
Abstract
The National Institute for Health and Care Excellence (NICE) invited the manufacturer of eribulin (Eisai Ltd) to submit evidence for the clinical and cost effectiveness of eribulin as treatment for patients with locally advanced or metastatic breast cancer (LABC/MBC) pre-treated with at least two chemotherapy regimens. This article summarizes the review of evidence by the Evidence Review Group (ERG) and provides a summary of the NICE Appraisal Committee’s (AC’s) decision. The clinical evidence was derived from a multi-centred, open-label, randomized, phase III study comparing eribulin with treatment of physician’s choice (TPC) in 762 patients with LABC/MBC. Clinical effectiveness results were submitted for two populations: the overall intention-to-treat (ITT) population and a subset (n = 488) that included only patients from North America, Western Europe and Australia (Region 1). For the primary endpoint of overall survival (OS), a primary analysis (after 55 % of patients had died) and an updated analysis (after 77 % of patients had died) were conducted. In the ITT population, treatment with eribulin was associated with a significant improvement in median OS compared with TPC in both primary [difference in median OS 2.5 months; hazard ratio (HR) 0.81, 95 % confidence interval (CI) 0.66–0.99] and updated analyses (2.7 months; HR 0.81, 95 % CI 0.67–0.96). A statistically significant improvement in progression-free survival (PFS) was reported for eribulin compared with TPC when assessed by the investigator (difference in median PFS 1.48 months; HR 0.76, 95 % CI 0.64–0.90), but not when assessed by the ERG (1.44 months; HR 0.87, 95 % CI 0.71–1.05). Gains in OS were greater for Region 1 patients than for the ITT population (3.1 vs. 2.7 months). Health-related quality of life (HRQoL) data suggested a benefit for eribulin responders, but was based on phase II studies. In the eribulin arm, serious adverse events included febrile neutropenia (4.2 %) and neutropenia (1.8 %), with peripheral neuropathy being the most common reason for treatment discontinuation. The manufacturer’s economic evaluation using Patient Access Scheme costs reported a base-case incremental cost-effectiveness ratio (ICER) for eribulin versus TPC (Region 1) of £46,050 per quality-adjusted life year gained (corrected to £45,106 when an erroneous data entry was removed). The ERG’s revised ICERs were £61,804 for Region 1 and £76,110 for the overall population. The AC concluded that the evidence had not demonstrated sufficient benefit in OS, cost effectiveness or HRQoL and that eribulin was not recommended for use in this patient group.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Selection and definition of the most appropriate population can have a major influence in arriving at reliable estimates of clinical effectiveness and cost effectiveness. Prior treatments, the intended treatment, the actual treatment and the geographical region of patients were all shown to be important considerations in this appraisal |
Accurate costing of drug acquisition and drug delivery was shown to be the single most important factor in determining cost effectiveness, and warrants close attention in any appraisal of a novel drug treatment |
The use of trial duration approach (truncation) to modelling survival outcomes may appear to be ‘conservative’, but can lead to significant bias. Therefore, the National Institute for Health and Care Excellence (NICE) correctly requires that decision modelling should encompass all relevant costs and effects to the end of life. In this instance, the re-analysis of trial data limiting heterogeneity revealed that outcome gains were probably restricted to a specific time period, so projective modelling was not required to obtain reliable estimates of treatment benefits |
Care is required in using utility valuation study results unrelated to trial data to provide health state model parameter values. Misapplied values may violate key assumptions in published results, with substantial effects on cost-effectiveness estimates |
1 Introduction
The National Institute for Health and Care Excellence (NICE) is an independent organization responsible for providing national guidance to the NHS in England and Wales on a range of clinical and public health issues, as well as appraisal of new health technologies. The NICE Single Technology Appraisal (STA) process is specifically designed for the appraisal of a single health technology for a single indication, where most of the relevant evidence lies with one manufacturer or sponsor [1]. Typically, the process is used for new pharmaceutical products close to launch. The evidence for an STA is principally derived from a submission by the manufacturer/sponsor of the technology, which should be based on a specification developed by NICE. The manufacturer’s submission is critiqued by members of the independent Evidence Review Group (ERG), who produce a report to be considered by the NICE Appraisal Committee (AC).
The NICE AC then considers the submissions from the manufacturer and the ERG alongside testimony from experts, patients and other stakeholders to formulate preliminary guidance. All stakeholders have an opportunity to comment on this preliminary guidance, after which the AC meets again to produce the final guidance (final appraisal determination). This article presents a summary of the ERG report for the STA of eribulin as treatment for patients with locally advanced or metastatic breast cancer (LABC/MBC) whose previous treatments included at least two chemotherapy (CTX) regimens. Full details of all the relevant appraisal documents (including the appraisal scope, ERG report, manufacturer and consultee submissions, appraisal consultation document, final appraisal determination and comments on each of these) can be found on the NICE website [2].
