
Abstract
Hyperuricaemia is an independent risk factor for renal function decline. Evidence is emerging that urate-lowering therapy might be beneficial in subjects with renal impairment. We review the association between renal impairment and gout, some of the related pathogenic processes and the possible impact of gout treatment on the progression of renal impairment. Nevertheless, the management of gout is more complex in the presence of chronic kidney disease. The main aim of gout therapy is to fully dissolve the urate crystals, thus curing the disease. Avoidance of attacks—prophylaxis—and their prompt treatment if they occur, along with accurate information to patients, completes the treatment strategy. This article provides a practical guide to managing gout in older patients and in those with renal impairment. We highlight the shortcomings in our current treatment options and strategies.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Silent crystal deposits in hyperuricemic patients and microtophi in the renal medulla possibly contribute to declining renal function. |
Gout is a urate crystal deposit disease; the main aim of gout treatment is to eliminate the crystals by reducing serum uric acid levels. The lower serum uric acid levels are reduced, the faster the deposited urate crystals are dissolved. |
If required, allopurinol can be used at higher doses than those recommended by the Hande guidelines based on creatinine clearance. |
If required, a uricosuric drug (by increasing uric acid renal clearance) added to a xanthine oxidase inhibitor (that reduces the load of uric acid produced) results in important further reductions of serum uric acid levels. |
1 Introduction
Gout is a frequent companion of renal impairment (RI). As renal function declines, the elimination of uric acid through the urine decreases, leading to hyperuricaemia. Persistent hyperuricaemia facilitates the formation of monosodium urate (MSU) crystals in joints and tissues [1]; this crystal deposit is core to the definition of gout. Patients with long-standing untreated, or poorly treated, gout may show renal disease. The mechanisms of RI are manifold and not completely understood. Monosodium urate crystals are recognised by the native immune system, thereby activating the NALP-3 inflammasome. This results in the activation and release of interleukin (IL)-1 (a pro-inflammatory cytokine) and the subsequent trigger of an inflammatory cascade [2] that is persistent and continuous [3, 4]. A chronic inflammatory infiltrate surrounds the crystal deposits in tophi [5]. Persistent subclinical inflammation is also noted at the joints (and at other crystal deposit sites) during inter-critical periods [3]. Acute gouty flares can occur at different time intervals, alerting the clinician about the presence of the crystals. However, as shown over 60 years ago [6], when serum urate (SU) normalises, crystals dissolve [7]. Thus, as a crystal deposit is fully reversible, gout is currently considered a curable disease [8].
Current European League Against Rheumatism (EULAR) [8] and American College of Rheumatology [9, 10] recommendations do not include specific management considerations for the elderly, although both outline that special care should to be taken in patients with co-morbidities. Many of these comorbidities are very common in this age group. This review outlines the possible connections between hyperuricaemia, gout and renal function decline. Furthermore, we discuss how this decline modifies the approach to gout treatment.
Gout is more common in older adults, with an incidence of 8% in those aged 70–79 years compared with only 1.7% in those aged < 50 years. Of note, an increase in gout prevalence has occurred in patients aged over 65 years, which is even more striking in those aged over 75 years [11]. The increased use of drugs that increase serum uric acid levels, such as thiazide, loop diuretics, low-dose aspirin or niacin [12], and the higher prevalence of RI are two of the reasons behind this increase. In women, a markedly higher frequency of gout is seen after menopause, which is related to an increase in tubular reabsorption of urate (5% in those aged ≥ 70 years, < 1% in those aged < 50 years) as a result of the decrease in oestrogen levels [13, 14].
Renal impairment is a frequent comorbidity of gout. A recent meta-analysis of eight studies estimated that 24% (95% confidence interval 15–28) of patients with gout presented with chronic kidney disease above stage 3 [15]. Conversely, RI decreases urate excretion, thereby raising the risk of gout. In a cohort of patients with renal disease, 16% of those with an estimated creatinine clearance (CrCL) > 60 mL/min experienced gout, a ratio that doubled in those with an estimated CrCL < 30 mL/min [16]. Furthermore, the risk of end-stage renal failure is increased in patients with gout [17]. The reasons for this association remain poorly understood. In any case, when considering gout treatment in patients with RI, one must consider: (1) the limitations in drug selection and dose modification related to RI, and (2) whether (and why) the treatment of concomitant gout (or hyperuricaemia) can modify the prognosis of the associated renal disease. Though related, these points should be considered separately.
Increasing evidence supports the association between hyperuricaemia and incident RI. It has become common practice among nephrologists to treat hyperuricaemia in patients with RI with the objective of stopping or slowing the renal function decline. A recent paper found that the risk of progression to renal failure increased 7% (hazard ratio 1.07, 95% confidence interval 1.00–1.14) for each 1-mg/dL increase in baseline SU level [18]. An earlier review of clinical trials reported that allopurinol seems to delay the progression of kidney disease [19]; similar data have also been suggested after treatment with febuxostat [20]. A recent comparative effectiveness study has shown that allopurinol is associated with a greater reduction in the risk of incident kidney disease than febuxostat in a representative sample of older Americans (≥ 65 years of age); the association of allopurinol with renal protection was dose related, and possibly duration related [21].
Soluble uric acid is recognised by the innate immune system [22, 23] and primes Toll-like receptors, inducing pro-inflammatory cytokine production [24]. In patients with gout, higher SU levels are associated with higher levels of tumour necrosis factor [25]. These data suggest that hyperuricaemia is associated with a more pro-inflammatory status; the consequences of this inflammatory state on the kidneys remain undetermined. However, hyperuricaemia can result in gout in a higher proportion of patients than previously considered. The first gout attack is often considered as the start of the disease; before it, subjects are diagnosed as having asymptomatic hyperuricaemia. Newer imaging techniques, such as an ultrasound or double-energy computed tomography scan, have shown that a considerable proportion of subjects with asymptomatic hyperuricaemia already have MSU crystal deposits in their joints and tendons, including tophi [26,27,28]. It remains unclear for how long crystal deposition precedes the initial symptoms. Presentations such as polyarticular gout or tophi at diagnosis indicate that a silent MSU crystal deposit build-up can be very prolonged. Although debate about when gout starts persists, it appears difficult to maintain the label of ‘asymptomatic hyperuricaemia’ in those patients in whom MSU crystal deposits have been demonstrated. Thus, when considering the consequences and associations of asymptomatic hyperuricaemia, the possibility that they are—at least partially—related to MSU crystal deposits and their associated low-grade inflammation has to be considered.
