
Abstract
Delandistrogene moxeparvovec (delandistrogene moxeparvovec-rokl; ELEVIDYS®) is an adeno-associated virus (AAV) vector-based gene therapy designed to deliver a gene encoding a micro-dystrophin protein [i.e. a shortened (138 kDa) version of the dystrophin protein expressed in normal muscle cells (427 kDa)] to all muscles involved in the pathology of Duchenne muscular dystrophy (DMD). Developed by Sarepta Therapeutics, it is the first gene therapy to be approved (in June 2023 under the Accelerated Approval pathway) for the treatment of DMD in the USA, where it is indicated for ambulatory paediatric patients aged 4 through 5 years with DMD and a confirmed mutation in the dystrophin (DMD) gene. The recommended dose of delandistrogene moxeparvovec is 1.33 × 1014 vector genomes per kg of body weight or 10 mL/kg body weight, administered as a single intravenous infusion. Delandistrogene moxeparvovec is undergoing clinical development in several countries/regions, including the EU and Japan. This article summarizes the milestones in the development of delandistrogene moxeparvovec leading to this first approval in the USA for the treatment of ambulatory paediatric patients aged 4 through 5 years with DMD and a confirmed mutation in the DMD gene.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Digital Features for this AdisInsight Report can be found at https://doi.org/10.6084/m9.figshare.23740191. |
AAV vector-based gene therapy developed by Sarepta Therapeutics for the treatment of DMD |
Received its first approval on 22 June 2023 in the USA under the Accelerated Approval pathway |
Approved for use in ambulatory paediatric patients aged 4 through 5 years with DMD and a confirmed mutation in the DMD gene |
1 Introduction
The X-linked recessive disorders Becker muscular dystrophy (BMD) and Duchenne muscular dystrophy (DMD) result from mutations in the dystrophin (DMD) gene (which spans > 2000 kb) [1,2,3]. DMD encodes dystrophin, a protein critical for maintaining muscle fibre integrity and protecting skeletal and cardiac muscle cells from mechanical stress during contraction [3, 4]. BMD is milder and has a later onset and slower progression than DMD, reflecting the differing effects of DMD mutations on the reading frame [1, 5]. In BMD, the reading frame is maintained despite the mutations, thereby permitting the production of partially functional dystrophin [1, 5]. In DMD, frameshift or nonsense mutations result in non-functional dystrophin; muscle cells lacking dystrophin are susceptible to damage from mechanical stress, engendering a progressive loss of muscle tissue and function [1, 2, 5]. While the rate of functional decline in patients with DMD is generally not linear, a recent natural history study determined that North Star Ambulatory Assessment (NSAA; which assesses motor function) total scores peak at ≈ 6.3 years of age and then decline [5, 6].
Among the emerging approaches to DMD management are genetic and molecular therapies focused on restoring the production or compensating for the dysfunction of dystrophin [1, 3]. Given DMD is caused by mutations in a single gene (DMD), it is well suited to gene transfer therapy [4]. Such treatment aims to ameliorate disease progression by providing a gene encoding a functional version of dystrophin to all muscles involved in DMD pathology [4]. Adeno-associated viruses (AAV) can efficiently transduce skeletal and heart muscle but have a limited carrying capacity (≈ 4.7 kb versus the ≈ 11.4 kb cDNA of dystrophin) [1]. As naturally occurring shortened dystrophin (as seen in patients with BMD) can be partially functional, shortened versions of the dystrophin gene (encoding only the most crucial domains) have been constructed [1, 4]. The transgenes of these micro-dystrophin constructs fit in AAV vectors [1, 4]. The potential for outcomes to decline over time is, however, of concern in gene therapy [7].
Delandistrogene moxeparvovec (delandistrogene moxeparvovec-rokl; ELEVIDYS®) is an AAV-based gene therapy designed to deliver a gene encoding a micro-dystrophin protein [i.e. a shortened (138 kDa) version of the dystrophin protein expressed in normal muscle cells (427 kDa)] [8, 9]. Developed by Sarepta Therapeutics, it is the first gene therapy to be approved (in June 2023 under the Accelerated Approval pathway) for the treatment of DMD in the USA, where it is indicated for ambulatory paediatric patients aged 4 through 5 years with DMD and a confirmed mutation in DMD [8,9,10]. This US accelerated approval was based on the presence of the micro-dystrophin protein expressed by delandistrogene moxeparvovec in the skeletal muscle of treated patients [8]. Continued approval may be contingent upon verification and description of clinical benefit in a confirmatory trial(s) [8].