2 The Decision Problem
Breast cancer is the most commonly diagnosed cancer in women across the world, with an estimated 1.7 million new diagnoses in 2012. This represents a 20 % increase in breast cancer incidence since 2008. The 522,000 deaths across the world in 2012 confirm breast cancer as the most common cause of cancer death among women [3].
The highest rates of female breast cancer occur in Western Europe and the lowest occur in East Africa. In the UK, breast cancer accounts for about one in three cases of cancer in women; the lifetime risk of developing breast cancer is one in eight [4]. Approximately 44,000 women were diagnosed with breast cancer in England and Wales during 2010 [4]. The risk of developing breast cancer is strongly correlated with age; 81 % of cases in the UK occur in women aged 50 years and over [4].
LABC and MBC are the most advanced forms of breast cancer, where the cancer is no longer localized to the breast and has spread to other parts of the body, commonly the lungs, liver, brain and bone [5]. Few patients (approximately 5 %) are diagnosed with MBC [5]; however, the risk of recurrence persists for many years following remission of non-metastatic disease. It is estimated that 30, 46 and 71 % of patients initially diagnosed with stages I, II and III disease, respectively, will eventually progress to metastatic disease [6].
The specific population considered in this appraisal comprises patients presenting with recurrent disease (whether or not metastatic) and those newly presenting with metastatic disease. The prognosis for these patients is very poor; the average length of survival following diagnosis of MBC is estimated to be 12 months for those receiving no treatment, compared with 18–24 months for those receiving CTX [7]. At the point in therapy where eribulin is proposed, survival would be expected to be even less.
Pre-treated patients are a particularly challenging subgroup to manage effectively because by this stage patients will have progressed despite treatment, and further treatment options will have limited effectiveness. Treatment is focused on prolonging survival, while controlling the symptoms and improving quality of life (QoL) [8]. Many patients gain significant benefit from continuing treatment through several lines of CTX; however, there is minimal high-quality evidence about the relative clinical effectiveness of current treatments [8] and none have demonstrated a survival benefit over any other [8, 9]. The proposed place for eribulin is as third-line CTX.
Eribulin is a monotherapy that is administered intravenously over 2–5 min on days 1 and 8 of every 21-day cycle [10]. It is licensed in Europe [11] for the treatment of patients with LABC/MBC who have progressed after at least two CTX regimens for advanced disease. Prior CTX should have included an anthracycline and a taxane unless patients were not suitable for these treatments.
NICE developed a scope [12] for the assessment of eribulin, which specified that the clinical and cost effectiveness of this drug should be established within its licensed indication relative to vinorelbine, capecitabine and gemcitabine. Five measures of clinical effectiveness were considered relevant for this appraisal: overall survival (OS), progression-free survival (PFS), response rate, adverse events (AEs) and health-related quality of life (HRQoL). The time horizon of analysis was specified as the remaining lifetime of patients.
3 The Independent Evidence Review Group Report
The manufacturer provided a submission to NICE on the use of eribulin (within the context of its licensed indication) in adults with LABC/MBC who had received at least two prior treatments with CTX. The submission included data from a comparison of eribulin with a ‘treatment of physician’s choice’ (TPC). The TPCs included, but were not limited to, vinorelbine, capecitabine and gemcitabine. The ERG examined and critiqued both the initial and subsequent evidence submissions from the manufacturer, taking into consideration the manufacturer’s response to their request for clarification on a number of issues. The ERG report comprised a critical review of the evidence for the clinical and cost effectiveness of the technology based upon the manufacturer’s submission to NICE. The review embodied three aims:
-
To assess whether the manufacturer’s submission conformed to the methodological guidelines issued by NICE;
-
To assess whether the manufacturer’s interpretation and analysis of the evidence were appropriate;
-
To indicate the presence of other sources of evidence or alternative interpretations of the evidence that could help inform NICE guidance.
In addition to providing this detailed critique, the ERG modified a number of key assumptions and parameters within the manufacturer’s economic model to examine the impact of such changes. The ERG had the opportunity to obtain clarification on specific points in the manufacturer’s submission, resulting in the manufacturer providing additional evidence. This section summarizes the submitted evidence and the ERG’s review of that evidence.
3.1 Clinical Evidence
The clinical effectiveness evidence was derived from a single trial known as the EMBRACE trial [13]. The EMBRACE trial [13] was an international, multi-centred, open-label randomized, phase III study (n = 762) designed to evaluate the efficacy of eribulin treatment compared with TPC for patients with LABC/MBC who had previously undergone treatment with at least two CTX regimens including an anthracycline and a taxane. Patients were stratified according to geographical region, human epidermal growth factor receptor 2 (HER2) status and prior capecitabine treatment and were randomized 2:1 to receive either eribulin or TPC.