Tophi in the renal medulla of patients with gout were already described at autopsies in the nieteenth century (reviewed in ref [29]). They have been found in 38% of autopsies of patients with gout, 17% of autopsies of patients who died from RI of different origins [30] and in 8% of all the autopsies carried out during a 1-year period. Furthermore, a significantly higher frequency of prior diagnosis of renal disease and of gout was found in those patients who showed medullary tophi on autopsy [31]. In a more recent retrospective study of 572 renal biopsies showing medullary tissue, 36 had tophi. Medullary tophi occurred in patients both with and without hyperuricaemia or a prior gout diagnosis [32]. Histopathology studies mention the presence of an inflammatory infiltrate surrounding the tophi, with some even using the term pyelonephritis. These crystal deposits have received little attention. Attempts to detect them by double-energy computed tomography in patients with severe gout have failed [33]; however, the limited sensibility of this technique for detecting smaller MSU crystal deposits should be kept in mind [34]. Monosodium urate crystals are dissolved by formaline and are therefore absent in routinely processed biopsies [35] unless they are cut unfixed by freezing [1]. Currently, when planning the treatment for patients with gout with RI—and likely also those with so-called asymptomatic hyperuricaemia—the possibility that its progress may be influenced by medullary tophi should be considered.
2 Treatment
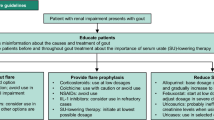
The primary aim of gout treatment is to eliminate all MSU crystals by lowering SU levels to at least below 6 mg/dL. This aim is maintained in the elderly and in patients with RI. These deposits are the cause of the disease and with their disappearance the associated inflammation is no longer possible. Monosodium urate crystal depositing is a reversible process; crystals continue to form in patients with gout while hyperuricaemia persists, but dissolve when SU is reduced below the saturation point. Secondary aims include: (1) the avoidance of gout attacks (prophylaxis), which are frequent at treatment initiation and a common cause of treatment discontinuation, (2) prompt treatment of attacks and (3) patient education on the nature and consequences of gout-related disease and the treatment aims and strategies. This education along with a proper follow-up greatly increases the patient’s compliance [36, 37]. Especially in patients with RI and until more definitive data are available, we recommend starting urate-lowering treatment on diagnosis, as the normalisation of SU levels and prompt elimination of MSU crystals could have a favourable impact on the evolution of RI (Fig. 1).

Algorithm of gout treatment in patients with renal impairment (RI). CKD chronic kidney disease, IL interleukin, IV intravenous, MSU monosodium urate, NSAIDs nonsteroidal anti-inflammatory drugs, SU serum urate, ULT urate-lowering therapy
2.1 Eliminating the Urate Crystals: Reducing Serum Urate Levels
Monosodium urate crystal depositing is a reversible process [6, 7, 38]. Importantly, lower SU levels result in a faster dissolution of crystals: SU values under 4 mg/dL reduce the tophus diameter at twice the rate of SU values over 5 mg/dL [39]. Recent EULAR recommendations [8] advise achieving a SU target of under 5 mg/dL in “patients with severe gout (tophi, chronic arthropathy, frequent attacks)”. The recommendations then state a “SU level < 3 mg/dL is not recommended in the long term” (“that is, for several years”), as the association between very low levels of SU and neurological diseases is still controversial. Crystal dissolution with such very low levels of SU is, however, more rapid. The 2012 American College of Rheumatology guidelines also recommend reducing SU levels below 5 mg/dL for severe gout [9]. Whether any of the different causes of gout-associated RI are particularly prone to improvement with a quick MSU crystal dissolution remains unchecked. Those patients with severe (tophaceous or having affected several joints) long-standing gout appear as reasonable candidates [40]. Additionally, slow crystal dissolution in patients with high crystal loads may reaffirm the belief of gout as an untreatable chronic disease in patients and physicians alike.
Published studies on the effect of urate-lowering therapy on RI progression are still controversial. In one study, renal function in patients with gout improved after appropriate urate-lowering treatment, although patients also discontinued nonsteroidal anti-inflammatory drugs, a potential confounder [41]. Although speculative, urate-lowering treatment may eliminate renal medullary tophi—as it does with tophi elsewhere—and perhaps under these circumstances, rapid crystal dissolution may be advantageous. Finally, an unresolved difficulty is to determine when the MSU crystals have finally been fully dissolved. Crystal disappearance from synovial fluid (SF) under treatment relates to the duration of the clinical disease [7]. An ultrasound can offer useful information about remaining deposits. After the deposits are considered as dissolved, SU should be maintained at a level that new crystals will not form (< 6 mg/dL) [42].
The most commonly used drugs are the xantine-oxidase inhibitors (XOIs), allopurinol and febuxostat, which lower uricaemia by reducing the amount of uric acid formed. The so-called uricosuric drugs (probenecid, benzbromarone, sulfinpyrazone and lesinurad) decrease the tubular reabsorption of uric acid, thereby increasing its renal clearance. In patients in whom XOIs may be insufficient to reach the desired target, their combination with an uricosuric drug is highly effective by reducing formation and easing excretion of SU. Finally, uricase and pegloticase (a pegylated uricase) transform UA to allantoin, sharply reducing uricaemia.
Haemodialysis results in proper clearance of SU and a recent study showed that the mean uricaemia in haemodialysed patients is lower than 5 mg/dL [43], with a mean post-dialysis SU of under 1 mg/dL. Older published literature, though meagre, indicates that the initiation of haemodialysis results in the clearance of tophi [44,45,46]. The diagnosis of hypeuricaemia in patients undergoing dialysis is many times based in pre-dialysis measurements of SU. However, before starting (or continuing) any urate-lowering drug, it appears reasonable to assess also measurements of SU post-dialysis—and perhaps also at 24 h post-dialysis—to have a clearer estimation of the mean uricaemia and the appropriateness of SU-lowering drugs. Attacks of gout following the initiation of haemodialysis probably result from the sharp drop in SU levels—one of the best-established triggers of gout flares—produced by the dialysis. If needed, allopurinol has been shown to effectively reduce uricaemia in patients undergoing haemodialysis [47, 48]. It is generally recommended to reduce the dose in patients undergoing dialysis, with a maximum dose of 100 mg daily. Higher doses have been used with more effective urate lowering, but should be used with caution [49]. Plasma oxipurinol concentrations are reduced by 50% during the dialysis process. Consideration should be given to administering the drug post-dialysis [50].