The recommended dose of delandistrogene moxeparvovec is 1.33 × 1014 vector genomes per kg of body weight (vg/kg) or 10 mL/kg body weight, administered as a single intravenous infusion [8]. The US prescribing information carries warnings and precautions regarding acute serious liver injury, immune-mediated myositis, myocarditis and pre-existing immunity against AAV serotype rh74 (AAVrh74; which may impede transgene transduction at desired therapeutic levels). Delandistrogene moxeparvovec is contraindicated in patients with any deletion in exon 8 and/or exon 9 in DMD (due to the increased risk of a severe immune-mediated myositis reaction) and its use is not recommended in patients with an anti-AAVrh74 total binding antibody titre of ≥ 1:400. Systemic corticosteroid treatment is recommended for patients before and after the administration of delandistrogene moxeparvovec (to reduce the risk of an immune response); the regimen should be modified in patients with liver function abnormalities following infusion [8]. Delandistrogene moxeparvovec is undergoing clinical development in several countries/regions, including the EU and Japan.
1.1 Company Agreements
In December 2019, Sarepta Therapeutics and Roche entered into a licensing agreement under which Roche obtained the exclusive right to launch and commercialize delandistrogene moxeparvovec outside the USA [11]. Under the terms of the agreement, Sarepta will continue to be responsible for the clinical development and manufacturing of delandistrogene moxeparvovec; global clinical development costs will be shared equally between Sarepta and Roche [11]. In June 2020, Sarepta entered into a research license and option agreement with Selecta Biosciences [12]. Under the terms of this agreement, Sarepta was granted an option to license the rights to develop and commercialize Selecta’s immune tolerance platform for use in DMD and certain limb-girdle muscular dystrophies [12]. In December 2021, Chugai Pharmaceutical and Roche entered into a licensing agreement under which the former obtained the exclusive marketing rights for delandistrogene moxeparvovec in Japan [13]. In June 2022, Sarepta extended the options under its research license and option agreement with Selecta [14].

Key milestones in the development of delandistrogene moxeparvovec for the treatment of Duchenne muscular dystrophy. BLA Biologic License Application, IND Investigational New Drug
2 Scientific Summary
2.1 Pharmacodynamics
Delandistrogene moxeparvovec consists of a non-replicating, recombinant AAVrh74 capsid containing a transgene encoding an engineered micro-dystrophin protein under the control of a muscle-specific promoter/enhancer (MHCK7) [4, 8]. Clinical and/or nonclinical studies have demonstrated AAVrh74 transduction in skeletal and cardiac muscle cells (including those of the diaphragm) [8].
A single intravenous infusion of delandistrogene moxeparvovec (equivalent to 1.33 × 1014 vg/kg) was associated with robust micro-dystrophin protein expression (as measured by Western blot analysis) and correct localization of the protein to the sarcolemma (as assessed by immunofluorescence) in ambulatory males aged ≥ 4 to < 8 years participating in three studies [a two-part (48 weeks per part) phase II crossover study (NCT03769116) [15], a phase I study (NCT04626674; ENDEAVOR) [16] and a phase I/IIa pilot study (NCT03375164) [7]]. In NCT03769116, patients who received delandistrogene moxeparvovec in either part 1 or 2 of the double-blind period demonstrated a significant (p < 0.01) % normal mean increase from baseline at 12 weeks’ post-treatment in micro-dystrophin protein expression (co-primary endpoint) relative to those who received placebo [15]. Patients treated in part 1 continued to show a significant (p < 0.01) mean increase from baseline in this endpoint at 60 weeks’ post-treatment [15]. In patients from cohort 1 of ENDEAVOR, the mean change from baseline in micro-dystrophin protein expression (primary endpoint) at 12 weeks’ post-treatment was 54.2% of the normal dystrophin expression level [16]. In NCT03375164, the mean micro-dystrophin expression at 12 weeks’ post-treatment was 95.8% and 74.3% of the normal dystrophin expression level with or without adjusting for fat or fibrosis [7].