Clinical effectiveness results (Tables 1, 2) were reported for two populations: the overall intention-to-treat (ITT) population and a subset (n = 488) of the overall ITT population that included only patients from Region 1 (North America, Western Europe and Australia). For the primary endpoint of OS, clinical effectiveness results were reported at two time points: the primary analysis (protocol specified after 55 % of patients had died) and an updated analysis (requested by regulatory authorities and conducted after 77 % of patients had died). All secondary endpoints were reported for the time of the primary analysis.
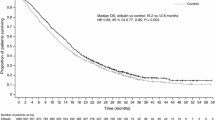
In the overall ITT population, treatment with eribulin was associated with a statistically significant improvement in OS compared with TPC in both the primary analysis [difference in median OS 2.5 months/75 days; hazard ratio (HR) 0.81, 95 % confidence interval (CI) 0.66–0.99] and the updated analysis (difference in median OS 2.7 months/82 days; HR 0.81, 95 % CI 0.67–0.96). Statistically significant improvement in PFS was reported for eribulin compared with TPC when assessed by the investigator (difference in median PFS 1.48 months/45 days; HR 0.76, 95 % CI 0.64–0.90), but not for data independently assessed by the ERG (difference in median PFS 1.44 months/44 days; HR 0.87, 95 % CI 0.71–1.05).
For Region 1 only patients, treatment with eribulin was associated with a statistically significant improvement in OS compared with TPC in both the primary analysis (difference in median OS 3.06 months/93 days; HR 0.72, 95 % CI 0.57–0.92) and the updated analysis (difference in median OS 3.09 months/94 days; HR 0.79, 95 % CI 0.64–0.98). The results of post hoc subgroup analyses of median OS by TPC subgroup for both the overall ITT population and the Region 1 patient subset were reported in confidence. The HRQoL data (derived from phase II trial data [14, 15]) suggested that QoL may be improved in patients whose tumour responds to eribulin treatment.
The most frequently reported serious AEs in the eribulin arm were febrile neutropenia (4.2 %) and neutropenia (1.8 %), the most common AE leading to treatment discontinuation in the eribulin arm was peripheral neuropathy.
3.1.1 Critique of Clinical Evidence and Interpretation
The ERG considered the EMBRACE trial [13] to be a large and well-designed trial, with a robust primary endpoint (OS) and safeguards to mitigate against possible bias in monitoring and assessment (particularly important as the trial was open label). A number of issues relating to the clinical effectiveness results from the EMBRACE trial [13] were noted.
-
The use of TPC is pragmatic and reflects patient experience in England and Wales; however, averaging the effects of a range of diverse treatments will obscure patient responses to individual treatments.
-
Patients in the trial were younger and fitter than patients typically seen in UK clinical practice.
-
Subgroup analyses comparing TPC patient outcomes with the outcomes of the individual CTX comparators (i.e. vinorelbine, capecitabine and gemcitabine) specified in the scope issued by NICE [12] were of questionable reliability as they were based on small patient numbers, and the trial was not powered to detect differences between individual treatment subgroups.
-
Data relating to HRQoL were not collected during the trial, and the evidence presented was weak because it was based on data derived from small, phase II, single-arm trials [14, 15].
-
The manufacturer had submitted clinical effectiveness data for both the overall trial population and for the Region 1 only population. It was debateable as to which data were more appropriate for this appraisal. The European marketing authorization for eribulin was based on the results of the overall trial population.
3.2 Cost-Effectiveness Evidence
3.2.1 Overview of Manufacturer’s Economic Evidence
The manufacturer’s literature search to identify papers that evaluated the cost effectiveness of eribulin as a third-line treatment for MBC identified no relevant published economic evaluations for consideration.
The manufacturer undertook a de novo economic evaluation of eribulin for the treatment of patients with LABC/MBC whose disease had progressed after at least two prior CTX regimens for advanced disease. A semi-Markov state transition model was constructed to model the lifetime clinical and economic outcomes for a hypothetical cohort of patients. The model consisted of three main health states: treated, progressive and dead. Clinical effectiveness data from Region 1 of the EMBRACE trial [13] were used to populate the base-case analysis. The HRQoL data were extracted from published literature [14, 15]. The economic evaluation was undertaken from the perspective of the NHS and Personal Social Services in England and Wales. The price of eribulin used in the model was the Department of Health-approved Patient Access Scheme (PAS) price.
The manufacturer’s base-case incremental cost-effectiveness ratio (ICER) for eribulin versus TPC (Region 1) was £46,050 per quality-adjusted life year (QALY) gained. In line with the NICE scope, the manufacturer also presented ICERs for eribulin versus gemcitabine, vinorelbine and capecitabine. In these comparisons, ICERs per QALY gained were £27,183, £35,602 and £47,631, respectively. Probabilistic sensitivity analyses carried out by the manufacturer demonstrated a low level of uncertainty around the base-case results.
In response to the points of clarification put to them by the ERG regarding the initial submission, the manufacturer provided additional evidence relating to the sensitivity of model results to different assumptions about the definition of disease progression, the duration of treatment and the timing of treatment response.