2.1.1 Allopurinol
Introduced over 40 years ago, allopurinol has been the mainstay drug for the treatment of gout. It is the most widely available drug, and the only urate-lowering therapy available in a number of countries. It is commercialised in 100- and 300-mg grooved tablets. A purine itself, it is degraded by xanthine-oxidase to its active metabolite oxypurinol, competitively inhibiting xanthine-oxidase and thereby reducing the amount of xanthine and hypoxanthine transformed to uric acid. The half-life of allopurinol is around 1–2 h, but the half-life of oxypurinol, which is largely excreted unchanged by the kidneys, is determined by renal function: in patients with normal renal function, the half-life of oxypurinol is less than 30 h, but can extend to 1 week in those with severe RI [51]. When CrCL is less than 10 mL/min, there is virtually no renal clearance of oxypurinol, and it may accumulate. With normal renal function, allopurinol can be used up to 800–900 mg/day (depending on the national legislation). The most common reason for failure to achieve target SU levels is under-dosing. A fixed dose of 300 mg/day is commonly taken as standard [52] and considered by many as appropriate, although it fails to reach the conservative SU target (< 6 mg/dL) in 47–79% of patients [53,54,55].
Most importantly, allopurinol hypersensitivity syndrome (AHS) has been associated with the starting dosage. Initial doses over 400 mg/day are 23 times more likely to produce AHS than starting doses of 100 mg/day. Therefore, all patients should be started with no more than a 100-mg/day dose [8, 9, 55] or, in those with a CrCL < 60 mL/min, a 50-mg daily dose [51]. Such low initial doses and gentle dose titration also help avoid gout attacks triggered by sharp drops of SU levels induced by higher initial doses or increments. The risk of developing AHS is increased in patients with RI and those receiving thiazide diuretics [56].
Maximal dosing of allopurinol in patients with RI remains an open question [53]; specific dosing for the elderly have not been published, but minor degrees of RI are common. With the aim of reducing AHS, it was proposed to adjust the allopurinol dose to the CrCL calculated through the Cockcroft and Gault formula (Table 1) [52]. However, this frequently results in under-treatment. This proposal recommends a maximal dose of 400 mg/day of allopurinol for those with a CrCL of 140 mL/min and 300 mg/day when the CrCL is 100 mL/min, well below the maximal dose of allopurinol 800–900 mg/day included in the package insert. In a study, only 19% of the patients receiving allopurinol at the CrCL-adapted doses achieved SU levels below 6 mg/dL compared with 38% of those receiving higher doses [57, 58]. There is a growing body of evidence that slowly escalating the dose of allopurinol above the CrCL-based dose is a reasonable option. No increase in the incidence of adverse reactions to allopurinol was seen in patients who received higher allopurinol maintenance doses than those recommended according to CrCL [59]. The need for escalating the dose should be considered even in patients with RI [60, 58], especially in countries where no other therapeutic alternatives are available, or where economic limitations are paramount.
2.1.2 Febuxostat
Febuxostat is a non-purine selective XOI commercialised as 40- and 80-mg tablets in USA and as 80- and 120-mg tablets in Europe. The main advantage of febuxostat is its predominant hepatic metabolism, around 70%, that has allowed adequate SU level reductions in patients with gout and RI since its licensing. Among subjects with a baseline serum urate level > 8.0 mg/dL, 36, 52 and 66% of subjects achieved target SU levels (< 6.0 mg/dL) in the last 3-month measurements while receiving febuxostat 80 mg, 120 mg and 240 mg, respectively. Pivotal trials have included patients with mild or moderate RI but have excluded those with a severe disease (CrCL < 30 mL/min) [61]. Recent data coming from a case series [62, 63] and an exploratory trial [64] suggest that safety and effectiveness is maintained in patients with reduced CrCL, as low as 15 mL/min (end-stage disease). Cautious use is recommended in patients at advanced stages of chronic kidney disease. An increased risk of myopathy has been recently reported [65, 66]. Proper dosage of febuxostat in patients with severe RI remains unclear. Starting with a reduced dose (40 or 80 mg/day in Europe) to check tolerance seems appropriate. In the case of insufficient SU reduction, febuxostat dosage may be increased to 80 mg/day (or 120 mg/day in Europe) to achieve further SU reductions and urate crystal dissolution.
Febuxostat is also especially helpful in cases of allopurinol allergy or intolerance [67]. Despite inhibiting the same enzyme, allopurinol and febuxostat are structurally different as is the mechanism of xanthine-oxidase inhibition: allopurinol is a purine analogue that exerts a competitive inhibition, while febuxostat directly blocks the enzyme [68]. However, caution is recommended in those with a prior allergy to allopurinol, as cross-over reactions—including severe reactions—have occasionally been reported [69, 70]. Limited data show that in older patients, even those with comorbidities, febuxostat has been found to be equally safe [71].
2.1.3 Uricosuric Drugs
Uricosuric drugs act by reducing urate reabsorption at the renal tubules, thereby increasing its renal clearance. These drugs increase the renal clearance of uric acid mainly through the inhibition of URAT1, a tubular transporter that ferries uric acid from the tubular lumen towards the proximal tubule cell [72]. Their mechanism of action implies an increase in the urinary level of urate especially at the initiation of treatment and care should be taken in patients with an already increased urate urinary output (urate > 700 mg/24 h of urine) or with pre-existing kidney stones. Benzbromarone and probenecid are still in use but availability is limited in a number of countries; sulfinpyrazone is another alternative in countries where available. Lesinurad has been recently introduced to be used along with a XOI.
2.1.3.1 Benzbromarone
Benzbromarone is a highly potent URAT1 inhibitor with limited availability in a number of countries because of tenuous links with idiosyncratic but severe hepatotoxicity. Dosing ranges from 50 up to 200 mg daily; most of the excretion takes place via faeces; therefore, no dose adjustment is needed in RI. However, its use is not recommended in patients with CrCL under 20 mL/min, given the limited efficacy of all uricosuric drugs in the setting of such limited renal reserve. A single study including 36 patients has explored the use of benzbromarone in patients with RI [73]; 94% of patients with benzbromarone achieved target serum uric acid levels compared with 63% of patients receiving allopurinol. No undue adverse events were observed with benzbromarone. In patients with severe gout and limited treatment options, a uricosuric drug trial is warranted [74].