Creatine kinase levels are usually high in patients with DMD [7]. Following the administration of delandistrogene moxeparvovec in NCT03375164, creatine kinase levels reduced by 46.5–85.8% from a mean baseline value of 27,064 U/L [7].
2.2 Pharmacokinetics
Following a single intravenous infusion of delandistrogene moxeparvovec, the vector DNA is distributed systemically into target muscle tissues [8]. In the serum, salvia, urine and faeces, respectively, of patients in cohort 1 of ENDEAVOR, mean maximum concentration (Cmax) values of the vector DNA were 0.0049 × 1013 copies/mL, 4.72 × 107 copies/mL, 4.11 × 105 copies/mL and 2.32 × 107 copies/µg and median time to Cmax values were 5.3 h, 6.7 h, 6.4 h and 13.5 days post-dose. The estimated elimination half-life of vector DNA in the serum, saliva, urine and faeces, respectively, was ≈ 12 h, 60 h, 40 h and 55 h; most vector DNA is expected to be cleared from the serum by 1-week post-dose. The median time to achieve complete elimination in saliva, urine and faeces, respectively, was 49.8 days, 123 days and 162 days post-treatment; elimination was confirmed by two consecutive measurements below the limit of detection [8]. Peak vector shedding typically occurs in the first few days post-administration (day 1 in saliva and urine and week 2 in faeces) and exponentially declines to insignificant levels by week 4, according to an analysis of data from 20 ambulatory males aged ≥ 4 to < 8 years from cohort 1 of ENDEAVOR [17].
Delandistrogene moxeparvovec is unlikely to exhibit drug–drug interaction potential as the capsid proteins are broken down via proteasomal degradation following entry of the AAV into target cells [8].
2.3 Therapeutic Trials
2.3.1 Phase II Study
A single intravenous infusion of delandistrogene moxeparvovec (equivalent to 1.33 × 1014 vg/kg) was associated with promising functional outcomes in ambulatory males aged ≥ 4 to < 8 years (over half were aged ≥ 6 years at baseline) with DMD participating in a randomized, double-blind, placebo-controlled, two-part (48 weeks per part) phase II crossover study with an ongoing open-label extension (NCT03769116) [15]. In part 1 of the double-blind period, the least-squares mean (LSM) change from baseline to week 48 in the NSAA total score (co-primary endpoint) did not significantly differ between the delandistrogene moxeparvovec group (n = 20) and the placebo group (n = 21) [+ 1.7 vs + 0.9 points; p = 0.37]. The lack of statistical significance for this endpoint may be reflective of the disparity in baseline motor function between the respective groups (i.e. mean NSAA total score of 19.8 and 22.6 points and mean time to rise of 5.1 and 3.6 s), suggesting that the delandistrogene moxeparvovec group in part 1 had more advanced DMD relative to the placebo group. In subgroup analyses, the between-group difference in the LSM change from baseline to week 48 in the NSAA total score significantly favoured delandistrogene moxeparvovec over placebo in patients aged 4 to 5 years with matched baseline motor function (+ 2.5 points; p = 0.0172) but not in patients aged 6 to 7 years with imbalanced baseline motor function (− 0.7 points). In part 2 of the double-blind period, patients treated with delandistrogene moxeparvovec (n = 20) achieved a mean change from baseline (of part 2) to week 48 in the NSAA total score of + 1.3 points. When compared with data from a prespecified, propensity-score-weighted external control cohort (n = 103), the LSM between-group difference in the NSAA total score (+ 2.0 points) was statistically significant (p = 0.0009). The motor function of patients who received placebo (n = 19) in part 2 (having received delandistrogene moxeparvovec in part 1) appeared stable, with a LSM change from baseline of part 1 to the end of part 2 (total duration 96 weeks) in the NSAA total score of 0.1 points) [15].