3.2.2 Critique of the Manufacturer’s Cost-Effectiveness Evidence
The ERG considered that the databases searched and the search terms used by the manufacturer were reasonable and both inclusion and exclusion criteria were explicitly stated. The ERG was confident that no relevant published studies were available for inclusion in the review.
The baseline ICERs generated by the submitted model for eribulin versus TPC for the Region 1 and ITT population using the PAS price of eribulin were £45,106 and £48,536, respectively (after an erroneous sensitivity value for cost per vial of vinorelbine had been deleted from the model, restoring the manufacturer’s original base-case cost). The issues found by the ERG to have the largest impact on the base-case ICERs related to the cost of CTX drugs, health state-based costs and the modelling of utility values.
3.2.2.1 Cost of Chemotherapy Drugs
All CTX treatments currently recommended for treatment of LABC/MBC are dosed on the basis of the body surface area (BSA) of the individual patient. The submitted model did not take account of BSA differences between patients, but used a fixed average value for all patients (1.74 m2) sourced from a UK survey of CTX patients. The costs of CTX drugs per cycle in nine regimens were re-estimated by the ERG using BSA values from the Sacco et al. [16] study in the population of patients receiving palliative CTX. For all regimens but one (nab-paclitaxel), the ERG estimated cost (including wastage) was lower than that used in the manufacturer’s model, in several cases by a very substantial amount.
Three aspects of the method used by the manufacturer to cost the administration of CTX were found to require correction, namely, (1) the unit costs of administration used in the model were extracted from 2008/2009 NHS reference costs [17], rather than the most recent figures (2009/2010) [18]; (2) all CTX administration was allocated costs appropriate to an out-patient department, but ERG clinical advice was that such therapy will normally be administered in a designated CTX day-case unit; and (3) the manufacturer ignored the different healthcare resource group costs appropriate to the first administration of a course of therapy (using the ‘subsequent cycles’ costs instead).
The impact of modelling changes to address these issues was to increase the base-case ICERs in the Region 1 and ITT populations, respectively.
3.2.2.2 Costs of Supportive Care
The manufacturer’s model included no provision for the cost of primary- and community-based services received prior to disease progression. Additionally, in the post-progression survival state, the model appeared to be based on a hospital-centric pattern of care, whereas the most appropriate basis for cost estimation is that used in the NICE guideline [8], based on a package of care provided by community nurses, therapists and GP home visits. Furthermore, during the terminal care state, the cost in the manufacturer’s model was dominated by hospice care, whereas evidence suggests [8] that only 10 % die in a hospice, with 50 % dying at home and the remaining 40 % dying in hospital.
The impact of making modelling changes to address these issues was to increase the baseline ICERS in the Region 1 and ITT populations, respectively.
3.2.2.3 Health State and Adverse Event Utility Values
A number of issues were raised by the ERG with respect to the calculation of utility values used within the model, namely, (1) the manufacturer employed the Lloyd et al. [19] mixed model analysis results but took no account of the non-linear nature of the analysis; (2) the Lloyd model employs an age adjustment based on the age of 100 health state valuers in their study; for consistency with EQ-5D utility values, the correction should use the mean age of participants in the original UK evaluation study, leading to increased estimates of all health state utility values; (3) average utility values were applied to AEs not considered by Lloyd et al. [19]. This was inappropriate as some of the AEs featuring in the EMBRACE trial [13] have been found in other studies to have larger disutility values than this average.
The ERG noted the limited consideration of AEs to those that feature in 10 % (or 5 %) more of patients and considered that this restriction risked excluding small events of great importance, for example, febrile neutropenia. Additionally, the ERG noted that the methods of calculating costs and loss of utilities were flawed in that (1) they were limited to those that experienced a Grade 3 or Grade 4 event; (2) there was no recognition that a serious clinical event may involve several important AEs occurring simultaneously; and (3) costs were based on typical episode descriptions from clinical opinion and appeared to be very low.
The impact of making modelling changes to address these issues was to increase the base-case ICERs in the Region 1 and ITT populations, respectively.
3.2.2.4 Additional Corrections and Amendments
The ERG made amendments to the model to address the method of discounting costs and outcomes, problems with the calculations relating to the terminal period, the incorrect use of a mid-cycle correction, and use of investigator PFS data rather than those of an independent assessor, and to include consideration of febrile neutropenia. Individually, each of these changes had an impact on the baseline ICER of less than £1,000.
The model revisions carried out by the ERG had the impact of increasing the baseline ICERs generated by the submitted model for eribulin versus TPC for Region 1 from £45,106 to £61,804 and of increasing estimates for the ITT population from £48,536 to £76,110 (Table 3).