2.1.3.2 Probenecid
Probenecid is available in USA but its use is limited by its short half-life, which implies the need to administer 2–4 times a day.
2.1.3.3 Sulfinpyrazone
Sulfinpyrazone can be started at a dose of 50 mg twice a day and gradually increased up to 200–400 mg in two divided doses [75].
2.1.3.4 Lesinurad
Lesinurad has recently been approved as combination therapy with a XOI in patients with gout refractory to adequate doses of a prior XOI. It has been approved at a daily maximal dose of 200 mg because of safety concerns regarding higher doses. Phase I studies [76] have shown that RI decreases lesinurad clearance. However, and in keeping with other uricosuric drugs, the serum uric acid-lowering effect of a single dose of lesinurad was reduced in moderate RI, and greatly diminished in severe RI. In the two pivotal CLEAR studies [77, 78], patients with severe RI (CrCl < 30 mL/min) were excluded but around 20% of randomised patients presented with moderate RI. We have currently found no published sub-analysis for this group. Dose adjustment is not required in mild-to-moderate RI. However, lesinurad has been associated with an increase in serum creatinine levels; at a dose of 200 mg daily, 3% of patients (when used in combination with febuxostat) and 2% (when used in combination with allopurinol) have shown an increase over twice its baseline value. These increases have proven largely reversible but are still of uncertain relevance. A recent trial of lesinurad 400 mg/day as monotherapy in patients with a prior XOI intolerance has resulted in a high incidence of serum creatinine elevations and renal-related adverse events, including serious adverse events; lesinurad should not be used as monotherapy [79]. Until more data are available, caution is warranted for the use of lesinurad in patients with RI.
2.1.4 Drug Combinations
Drug combinations can be considered when the desired SU levels are not achieved with XOI monotherapy. Addition of an uricosuric drug further reduces SU levels and often results in appropriate SU levels [54, 80] even in patients with severe gout [40]. Both probenecid and benzbromarone can be used combined with a XOI; the choice is often based on their availability within different countries. A detailed comparison of each drug with the different XOI is not available. The newly marketed lesinurad is to be used only alongside a XOI. In selected patients in whom a rapid debulking of MSU deposits was highly desirable, uricase has been added to the XOI–uricosuric drug combination with success [81]. High immunogenicity associated with uricase and the high rate of adverse infusion reactions and anaphylaxis seen in transient responders are important limitations. Experience with the use of these combinations is limited, though they appear as promising tools to reduce SU to extremely low levels.
2.2 Prophylaxis: Prevention of Attacks
The drugs used in prophylaxis—colchicine, nonsteroidal anti-inflammatory drugs, corticosteroids or anti-IL-1—work by stabilising the subclinical inflammation [4]. Importantly, after SU levels are reduced by treatment, attacks become less frequent [82]; thus, in the long term, adequate reduction of uricaemia is a key part of flare prophylaxis. The larger and sharper the reduction in SU levels at treatment initiation, the higher the chance of an attack and the greater its severity. In the treatment strategies, flare prophylaxis should be given when treatment aimed at crystal dissolution is started (and patients should be warned about the possibility of flares at this time). Prophylaxis in the absence of SU-lowering treatment allows the continuous formation of MSU crystals, without flares warning about their presence: an undesirable situation. Prophylaxis can occasionally be omitted when urate-lowering therapy is started at low doses followed by gradual increases, which usually avoids attacks; however, not all urate-lowering therapies are manufactured in tablet dosages permitting a low-dose start. The duration of prophylaxis remains undefined. Published data support a duration of more than 6 months [83], although a longer duration may be required. If the possibility of severe attacks is foreseen, as in frequently flaring severe gout, or treatment with uricase, the combination of more than one drug (such as colchicine and glucocorticoids) might be necessary for a limited time.
The standard drug for flare prophylaxis is colchicine. It reduces the mild persistent inflammation present during inter-critical periods [4], thus reducing the likelihood of a gout flare. In patients with gout with normal renal function, colchicine is given at 0.5–1.2 mg/day. Recommendations for low-dose colchicine prophylaxis [75] in patients with RI are shown in Table 2. Creatinine clearance should be estimated before starting prophylactic treatment. Normal serum creatinine values in the elderly owing to a decreased muscular mass may result in overestimation of the renal function and if this possibility arises, CrCL must be estimated before starting prophylactic colchicine. Drug–drug interaction studies of concomitant treatment with colchicine and known inhibitors of cytochrome P450 3A4/P-glycoprotein show that the colchicine dose should be reduced if used in combination with calcium channel blockers (verapamil or diltiazem), but not with azithromycin [84]. In sporadic cases, a course of low-dose prednisone of longer duration, 5–10 mg/day, might be needed. This poses no specific problem in patients with RI. However, low-dose prednisone must always be accompanied by an effective SU-lowering treatment to reduce the likelihood of attacks curtailing the time of such prophylaxis. Canakinumab, an anti IL-1 agent, is an effective treatment for gout flares, offering effective prophylaxis for a limited time in difficult-to-treat patients. In a controlled study, a single dose of ≥ 50 mg of canakinumab was more effective at prophylaxis than daily colchicine 0.5 mg [85]. On-demand, subcutaneous canakinumab 150 mg compared favourably to on-demand, intramuscular triamcinolone acetonide 40 mg [86]. Canakinumab can be considered for gout flare prophylaxis in difficult situations where alternatives are considered inappropriate.
2.3 Treatment of Gout Attacks
In most cases, gout inflammation takes the form of an acute monoarthritis, although episodes of oligo or (less frequently) polyarthritis can also occur. If untreated, most gout attacks will subside in about a fortnight. Although many anti-inflammatory mechanisms probably converge, a switch in the secretion of cytokines by the monocytic cells helps prompt the subsidence of the attack [87, 88]. Early treatment of the attacks results in easier resolution. Administration of nonsteroidal anti-inflammatory drugs is a very common treatment of gout flares, but inappropriate for patients with RI and many elderly individuals with comorbidities. In patients with normal renal function, colchicine is a classic treatment for gout attacks. Lower doses (1.2 mg followed 1 h later by 0.6 mg) than those previously used are effective with fewer side effects [89]. However, patients with RI have been excluded from colchicine trials and colchicine is not free of side effects [90]. There are no trials comparing colchicine with other options in patients with RI or the elderly [91]. The use of colchicine for the treatment of gout flares in patients with RI has been discouraged [92].