Features and properties of delandistrogene moxeparvovec
Alternative names | Delandistrogene moxeparvovec-rokl; Delandistrogenum moxeparvovecum - Roche/Sarepta Therapeutics; ELEVIDYS; rAAVrh74.MHCK7.micro-dystrophin; RG 6356; RO-7494222; SRP-9001 |
Class | Gene therapies |
Mechanism of action | Dystrophin replacements; Gene transference |
Route of administration | Intravenous infusion |
Pharmacodynamics | Transduces skeletal and cardiac muscle cells, resulting in robust and sustained micro-dystrophin protein expression |
Pharmacokinetics | Vector DNA identified in serum, salvia, urine and faeces after infusion |
Most frequent treatment-emergent adverse events | Vomiting, decreased appetite, nausea, upper respiratory tract infection, pain in extremity, upper abdominal pain, irritability and procedural pain |
ATC codes | |
WHO ATC code | M09A-X15 (Delandistrogene moxeparvovec) |
EphMRA ATC code | M5 (Other Drugs for Disorders of the Musculo-Skeletal System) |
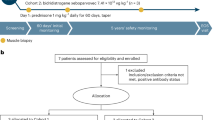
In NCT03769116 [15], eligible patients were ambulatory males aged ≥ 4 to < 8 years with confirmed DMD mutations (including frameshift or premature stop codon mutations between exons 18 and 58), a creatine kinase level of > 1000 U/L and a below average 100 m walk/run test who had received a stable dose of corticosteroids for ≥ 12 weeks before study entry. Patients were excluded if they had an rAAVrh74 binding antibody titre > 1:400 and randomization was stratified by age at baseline (4 to 5 years vs 6 to 7 years). In part 1 of the double-blind period of the study, patients received a single intravenous infusion of delandistrogene moxeparvovec or placebo; upon completion of part 1, participants were crossed over to the corresponding treatment group for part 2. Following completion of part 2 of the double-blind period, patients entered the open-label extension period; the final study visit will be at week 212 for patients who received delandistrogene moxeparvovec in part 1 and week 260 for those who received it in part 2. At baseline in the delandistrogene moxeparvovec and placebo groups, respectively, in part 1 of the double-blind period, patients had a mean age of 6.3 and 6.2 years [15]. NSAA is a 17-item measure of ambulatory function; scores range from 0 to 34, with the highest score indicating perfect [7]. The variability in the dose of delandistrogene moxeparvovec administered in part 1 of the study was determined via a retrospective analysis: of the 20 patients, 40% received 1.33 × 1014 vg/kg, 30% received 8.94 × 1013 vg/kg and 30% received 6.29 × 1013 vg/kg [15]. All 21 patients in part 2 received the 1.33 × 1014 vg/kg dose [15].
In addition to this study, a cost-effectiveness model conducted from a US healthcare system perspective using a simulated cohort of 4-year-old males with DMD suggests that delandistrogene moxeparvovec plus standard of care (SOC) has the potential to be cost-effective up to US$5.1 million compared with SOC alone at a willingness-to-pay threshold of US$500,000/equal value of life-years gained. SOC consisted of corticosteroid and physical/occupational therapy, multidisciplinary assessments and gastrointestinal/respiratory/cardiac management) [18].
2.3.2 Phase I/IIa Study
Promising functional outcomes with delandistrogene moxeparvovec (equivalent to 1.33 × 1014 vg/kg, infused over 1.25–1.5 h into a peripheral limb vein) were displayed in four ambulatory males aged 4–7 years with DMD participating in a nonrandomized, open-label, phase I/IIa pilot study (NCT03375164) [7, 19, 20]. At 52 weeks’ post treatment, the mean change from baseline in muscle function [as assessed by the NSAA total score; exploratory outcome] was + 5.5 points (range 2–8 points); an ≈ 2-point improvement in the NSAA total score over a 48-week period is considered to be clinically relevant. A clinically relevant improvement from baseline in this outcome was seen as early as day 90. Other functional outcomes, including time to rise, four-stair climb, 100 m walk/run test and handheld dynamometry (for knee and elbow extensors and flexors) showed varying magnitudes of improvement. Such variability can be associated with multiple factors, including age and disease severity [7]. The benefits of delandistrogene moxeparvovec on functional outcomes appear to be sustained over the longer term [20]. At 2 [19] and 4 [20] years’ post treatment, the mean change from baseline in muscle function (as assessed by the NSAA total score) was + 7.0 and + 7.0 points. Moreover, at 4 years’ post treatment, the mean improvement from baseline in the mean time to rise, the mean four-stair climb, the mean 100 m walk/run test, and the mean 10 m walk/run test was 0.1 s, 1.1 s, 7.0 s and 0.3 s, respectively [20].