3.2.2.5 Survival Estimation
Instead of employing projective modelling of patient survival, the manufacturer’s model used the EMBRACE data [13] directly, with the assumption that all patients alive at the time of cut-off die at this point. Two aspects of this approach were concerning.
-
There is potential for the introduction of bias which can significantly impact on the incremental survival (survival gain); Kaplan–Meier plots can become unstable when only small numbers of patients remain alive and uncensored.
-
The NICE reference case [1] requires decision analysis to take account of costs and outcomes that are likely to be affected by the choice of treatment at any subsequent time, and in the case of advanced or metastatic cancers, this is generally interpreted as the whole of the remaining lifetime of patients.
It is therefore likely that, in some model scenarios, OS may be either over- or underestimated by the manufacturer’s model. The ERG tested the potential size and importance of this problem via a revised survival analysis of both the ITT and Region 1 only populations. This involved truncating the accumulation of survival time at a common time in both trial arms, to eliminate the effect of residual ‘tails’ of different sizes and durations. The estimated mean gain in OS from the use of eribulin was reduced by 10–14 days (14–15 %), which alone could increase the size of the estimated ICER by approximately 18–19 %. Using clinical data from Region 1 only at the point when 77 % of deaths had occurred, the ERG’s revised base-case ICER (including projection) for the comparison of eribulin versus TPC was £68,590 per QALY gained.
The ERG estimated the OS gain with eribulin compared with TPC to be 2.69 months for the overall ITT population and 3.25 months for the Region 1 population. The ERG was unable to amend the submitted model directly to incorporate the effects of using projected OS estimates without a major restructuring of the model architecture. However, a good approximation was made by increasing the aggregated post-progression survival and adjusting post-progression costs and post-progression utility values in parallel.
The ERG concluded that if the whole population of the EMBRACE [13] trial was considered sufficiently representative of UK patients and clinical practice, then the best estimated ICER for eribulin exceeded £76,000 per QALY gained, but may fall to about £68,000 if projected lifetime estimates of OS are preferred to truncated estimates (Table 3). If only Region 1 patients are deemed representative of the UK NHS context, then the ERG estimated ICER exceeded £61,000 per QALY gained, but reduced to almost £56,000 if survival projections were preferred (Table 4).
3.3 End of Life Guidance Criteria
The NICE ‘End of Life’ treatment criteria [20] have three key points:
-
Treatment is indicated for patients with a short life expectancy, normally less than 24 months.
-
There is sufficient evidence to indicate that the treatment offers an extension to life, normally of at least an additional 3 months, compared with NHS treatment.
-
The treatment is licensed or otherwise indicated for small patient populations.
For the comparison of eribulin versus TPC for patients with LABC/MBC, the requirements of short life expectancy and small patient populations appeared to be met. The estimated OS data from Region 1 (median 3.1 months, mean 2.8 months, ERG projected mean 3.25 months) appeared to meet the life extension of 3 months criterion, whereas OS data from the overall (ITT) population (median 2.7 months, mean 2.33 months, ERG projected mean 2.69 months) appeared to be less than 3 months.
3.4 Conclusions of the ERG Report
The manufacturer’s evidence of the clinical benefit of eribulin versus TPC as a treatment for LABC/MBC following treatment failure with an anthracycline and a taxane was derived from a large, multi-centred, international trial. The EMBRACE trial [13] was well designed, with a robust primary outcome of OS. The submitted HRQoL data were considered to be weak as they were derived from phase II studies. The subgroup analyses comparing eribulin with individual CTX treatments did not provide convincing evidence of clinical or cost-effectiveness differences between eribulin and the three individual comparators.
The main weakness in the economic model was related to inaccurate costings of comparators compared with eribulin. The ERG also considered that OS estimates should be projected beyond the trial data; projection led to a gain in OS for all patients, especially for patients in Region 1. The ERG’s estimates of OS were greater than the estimates submitted by the manufacturer.
A key area of uncertainty was whether or not the clinical effectiveness data from Region 1 patients only should be preferred to data from the ITT population. The incremental OS gain was higher in Region 1 patients compared with the incremental OS gain in patients in the ITT population. As a result, the ICER in the Region 1 population was lower than the ICER in the ITT population. The ERG considered that eribulin compared with TPC met the NICE ‘End of Life’ criteria only when data for Region 1 were employed.
4 Key Methodological Issues
The ERG considered that the manufacturer’s ‘conservative’ approach to modelling whereby patients who remained alive beyond the end of the trial were considered to be dead in the model was problematic in two ways: (1) the censoring method used was open to potential bias and (2) the true lifetime experience of the patients was not captured, possibly leading to an underestimation of the costs of patient support and any additional treatment. In the view of the ERG, projective modelling of OS outcomes was appropriate to gain a more accurate picture of costs.
The analyses of eribulin versus the individual CTX treatments involved very small numbers of patients and were of a post hoc nature. These comparisons were considered to be wholly unreliable to aid decision making.
