice. 2010;16:39–44.
Niemi M, Pasanen MK, Neuvonen PJ. Organic anion transporting polypeptide 1B1: a genetically polymorphic transporter of major importance for hepatic drug uptake. Pharmacol Rev. 2011;63:157–81.
Oshiro C, Mangravite L, Klein T, et al. PharmGKB very important pharmacogene: SLCO1B1. Pharmacogenet Genomics. 2010;20:211–6.
Link E, Parish S, Armitage J, et al. SLCO1B1 variants and statin-induced myopathy—a genome wide study. N Engl J Med. 2008;359:789–99.
Tirona RG, Leake BF, Merino G, et al. Polymorphisms in OATP-C: identification of multiple allelic variants associated with altered transport activity among European– and African–Americans. J Biol Chem. 2001;276:35669–75.
National Cholesterol Education Program (NCEP) Expert Panel on Detection, Evaluation, and Treatment of High Blood Cholesterol in Adults (Adult Treatment Panel III). Third report of the National Cholesterol Education Program (NCEP) Expert Panel on Detection, Evaluation, and Treatment of High Blood Cholesterol in Adults (Adult Treatment Panel III) final report. Circulation. 2002;106:3143–421.
Friedewald WT, Levy RI, Fredrickson DS. Estimation of the concentration of low-density lipoprotein cholesterol in plasma, without use of the preparative ultracentrifuge. Clin Chem. 1972;18:499–502.
Cascorbi I, Gerloff T, Johne A, et al. Frequency of single nucleotide polymorphisms in the P-glycoprotein drug transporter MDR1 gene in white subjects. Clin Pharmacol Ther. 2001;69:169–74.
Deeken JF, Figg WD, Bates SE, et al. Toward individualized treatment: predication of anticancer drug disposition and toxicity with pharmacogenetics. Anticancer Drugs. 2007;18:111–26.
Evans WE, McLeod HL. Pharmacogenomics–drug disposition, drug targets, and side effects. N Engl J Med. 2003;348:538–49.
Voora D, Shah SH, Reed CR, et al. Pharmacogenetic predictors of statin-mediated low-density lipoprotein cholesterol reduction and dose response. Circ Cardiovasc Genet. 2008;1(2):100–6.
Rebecchi MM, Rodrigues AC, Arazi SS, et al. ABCB1 and ABCC1 expression in peripheral mononuclear cells is influenced by gene polymorphisms and atorvastatin treatment. Biochem Pharmacol. 2009;77:66–75.
Rodrigues AC, Perin PM, Purim SG, et al. Pharmacogenetics of OATP transporters reveals that SLCO1B1 c.388A > G variant is determinant of increased atorvastatin response. Int J Mol Sci. 2011;12:5815–27.
Pedro-Botet J, Schaefer EJ, Arkema RB, et al. Apolipoprotein E genotype affects plasma lipid response to atorvastatin in a gender specific manner. Atherosclerosis. 2001;158:183–93.
Kajinami K, Brousseau ME, Lamon-Fava S, et al. Gender-specific effects of estrogen receptor alpha gene haplotype on high-density lipoprotein cholesterol response to atorvastatin: interaction with apolipoprotein AI gene polymorphism. Atherosclerosis. 2005;178:331–8.
Sakabe K, Fukuda N, Fukuda Y, et al. Gender differences in short-term effects of atorvastatin on lipid profile, fibrinolytic parameters, and endothelial function. Nutr Metabol Cardiovasc Dis. 2008;18:182–8.
Lennernäs H. Clinical pharmacokinetics of atorvastatin. Clin Pharmacokinet. 2003;42(13):1141–60.
Rosales A, Alvear M, Cuevas A, et al. Identification of pharmacogenetic predictors of lipid-lowering response to atorvastatin in Chilean subjects with hypercholesterolemia. Clin Chim Acta. 2012;413(3–4):495–501.
Elise Jeannesson, Gérard Siest, Bérangère Bastien, et al. Association of ABCB1 gene polymorphisms with plasma lipid and apolipoprotein concentrations in the STANISLAS cohort. Clin Chim Acta. 2009;403:198–202.
Garrigues A, Escargueil AE, Orlowski S. The multidrug transporter, P-glycoprotein, actively mediates cholesterol redistribution in the cell membrane. Proc Natl Acad Sci USA. 2002;99:10347–52.
Kajinami K, Brousseau ME, Ordovas JM, et al. Polymorphisms in the multidrug resistance-1 (MDR1) gene influence the response to atorvastatin treatment in a gender-specific manner. Am J Cardiol. 2004;93:1046–50.
Hoffmeyer S, Burk O, von Richter O, et al. Functional polymorphisms of the human multidrug-resistance gene: multiple sequence variations and correlation of one allele with P-glycoprotein expression and activity in vivo. Proc Natl Acad Sci USA. 2000;97:3473–8.
Wang D, Johnson AD, Papp AC, et al. Multidrug resistance polypeptide 1 (MDR1, ABCB1) variant 3435C > T affects mRNA stability. Pharmacogenet Genom. 2005;15:693–704.
