
Abstract
Drug-induced parkinsonism (DIP) and tardive dyskinesia (TD) are iatrogenic consequences of antidopaminergic drugs. Both are particularly prevalent among the elderly and those with dementia. However, despite their prevalence, these disorders are often overlooked. Both entities share risk factors, physiopathological mechanisms and, to some degree, therapeutic approaches. Withdrawing the causal agent, reducing the dose or switching to a less potent antidopaminergic drug should be the first therapeutic options. Here we review both entities and emerging therapies including the recently approved drugs deutetrabenazine and valbenazine. We discuss relevant aspects for clinical practice such as new diagnostic techniques and the latest advances in the understanding of DIP and TD.
Similar content being viewed by others
Avoid common mistakes on your manuscript.
Drug-induced parkinsonism (DIP) and tardive dyskinesia (TD) are iatrogenic consequences of antidopaminergic drugs. |
DIP is considered a direct consequence of the blockage of dopamine D2 receptors while TD has a more complex pathophysiology, including enhanced sensitivity of dopamine receptors. |
Both entities are more frequent in older people due to a progressive loss of dopaminergic neurons with age. |
New therapies are available for TD, including deutetrabenazine and valbenazine. |
1 Introduction
Tardive dyskinesia (TD) and drug-induced parkinsonism (DIP) are iatrogenic disorders caused by dopamine receptor blockers (DRBs) [1]. They both share risk factors, pathological mechanisms and management, and can coexist in one subject. The first reports of these disorders came early after the introduction of antipsychotics [2] in clinical practice, leading to the finding that DIP caused by reserpine, a dopamine depleter, was related to dopamine deficiency [3].
Elderly patients are more susceptible to these adverse effects, probably due to an age-related decrease in nigral neurons and dopamine [4]. The introduction of newer second-generation antipsychotics (SGAs) has reduced the frequency and severity of these adverse effects [5,6,7,8] but these conditions remain problematic in clinical practice [9, 10].
The latest available evidence has clarified some intriguing features of TD and DIP. Meanwhile, treatment options have increased with the addition of the recently approved agents deutetrabenazine and valbenazine. This article offers a comprehensive review of the incidence, clinical features and mechanisms of DIP and TD. The benefits and risks of previous available drugs and those that have been recently approved by regulatory agencies are also discussed.
2 Drug-Induced Parkinsonism
2.1 Definition
DIP occurs when characteristic symptoms emerge after exposure to offending drugs, usually those that either deplete dopamine stores [11] or block dopamine receptors [12]. By definition, symptoms should be reversible within 6 months after the withdrawal of the offending agent [13], but up to 20% of patients can develop persistent deficits despite drug discontinuation [14]. It is speculated that patients in this subgroup might have subclinical Parkinson’s disease (PD) unmasked by neuroleptic exposure (umPD), complicating the differential diagnosis [15]. This is particularly likely with older patients, who are more prone to both PD and DIP.
2.2 Epidemiology and Risk Factors
After PD, DIP is the most common cause of parkinsonism [16]. Prevalence and incidence rates of DIP vary depending on the population studied and the ascertainment method [17]. A door-to-door study in Spain [18] found DIP to be the third most common cause of parkinsonism, with a global prevalence rate of 2% in patients older than 65 years. The Rotterdam Study, a prospective, population-based cohort study, concluded that DIP was responsible for 12% of all causes of parkinsonism [19], and another cohort study found that DIP was the second most common cause of parkinsonism (32.3%) after PD [20]. Recently, Savica et al. [21] analysed the incidence and time trends of DIP over 30 years in Olmsted County, Minnesota, USA. The authors found an annual incidence rate of 3.3 per 100,000 person-years, a decrease of 68% over the 30 years of the study.
Globally, about 15% of patients on antipsychotics develop DIP after long-term therapy and this proportion exceeds 50% among subjects over 60 years [22, 23]. The main risk factors predisposing to DIP are either related to the patient or to the drug that is responsible for the symptoms.
Across studies, age has been shown to be a consistent risk factor. Prevalence increases from 9.4 in patients between 60 and 69 years to 29.3 in those between 80 and 99 years [21, 24], possibly driven by an age-related decline in the number of nigrostriatal neurons [25]. Another individual-related risk factor is the presence of dementia, a condition that affects mostly old populations, which is found in 67% of DIP [26]. Likewise, sex has a modifying effect on DIP prevalence, with a higher incidence in women [27, 28].
Drug and patient risk factors only partially account for all cases of DIP. This suggests that genetic predisposition might play a role in the development of DIP. Shiroma et al. [29] showed genetic polymorphisms in genes involved in dopamine transmission, whereas Metzer et al. [30] found a higher prevalence of HLA antigen B44 in DIP patients and families have been described with a hereditary predisposition to DIP [31].
Many drugs have been described as causing parkinsonism. Antipsychotics are well-known causes of DIP, with potency and dose being unequivocal risk factors [23]. First-generation antipsychotics (FGAs) might trigger symptoms even at low doses, while SGAs rarely cause symptoms.
2.3 Pathophysiology and Causal Agents
Virtually any agent that blocks the postsynaptic dopamine D2 receptors or depletes presynaptic dopamine has the potential to cause parkinsonism [32, 33]. The most common causal agents are antipsychotics and dopamine-depleting drugs, but other medications with antidopaminergic effects such as antiemetics, antihistaminics and calcium channel antagonists can also cause DIP (Table 1).
In fact, calcium channel antagonists are among the commonest drugs related to DIP, these agents being the cause of up to 40% of DIP in some series [34]. They possibly affect elderly patients through their affinity for blocking the D2 receptor, which is similar to that of atypical antipsychotics [35, 36]
Substances such as lithium, valproic acid or amiodarone cause parkinsonism due to unknown mechanisms [23, 37]. Selective serotonin reuptake inhibitors (SSRIs) might rarely produce DIP, but due to the frequency of their use in general medical practice their contribution should always be considered [38]. Strikingly, not all antipsychotics cause DIP. Quetiapine does not appear to worsen parkinsonism [39], whereas clozapine has even been reported to improve PD motor symptoms, possibly through its serotonergic and anticholinergic effects [40]. Furthermore, dopamine blockade itself does not completely account for DIP since symptoms last for weeks to months, whereas the effect of antipsychotics on psychosis last only for several hours [41].
Reserpine was the first presynaptic vesicular monoamine transporter (VMAT) inhibitor; it acts on both the central and peripheral isoforms, producing marked adverse effects [42]. Tetrabenazine was the first selective VMAT2 inhibitor. It was initially developed as an antipsychotic to treat schizophrenia [43]; however, it was rapidly recognised to be effective against hyperkinetic movements and, in 2006, following the publication of the TETRA-HD [tetrabenazine in Huntington’s Disease (HD)] study, it was approved for the treatment of chorea associated with HD [44]. Depression, present in 15% of the patients, and parkinsonism, present in 12%, are dose-dependent and reversible adverse effects [44].
Recently, new highly selective VMAT2 inhibitors have been developed. Valbenazine has been shown to improve TD in the KINECT3 trial [45]. Parkinsonism was an exclusion criteria in this study, and the results revealed no differences between treatment groups at both 6 weeks and after a 1-year period in the rates of new-onset parkinsonism, as assessed by the Simpson-Angus Scale [46]. With a similar mechanism of action, deutetrabenazine has shown similar DIP rates in both treatment and placebo arms in two studies, one in a population with chorea associated with HD and the other in patients with TD. Outcome measures were the parkinsonism items of the Unified Huntington’s Disease Rating Scale (UHDRS) and the Unified Parkinson’s Disease Rating Scale (UPDRS) [47, 48]. However, some methodologic concerns have been raised regarding published post hoc comparisons with tetrabenazine [49].