Oral, parenteral or intra-articular corticosteroids result in an effective subsidence of gout attacks, and are a good and safe alternative for most patients with RI and many elderly individuals. Different dose schedules have been proposed for oral glucocorticoids in gout attacks: 30–35 mg/day of prednisone or equivalent is recommended by the 2016 EULAR guidelines [8], while the American College of Rheumatology 2012 guidelines [10] suggest prednisone or prednisolone at a starting dose of at least 0.5 mg/kg/day for 5–10 days, followed by discontinuation or alternatively 2–5 days at full dose, and tapering for 7–10 days. Severe polyarticular gout flares or flares of long duration may benefit from treatment for a full 15 days. In the authors’ experience, for regular flares, 30 mg of prednisone the first day or two, followed by 1 day of 20 mg and 2–4 days of 10 mg is enough in most cases. Prednisone should also be accompanied by prophylactic colchicine from the first day to avoid rebound attacks after stopping prednisone. In patients with RI, colchicine prophylaxis can be substituted by a small daily dose of prednisone (such as 5 or 7.5 mg) for a few extra days. An alternative is 40–60 mg of triamcinolone acetonide administered by an intramuscular route and followed or not by a short course of prednisone, especially for those not tolerating oral medication [10]. Parenteral adrenocorticotropic hormone is another possibility [93] but its advantages are uncertain. Intra-articular glucocorticoids require a prior exclusion of septic arthritis, but are highly effective and convenient when only a few accessible joints are inflamed. Smaller doses of triamcinolone acetonide tailored to the joint size were found to be effective if the glucocorticoid dose is a concern [94]. Of note, if mepivacaine is added to the intra-articular glucocorticoid, it results in very rapid pain relief that tends to persist even after the effect of the anaesthetic is over [95]. Such rapid pain relief is very appreciated by patients. This can also be performed in elderly patients or in those with RI.
Finally, gout inflammation occurs through the native immune system and IL-1 is the initial trigger. The anti-IL-1 monoclonal antibody canakinumab [96] has been found to be effective in complex or refractory situations. This option appears especially appropriate if attacks occur in patients with RI and unstable diabetes mellitus. Canakinumab has the advantage of being long lasting, avoiding new flares for a prolonged period; however, its cost is an important constraint. Although no trials support the use of anakinra and it has not been approved for gout, its mode of action as an anti-IL-1 drug and a short case series [97,98,99] suggest that it is also useful. Of note, only a few doses of anakinra are usually required, thus the price is most often affordable.
2.4 Education of Patients
Gout is a well-known disease whose pathogenesis is understood and very effective treatments are available. It is difficult to fathom how such a disease can be managed so poorly by many physicians [100]. The majority of patients experiencing gout long term report the experience of poor treatment and poor explanations by physicians and medical personnel. This supports the notion of gout as a chronic and apparently untreatable disease. In this setting, dealing with a physician who has knowledge and understanding of gout, and the time and will to explain, offers a unique opportunity for patients. A recent trial of individualised education to patients by a knowledgeable physician and nurses [36] showed that 90% of them were adherent to urate-lowering treatment at 5 years, with 85% of them taking the medication at least 6 days per week. Education in gout is considered an essential part of treatment both by the American College of Rheumatology [9] and EULAR [8]. Because of the misinformation that surrounds gout, this is likely the key to the success of gout treatment.
3 Conclusion
Gout and RI have a bidirectional link; gout can result in RI, which by producing hyperuricemia facilitates the formation of MSU crystals resulting in gout. The general scheme of treatment in gout associated with renal impairment is similar to that of uncomplicated gout, but most drugs have to be used with modified dosages and some are better avoided. However, emerging evidence also suggests that RI is likely influenced by gout, and the presence of tophi in the renal medulla appears to be a direct link, though so far poorly analyzed. Hence, when gout is diagnosed in patients with RI, it should be treated upon diagnosis, and since achieving lower levels of uricemia results in faster dissolution of MSU crystal deposits, targeting these merits consideration, at least for the more severe cases.
References
Pascual E, Addadi L, Andrés M, et al. Mechanisms of crystal formation in gout: a structural approach. Nat Rev Rheumatol. 2015;11:725–30.
Martinon F, Petrilli V, Mayor A, et al. Gout associated uric acid crystals activate the NALP3 inflammasome. Nature. 2006;440:237–41.
Pascual E. Persistence of monosodium urate crystals and low grade inflammation in the synovial fluid of patients with untreated gout. Arthritis Rheum. 1991;34:141–5.
Pascual E, Castellano JA. Treatment with colchicine decreases the white cell counts in the synovial fluid of asymp-tomatic knees that contain monosodium urate crystals. J Rheuma-tol. 1992;19:600–3.
Dalbeth N, Pool B, Gamble GD, et al. Cellular characterization of the gouty tophus: a quantitative analysis. Arthritis Rheum. 2010;62:1549–56.
Yu TF, Gutmann AB. Mobilization of gouty tophi by protracted use of uricosuric agents. Am J Med. 1951;11:765–9.
Pascual E, Sivera F. The time required for disappearance of urate crystals from synovial fluid after successful hypouricemic treatment relates to the duration of gout. Ann Rheum Dis. 2007;66:1056–8.
Richette P, Doherty M, Pascual E, et al. 2016 updated EULAR evidence-based recommendations for the management of gout. Ann Rheum Dis. 2017;76:29–42.
Khanna D, Fitzgerald JD, Khanna PP, American College of Rheumatology, et al. 2012 American College of Rheumatology guidelines for management of gout. Part 2: therapy and antiinflammatory prophylaxis of acute gouty arthritis. Arthritis Care Res (Hoboken). 2012;64(10):1431–46.
Khanna D, Khanna PP, Fitzgerald JD, American College of Rheumatology, et al. 2012 American College of Rheumatology guidelines for management of gout. Part 2: therapy and antiinflammatory prophylaxis of acute gouty arthritis. Arthritis Care Res (Hoboken). 2012;64:1447–61.
Wallace KL, Riedel AA, Joseph-Ridge N, Wortmann R. Increasing prevalence of gout and hyperuricemia over 10 years among older adults in a managed care population. J Rheumatol. 2004;31:1582–7.
Bieber JD, Terkeltaub RA. Gout: on the brink of novel therapeutic options for an ancient disease. Arthritis Rheum. 2004;50:2400–14.