Eligible patients in NCT03375164 were ambulatory males aged 4–7 years with confirmed DMD mutations (including frameshift or premature stop codon mutations between exons 18 and 58), a creatine kinase level of > 1000 U/L and a below average 100 m walk/run test who had received a stable dose of corticosteroids for ≥ 12 weeks before study entry [7]. Patients were excluded if rAAVrh74 binding antibody was detected at a > 1:400 dilution. Prior to and following delandistrogene moxeparvovec administration, patients received prednisolone therapy. At enrolment, patients had a mean creatine kinase level of 27,064.3 U/L and a mean NSAA total score of 20.5 points [7]. At baseline, mean time to rise, mean four-stair climb, mean 100 m walk/run and mean 10 m walk/run values were 3.7 s, 3.5 s, 56.4 s and 4.9 s, respectively [20].
2.3.3 Phase Ib Study
Encouraging clinical activity [improvements from baseline to week 52 post-treatment in the NSAA total score, time to rise, 10 m walk/run time, four-stair climb and 100 m walk/run time (exploratory endpoints)] with delandistrogene moxeparvovec was displayed in 20 ambulatory males aged ≥ 4 to < 8 years with DMD participating in an ongoing, open-label phase Ib study (NCT04626674; ENDEAVOR) [16]. When compared with data from a propensity-score-weighted external control cohort (n = 91), the between-group difference in the LSM change from baseline to week 52 post-treatment in the NSAA total score (+ 3.2 points) was statistically significant (p = 0.0001) [16].
ENDEAVOR is a two-part study (part 1: 12 weeks; part 2: up to 5 years) with five patient cohorts: ambulatory males aged ≥ 4 to < 8 years (cohort 1); ambulatory males aged ≥ 8 to < 18 years (cohort 2); non-ambulatory males (cohort 3); ambulatory males aged ≥ 3 to < 4 years (cohort 4); ambulatory males aged ≥ 4 to < 9 years (cohort 5a); and non-ambulatory males (cohort 5b) [16]. Genetic mutation criteria varied by cohort. Patients in the study received a single intravenous infusion of delandistrogene moxeparvovec (equivalent to 1.33 × 1014 vg/kg) [16].
Key clinical trials of delandistrogene moxeparvovec
Drug | Indication | Phase | Status | Location | Identifier | Sponsor (Collaborator) |
|---|---|---|---|---|---|---|
Delandistrogene moxeparvovec | Duchenne muscular dystrophy | III | Active, not recruiting | Multinational | NCT05096221 (EMBARK) | Sarepta Therapeutics (Hoffmann-La Roche) |
Delandistrogene moxeparvovec | Duchenne muscular dystrophy | III | Not yet recruiting | USA | NCT05881408 (ENVISION) | Sarepta Therapeutics (Hoffmann-La Roche) |
Delandistrogene moxeparvovec | Duchenne muscular dystrophy | II | Active, not recruiting | USA | NCT03769116 | Sarepta Therapeutics |
Delandistrogene moxeparvovec | Duchenne muscular dystrophy | II | Ongoing | Multinational | EudraCT 2022-000691-19 (ENVOL) | Hoffmann-La Roche |
Delandistrogene moxeparvovec | Duchenne muscular dystrophy | I/IIa | Completed | USA | NCT03375164 | Sarepta Therapeutics |
Delandistrogene moxeparvovec | Duchenne muscular dystrophy | I | Enrolling by invitation | USA | NCT04626674 (ENDEAVOR) | Sarepta Therapeutics (Hoffmann-La Roche) |
2.4 Adverse Events
Delandistrogene moxeparvovec, administered as a single intravenous infusion, demonstrated a consistent and manageable safety profile in ambulatory and non-ambulatory males of different ages with DMD [7, 15, 16, 21, 22].