Antipsychotics might possibly be intrinsically neurotoxic to dopaminergic neurons instead of merely unmasking pre-existing PD by means of dopamine blockade [15]. This is supported by the increased long-term risk of incident PD after past exposure to antipsychotics, whereas only 30% of patients develop parkinsonism during exposure [50]. Animal models also suggest that exposure to antipsychotics induces dopaminergic neuron death through inhibition of the mitochondrial respiratory chain, increased dopamine receptor turnover and free radical production [15].
There are also data suggesting that parkinsonism developing shortly after exposure to antipsychotics is more likely to be associated with subtle underlying nigrostriatal dysfunction. Chung et al. [51] performed a quantitative analysis of DaTscan (Dopamine agonist Transporter scan) results from 71 patients from South Korea with normal scans on visual inspection and found a statistically significant reduction in dopamine transporter availability in those who presented parkinsonism less than 6 months after initiation of antipsychotics. In contrast, no alterations were found in subjects with late-onset parkinsonism, suggesting that early-onset DIP might in fact be umPD [51]. In addition, one clinico-pathological study in seven patients with DIP did not report any relationship between presence of Lewy bodies and symptom reversal [52].
In conclusion, DIP is likely driven by complex mechanisms involving an interaction between predisposing patient factors and biochemical properties of the offending agent. Whether or not long-term use of antipsychotics may cause morphological changes in the brain is still a matter of debate.
2.4 Clinical Features and Diagnosis
DIP is frequently under-recognised. Common aetiological agents such as SGAs and antiemetics do not appear to be well-known causes of DIP [53]. Although current treatment with antipsychotics is an exclusion diagnostic criteria for PD [54], there are no officially established time periods for drug washout after detection of DIP. However, it is generally accepted that symptoms should recover within 6 months after drug withdrawal [13].
There are some clinical features and ancillary tests that might help differentiate DIP from PD (Table 2). Asymmetry of symptoms, typical of PD, is usually absent in patients with DIP and is possibly the most informative sign in the differential diagnosis. Other features in the clinical history and physical examination such as acute onset, absence of rest tremor, presence of orolingual dyskinesias or akathisia also point towards DIP. Moreover, a recent literature review by Brigo et al. [55] suggests that non-motor symptoms, especially anosmia, urinary and sleep problems, if assessed systematically, help in establishing a diagnosis.
Sustantia nigra hyperechogenicity, as assessed by transcranial sonography, could be a useful prognostic marker [56] but an abnormal DaTscan, is the best predictor for the differential diagnosis of PD and DIP, with a higher proportion of positive DaTscan patients developing persistent symptoms [57,58,59,60]. Furthermore, cardiac 123I-MIBG (iodine-123 meta-iodobenzylguanidine) scintigraphy [61] has been reported to be altered in patients with persistent parkinsonism [62], those with abnormal smell function at baseline [63] and in those who develop parkinsonism years later [62], suggesting a diagnosis of umPD rather than DIP in those with these features (Table 2).
Therefore, meticulous physical examination and clinical history is warranted in order to assess patients, while ancillary testing might be requested when in doubt following cessation of the offending drugs.
2.5 Prevention and Treatment
2.5.1 Prevention
Prevention of DIP is the most important factor. Using SGAs only when needed, at the lowest effective doses and always monitoring for signs of parkinsonism, especially in high-risk patients, is essential [17]. Prophylaxis with anticholinergics has reported conflicting results and it is not recommended [64, 65].
2.5.2 Management
DIP should be treated only if it affects patients in their activities of daily living. If possible, the first option for management should be either lowering the dose or switching to a less potent antipsychotic. If the clinical scenario does not allow these changes, or they are ineffective, anticholinergic drugs should be the next choice. Despite most evidence coming from low-quality studies with small numbers of patients and subjective methods of assessment where efficacy has been possibly overestimated [32, 66], positive results and wide experience favours the use of anticholinergics in DIP [67, 68].
Immediate-release amantadine has been during several years the main drug with an antidyskinetic effect in PD [69, 70]. It has been tested in several small clinical trials suggesting benefit in patients with DIP [71]. More recently, extended-release amantadine received FDA approval for PD dyskinesia [72] but there are yet no reports regarding its use in DIP.
Levodopa and dopamine agonists are usually ineffective for DIP. However, parkinsonism in the context of presynaptic dopamine depleters or in umPD is expected to improve with levodopa [73].
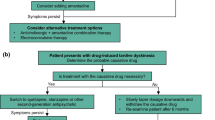
Electroconvulsive therapy (ECT) can achieve rapid improvement of symptoms in both DIP and PD and might be of benefit when there are coexistent mood disorders or psychosis [74]. An increase in the sensitivity of dopamine receptors and potentiating dopamine transmission are proposed mechanisms of the antiparkinsonian effects of ECT in most empirical studies [75, 76] (Fig. 1).

Algorithm for the treatment of drug-induced parkinsonism. ADL activities of daily living, ECT electroconvulsive therapy
3 Tardive Dyskinesia
3.1 Definition
Tardive syndromes (TSs) are characterised by abnormal involuntary movements caused by long-term exposure to DRBs [77]. The movements typically appear later during the treatment course and tend to persist for long periods of time, occurring in a variety of phenomenologies, with rhythmic oral–buccal–lingual (OBL) chewing movements being the most typical presentation [78]. Therefore, the term ‘tardive dyskinesia’ has been reserved by some for this type of movement, with other syndromes being named based on the specific phenomenology and using TS to describe the frequent combination of different movement disorders. However, most studies refer to TD without further specification.
3.2 Epidemiology and Risk Factors
The prevalence of TD varies from 0.5% to 65% [79, 80] of patients treated with antipsychotics. These differences account for the heterogeneity of the different populations studied, as well as the intrinsic variability of the disorder. Most authors estimate the mean prevalence to be between 20% and 50% based on large prospective cohort studies [80]. The incidence of TD seems to increase with longer exposure times. Caligiuri et al. [81, 82] showed a cumulative incidence of TD from 2.5% after 1 year of treatment, rising to 22.9% after 3 years. In addition, the Hillside study found that 43% of patients treated with FGAs developed TD after 10 years [83].
A recent meta-analysis across 41 studies, including a total of 11,493 psychiatric patients, showed that global TD prevalence was 25.3%. Prevalence rates varied according to antipsychotic type, being lower with SGA (20%) than FGA (30%) use associated with TD [84]. However, in a separate study of 352 schizophrenic patients over a 4-year period, the annual incidence of TD was 0.056 in patients with FGAs compared with 0.059 in patients with SGAs [85]. Globally, despite the increasing use of SGAs [6], recent prevalence studies have yielded conflicting results [9, 86, 87].
In addition, there are individual factors that increase the risk for TD. Similar to DIP, age is the most consistent risk factor for the development and persistence of dyskinesia, with risk rates increasing 3- to 5-fold when compared with younger patients [88, 89].
Female sex increases the risk of TD [79], possibly through the antioxidant and dopamine modulating properties of oestrogen [90]. Non-white ethnicity also predisposes for the development of TD [91, 92]. Genetic polymorphisms in genes related to the dopamine pathway also influence the risk of TD [93].
Besides antipsychotics, other antidopaminergic agents may cause TD. Metoclopramide, an antiemetic, and veralapride, used to treat perimenopausal syndromes, are major causes of TD, especially in higher doses [94]. Although rare, other non-antidopaminergic drugs have been described as producing TD, such as antidepressants (SSRIs and tricyclic antidepressants) [95, 96], antiepileptic drugs [97], antihistaminics [98], oral contraceptives and amphetamines [99].