Lawrence RC, Felson DT, Helmick CG, et al. Estimates of the prevalence of arthritis and other rheumatic conditions in the United States. Arthritis Rheum. 2008;58:26–35.
Terkeltaub RA. Gout: epidemiology, pathology, and pathogenesis. In: Klippel JH, Crofford LJ, Stone JH, et al., editors. Primer on the rheumatic diseases. 12th ed. Atlanta: Arthritis Foundation; 2001. p. 307–12.
Roughley MJ, Belcher J, Mallen CD, et al. Gout and risk of chronic kidney disease and nephrolithiasis: meta-analysis of observational studies. Arthritis Res Ther. 2015;1(17):90.
Jing J, Kielstein JT, Schultheiss UT, et al. Prevalence and correlates of gout in a large cohort of patients with chronic kidney disease: the German Chronic Kidney Disease (GCKD) study. Nephrol Dial Transpl. 2015;30:613–21.
Yu KH, Kuo CF, Luo SF, et al. Risk of end stage renal disease associated with gout: a nationwide population study. Arthritis Res Ther. 2012;14:R83.
Tsai CW, Lin SY, Kuo CC, et al. Serum uric acid and progression of kidney disease: a longitudinal analysis and mini-review. PLoS One. 2017;12:e0170393.
Bose B, Badve SV, Hiremath SS, et al. Effects of uric acid-lowering therapy on renal outcomes: a systematic review and meta-analysis. Nephrol Dial Transpl. 2014;29:406–13.
Sircar D, Chatterjee S, Waikhom R, et al. Efficacy of febuxostat for slowing the GFR decline in patients with CKD and asymptomatic hyperuricemia: a 6-month, double-blind, randomized, placebo-controlled trial. Am J Kidney Dis. 2015;66:945–50.
Singh JA, Cleveland JD. Comparative effectiveness of allopurinol versus febuxostat for preventing incident renal disease in older adults: an analysis of medicare claims data. Ann Rheum Dis. 2017;76(10):1669–78.
Shi Y, Evans JE, Rock KL. Molecular identification of a danger signal that alerts the immune system to dying cells. Nature. 2003;425:516–21.
Ghaemi-Oskouie F, Shi Y. The role of uric acid as an endogenous danger signal in immunity and inflammation. Curr Rheumatol Rep. 2011;13:160–6.
Crişan TO, Cleophas MC, Oosting M, et al. Soluble uric acid primes TLR-induced proinflammatory cytokine production by human primary cells via inhibition of IL-1Ra. Ann Rheum Dis. 2016;75:755–62.
Andrés M, Francés R, Pascual E. Uric acid enhances monosodium urate induced pro-inflammatory response in gouty patients: a basic and translational research study. Ann Rheum Dis. 2015;74(Suppl. 2):777.
De Miguel E, Puig JG, Castillo C, et al. Diagnosis of gout in patients with asymptomatic hyperuricaemia: a pilot ultrasound study. Ann Rheum Dis. 2012;71:157–8.
Pineda C, Amezcua-Guerra LM, Solano C, et al. Joint and tendon subclinical involvement suggestive of gouty arthritis in asymptomatic hyperuricemia: an ultrasound controlled study. Arthritis Res Ther. 2011;17(13):R4.
Dalbeth N, House ME, Aati O, et al. Urate crystal deposition in asymptomatic hyperuricaemia and symptomatic gout: a dual energy CT study. Ann Rheum Dis. 2015;74:908–11.
Talbott JH, Terplan KL. The kidney in gout. Medicine (Baltimore). 1960;39:405–67.
Verger D, Leroux-Robert C, Ganter P, et al. Gouty tophi in the renal medulla in chronic uremia: study of 17 cases discovered from among 62 autopsies. Nephron. 1967;4(6):356–70.
Linnane JW, Burry AF, Emmerson BT. Urate deposits in the renal medulla: prevalence and associations. Nephron. 1981;29:216–22.
Ayoub I, Almaani S, Brodsky S, et al. Revisiting medullary tophi: a link between uric acid and progressive chronic kidney disease? Clin Nephrol. 2016;85:109–13.
Carr A, Doyle AJ, Dalbeth N, et al. Dual-energy CT of urate deposits in costal cartilage and intervertebral disks of patients with tophaceous gout and age-matched controls. Am J Roentgenol. 2016;206:1063–7.
Baer AN, Kurano T, Thakur UJ, et al. Dual-energy computed tomography has limited sensitivity for non-tophaceous gout: a comparison study with tophaceous gout. BMC Musculoskelet Disord. 2016;18(17):91.
Simkin PA, Bassett JE, Lee QP. Not water, but formalin, dissolves urate crystals in tophaceous tissue samples. J Rheumatol. 1994;21:2320–1.
Rees F, Jenkins W, Doherty M. Patients with gout adhere to curative treatment if informed appropriately: proof-of-concept observational study. Ann Rheum Dis. 2013;72:826–30.
Abhishek A, Jenkins W, La-Crette J, et al. Long-term persistence and adherence on urate-lowering treatment can be maintained in primary care-5-year follow-up of a proof-of-concept study. Rheumatology (Oxford). 2017;56:529–33.
Araujo EG, Bayat S, Petsch C, et al. Tophus resolution with pegloticase: a prospective dual-energy CT study. RMD Open. 2015;1:1–e000075.
Perez-Ruiz F, Calabozo M, Pijoan JI, et al. Effect of urate-lowering therapy on the velocity of size reduction of tophi in chronic gout. Arthritis Rheum. 2002;47:356–60.
Pascual E, Andrés M, Vázquez-Mellado J, et al. Severe gout: strategies and innovations for effective management. Jt Bone Spine. 2017;84:541–6.
Perez-Ruiz F, Calabozo M, Herrero-Beites AM, et al. Improvement of renal function in patients with chronic gout after proper control of hyperuricemia and gouty bouts. Nephron. 2000;86:287–91.
Perez-Ruiz F, Herrero-Beites AM, Carmona L. A two-stage approach to the treatment of hyperuricemia in gout: the “dirty dish” hypothesis. Arthritis Rheum. 2011;63:4002–6.
Andrés M, Soriano R, Oliveira E, et al. Serum uric acid lowering treatment appears unnecessary during hemodialysis [abstract]. Ann Rheum Dis. 2017;76(Suppl. 2):361.