In an integrated analysis [22] (n = 85; 73 patients received the recommended dose of delandistrogene moxeparvovec [8]) of data from three studies (NCT03769116 [15], NCT03375164 [7] and multiple cohorts of ENDEAVOR [21]; data cut-off date 1 April 2022, 17 October 2022 and 19 September 2022, respectively), almost all (96.5%) patients experienced a treatment-emergent adverse event (TEAE). The most frequently reported (incidence of > 25%) TEAEs were vomiting (61.2% of patients), decreased appetite (47.1%), upper respiratory tract infection (42.4%), nausea (40.0%), pain in extremity (32.9%), upper abdominal pain (27.1%), procedural pain (27.1%) and irritability (25.9%) [22]. Most adverse events (AEs) were mild to moderate in severity and were generally observed within the first 2 weeks (nausea, vomiting, thrombocytopenia, pyrexia) or first 2 months (immune-mediated myositis, elevated liver function tests) of treatment [8, 22]. Vomiting, however, may occur as early as on the day of the infusion [8]. Treatment-related AEs (TRAEs) and serious AEs were reported in 85.9% and 12.9% of patients, with 8.2% experiencing treatment-related serious AEs [22]. Most TRAEs occurred within 90 days of treatment and resolved. The treatment-related serious AEs were vomiting, increased transaminase levels and rhabdomyolysis (two events each) and liver injury, immune-mediated myositis and myocarditis (one event each). There were no AEs that led to study discontinuation, and no deaths [22].
No new safety signals emerged during part 2 of NCT03769116 in patients treated with delandistrogene moxeparvovec in part 1 (i.e. 96 weeks’ post treatment) [15] nor at 4 years’ post treatment in NCT03375164 (for which safety was the primary outcome) [7, 20].
Patients participating in the delandistrogene moxeparvovec clinical studies were required to have a baseline anti-AAVrh74 total binding antibody titre of ≤ 1:400 [8]. In clinical studies, anti-AAVrh74 total binding antibody titres were ≥ 1:409,600 in every patient following a single intravenous infusion of delandistrogene moxeparvovec (n = 84) and > 1:26,214,400 in some patients. The safety of readministering delandistrogene moxeparvovec (or any other AAVrh74 vector-based gene therapy) in the presence of a high anti-AAVrh74 total binding antibody titre has not been assessed [8].
2.5 Ongoing Clinical Trials
In addition to the ongoing studies discussed in Sect. 2.3 [NCT03769116 and ENDEAVOR (NCT04626674)], the phase II ENVOL (EudraCT 2022-000691-19) study and the phase III EMBARK (NCT05096221) and ENVISION (NCT05881408) studies are underway to evaluate the efficacy and safety of delandistrogene moxeparvovec in male patients (ENVISION) aged 4 to < 8 years (EMBARK) or < 4 years (ENVOL) with DMD.
References
Duan D, Goemans N, Takeda S, et al. Duchenne muscular dystrophy. Nat Rev Dis Primers. 2021;7(1):13.
Chang M, Cai Y, Gao Z, et al. Duchenne muscular dystrophy: pathogenesis and promising therapies. J Neurol. 2023;270(8):3733–49.
Birnkrant DJ, Bello L, Butterfield RJ, et al. Cardiorespiratory management of Duchenne muscular dystrophy: emerging therapies, neuromuscular genetics, and new clinical challenges. Lancet Respir Med. 2022;10(4):403–20.
Asher DR, Thapa K, Dharia SD, et al. Clinical development on the frontier: gene therapy for Duchenne muscular dystrophy. Expert Opin Biol Ther. 2020;20(3):263–74.
Schneider AE, Aartsma-Rus A. Developments in reading frame restoring therapy approaches for Duchenne muscular dystrophy. Expert Opin Biol Ther. 2021;21(3):343–59.
Muntoni F, Domingos J, Manzur AY, et al. Categorising trajectories and individual item changes of the North Star Ambulatory Assessment in patients with Duchenne muscular dystrophy. PLoS One. 2019;14(9): e0221097.
Mendell JR, Sahenk Z, Lehman K, et al. Assessment of systemic delivery of rAAVrh74.MHCK7.micro-dystrophin in children with Duchenne muscular dystrophy: a nonrandomized controlled trial. JAMA Neurol. 2020;77(9):1122–31.
Sarepta Therapeutics Inc. ELEVIDYS (delandistrogene moxeparvovec-rokl) suspension, for intravenous infusion: US prescribing information; 2023. https://www.fda.gov/. Accessed 23 Jun 2023.
Sarepta Therapeutics. Sarepta Therapeutics announces FDA approval of ELEVIDYS, the first gene therapy to treat Duchenne muscular dystrophy [media release]; 22 Jun 2023. https://www.sarepta.com/.
US Food and Drug Administration. FDA approves first gene therapy for treatment of certain patients with Duchenne muscular dystrophy [media release]; 22 Jun 2023. https://www.fda.gov/.
Roche. Roche enters licensing agreement with Sarepta Therapeutics to improve the lives of patients living with Duchenne muscular dystrophy [media release]; 23 Dec 2019. http://www.roche.com.