3.3 Pathophysiology
Despite the early development of animal models such as rodents showing vacuous chewing movements (VCMs) after exposure to haloperidol [100, 101] or non-human primates, such as the Cebus monkey, with orofacial dyskinetic movements notably similar to those found in humans [102], the pathophysiology of TD remains obscure. TD is believed to be a consequence of the hypersensitivity and upregulation of D2 receptors due to their chronic blockade [103] by antidopaminergics. Since D2 receptors inhibit the indirect pathway, such upregulation could produce hyperkinesia. This hypothesis is also supported by the observation that increasing blockade can alleviate symptoms of TD, whereas abrupt withdrawal might prompt the appearance of symptoms. However, this cannot explain its persistence for up to years after drug withdrawal [90]. One possible explanation is that sustained DRB use increases free radical formation, leading to increased dopamine turnover [104], neuronal loss and gliosis in the basal ganglia, which could account for the persistence of symptoms [105]. Post-mortem animal studies have shown data consistent with this hypothesis [106]. Following this theory, the chronic blockade of D2 and hypersensitisation may be followed by maladaptive plasticity in cortico-striatal pathways due to oxidative stress. This alteration could then result in an imbalance between direct and indirect pathways that contributes to the abnormal output to the sensorimotor cortex perpetuating TD [107].
Since most patients taking antipsychotics do not develop TD, genetic susceptibility may contribute to the development of TD [90]. Steen et al. [108] have shown that homozygosity for the Ser9Gly variant in the D3 receptor is present in 22–24% of patients with TD compared with 5% of controls [108]. A meta-analysis of genetic studies indicates that polymorphisms in COMT and mnSOD genes confer a protective effect [109].
More recently, optogenetic-stimulating techniques have shown the involvement of striatal cholinergic neurons and GABAergic D2 medium spiny neurons in modulating VCM in mice, suggesting involvement of nicotinic receptors [110, 111] in the pathophysiology of TD.
3.4 Clinical Features
The symptoms of TD typically arise more than 1 year after DRB exposure. Its emergence frequently follows abrupt discontinuation of the drug, a switch of DRB or reduction in dose [90]. TD typically has an insidious onset over days to weeks followed by plateau of symptoms, and thereafter a waxing and waning course but generally persisting over decades. Chronicity depends on a prompt detection and management, with remission rates varying between 5 and 90% [112,113,114] depending on the date of DRB withdrawal. However, once it appears, TD does not become more severe if DRB is continued [115].
The phenomenology of TD is highly diverse (Table 3). In classic TD, the mouth adopts repetitive chewing movements with occasional lip smacking, opening of the mouth and tongue protrusion. Movements are usually repetitive and coordinated but they can be suppressed if asked to do so. Despite its striking appearance, patients are often unaware of their symptoms [78].
Akathisia, another form of TD, is more common in younger patients and consists of a feeling of inner restlessness resulting in an inability to sit or stand still. Patients are seen constantly moving with crossing and uncrossing of the legs, trunk rocking, moaning and groaning [77, 116, 117]. Tardive dystonia also tends to occur during early adulthood. Retrocollis, trunk arching with internal rotation of the arms with extension of elbows and flexion of the wrists are key features for differentiating tardive dystonia from primary dystonias [118].
Chorea as a TS usually accompanies OBL dyskinesia, but when in isolation it is known as ‘tardive chorea’. ‘Tardive tremor’ has also been described, presenting as a slow-frequency and low-amplitude tremor [119]. ‘Tardive tics’, indistinguishable from Tourette’s syndrome [120], and ‘tardive myoclonus’ affecting upper extremities [121] have been described as well. Pain in oral and genital regions, which sometimes becomes a source of profound distress, known as ‘tardive pain’, is another complication of treatment with DRBs [122]. Tardive parkinsonism is a term used when parkinsonism lasts for years after drug discontinuation coupled with a normal DaTscan, with one report disclosing absence of PD pathology [123]. However, most cases tend to coincide with abnormal functional neuroimaging [51].
Usually a combination of different phenomenologies is present; therapy should be targeted towards the main movement disorder. In addition, differential diagnosis should always take into account the possibility of primary neurological diseases with prominent dyskinetic manifestations and psychiatric symptoms such as Wilson’s disease, chorea-acanthocytosis or HD. Hence, a thorough clinical history and examination is important.
3.5 Prevention and Treatment
The iatrogenic nature of TD mandates that clinicians should avoid treatment with DRBs where possible, regularly monitor for the presence of TD and use the smallest effective doses of DRBs. This applies especially in those at higher risk for TD such as elderly patients [88]. Once TD has been identified, the first step should be lowering the dose and eventually removing the causative agent [78, 106, 112, 124]. Rapid withdrawal may produce a transient increase in the severity of dyskinesias; therefore, slow tapering is strongly encouraged [125]. Restarting or increasing the DRB dose might lead to transient improvement of symptoms, but this approach should be used only in emergency situations.
The approach of discontinuation of DRBs has been questioned in a recent meta-analysis, which included two very-low-quality trials involving 17 patients with unclear results [126]. However, in the absence of solid evidence, withdrawal of the offending drug seems reasonable in a drug-induced condition.
However, even after discontinuation of DRBs, symptoms last more than 10 years in the majority of patients [114]. Long-term prevalence studies have also shown that decreased scores on the Abnormal Involuntary Movements Scale (AIMS) are linked to increasing scores in parkinsonism scales [127], meaning that an augmented DRB dosage may be masking symptoms of TD with DIP.
If the patient requires continuous treatment with antipsychotics, switching to an SGA, especially clozapine or quetiapine, should be the first option, since high doses might be effective for the treatment of TD [128, 129], despite most evidence coming from small, low-quality studies [126]. In addition, it is possible that the antidyskinetic effect of those drugs at high doses is caused by D2 receptor blockade.
If further treatment is needed, VMAT2 inhibitors should be the first option. Tetrabenazine studies have provided positive results, with improvements in up to 95% of patients treated [130]. Nevertheless, most studies were small and had methodological flaws [131]. Tetrabenazine may have dose-limiting adverse effects such as depression, akathisia and parkinsonism [130].
In the KINECT3 study, a double-blind trial in 205 psychiatric patients, valbenazine showed statistically significant changes from baseline in the AIMS scores, from − 3.2 in the 80 mg/day group to − 0.1 in controls, and was well-tolerated [45]. Following these results, US Food and Drug Administration (FDA) approval was gained during the same year [132]. The 1-year follow-up study showed no risk of suicidal ideation, worsening akathisia or parkinsonism [46].
The ARM-TD (Aim to Reduce Movements in Tardive Dyskinesia) study investigating the role of deutetrabenazine was also published in 2017. Deutetrabenazine has a similar structure to tetrabenazine, but the addition of deuterium leads to decreased plasma fluctuations and so requires less frequent dosing. This randomised double-blind study during a 12-week period, which included 117 psychiatric patients, reported a statistically significant reduction in AIMS scores from − 3.0 in the treatment group to − 1.6 in the placebo arm without substantial adverse effects [41]. However, despite the positive outcomes in both KINECT3 and ARM-TD, the results should be taken with caution. The chronic course of TD requires longer follow-up in order to stablish the safety and efficacy of novel VMAT2 inhibitors.
Anticholinergics have been used in the treatment of TS, but they might have opposite effects on tardive dystonia and classic TD [90], improving the former but worsening OBL dyskinesias. Moreover, a recent Cochrane review could not make a confident statement about the effectiveness of anticholinergics (or its withdrawal) in the treatment of antipsychotic-induced TD [133]. Frequent adverse effects are markedly disabling in the elderly, precluding its use in patients over 60 years, in particular among those with dementia.
