
Abstract
Avalglucosidase alfa (NEXVIAZYME™; avalglucosidase alfa-ngpt) is a hydrolytic lysosomal glycogen-specific recombinant human α-glucosidase product being developed by Sanofi Genzyme (formerly Genzyme Corporation) for the treatment of Pompe disease. Pompe disease is an autosomal recessive lysosomal storage disease caused by a deficiency of the lysosomal enzyme acid α-glucosidase (GAA), which results in intralysosomal accumulation of glycogen in various tissues. In August 2021, avalglucosidase alfa received its first approval in the USA for the treatment of patients 1 year of age and older with late-onset Pompe disease (GAA deficiency). In July 2021, avalglucosidase alfa received a positive opinion in the EU for long-term enzyme replacement therapy for the treatment of patients with Pompe disease. The drug is under regulatory review in the UK and Japan, and clinical studies are underway in several countries worldwide. This article summarizes the milestones in the development of avalglucosidase alfa leading to this first approval for late-onset Pompe disease.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Digital Features for this AdisInsight Report can be found at https://doi.org/10.6084/m9.figshare.16531248. |
A hydrolytic lysosomal glycogen-specific recombinant human acid α-glucosidase product being developed by Sanofi Genzyme for the treatment of Pompe disease. |
Received its first approval on 6 August 2021 in the USA. |
Approved for the treatment of patients 1 year of age and older with late-onset Pompe disease. |
1 Introduction
Pompe disease is an autosomal recessive lysosomal storage disease caused by a deficiency of the lysosomal enzyme acid α-glucosidase (GAA) that results in intralysosomal accumulation of glycogen in various tissues (particularly cardiac and skeletal muscle cells) and impairment of cellular function [1,2,3]. Pompe disease is classified according to age of onset into infantile- and late-onset disease [1, 3]. Infantile-onset disease is characterized by an almost total lack of GAA and patients invariably die around 1 year of age, mainly due to cardiac and/or respiratory failure. Patients with late-onset Pompe disease have varying levels of residual GAA activity and present with a wide range of symptoms and disease severity and a later onset of disease. However, all patients with late-onset Pompe disease will experience impaired motor function, decline in respiratory function and progressive muscle weakness, resulting in significant morbidity and mortality, ultimately leading to death [1, 3].
Until recently alglucosidase alfa (recombinant human GAA; rhGAA) enzyme replacement therapy was the only approved treatment for Pompe disease in the USA [1, 2]. However, the effectiveness of alglucosidase alfa is limited by its poor drug targeting to skeletal muscles, resulting in the need for higher drug doses [1]. Although the exact reason for the requirement of higher drug doses is unclear, it may be related to the low density of the mannose 6-phosphate receptor (CI-MPR) in muscle tissue and the relatively low affinity of alglucosidase alfa for the CI-MPR.

Key milestones in the development of avalglucosidase alfa for the treatment of Pompe disease. CHMP Committee for Medicinal Products for Human Use, EMA European Medicines Agency, FDA Food and Drug Administration, MHRA Medicines and Healthcare Products Regulatory Agency, MAA Marketing Authorisation Application, PIM Promising Innovative Medicine
The CI-MPR-mediated pathway is thought to be the major route for internalization of alglucosidase alfa into cells. To address this limitation, novel treatments are being developed with the aim of improving enzyme uptake into cells [1].
Avalglucosidase alfa (NEXVIAZYME™; avalglucosidase alfa-ngpt) is a next-generation hydrolytic lysosomal glycogen-specific recombinant human α-glucosidase (neo-rhGAA) being developed by Sanofi Genzyme (formerly Genzyme Corporation) for the treatment of Pompe disease. On 6 August 2021 [4], avalglucosidase alfa received its first approval in the USA for the treatment of patients 1 year of age and older with late-onset Pompe disease (GAA deficiency) [5]. The recommended dosage of avalglucosidase alfa for patients with bodyweight ≥ 30 kg is 20 mg/kg (of actual bodyweight) every two weeks, and for patients with bodyweight < 30 kg is 40 mg/kg (of actual bodyweight) every 2 weeks. The recommended initial intravenous infusion rate of avalglucosidase alfa is 1 mg/kg/h, which can be gradually increased every 30 min if there are no signs or symptoms of infusion-rate reactions. Prior to treatment with avalglucosidase alfa, pre-treatment with antihistamines, antipyretics and/or corticosteroids should be considered. The US prescribing information for avalglucosidase alfa carries warnings regarding the potential for severe hypersensitivity reactions (including anaphylaxis), infusion-site reactions and the risk of acute cardiorespiratory failure in susceptible patients [5]. Avalglucosidase alfa received a positive opinion in the EU on 23 July 2021 for long-term enzyme replacement therapy for the treatment of patients with Pompe disease [6].The drug is under regulatory review in the UK and Japan, and clinical studies are underway in several countries worldwide.
2 Scientific Summary
2.1 Pharmacodynamics
Avalglucosidase alfa is a hydrolytic lysosomal glycogen-specific rhGAA enzyme conjugated with multiple synthetic bis-mannose-6-phosphate (bis-M6P)-tetra-mannose glycans (approximately 15 moles of M6P per mole of enzyme) [3, 5]. It was designed to increase bis-M6P levels on the molecule to enhance receptor targeting and enzyme uptake, thereby increasing glycogen clearance [3, 5, 7, 8]. In vitro and in vivo (Pompe disease mouse model) studies found that avalglucosidase alfa had increased binding affinity for the M6P receptor compared with alglucosidase alfa (> 95% binding vs 15–30% binding, respectively), and its uptake into muscle cells was increased approximately five-fold (approaching saturation at 100 nmol/L vs approximately 500 nmol/L) [7, 8]. Clearance of glycogen from Pompe mouse tissues was also improved with avalglucosidase alfa versus alglucosidase alfa, with approximately fivefold greater glycogen clearance in cardiac and skeletal muscles, including the heart, diaphragm, quadriceps, and triceps [8].