5 NICE Guidance
The final appraisal determination issued by NICE did not recommend the use of eribulin in this patient population.
5.1 Consideration of Clinical and Cost-Effectiveness Issues
5.1.1 First Appraisal Committee (AC) Meeting
5.1.1.1 Clinical Benefit and Quality of Life
The AC considered that the trial broadly reflected UK clinical practice and that eribulin was associated with a greater OS than TPC; however, lack of QoL data from the trial was an important omission. The clinical experts advised that (1) it is unusual for a technology to show an OS benefit at this stage of the clinical pathway; (2) a further treatment option for patients whose previous CTX has failed is important; and (3) eribulin may be less well tolerated than capecitabine and vinorelbine, and is associated with peripheral neuropathy and alopecia. The AC concluded that eribulin was associated with a greater overall OS benefit compared with TPC, but with a less favourable toxicity profile, and its effects on QoL had not been adequately captured.
5.1.1.2 Region 1 Versus Intention-to-Treat Population
The AC acknowledged the increased OS gain for patients treated with eribulin compared with TPC for patients from Region 1 compared with the overall ITT population (3.1 months vs. 2.7 months). However, the AC took into account that (1) differences in OS between Region 1 and overall ITT populations were only evident for the comparator arm and not for the eribulin-treated group (possibly the result of small numbers in the Region 1 group); (2) analysis by the ERG comparing the mean OS for Region 1 with that for Regions 2 and 3 combined suggested that patients in Region 1 did not differ in terms of prognosis from the patients in the remainder of the trial population; and (3) UK practice differs considerably from some areas of Region 1. The AC was also aware that the European marketing authorization for eribulin was based on the results of the overall EMBRACE [13] population. The AC concluded that it would be most appropriate to base its recommendations on the results from the overall ITT population.
5.1.1.3 Eribulin Versus Individual Treatments
The AC agreed that it was inappropriate to consider the results from individual TPC comparisons because they were post hoc, based on small numbers, had wide CIs and did not include appropriate adjustment for multiple testing. In addition, the trial was not powered to detect differences between individual treatment groups.
5.1.1.4 Pre-treatment with Capecitabine
From the European Public Assessment Report for eribulin [11], which was produced by the European Medicines Agency, the AC noted that a major stratification factor in the trial was pre-treatment with capecitabine (73.4 % of patients). The AC considered that this was potentially relevant to clinical practice; however, the manufacturer had not submitted evidence on clinical or cost effectiveness for this subgroup.
5.1.1.5 Cost-Effectiveness Evidence
The AC agreed that the manufacturer’s model was generally well constructed and in accordance with the scope issued by NICE. The AC noted, however, that HRQoL data from the trial had not been recorded to inform the modelling. The AC acknowledged that data from the Region 1 population were used in the manufacturer’s base-case economic evaluation, but that data from the overall ITT population were used in a sensitivity analysis. The AC further noted that the manufacturer’s estimate of the incremental cost per QALY gained of eribulin compared with TPC in the overall ITT population was largely unchanged by the ERG’s minor corrections (a reduction of only £1,500).
5.1.1.6 Approaches to Costs
The AC preferred the ERG’s approach to estimating the costs of CTX drugs, costs of supportive care and state-based costs, day-case CTX costs and the use of up-to-date NHS reference costs. The clinical specialist stated that intravenous vinorelbine (generically available) is more frequently used in UK clinical practice in contrast to the branded, oral vinorelbine assumed in the manufacturer’s base-case model. Moreover, vinorelbine tends to be given on day 1 and day 8 of a 21-day cycle rather than weekly, as this is better tolerated. The AC considered this issue to be important as vinorelbine is the most commonly used comparator and accounted for 24 % of the comparators used in the TPC analysis. The AC concluded that the effect of these factors in the manufacturer’s model was to overestimate the costs of the comparators and to underestimate administration, supportive care and state-based costs.
5.1.1.7 Utility Values
The AC noted that in the absence of data on HRQoL derived from the EMBRACE trial [13], the manufacturer’s model incorporated utility values from previously published studies. The AC also had concerns that the manufacturer’s base-case model included only Grade 3 and 4 AEs that occurred in at least 10 % of patients; this has the potential for the exclusion of important AEs such as febrile neutropenia and peripheral neuropathy. It was further observed that the disutility associated with alopecia had been omitted from the manufacturer’s model.
5.1.1.8 Projection
The AC agreed that (1) it was more appropriate to use the ERG’s exploratory analysis that projected survival trends to the end of life in line with the lifetime time horizon recommended in the NICE methods guide; (2) the ERG’s exploratory analysis of the manufacturer’s model for the overall ITT population, which included projection of OS and re-estimation of costs, was a more plausible estimate for the cost effectiveness of eribulin compared with TPC than the manufacturer’s estimate. However, the AC considered that this was likely to underestimate the true cost per QALY gained from eribulin relative to TPC because it did not incorporate the full toxicity profile of eribulin, including the disutility associated with alopecia. In addition, there remained significant uncertainties about HRQoL associated with eribulin. Furthermore, the AC was aware that some of its concerns about costs were not accounted for in the ERG’s exploratory analyses (less frequent administration of vinorelbine and the use of generic prices as an estimate of the price of the comparators).