Other less studied medications may provide some degree of symptomatic improvement in TD and have been included in the American Academy of Neurology (AAN) evidence-based guidelines published in 2013 [134]. They found that clonazepam and Ginkgo biloba are probably effective for TD. Amantadine, a glutamate receptor antagonist might be considered for the treatment of TD when used conjointly with an antipsychotic during the first 7 weeks [134]. Extended-release amantadine remains yet to be studied in TD [135]. Other drugs such as levetiracetam, acetazolamide, baclofen, vitamin E, cholinergic agents and dopamine agonists have insufficient data to support or refute their role in TD according to the AAN guidelines [134].
Despite insufficient evidence, botulinum toxin injections into the muscles causing focal dyskinesia are recommended in the treatment of TD [136]. There are also case reports supporting the use of globus pallidus interna (GPi) deep brain stimulation (DBS) in the treatment of severe, refractory TD, with up to 50% improvement in symptoms in the majority of cases [137].
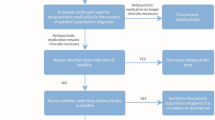
Additionally, some exciting new therapeutic approaches are in the pipeline. One of the most appealing is nicotine, which itself has been shown to decrease VCM in rodents, whereas the nicotinic acetylcholine receptor (nAChR) agonist varenicline demonstrated reductions in VCM in a dose-dependent fashion [138] (Fig. 2).

Algorithm for the treatment of tardive dyskinesia. BTX botulinum toxin, DBS deep brain stimulation, SGA second-generation antipsychotic, VMAT2 vesicle monoamine transporter 2
4 Discussion
DIP and TD are phenomenologically opposing disorders presenting with hypo- and hyperkinesia, respectively. However, they share risk factors, therapeutic approaches and, most importantly, the same causative drugs. Moreover, they frequently coexist within the same patient. Advanced age is the main predisposing factor related to the patient. Other risk factors are associated with the drug, such as the potency, dose and duration of treatment with DRBs. Prevention and a low threshold for diagnosis are essential in order to avoid and treat both disorders.
The first step after diagnosis should be the gradual withdrawal of the offending agent. If this is not possible, dose reduction or switching to another antipsychotic with lower D2 receptor affinity should be considered. In patients with DIP, if symptoms are still troublesome, other diagnostic possibilities, such as PD or other neurodegenerative parkinsonian disorders, should then be evaluated and a DaTscan is recommended. If symptoms persist in absence of neurodegenerative conditions, anticholinergics or amantadine are the first options for DIP, but signs of primary parkinsonism should be evaluated at every visit.
In TD, symptoms unfortunately usually become chronic. VMAT2 inhibitors should be the first treatment option, followed by other alternative drugs, and DBS may be considered in refractory cases. Two new VMAT2 inhibitors, deutetrabenazine and valbenazine, are already available for the treatment of TD [48, 132]. Although their lower propensity to trigger parkinsonism and longer half-lives make them theoretically attractive options [43], some methodological concerns [49] and the lack of long-term data mean they should be prescribed and monitored with caution.
There are some key issues that need to be assessed. An official definition of TD should be adopted in order to resolve fundamental issues in reporting regarding epidemiology, pathophysiology or response to treatment [139, 140]. In addition, better diagnostic methods need to be developed in DIP to avoid treatment delay in patients with umPD. There is also a surprising lack of knowledge about pathophysiology that should be addressed. Evidence regarding treatments for both disorders comes from small, low-quality studies that may lack power to find differences or could even be biased.
Thorough knowledge of TD and DIP is essential for newer preventive and therapeutic strategies and is central to the understanding of the physiology of the basal ganglia and, hence, the development of new drugs for other movement disorders.
5 Conclusion
With an aging population and increasing prevalence of dementia, the use of antipsychotic treatments is likely to rise [141]. Despite recent advances in the understanding of both TD and DIP, these conditions are frequently a puzzling problem for clinicians. Although newer treatments are available, only prevention and early detection guarantee a favourable prognosis. It is therefore essential to be aware of the adverse effects of these commonly prescribed drugs. The latest evidence shines a light on the complex mechanisms of TD and DIP, but our understanding of these conditions remains incomplete. Further research is required to develop a better understanding of the role of the basal ganglia in physiology and pathophysiology and for the development of more targeted and successful therapeutic strategies.
References
Van Gerpen JA. Drug-induced Parkinsonism. Neurologist. 2002;8:363–70. https://doi.org/10.1097/01.nrl.000003012.85777.f1.
Steck H. Extrapyramidal and diencephalic syndrome in the course of largactil and serpasil treatments. Ann Med Psychol. 1954;112:737–44.
Carlsson A. The occurrence, distribution and physiological role of catecholamines in the nervous system. Pharmacol Rev. 1959;11:490–3.
de la Fuente-Fernandez R, Schulzer M, Kuramoto L, Cragg J, Ramachandiran N, Au WL, et al. Age-specific progression of nigrostriatal dysfunction in Parkinson’s disease. Ann Neurol. 2011;69:803–10. https://doi.org/10.1002/ana.22284.
Caroff SN, Mann SC, Campbell EC, Sullivan KA. Movement disorders associated with atypical antipsychotic drugs. J Clin Psychiatry. 2002;63(Suppl 4):12–9.
Correll CU, Leucht S, Kane JM. Lower risk for tardive dyskinesia associated with second-generation antipsychotics: a systematic review of 1-year studies. Am J Psychiatry. 2004;161:414–25. https://doi.org/10.1176/appi.ajp.161.3.414.
Kane JM. Tardive dyskinesia rates with atypical antipsychotics in adults: prevalence and incidence. J Clin Psychiatry. 2004;65(Suppl 9):16–20.
Marder SR, Meibach RC. Risperidone in the treatment of schizophrenia. Am J Psychiatry. 1994;151:825–35. https://doi.org/10.1176/ajp.151.6.825.
de Leon J. The effect of atypical versus typical antipsychotics on tardive dyskinesia: a naturalistic study. Eur Arch Psychiatry Clin Neurosci. 2007;257:169–72. https://doi.org/10.1007/s00406-006-0705-z.
Correll CU, Schenk EM. Tardive dyskinesia and new antipsychotics. Curr Opin Psychiatry. 2008;21:151–6.
Freyhan FA. Psychomotility and Parkinsonism in treatment with neuroleptic drugs. AMA Arch Neurol Psychiatry. 1957;78:465–72.
Rajput AH, Rozdilsky B, Hornykiewicz O, Shannak K, Lee T, Seeman P. Reversible drug-induced Parkinsonism: clinicopathologic study of two cases. Arch Neurol. 1982;39:644–6.
Bower JH, Maraganore DM, McDonnell SK, Rocca WA. Incidence and distribution of Parkinsonism in Olmsted County, Minnesota, 1976–1990. Neurology. 1999;52:1214.
Llau ME, Nguyen L, Senard JM, Rascol O, Montastruc JL. Drug-induced Parkinson syndrome: 10 years of drug vigilance. Therapie. 1994;49:459–60 (in French).
Erro R, Bhatia KP, Tinazzi M. Parkinsonism following neuroleptic exposure: a double-hit hypothesis? Mov Disord. 2015;30:780–5. https://doi.org/10.1002/mds.26209.
Stephen P, Williamson J. Drug-induced Parkinsonism in the elderly. Lancet. 1984;324:1082–3. https://doi.org/10.1016/S0140-6736(84)91516-2.
López-Sendón JL, Mena MA, de Yébenes JG. Drug-induced Parkinsonism in the elderly. Drugs Aging. 2012;29:105–18.
Seijo-Martinez M, Del Rio MC, Alvarez JR, Prado RS, Salgado ET, Esquete JP, et al. Prevalence of Parkinsonism and Parkinson’s disease in the Arosa Island (Spain): a community-based door-to-door survey. J Neurol Sci. 2011;304:49–54.