Rodrigues AC, Curi R, Britto LR, et al. Down-regulation of ABCB1 transporter by atorvastatin in a human hepatoma cell line and in human peripheral blood mononuclear cells. Biochim Biophys Acta. 2006;1760:1866–73.
Mega JL, Morrow DA, Brown A, et al. Identification of genetic variants associated with response to statin therapy. Arterioscler Thromb Vasc Biol. 2009;29:1310–5.
Miao M, Mak, Valiant WL, Tomlinson B. Intronic variants in SLCO1B1 related to statin-induced myopathy are associated with the low-density lipoprotein cholesterol response to statins in Chinese patients with hyperlipidemia. Pharmacogenet Genomics. 2012;22(11):803–6. doi:10.1097/FPC.0b013e3283557c98.
Donnelly LA, Doney ASF, Tavendale R, et al. Common non-synonymous substitutions in SLCO1B1 predispose to statin intolerance in routinely treated individuals with type 2 diabetes: a Go-DARTS study. Clin Pharmacol Ther. 2011;89(2):210–6.
Heart Protection Study Collaborative Group. MRC/BHF heart protection study of cholesterol lowering with simvastatin in 20,536 high-risk individuals: a randomized placebo-controlled trial. Lancet. 2002;360:7–22.
SEARCH Collaborative Group, Link E, Parish S, Armitage J, Bowman L, Heath S, Matsuda F, Gut I, Lathrop M, Collins R. SLCO1B1 variants and statin-induced myopathy–a genomewide study. N Engl J Med. 2008;359(8):789–99.
Thompson JF, Man M, Johnson KJ, et al. An association study of 43 SNPs in 16 candidate genes with atorvastatin response. Pharmacogenomics J. 2005;5:352–8.
Han YH, Busler D, Hong Y, et al. Transporter studies with the 3-O-sulfate conjugate of 17alpha-ethinylestradiol: assessment of human liver drug transporters. Drug Metab Dispos. 2010;38:1072–82.
Kokaze A, Ishikawaa M, Matsunaga N, et al. Longevity-associated mitochondrial DNA 5178 A/C polymorphism modulates effects of daily drinking and cigarette consumption on serum triglyceride levels in middle-aged Japanese men. Exp Gerontol. 2003;38:1071–6.
Hoenig MR, Sellke FW. Insulin resistance is associated with increased cholesterol synthesis, decreased cholesterol absorption and enhanced lipid response to statin therapy. Atherosclerosis. 2010;211:260–5.
Mitsios JV, Papathanasiou AI, Rodis FI. Atorvastatin does not affect the antiplatelet potency of clopidogrel when it is administered concomitantly for 5 weeks in patients with acute coronary syndromes. Circulation. 2004;109:1335–8.
Ozbilen S, Eren MA, Turan MN, et al. The impact of carvedilol and metoprolol on serum lipid concentrations and symptoms in patients with hyperthyroidism. Endocr Res. 2012;37(3):117–23.
Wen J, Xiong Y. OATP1B1 388A > G polymorphism and pharmacokinetics of pitavastatin in Chinese healthy volunteers. J Clin Pharm Ther. 2010;35:99–104.
Akaoa H, Polisecki E, Kajinami K, et al. Genetic variation at the SLCO1B1 gene locus and low density lipoprotein cholesterol lowering response to pravastatin in the elderly. Atherosclerosis. 2012;220:413–7.
Hamilton RJ, Goldberg KC, Platz EA, et al. The influence of statin medications on prostate specific antigen levels. J Natl Cancer Inst. 2008;100:1511–8.
Paulo CJ, Santos L, Gagliardi AM, et al. SLCO1B1 haplotypes are not associated with atorvastatin-induced myalgia in Brazilian patients with familial hypercholesterolemia. Eur J Clin Pharmacol. 2012;68:273–9.
Disclosure statement
The authors have no conflicts of interest that are directly relevant to the content of this article.
Author information
Authors and Affiliations
Corresponding author
Electronic supplementary material
Below is the link to the electronic supplementary material.
40291_2013_38_MOESM2_ESM.docx
Supplementary Fig. II. Agarose gel electrophoresis showing solute carrier organic anion transporter 1B1 (SLCO1B1) A388G genotypes (DOCX 167 kb)
40291_2013_38_MOESM4_ESM.docx
Supplementary Table II. Percentage change in lipid profile after atorvastatin treatment according to patients’ characteristics and concomitant medication (DOCX 18 kb)
Rights and permissions
About this article
Cite this article
Shabana, M.F., Mishriki, A.A., Issac, M.S.M. et al. Do MDR1 and SLCO1B1 Polymorphisms Influence the Therapeutic Response to Atorvastatin? A Study on a Cohort of Egyptian Patients with Hypercholesterolemia. Mol Diagn Ther 17, 299–309 (2013). https://doi.org/10.1007/s40291-013-0038-3
Published:
Issue Date:
DOI: https://doi.org/10.1007/s40291-013-0038-3