5.1.1.9 Conclusion
The AC concluded that eribulin did not fulfil the entire end of life criteria because it had not demonstrated an extension to life in the overall ITT population of at least an additional 3 months compared with TPC. Furthermore, given that the ICER was likely to exceed £68,600 per QALY gained, eribulin could not be considered a cost-effective use of resources for NHS use even if all of the criteria for being a life-extending, end of life treatment had been met.
The first appraisal consultation document issued in July 2011 concluded that eribulin should not be recommended for the treatment of LABC/MBC in people whose disease has progressed after at least two CTX regimens for advanced disease.
5.1.2 Second AC Meeting
A supplementary evidence submission from the manufacturer was considered. This comprised data from the subgroup of Region 1 only patients from the EMBRACE trial [13] who were pre-treated with capecitabine. The comparator technology was vinorelbine rather than TPC. The manufacturer’s projective modelling approach utilized proportional hazards methodology; the produced ICERs ranged between £26,000 and £41,000 without adjustment for end of life, and below £30,000 when adjusted for end of life.
The ERG’s critique of the supplementary evidence noted that the narrowed decision problem (post-capecitabine treated patients and a single comparator) might be more reflective of the UK patient population; however, the number of patients in the analysis was considerably reduced. The ERG’s opinion was that (1) there was no support for excluding data on the basis of region and (2) the narrowing of the decision problem to a single comparator reduced heterogeneity and indicated that PFS and OS survival trends converged during the trial period so that the net outcome benefits attributable to the use of eribulin in terms of PFS and OS could be estimated without the need for projective modelling. Therefore, the most reliable estimates of benefit could be obtained directly from a non-parametric Kaplan–Meier analysis of the trial data. The ERG estimated that the most reliable estimated ICER for eribulin compared with vinorelbine in treating patients who had previously undergone treatment with capecitabine was £53,000 per QALY gained.
The AC preferred the ERG’s straightforward approach to modelling. The AC also considered that the manufacturer had failed to adequately capture the toxicity profile of eribulin, particularly in relation to AEs. In the view of the AC, the manufacturer had not presented any more compelling evidence than was in their original submission.
6 Conclusion
The main evidence for this STA was derived from a large randomized controlled trial that compared eribulin with a number of different CTX treatments (TPC). The use of TPC represented a pragmatic approach to the decision problem as it reflected the experience of most patients with LABC/MBC. However, this approach proved to be fraught with difficulties in the subsequent interpretation. At the outset, the manufacturer did not collect HRQoL data on the grounds that the results of any such exercise would be difficult to interpret (because of the number of different treatments) and also stated in the manufacturer’s submission that the analysis of trial-reported AEs was of limited value. In order to address NICE’s requirements (eribulin compared with vinorelbine, capecitabine and gemcitabine), the manufacturer was obliged to disaggregate the dataset, and this resulted in very small group sizes with unconvincing comparisons.
In terms of modelling, the manufacturer initially opted not to project outcomes beyond the duration of the trial. Although this may be considered a ‘conservative’ approach, careful consideration should be given to the methods used to censor data in order to avoid potential biases. The approach will also fail to capture the lifetime experiences of all patients and therefore underestimate costs associated with treatment. In the supplementary evidence submission, the manufacturer did extrapolate patient outcomes beyond the end of the trial; however, the decision problem had then changed to assess the outcomes of a subset of patients with a single comparator only. This subsequent dataset was more homogeneous and the survival outcomes could be predicted from within the lifetime of the trial, obviating the need for further projection.
The manufacturer chose to present data from a subset of the overall dataset, patients from Region 1 only. The OS data for the Region 1 subgroup were longer and extended to beyond 3 months, with smaller associated ICERs. However, it is generally agreed that the complete dataset from any trial is preferred unless there are compelling reasons to use a smaller subset, and in this case, the manufacturer failed to convince the ERG and the AC of any rationale for accepting the Region 1 dataset over that of the overall trial population.
References
National Institute for Health and Clinical Excellence. Guide to the methods of technology appraisal. 2013. http://publications.nice.org.uk/guide-to-the-methods-of-technology-appraisal-2013-pmg9/the-appraisal-of-the-evidence-and-structured-decision-making.
National Institute for Health and Clinical Excellence. Breast cancer (advanced) eribulin: appraisal consultation document. 2011. Cited 2011 September 2011. http://www.nice.org.uk/guidance/TA250.
The International Agency for Research on Cancer. Latest world cancer statistics. 2013. www.iarc.fr/en/media-centre/pr/2013/pdfs/pr223_E.pdf.