De Lau LML, Giesbergen P, De Rijk MC, Hofman A, Koudstaal PJ, Breteler MMB. Incidence of Parkinsonism and Parkinson disease in a general population the Rotterdam Study. Neurology. 2004;63:1240–4.
Benito-Leon J, Bermejo-Pareja F, Morales-Gonzalez JM, Porta-Etessam J, Trincado R, Vega S, et al. Incidence of Parkinson disease and Parkinsonism in three elderly populations of central Spain. Neurology. 2004;62:734–41.
Savica R, Grossardt BR, Bower JH, Ahlskog JE, Mielke MM, Rocca WA. Incidence and time trends of drug-induced parkinsonism: a 30-year population-based study. Mov Disord. 2017;32:227–34. https://doi.org/10.1002/mds.26839.
Wenning GK, Kiechl S, Seppi K, Müller J, Högl B, Saletu M, et al. Prevalence of movement disorders in men and women aged 50–89 years (Bruneck Study cohort): a population-based study. Lancet Neurol. 2005;4:815–20. https://doi.org/10.1016/S1474-4422(05)70226-X.
Friedman JH, Trieschmann ME, Fernandez HH. Drug-induced Parkinsonism. In: Factor SA, Lang AE, Weiner WJ, editors. Drug induced movement disorders. Malden: Blackwell Publishing Inc; 2008. p. 103–39. https://doi.org/10.1002/9780470753217.ch6.
Rajput AH, Rozdilsky B, Ang L, Rajput A. Significance of Parkinsonian manifestations in essential tremor. Can J Neurol Sci. 1993;20:114–7.
Kish SJ, Shannak K, Rajput A, Deck JHN, Hornykiewicz O. Aging produces a secific pattern of striatal dopamine loss. Implication for the etiology of idiopathic Parkinson’s disease. J Neurochem. 1992;58:642–8.
Sweet RA, Pollock BG, Rosen J, Mulsant BH, Altieri LP, Perel JM. Early detection of neuroleptic-induced Parkinsonism in elderly patients with dementia. J Geriatr Psychiatry Neurol. 1994;7:251–3. https://doi.org/10.1177/089198879400700411.
Rajput AH, Rajput EF. Octogenarian Parkinsonism–clinicopathological observations. Parkinsonism Relat Disord. 2017;37:50–7. https://doi.org/10.1016/j.parkreldis.2017.01.009.
Rajput AH, Offord KP, Beard CM, Kurland LT. Epidemiology of Parkinsonism: incidence, classification, and mortality. Ann Neurol. 1984;16:278–82. https://doi.org/10.1002/ana.410160303.
Shiroma PR, Geda YE, Mrazek DA. Pharmacogenomic implications of variants of monoaminergic-related genes in geriatric psychiatry. Pharmacogenomics. 2010;11:1305–30. https://doi.org/10.2217/pgs.10.118.
Metzer WS, Newton JEO, Steele RW, Claybrook M, Paige SR, McMillan DE, et al. HLA antigens in drug-induced Parkinsonism. Mov Disord. 1989;4:121–8. https://doi.org/10.1002/mds.870040203.
Assmann BE, Robinson RO, Surtees RAH, Bräutigam C, Heales SJR, Wevers RA, et al. Infantile Parkinsonism-dystonia and elevated dopamine metabolites in CSF. Neurology. 2004;62:1872–4.
Friedman JH. Viewpoint: Challenges in our understanding of neuroleptic induced parkinsonism. Parkinsonism Relat Disord. 2014;20:1325–8. https://doi.org/10.1016/j.parkreldis.2014.09.030.
Yang S-Y, Kao Yang Y-H, Chong M-Y, Yang Y-H, Chang W-H, Lai C-S. Risk of extrapyramidal syndrome in schizophrenic patients treated with antipsychotics: a population-based study. Clin Pharmacol Ther. 2007;81:586–94. https://doi.org/10.1038/sj.clpt.6100069.
Cardoso F, Camargos ST, Silva GA Jr. Etiology of parkinsonism in a Brazilian movement disorders clinic TT [Etiologia de parkinsonismo em uma clínica brasileira de distúrbios do movimento]. Arq Neuropsiquiatr. 1998;56:171–5. https://doi.org/10.1590/S0004-282X1998000200001.
Dall’Igna OP, Tort ABL, Souza DO, Lara DR. Cinnarizine has an atypical antipsychotic profile in animal models of psychosis. J Psychopharmacol. 2005;19:342–6. https://doi.org/10.1177/0269881105053284.
Brücke T, Wöber C, Podreka I, Wöber-Bingöl C, Asenbaum S, Aull S, et al. D2 receptor blockade by flunarizine and cinnarizine explains extrapyramidal side effects. A SPECT study. J Cereb Blood Flow Metab. 1995;15:513–8. https://doi.org/10.1038/jcbfm.1995.63.
Easterford K, Clough P, Kellett M, Fallon K, Duncan S. Reversible Parkinsonism with normal beta-CIT-SPECT in patients exposed to sodium valproate. Neurology. 2004;62:1435–7.
Hawthorne JM, Caley CF. Extrapyramidal reactions associated with serotonergic antidepressants. Ann Pharmacother. 2015;49:1136–52. https://doi.org/10.1177/1060028015594812.
Fernandez HH, Trieschmann ME, Burke MA, Friedman JH. Quetiapine for psychosis in Parkinson’s disease versus dementia with Lewy bodies. J Clin Psychiatry. 2002;63:513–5.
Yaw TK, Fox SH, Lang AE. Clozapine in Parkinsonian rest tremor: a review of outcomes, adverse reactions, and possible mechanisms of action. Mov Disord Clin Pract. 2016;3:116–24. https://doi.org/10.1002/mdc3.12266.
Fernandez HH, Factor SA, Hauser RA, Jimenez-Shahed J, Ondo WG, Jarskog LF, et al. Randomized controlled trial of deutetrabenazine for tardive dyskinesia: the ARM-TD study. Neurology. 2017;88:2003–10. https://doi.org/10.1212/wnl.0000000000003960.
Belleau B, Burba J, Pindell M, Reiffenstein J. Effect of deuterium substitution in sympathomimetic amines on adrenergic responses. Science. 1961;133:102–4. https://doi.org/10.1126/science.133.3446.102.
Jankovic J. Dopamine depleters in the treatment of hyperkinetic movement disorders. Expert Opin Pharmacother. 2016;17:2461–70. https://doi.org/10.1080/14656566.2016.1258063.
Kenney C, Hunter C, Jankovic J. Long-term tolerability of tetrabenazine in the treatment of hyperkinetic movement disorders. Mov Disord. 2007;22:193–7.
Hauser RA, Factor SA, Marder SR, Knesevich MA, Ramirez PM, Jimenez R, et al. KINECT 3: a phase 3 randomized, double-blind, placebo-controlled trial of valbenazine for tardive dyskinesia. Am J Psychiatry. 2017;174:476–84. https://doi.org/10.1176/appi.ajp.2017.16091037.
Factor SA, Remington G, Comella CL, Correll CU, Burke J, Jimenez R, et al. The effects of valbenazine in participants with tardive dyskinesia: results of the 1-year KINECT 3 extension study. J Clin Psychiatry. 2017;78:1344–50. https://doi.org/10.4088/JCP.17m11777.
Frank S, Testa CM, Stamler D, Kayson E, Davis C, Edmondson MC, et al. Effect of deutetrabenazine on chorea among patients with Huntington disease: a randomized clinical trial. JAMA. 2016;316:40–50. https://doi.org/10.1001/jama.2016.8655.
Anderson KE, Stamler D, Davis MD, Factor SA, Hauser RA, Isojärvi J, et al. Deutetrabenazine for treatment of involuntary movements in patients with tardive dyskinesia (AIM-TD): a double-blind, randomised, placebo-controlled, phase 3 trial. Lancet Psychiatry. 2017;4:595–604. https://doi.org/10.1016/S2215-0366(17)30236-5.