Duncan H, Elliott W, Horn DB, et al. Haemodialysis in the treatment of gout. Lancet. 1962;1(7241):1209–11.
Johnson WJ, O’Duffy JD. Chronic gouty nephropathy treated by long-term hemodialysis and allopurinol. Mayo Clin Proc. 1979;54:618–20.
Ifudu O, Tan CC, Dulin AL, et al. Gouty arthritis in end-stage renal disease: clinical course and rarity of new cases. Am J Kidney Dis. 1994;23(3):347–51.
Rundles R. Allopurinol in gouty nephropathy and renal dialysis. Ann Rheum Dis. 1966;25:694–6.
Hayes C, Metz E, Robinson R, et al. The use of allopurinol to control hyperuricaemia in patients on chronic intermittent hemodialysis. Trans Am Soc Artif Intern Organs. 1965;11:247–54.
Day R, Kannangara D, Hayes J, et al. Successful use of allopurinol in a patient on dialysis. BMJ Case Rep. 2012. https://doi.org/10.1136/bcr.02.2012.5814.
Stamp LK, Chapman PT, Palmer SC. Allopurinol and kidney function: an update. Jt Bone Spine. 2016;83:19–24.
Murrell G, Rapeport W. Clinical pharmacokinetics of allopurinol. Clin Pharmacokinet. 1986;11:343–53.
Hande KR. NooneRM, Stone WJ. Severe allopurinol toxicity: description and guidelines for prevention in patients with renal insufficiency. Am J Med. 1984;76:47–56.
Becker MA, Schumacher HR Jr, Wortmann RL, et al. Febuxostat compared with allopurinol in patients with hyperuricemia and gout. N Engl J Med. 2005;353:2450–61.
Perez-Ruiz F, Alonso-Ruiz A, Calabozo M, et al. Efficacy of allopurinol and benzbromarone for the control of hyperuricaemia: a pathogenic approach to the treatment of primary chronic gout. Ann Rheum Dis. 1998;57:545–9.
Reinders MK, Haagsma C, Jansen TL, et al. A randomised controlled trial on the efficacy and tolerability with dose escalation of allopurinol 300–600 mg/day versus benzbromarone 100–200 mg/day in patients with gout. Ann Rheum Dis. 2009;68:892–7.
Chao J, Terkeltaub RA. Critical reappraisal of allopurinol dosing, safety, and efficacy for hyperuricemia in gout. Curr Rheumatol Rep. 2009;11:135–40.
Dalbeth N, Kumar S, Stamp L, et al. Dose adjustment of allopurinol according to creatinine clearance does not provide adequate control of hyperuricemia in patients with gout. J Rheumatol. 2006;33:1646–50.
Stamp LK, Chapman PT, Barclay M, et al. Allopurinol dose escalation to achieve serum urate below 6 mg/dL: an open-label extension study. Ann Rheum Dis. 2017;76(12):2065–70.
Vázquez-Mellado J, Morales EM, Pacheco-Tena C, et al. Relation between adverse events associated with allopurinol and renal function in patients with gout. Ann Rheum Dis. 2001;60:981–3.
Dalbeth N, Stamp L. Allopurinol dosing in renal impairment: walking the tightrope between adequate urate lowering and adverse events. Semin Dial. 2007;20:391–5.
Schumacher HR Jr, Becker MA, Wortmann RL, et al. Effects of febuxostat versus allopurinol and placebo in reducing serum urate in subjects with hyperuricemia and gout: a 28-week, phase III, randomized, double-blind, parallel-group trial. Arthritis Rheum. 2008;59:1540–8.
Quilis N, Andrés M, Gil S, Ranieri L, Vela P, Pascual E. E, et al. Febuxostat for patients with gout and severe chronic kidney disease: which is the appropriate dosage? Comment on the Article by Saag et al. Arthritis Rheumatol. 2016;68:2563–4.
Juge PA, Truchetet ME, Pillebout E, et al. Efficacy and safety of febuxostat in 73 gouty patients with stage 4/5 chronic kidney disease: a retrospective study of 10 centers. Jt Bone Spine. 2017;84:595–8.
Saag KG, Whelton A, Becker MA, et al. Impact of febuxostat on renal function in gout patients with moderate-to-severe renal impairment. Arthritis Rheumatol. 2016;68:2035–43.
Ghosh D, McGann PM, Furlong TJ, et al. Febuxostat-associated rhabdomyolysis in chronic renal failure. Med J Aust. 2015;203:107–8.
Liu CT, Chen CY, Hsu CY, et al. Risk of febuxostat-associated myopathy in patients with CKD. Clin J Am Soc Nephrol. 2017;12:744–50.
Chohan S. Safety and efficacy of febuxostat treatment in subjects with gout and severe allopurinol adverse reactions. J Rheumatol. 2011;38:1957–9.
Chinchilla SP, Urionaguena I, Perez-Ruiz F. Febuxostat for the chronic management of hyperuricemia in patients with gout. Expert Rev Clin Pharmacol. 2016;9:665–73.
Bardin T, Chalès G, Pascart T, et al. Risk of cutaneous adverse events with febuxostat treatment in patients with skin reaction to allopurinol: a retrospective, hospital-based study of 101 patients with consecutive allopurinol and febuxostat treatment. Jt Bone Spine. 2016;83:314–7.
Quilis N, Andrés M, Muñoz C, et al. Skin events with febuxostat in gout patients and previous skin reactions to allopurinol: a retrospective review [abstract]. Arthritis Rheumatol. 2016;68(Suppl. 10):273–4.
Becker MA, MacDonald PA, Hunt B, et al. Treating hyperuricemia of gout: safety and efficacy of febuxostat and allopurinol in older versus younger subjects. Nucleosides Nucleotides Nucleic Acids. 2011;30:1011–7.
Miner JN, Tan PK, Hyndman D, et al. Lesinurad, a novel, oral compound for gout, acts to decrease serum uric acid through inhibition of urate transporters in the kidney. Arthritis Res Ther. 2016;18:214.
Perez-Ruiz F, Calabozo M, Fernandez-Lopez JM, et al. Treatment of chronic gout in patients with renal function impairment: an open, randomized, actively controlled study. J Clin Rheumatol. 1999;5:49–55.
Reinders MK, van Roon EN, Jansen TL, et al. Efficacy and tolerability of urate-lowering drugs in gout: a randomized controlled trial of benzbromarone versus probenecid after failure of allopurinol. Ann Rheum Dis. 2009;68:51–6.