Cancer Research UK. Breast cancer incidence statistics. 2014. http://www.cancerresearchuk.org/cancer-info/cancerstats/types/breast/incidence/.
Cancer Research UK (CancerHelp UK). Number of stages of breast cancer. 2011 March 2011. http://www.cancerhelp.org.uk/type/breast-cancer/treatment/number-stages-of-breast-cancer.
Berkowitz N, Gupta S, Silberman G. Estimates of the lifetime direct costs of treatment for metastatic breast cancer. Value Health. 2000;3(1):23–30.
Liverpool Reviews and Implementation Group (LRiG). Lapatinib and trastuzumab in combination with an aromatase inhibitor for the first-line treatment of metastatic hormone receptor positive breast cancer which over-expresses HER2. 2010.
National Institute for Health and Clinical Excellence. Advanced breast cancer: NICE Clinical Guideline 81;2009.
Jassem J, Carroll C, Ward SE, Simpson E, Hind D. The clinical efficacy of cytotoxic agents in locally advanced or metastatic breast cancer patients pretreated with an anthracycline and a taxane: a systematic review. Eur J Cancer. 2009;45(16):2749–58.
European Medicines Agency. Halaven (eribulin). 2011.
European Medicines Agency. Assessment report for Halaven eribulin. London, 2011.
National Institute for Health and Clinical Excellence. Eribulin for the treatment of locally advanced and metastatic breast cancer: final scope. 2011. Cited 2011 February. http://www.nice.org.uk/guidance/TA250.
Cortes J, O’Shaughnessy J, Loesch D, Blum JL, Vahdat LT, Petrakova K, et al. Eribulin monotherapy versus treatment of physician’s choice in patients with metastatic breast cancer (EMBRACE): a phase 3 open-label randomised study. Lancet. 2011;377(9769):914–23.
Cortes J, Vahdat L, Blum JL, Twelves C, Campone M, Roche H, et al. Phase II study of the halichondrin B analog eribulin mesylate in patients with locally advanced or metastatic breast cancer previously treated with an anthracycline, a taxane, and capecitabine. J Clin Oncol. 2010;28(25):3922–8.
Iwata H, Aogi K, Masuda N, Mukai H, Yoshida M, Rai Y, et al. Efficacy and safety of eribulin in Japanese patients with advanced breast cancer. J Clin Oncol Conf. 2010;28(15 SUPPL. 1).
Sacco JJ, MacBeth F, Bagust A, Clark P. The average body surface area of adult cancer patients in the UK: a multicentre retrospective study PLoS ONE. 2010;5(1).
Department of Health. NHS reference costs (2008–2009). 2010.
Department of Health. NHS reference costs (2009–2010). 2011.
Lloyd A, Nafees B, Narewska J, Dewilde S, Watkins J. Health state utilities for metastatic breast cancer. British J Cancer. 2006;95:683–90.
National Institute for Health and Clinical Excellence. Supplementary advice to the Appraisal Committees—end of life treatments. 2009. http://www.nice.org.uk/aboutnice/howwework/devnicetech/endoflifetreatments.jsp?domedia=1&mid=88ACDAE5-19B9-E0B5-D422589714A8EC6D.
Acknowledgments
This project was funded by the National Institute of Health Research (NIHR) Health Technology Assessment (HTA) Programme (project number 10/19) and will be published on the HTA Programme website, with links to the relevant ERG reports on the NICE website. See the HTA Programme website for further project information: http://www.hta.ac.uk. This summary of the ERG report was compiled after the AC’s consideration of the evidence and incorporates additional information and comment from the authors on the STA process and iterations of the NICE guidance not covered by the HTA report.
The views and opinions expressed herein are those of the authors and do not necessarily reflect those of the National Institute for Health and Care Excellence or the Department of Health.
The authors have no competing interests.
Author contribution’s
Janette Greenhalgh: project lead; critical appraisal of the clinical evidence. Adrian Bagust: critical appraisal of the economic model. Angela Boland: critical appraisal of clinical and economic evidence. James Oyee: critical appraisal of the clinical statistical approach. Nicola Trevor: critical appraisal of economic evidence. Sophie Beale: critical appraisal of the economic evidence. Yenal Dundar: cross-checking of the manufacturer’s search strategies. Juliet Hockenhull: critical appraisal of the clinical evidence. Chris Proudlove: critical appraisal of the manufacturer’s submission. Susan O’Reilly: critical appraisal of the clinical sections of the manufacturer’s submission.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article

Cite this article
Greenhalgh, J., Bagust, A., Boland, A. et al. Eribulin for the Treatment of Advanced or Metastatic Breast Cancer: A NICE Single Technology Appraisal. PharmacoEconomics 33, 137–148 (2015). https://doi.org/10.1007/s40273-014-0214-2
Published:
Issue Date:
DOI: https://doi.org/10.1007/s40273-014-0214-2