Rodrigues FB, Duarte GS, Costa J, Ferreira JJ, Wild EJ. Tetrabenazine versus deutetrabenazine for Huntington’s disease: twins or distant cousins? Mov Disord Clin Pract. 2017;4:582–5. https://doi.org/10.1002/mdc3.12483.
Foubert-Samier A, Helmer C, Perez F, Le Goff M, Auriacombe S, Elbaz A, et al. Past exposure to neuroleptic drugs and risk of Parkinson disease in an elderly cohort. Neurology. 2012;79:1615–21. https://doi.org/10.1212/WNL.0b013e31826e25ce.
Chung SJ, Yoo HS, Moon H, Oh JS, Kim JS, Park YH, et al. Early-onset drug-induced Parkinsonism after exposure to offenders implies nigrostriatal dopaminergic dysfunction. J Neurol Neurosurg Psychiatry. 2018;89:169–74. https://doi.org/10.1136/jnnp-2017-315873.
Shuaib UA, Rajput AH, Robinson CA, Rajput A. Neuroleptic-induced Parkinsonism: clinicopathological study. Mov Disord. 2016;31:360–5. https://doi.org/10.1002/mds.26467.
Esper CD, Factor SA. Failure of recognition of drug-induced Parkinsonism in the elderly. Mov Disord. 2008;23:401–4. https://doi.org/10.1002/mds.21854.
Postuma RB, Berg D, Stern M, Poewe W, Olanow CW, Oertel W, et al. MDS clinical diagnostic criteria for Parkinson’s disease. Mov Disord. 2015;30:1591–601. https://doi.org/10.1002/mds.26424.
Brigo F, Erro R, Marangi A, Bhatia K, Tinazzi M. Differentiating drug-induced Parkinsonism from Parkinson’s disease: an update on non-motor symptoms and investigations. Parkinsonism Relat Disord. 2014;20:808–14. https://doi.org/10.1016/j.parkreldis.2014.05.011.
López-Sendón Moreno JL, Alonso-Cánovas A, Buisán Catevilla J, García Barragán N, Corral Corral I, de Felipe Mimbrera A, et al. Substantia nigra echogenicity predicts response to drug withdrawal in suspected drug-induced Parkinsonism. Mov Disord Clin Pract. 2016;3:268–74. https://doi.org/10.1002/mdc3.12281.
Tinazzi M, Ottaviani S, Isaias IU, Pasquin I, Steinmayr M, Vampini C, et al. [123I]FP–CIT SPET imaging in drug-induced Parkinsonism. Mov Disord. 2008;23:1825–9. https://doi.org/10.1002/mds.22098.
Tinazzi M, Geroin C, Gandolfi M, Smania N, Tamburin S, Morgante F, et al. Pisa syndrome in Parkinson’s disease: an integrated approach from pathophysiology to management. Mov Disord. 2016;31:1785–95. https://doi.org/10.1002/mds.26829.
Cuberas-Borrós G, Lorenzo-Bosquet C, Aguadé-Bruix S, Hernández-Vara J, Pifarré-Montaner P, Miquel F, et al. Quantitative evaluation of striatal I-123-FP-CIT uptake in essential tremor and Parkinsonism. Clin Nucl Med. 2011;36:991–6. https://doi.org/10.1097/RLU.0b013e3182291a7b.
Diaz-Corrales FJ, Sanz-Viedma S, Garcia-Solis D, Escobar-Delgado T, Mir P. Clinical features and 123I-FP-CIT SPECT imaging in drug-induced Parkinsonism and Parkinson’s disease. Eur J Nucl Med Mol Imaging. 2010;37:556–64. https://doi.org/10.1007/s00259-009-1289-4.
Rascol O, Schelosky L. 123I-metaiodobenzylguanidine scintigraphy in Parkinson’s disease and related disorders. Mov Disord. 2009;24:S732–41. https://doi.org/10.1002/mds.22499.
Lee PH, Kim JS, Shin DH, Yoon S-N, Huh K. Cardiac 123I-MIBG scintigraphy in patients with drug induced Parkinsonism. J Neurol Neurosurg Psychiatry. 2006;77:372–4.
Chaudhuri KR, Healy DG, Schapira AH, National Institute for Clinical Excellence. Non-motor symptoms of Parkinson’s disease: diagnosis and management. Lancet Neurol. 2006;5:235–45. https://doi.org/10.1016/S1474-4422(06)70373-8.
Chien CP, Dimascio A, Cole JO. Antiparkinsonian agents and depot phenothiazine. Am J Psychiatry. 1974;131:86–90. https://doi.org/10.1176/ajp.131.1.86.
Keepers GA, Clappison VJ, Casey DE. Initial anticholinergic prophylaxis for neuroleptic-induced extrapyramidal syndromes. Arch Gen Psychiatry. 1983;40:1113–7.
Hardie RJ, Lees AJ. Neuroleptic-induced Parkinson’s syndrome: clinical features and results of treatment with levodopa. J Neurol Neurosurg Psychiatry. 1988;51:850–4.
Gerlach J. Relationship between tardive dyskinesia, l-dopa-induced hyperkinesia and Parkinsonism. Psychopharmacology (Berl). 1977;51:259–63.
Saltz BL, Woerner MG, Robinson DG, Kane JM. Side effects of antipsychotic drugs. Avoiding and minimizing their impact in elderly patients. Postgrad Med. 2000;107(169–72):175–8. https://doi.org/10.3810/pgm.2000.02.891.
Ory-Magne F, Corvol J-C, Azulay J-P, Bonnet A-M, Brefel-Courbon C, Damier P, et al. Withdrawing amantadine in dyskinetic patients with Parkinson disease: the AMANDYSK trial. Neurology. 2014;82:300–7. https://doi.org/10.1212/WNL.0000000000000050.
Wolf E, Seppi K, Katzenschlager R, Hochschorner G, Ransmayr G, Schwingenschuh P, et al. Long-term antidyskinetic efficacy of amantadine in Parkinson’s disease. Mov Disord. 2010;25:1357–63. https://doi.org/10.1002/mds.23034.
Silver H, Geraisy N, Schwartz M. No difference in the effect of biperiden and amantadine on Parkinsonian- and tardive dyskinesia-type involuntary movements: a double-blind crossover, placebo-controlled study in medicated chronic schizophrenic patients. J Clin Psychiatry. 1995;56:167–70.
Pahwa R, Hauser RA. ADS-5102 (Amantadine) extended release for levodopa-induced dyskinesia. JAMA Neurol. 2017;74:1507–8. https://doi.org/10.1001/jamaneurol.2017.3205.
Tinazzi M, Antonini A, Bovi T, Pasquin I, Steinmayr M, Moretto G, et al. Clinical and [123I]FP-CIT SPET imaging follow-up in patients with drug-induced Parkinsonism. J Neurol. 2009;256:910–5. https://doi.org/10.1007/s00415-009-5039-0.
Goswami U, Dutta S, Kuruvilla K, Papp E, Perenyi A. Electroconvulsive therapy in neuroleptic-induced Parkinsonism. Biol Psychiatry. 1989;26:234–8.
Berg JE. Electroconvulsive treatment of a patient with Parkinson’s disease and moderate depression. Ment Illn. 2011;3:8–10. https://doi.org/10.4081/mi.2011.e3.
Moellentine C, Rummans T, Ahlskog JE, Harmsen WS, Suman VJ, O’Connor MK, et al. Effectiveness of ECT in patients with Parkinsonism. J Neuropsychiatry Clin Neurosci. 1998;10:187–93. https://doi.org/10.1176/jnp.10.2.187.