Terkeltaub RA. Clinical practice. Gout N Engl J Med. 2003;349:1647–55.
Gillen M, Valdez S, Zhou D, et al. Effects of renal function on pharmacokinetics and pharmacodynamics of lesinurad in adult volunteers. Drug Des Dev Ther. 2016;10:3555–62.
Dalbeth N, Jones G, Terkeltaub R, et al. Lesinurad, a selective uric acid reabsorption inhibitor, in combination with febuxostat in patients with tophaceous gout: findings of a phase III clinical trial. Arthritis Rheumatol. 2017;69:1903–13.
Bardin T, Keenan RT, Khanna PP, et al. Lesinurad in combination with allopurinol: a randomised, double-blind, placebo-controlled study in patients with gout with inadequate response to standard of care (the multinational CLEAR 2 study). Ann Rheum Dis. 2017;76:811–20.
Tausche AK, Alten R, Dalbeth N, et al. Lesinurad monotherapy in gout patients intolerant to a xanthine oxidase inhibitor: a 6 month phase 3 clinical trial and extension study. Rheumatology (Oxford). 2017;56(12):2170–8.
Reinders M, van Roon E, Houtman P, et al. Biochemical effectiveness of allopurinol and allopurinol-probenecid in previously benzbromarone-treated gout patients. Clin Rheumatol. 2007;26:1459–65.
Mejía-Chew C, Torres RJ, de Miguel E, et al. Resolution of massive tophaceous gout with three urate-lowering drugs. Am J Med. 2013;126(11):e9–10.
Shoji A, Yamanaka H, Kamatani N. A retrospective study of the relationship between serum urate level and recurrent attacks of gouty arthritis: evidence for reduction of recurrent gouty arthritis with antihyperuricemic therapy. Arthritis Rheum. 2004;51:321–5.
Borstad GC, Bryant LR, Abel MP, et al. Colchicine for prophylaxis of acute flares when initiating allopurinol for chronic gouty arthritis. J Rheumatol. 2004;31:2429–32.
Terkeltaub RA, Furst DE, Digiacinto JL, et al. Novel evidence-based colchicine dose-reduction algorithm to predict and prevent colchicine toxicity in the presence of cytochrome P450 3A4/P-glycoprotein inhibitors. Arthritis Rheum. 2011;63:2226–37.
Schlesinger N, Mysler E, Lin HY, et al. Canakinumab reduces the risk of acute gouty arthritis flares during initiation of allopurinol treatment: results of a double-blind, randomised study. Ann Rheum Dis. 2011;70:1264–71.
Lyseng-Williamson KA. Canakinumab: a guide to its use in acute gouty arthritis flares. BioDrugs. 2013;27:401–6.
Yagnik DR, Hillyer P, Marshall D, et al. Non inflammatory phagocytosis of monosodium urate monohydrate crystals by mouse macrophages: implications for the control of joint inflammation in gout. Arthritis Rheum. 2000;43:1779–89.
Scanu A, Oliviero F, Ramonda R, et al. Cytokine levels in human synovial fluid during the different stages of acute gout: role of transforming growth factor β1 in the resolution phase. Ann Rheum Dis. 2012;71:621–4.
Terkeltaub RA, Furst DE, Bennett K, et al. High versus low dosing of oral colchicine for early acute gout flare: twenty-four-hour outcome of the first multicenter, randomized, double-blind, placebo-controlled, parallel-group, dose-comparison colchicine study. Arthritis Rheum. 2010;62:1060–8.
Medani S, Wall C. Colchicine toxicity in renal patients: are we paying attention? Clin Nephrol. 2016;86:100–5.
Van Echteld I, Wechalekar MD, Schlesinger N, et al. Colchicine for acute gout. Cochrane Database Syst Rev. 2014; (8):CD006190. https://doi.org/10.1002/14651858.cd006190.
Bardin T, Richette P. Impact of comorbidities on gout and hyperuricaemia: an update on prevalence and treatment options. BMC Med. 2017;15:123.
Janssens HJ, Lucassen PL, Van de Laar FA, Janssen M, Van de Lisdonk EH. Systemic corticosteroids for acute gout. Cochrane Database Syst Rev. 2008;16(2):CD005521
Fernández C, Noguera R, González JA, et al. Treatment of acute gouty attacks with a small dose of intra-articular triamcinolone acetonide. J Rheumatol. 1999;26:2285–6.
Andrés M, Begazo A, Sivera F. Intraarticular triamcinolone plus mepivacaine provides a rapid and sustained relief for acute gouty arthritis. Ann Rheum Dis. 2016;75(Suppl. 2):1182.
Bardin T. Canakinumab for the patient with difficult-to-treat gouty arthritis: review of the clinical evidence. Jt Bone Spine. 2015;82(Suppl. 1):eS9–16.
Thueringer JT, Doll NK, Gertner E. Anakinra for the treatment of acute severe gout in critically ill patients. Semin Arthritis Rheum. 2015;45(1):81–5.
Aouba A, Deshayes S, Frenzel L, et al. Efficacy of anakinra for various types of crystal-induced arthritis in complex hospitalized patients: a case series and review of the literature. Mediators Inflamm. 2015;2015:792173.
Petite SE. Effectiveness of anakinra in acute gout: a retrospective review of initial and refractory therapy. Am J Ther. 2017;24:e633–4.
Pascual E, Sivera F. Why should be gout so poorly treated? Ann Rheum Dis. 2007;66:1269–70.
Funding
No funding was received for the preparation of this article.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
Eliseo Pascual has participated in the boards of Astra Zeneca, Menarini, Grunenthal and Pegloticase (unrelated to this article), and has received speaking fees from Astra Zeneca and Grunenthal (unrelated to this article). Mariano Andrés has received fees for participation in review activities such as data monitoring boards from Grunenthal and Astra-Zeneca, and has received support for travel from Menarini, Grunenthal and Astra-Zeneca. Francisca Sivera has received speaking fees from Menarini.
Rights and permissions
About this article

Cite this article
Pascual, E., Sivera, F. & Andrés, M. Managing Gout in the Patient with Renal Impairment. Drugs Aging 35, 263–273 (2018). https://doi.org/10.1007/s40266-018-0517-7
Published:
Issue Date:
DOI: https://doi.org/10.1007/s40266-018-0517-7