Sarepta Therapeutics. Sarepta Therapeutics and Selecta Biosciences enter into research license and option agreement for Selectas ImmTOR immune tolerance platform in neuromuscular diseases [media release]; 18 Jun 2020. http://www.sarepta.com.
Chugai Pharmaceutical. Chugai in-licenses gene therapy delandistrogene moxeparvovec (SRP-9001) for Duchenne muscular dystrophy [media release]; 16 Dec 2021. https://www.chugai-pharm.co.jp/.
Selecta Biosciences. Selecta Biosciences announces partnership advancements and clinical trial updates [media release]; 13 Jun 2022. http://www.selectabio.com.
Mendell J, Shieh P, McDonald C, et al. Expression of SRP-9001 dystrophin and stabilization of motor function up to 2 years post-treatment with delandistrogene moxeparvovec gene therapy in individuals with Duchenne muscular dystrophy. Front Cell Dev Biol. 2023;11: 1167762.
Proud C, Zaidman C, McDonald C, et al. One-year data from ENDEAVOR, a phase 1b trial of delandistrogene moxeparvovec in patients with DMD [abstract no. 107 plus oral]. In: Muscular Dystrophy Association (MDA) Clinical and Scientific Conference. 2023.
Malhotra J, Lewis S, Zhang X, et al. Analysis of vector shedding following treatment with delandistrogene moxeparvovec, an investigational rAAVrh74-based gene therapy for DMD [abstract no. LSVP.35 plus poster]. Neuromuscul Disord. 2022;32(Suppl 1):47.
Klimchak AC, Sedita LE, Rodino-Klapac LR, et al. Assessing the value of delandistrogene moxeparvovec (SRP-9001) gene therapy in patients with Duchenne muscular dystrophy in the United States. J Mark Access Health Policy. 2023;11:1–14.
Sarepta Therapeutics. Sarepta Therapeutics reports sustained functional improvement two years after treatment with SRP-9001, its investigational micro-dystrophin gene therapy for Duchenne muscular dystrophy [media release]; 28 Sep 2020. http://www.sarepta.com.
Mendell J, Sahenk Z, Lehman K, et al. Phase 1/2a trial of delandistrogene moxeparvovec in patients with DMD: 4-year update [abstract no. 354 plus presentation]. Mol Ther. 2023;31(4 Suppl 1):190–1.
Zaidman C, Proud C, McDonald C, et al. One-year data from ENDEAVOR, a phase 1b trial of delandistrogene moxeparvovec in boys with DMD [abstract no. P.129 plus poster]. Neuromuscul Disord. 2022;32(Suppl 1):S101.
Goedeker NL, Aqul A, Butterfield RJ, et al. Management of patients following investigational delandistrogene moxeparvovec gene therapy for Duchenne muscular dystrophy: Delphi panel consensus considerations based on clinical trial experience [abstract no. 132 plus poster]. Mol Ther. 2023;31(4S1):71.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Funding
The preparation of this review was not supported by any external funding.
Authorship and Conflict of Interest
During the peer review process the manufacturer of the agent under review was offered an opportunity to comment on the article. Changes resulting from any comments received were made by the authors on the basis of scientific completeness and accuracy. Sheridan M. Hoy is a salaried employee of Adis International Ltd/Springer Nature, and declares no relevant conflicts of interest. All authors contributed to this article and are responsible for its content.
Ethics Approval, Consent to Participate, Consent to Publish, Availability of Data and Material, Code Availability
Not applicable.
Additional information
This profile has been extracted and modified from the AdisInsight database. AdisInsight tracks drug development worldwide through the entire development process, from discovery, through pre-clinical and clinical studies to market launch and beyond.
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
Springer Nature or its licensor (e.g. a society or other partner) holds exclusive rights to this article under a publishing agreement with the author(s) or other rightsholder(s); author self-archiving of the accepted manuscript version of this article is solely governed by the terms of such publishing agreement and applicable law.
About this article

Cite this article
Hoy, S.M. Delandistrogene Moxeparvovec: First Approval. Drugs 83, 1323–1329 (2023). https://doi.org/10.1007/s40265-023-01929-x
Published:
Issue Date:
DOI: https://doi.org/10.1007/s40265-023-01929-x