Aquino CCH, Lang AE. Tardive dyskinesia syndromes: current concepts. Parkinsonism Relat Disord. 2014;20:S113–7. https://doi.org/10.1016/s1353-8020(13)70028-2.
Fahn S, Jankovic J, Hallett M. The tardive syndromes: phenomenology, concepts on pathophysiology and treatment, and other neuroleptic-induced syndromes. Principles and practice of movement disorders. 2nd ed. Philadelphia: Elsevier; 2011. p. 415–46. https://doi.org/10.1016/b978-1-4377-2369-4.00019-6.
Jeste DV, Wyatt RJ. Therapeutic strategies against tardive dyskinesia. Two decades of experience. Arch Gen Psychiatry. 1982;39:803–16.
Kane JM, Smith JM. Tardive dyskinesia: prevalence and risk factors, 1959 to 1979. Arch Gen Psychiatry. 1982;39:473–81.
Caligiuri MP, Jeste DV, Lacro JP. Antipsychotic-induced movement disorders in the elderly. Drugs Aging. 2000;17:363–84.
Caligiuri MP, Lacro JP, Rockwell E, McAdams LA, Jeste DV. Incidence and risk factors for severe tardive dyskinesia in older patients. Br J Psychiatry. 1997;171:148–53.
Tarsy D, Baldessarini RJ. Epidemiology of tardive dyskinesia: is risk declining with modern antipsychotics? Mov Disord. 2006;21:589–98. https://doi.org/10.1002/mds.20823.
Carbon M, Hsieh C-H, Kane JM, Correll CU. Tardive dyskinesia prevalence in the period of second-generation antipsychotic use: a meta-analysis. J Clin Psychiatry. 2017;78:e264–78. https://doi.org/10.4088/JCP.16r10832.
Woods SW, Morgenstern H, Saksa JR, Walsh BC, Sullivan MC, Money R, et al. Incidence of tardive dyskinesia with atypical versus conventional antipsychotic medications. J Clin Psychiatry. 2010;71:463–74. https://doi.org/10.4088/JCP.07m03890yel.
van Os J, Fahy T, Jones P, Harvey I, Toone B, Murray R. Tardive dyskinesia: who is at risk? Acta Psychiatr Scand. 1997;96:206–16.
van Harten PN, Hoek HW, Matroos GE, Koeter M, Kahn RS. Intermittent neuroleptic treatment and risk for tardive dyskinesia: Curaçao Extrapyramidal Syndromes Study III. Am J Psychiatry. 1998;155:565–7. https://doi.org/10.1176/ajp.155.4.565.
Smith JM, Baldessarini RJ. Changes in prevalence, severity, and recovery in tardive dyskinesia with age. Arch Gen Psychiatry. 1980;37:1368–73.
Woerner MG, Alvir JM, Saltz BL, Lieberman JA, Kane JM. Prospective study of tardive dyskinesia in the elderly: rates and risk factors. Am J Psychiatry. 1998;155:1521–8. https://doi.org/10.1176/ajp.155.11.1521.
Waln O, Jankovic J. An update on tardive dyskinesia: from phenomenology to treatment. Tremor Other Hyperkinet Mov (N Y). 2013. https://doi.org/10.7916/D88P5Z71.
Wonodi I, Adami HM, Cassady SL, Sherr JD, Avila MT, Thaker GK. Ethnicity and the course of tardive dyskinesia in outpatients presenting to the motor disorders clinic at the Maryland psychiatric research center. J Clin Psychopharmacol. 2004;24:592–8.
Tenback DE, van Harten PN, van Os J. Non-therapeutic risk factors for onset of tardive dyskinesia in schizophrenia: a meta-analysis. Mov Disord. 2009;24:2309–15. https://doi.org/10.1002/mds.22707.
Ferentinos P, Dikeos D. Genetic correlates of medical comorbidity associated with schizophrenia and treatment with antipsychotics. Curr Opin Psychiatry. 2012;25:381–90. https://doi.org/10.1097/YCO.0b013e3283568537.
Ganzini L, Casey DE, Hoffman WF, McCall AL. The prevalence of metoclopramide-induced tardive dyskinesia and acute extrapyramidal movement disorders. Arch Intern Med. 1993;153:1469–75.
Dubovsky SL, Thomas M. Tardive dyskinesia associated with fluoxetine. Psychiatr Serv. 1996;47:991–3. https://doi.org/10.1176/ps.47.9.991.
Woogen S, Graham J, Angrist B. A tardive dyskinesia-like syndrome after amitriptyline treatment. J Clin Psychopharmacol. 1981;1:34–6.
Harrison MB, Lyons GR, Landow ER. Phenytoin and dyskinesias: a report of two cases and review of the literature. Mov Disord. 1993;8:19–27. https://doi.org/10.1002/mds.870080104.
Thach BT, Chase TN, Bosma JF. Oral facial dyskinesia accociated with prolonged use of antihistaminic decongestants. N Engl J Med. 1975;293:486–7. https://doi.org/10.1056/NEJM197509042931008.
Raja M, Azzoni A. Tardive dyskinesia after long-term veralipride treatment. J Neuropsychiatry Clin Neurosci. 2005;17:252–3. https://doi.org/10.1176/jnp.17.2.252-a.
Bordia T, McIntosh JM, Quik M. Nicotine reduces antipsychotic-induced orofacial dyskinesia in Rats. J Pharmacol Exp Ther. 2012;340:612–9. https://doi.org/10.1124/jpet.111.189100.
Waddington JL. Spontaneous orofacial movements induced in rodents by very long-term neuroleptic drug administration: phenomenology, pathophysiology and putative relationship to tardive dyskinesia. Psychopharmacology (Berl). 1990;101:431–47.
Blanchet PJ, Parent M-T, Rompré PH, Lévesque D. Relevance of animal models to human tardive dyskinesia. Behav Brain Funct. 2012;8:12. https://doi.org/10.1186/1744-9081-8-12.
Loonen AJM, Ivanova SA. New insights into the mechanism of drug-induced dyskinesia. CNS Spectr. 2013;18:15–20. https://doi.org/10.1017/S1092852912000752.
Lohr JB, Kuczenski R, Niculescu AB. Oxidative mechanisms and tardive dyskinesia. CNS Drugs. 2003;17:47–62.
Elkashef AM, Wyatt RJ. Tardive dyskinesia: possible involvement of free radicals and treatment with vitamin E. Schizophr Bull. 1999;25:731–40.
Kiriakakis V, Bhatia KP, Quinn NP, Marsden CD. The natural history of tardive dystonia. A long-term follow-up study of 107 cases. Brain. 1998;121(Pt 1):2053–66.
Teo JT, Edwards MJ, Bhatia K. Tardive dyskinesia is caused by maladaptive synaptic plasticity: a hypothesis. Mov Disord. 2012;27:1205–15. https://doi.org/10.1002/mds.25107.
Steen VM, Løvlie R, MacEwan T, McCreadie RG. Dopamine D3-receptor gene variant and susceptibility to tardive dyskinesia in schizophrenic patients. Mol Psychiatry. 1997;2:139–45.
Bakker PR, van Harten PN, van Os J. Antipsychotic-induced tardive dyskinesia and polymorphic variations in COMT, DRD2, CYP1A2 and MnSOD genes: a meta-analysis of pharmacogenetic interactions. Mol Psychiatry. 2008;13:544–56. https://doi.org/10.1038/sj.mp.4002142.
Bordia T, Perez XA, Heiss J, Zhang D, Quik M. Optogenetic activation of striatal cholinergic interneurons regulates l-dopa-induced dyskinesias. Neurobiol Dis. 2016;91:47–58. https://doi.org/10.1016/j.nbd.2016.02.019.
Salem H, Pigott T, Zhang XY, Zeni CP, Teixeira AL. Antipsychotic-induced Tardive dyskinesia: from biological basis to clinical management. Expert Rev Neurother. 2017;17:883–94. https://doi.org/10.1080/14737175.2017.1361322.
Tarsy D. Neuroleptic-induced extrapyramidal reactions: classification, description, and diagnosis. Clin Neuropharmacol. 1983;6(Suppl 1):S9–26.
Tarsy D, Baldessarini RJ. Tardive dyskinesia. Annu Rev Med. 1984;35:605–23. https://doi.org/10.1146/annurev.me.35.020184.003133.
Gardos G, Casey DE, Cole JO, Perenyi A, Kocsis E, Arato M, et al. Ten-year outcome of tardive dyskinesia. Am J Psychiatry. 1994;151:836–41. https://doi.org/10.1176/ajp.151.6.836.
Labbate LA, Lande RG, Jones F, Oleshansky MA. Tardive dyskinesia in older out-patients: a follow-up study. Acta Psychiatr Scand. 1997;96:195–8. https://doi.org/10.1111/j.1600-0447.1997.tb10151.x.
Burke RE, Kang UJ, Jankovic J, Miller LG, Fahn S. Tardive akathisia: an analysis of clinical features and response to open therapeutic trials. Mov Disord. 1989;4:157–75. https://doi.org/10.1002/mds.870040208.
Munetz MR, Roth LH, Cornes CL. Tardive dyskinesia and informed consent: myths and realities. Bull Am Acad Psychiatry Law. 1982;10:77–88.
Kang UJ, Burke RE, Fahn S. Natural history and treatment of tardive dystonia. Mov Disord. 1986;1:193–208. https://doi.org/10.1002/mds.870010305.
Stacy M, Jankovic J. Tardive tremor. Mov Disord. 1992;7:53–7. https://doi.org/10.1002/mds.870070110.
Fountoulakis KN, Samara M, Siapera M, Iacovides A. Tardive Tourette-like syndrome: a systematic review. Int Clin Psychopharmacol. 2011;26:237–42. https://doi.org/10.1097/YIC.0b013e32834aa924.
Tominaga H, Fukuzako H, Izumi K, Koja T, Fukuda T, Fujii H, et al. Tardive myoclonus [letter]. Lancet. 1987;1:322.
Ford B, Greene P, Fahn S. Oral and genital tardive pain syndromes. Neurology. 1994;44:2115–9.
Jankovic J, Casabona J. Coexistent tardive dyskinesia and parkinsonism. Clin Neuropharmacol. 1987;10:511–21.
Quitkin F, Rifkin A, Gochfeld L, Klein DF. Tardive dyskinesia: are first signs reversible? Am J Psychiatry. 1977;134:84–7. https://doi.org/10.1176/ajp.134.1.84.
Gardos G, Cole JO, Tarsy D. Withdrawal syndromes associated with antipsychotic drugs. Am J Psychiatry. 1978;135:1321–4. https://doi.org/10.1176/ajp.135.11.1321.
Bergman H, Rathbone J, Agarwal V, Soares-Weiser K. Antipsychotic reduction and/or cessation and antipsychotics as specific treatments for tardive dyskinesia. Cochrane Database Syst Rev. 2018. https://doi.org/10.1002/14651858.CD000459.pub3.
Fernandez HH, Krupp B, Friedman JH. The course of tardive dyskinesia and parkinsonism in psychiatric inpatients: 14-Year follow-up. Neurology. 2001;56:805–7. https://doi.org/10.1212/WNL.56.6.805.
Emsley R, Turner HJ, Schronen J, Botha K, Smit R, Oosthuizen PP. A single-blind, randomized trial comparing quetiapine and haloperidol in the treatment of tardive dyskinesia. J Clin Psychiatry. 2004;65:696–701.
Bassitt DP, Louzã Neto MR. Clozapine efficacy in tardive dyskinesia in schizophrenic patients. Eur Arch Psychiatry Clin Neurosci. 1998;248:209–11.
Kenney C, Jankovic J. Tetrabenazine in the treatment of hyperkinetic movement disorders. Expert Rev Neurother. 2006;6:7–17. https://doi.org/10.1586/14737175.6.1.7.
Chen JJ, Ondo WG, Dashtipour K, Swope DM. Tetrabenazine for the treatment of hyperkinetic movement disorders: a review of the literature. Clin Ther. 2012;34:1487–504. https://doi.org/10.1016/j.clinthera.2012.06.010.
Davis MC, Miller BJ, Kalsi JK, Birkner T, Mathis MV. Efficient trial design—FDA approval of valbenazine for tardive dyskinesia. N Engl J Med. 2017;376:2503–6. https://doi.org/10.1056/NEJMp1704898.
Bergman H, Soares-Weiser K. Anticholinergic medication for antipsychotic-induced tardive dyskinesia. Cochrane Database Syst Rev. 2018. https://doi.org/10.1002/14651858.CD000204.pub2.
Bhidayasiri R, Fahn S, Weiner WJ, Gronseth GS, Sullivan KL, Zesiewicz TA. Evidence-based guideline: treatment of tardive syndromes: report of the Guideline Development Subcommittee of the American Academy of Neurology. Neurology. 2013;81:463–9. https://doi.org/10.1212/WNL.0b013e31829d86b6.
Oertel W, Eggert K, Pahwa R, Tanner CM, Hauser RA, Trenkwalder C, et al. Randomized, placebo-controlled trial of ADS-5102 (amantadine) extended-release capsules for levodopa-induced dyskinesia in Parkinson’s disease (EASE LID 3). Mov Disord. 2017;32:1701–9. https://doi.org/10.1002/mds.27131.
Ramirez-Castaneda J, Jankovic J. Long-term efficacy and safety of botulinum toxin injections in dystonia. Toxins (Basel). 2013;5:249–66. https://doi.org/10.3390/toxins5020249.
Spindler MA, Galifianakis NB, Wilkinson JR, Duda JE. Globus pallidus interna deep brain stimulation for tardive dyskinesia: case report and review of the literature. Parkinsonism Relat Disord. 2013;19:141–7. https://doi.org/10.1016/j.parkreldis.2012.09.016.
Quik M, Zhang D, Perez XA, Bordia T. Role for the nicotinic cholinergic system in movement disorders; therapeutic implications. Pharmacol Ther. 2014;144:50–9. https://doi.org/10.1016/j.pharmthera.2014.05.004.
Bhidayasiri R, Jitkritsadakul O, Friedman JH, Fahn S. Updating the recommendations for treatment of tardive syndromes: a systematic review of new evidence and practical treatment algorithm. J Neurol Sci. 2018;389:67–75. https://doi.org/10.1016/j.jns.2018.02.010.
Meyer JM. Future directions in tardive dyskinesia research. J Neurol Sci. 2018;389:76–80. https://doi.org/10.1016/j.jns.2018.02.004.
Jorm AF, Jolley D. The incidence of dementia: a meta-analysis. Neurology. 1998;51:728–33.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Funding
No funding was received for this study.
Conflict of interest
Jose Lopez-Sendón has received honoraria from Zambon and travel grants from Lundbeck and KRKA pharmaceuticals. He has no other conflicts of interest. Carlos Estevez-Fraga and Paul Zeun have no conflicts of interest.
Rights and permissions
About this article

Cite this article
Estevez-Fraga, C., Zeun, P. & López-Sendón Moreno, J.L. Current Methods for the Treatment and Prevention of Drug-Induced Parkinsonism and Tardive Dyskinesia in the Elderly. Drugs Aging 35, 959–971 (2018). https://doi.org/10.1007/s40266-018-0590-y
Published:
Issue Date:
DOI: https://doi.org/10.1007/s40266-018-0590-y