Avalglucosidase alfa reduced glucose tetrasaccharide (Glc4) levels in the urine (a limit dextrin of glycogen), urinary excretion of which is elevated in patients with Pompe disease [5]. In the pivotal phase 3 COMET study in patients with late-onset Pompe disease, the mean change from baseline to week 49 in urinary Glc4 (normalised by urine creatinine and reported as mmol Glc4/mol creatinine) was – 54% with avalglucosidase alfa and – 11% with alglucosidase alfa (baseline values were 12.7 and 8.7 mmol/mol, respectively) [5]. In a phase 1 study and its extension (NEO1 and NEO-EXT), treatment with avalglucosidase alfa for up to 4.5 years resulted in persistent improvement from baseline in biomarker levels; across treatment groups, the mean changes from baseline in plasma creatine kinase (marker of muscle damage) were – 24% to – 55%, urinary Glc4 (disease substrate) were – 38% to – 44%, plasma aspartate aminotransferase (liver/muscle) were – 25% to – 39% and alanine aminotransferase (heart/liver/muscle) were – 21% to – 39% [9].
2.2 Pharmacokinetics
In patients with late-onset Pompe disease, exposure to avalglucosidase alfa increases in an approximately dose-proportional manner over a dose range of 5–20 mg/kg every 2 weeks (0.25–1.0 times the approved recommended dosage in patients weighing ≥ 30 kg, or 0.125–0.5 times the approved recommended dosage in patients weighing < 30 kg) [5, 7]. No accumulation of avalglucosidase alfa was observed after dosing every 2 weeks. The volume of distribution of avalglucosidase alfa was 3.4 L [5].
Features and properties of avalglucosidase alfa
Alternative names | Avalglucosidase alfa-ngpt; GZ 402666; neo rhGAA; neo-recombinant human acid alpha-glucosidase; Neo-rhGAA; Neo-rhGAA enzyme therapy; NeoGAA; Nexviadyme; Nexviazyme |
Class | Alpha-glucosidases, Recombinant proteins |
Mechanism of action | ERT, providing an exogenous source of α-glucosidase that exerts enzymatic activity in cleaving glycogen |
Route of administration | Intravenous infusion |
Pharmacodynamics | Higher binding affinity for the M6P receptor compared with alglucosidase alfa (> 95% binding vs 15–30% binding, respectively); uptake into muscle cells ≈ fivefold higher (approaching saturation at 100 nmol/L vs ≈ 500 nmol/L for alglucosidase alfa) |
Reduced biomarker levels in patients with late-onset Pompe disease, including urinary Glc4 levels | |
Pharmacokinetics | Approximately dose-proportional increase in exposure over a dose range of 5–20 mg/kg every 2 weeks |
No accumulation after dosing every 2 weeks | |
Mean elimination half-life 1.6 h; mean clearance 0.9 L/h | |
Adverse reactions (in LOPD) | |
Most frequent (> 5%) | Headache, diarrhoea, nausea, fatigue, arthralgia, myalgia, dizziness, rash, vomiting and pyrexia, abdominal pain, pruritus, erythema, abdominal pain upper, chills, cough, urticaria, dyspnea, hypertension and hypotension |
ATC codes | |
WHO ATC code | A16A-B07 (alglucosidase alfa) |
EphMRA ATC code | A16A (other alimentary tract and metabolism products) |
Although the metabolic pathway of avalglucosidase alfa has not been determined, it is expected that the protein portion of avalglucosidase alfa is metabolized into small peptides and amino acids via catabolic pathways [5]. The mean elimination half-life of avalglucosidase alfa in patients with late-onset Pompe disease was 1.6 h and mean clearance was 0.9 L/h.
In treatment-naïve patients with late-onset Pompe disease, 96% (49/51) of patients developed treatment emergent anti-drug antibodies (ADAs) after treatment with avalglucosidase alfa 20 mg/kg every 2 weeks [5]. The exposure to avalglucosidase alfa in patients who developed ADA was similar to exposure in the two ADA-negative patients. In patients who developed ADA, the median avalglucosidase alfa area under the concentration-time curve remained generally unchanged between weeks 1 and 49, regardless of ADA titres and neutralizing antibodies. Patients with sustained high ADA titres (> 12,800) had increased incidence of infusion-associated reactions (IARs) [5].
Age and sex did not significantly affect the pharmacokinetics of avalglucosidase alfa in patients aged 1–78 years with Pompe disease [5].
2.3 Therapeutic Trials
2.3.1 Late-onset Pome Disease
2.3.1.1 COMET Study
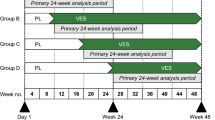
Avalglucosidase alfa provided clinically meaningful improvements compared with alglucosidase alfa in terms of respiratory function and movement endurance measures in treatment-naïve patients with late-onset Pompe disease participating in the phase 3, randomized, double-blind, multinational COMET study (NCT02782741) [5]. Key eligibility criteria were age ≥ 3 years, GAA deficiency, ambulation for 40 m (without stopping/using assistive device), not on invasive ventilation and upright forced vital capacity (FVC) ≥ 30 to ≤ 85% predicted [10]. Patients were stratified by baseline FVC, gender, age and country (Japan/ex-Japan), and randomized to receive intravenous avalglucosidase alfa 20 mg/kg (n = 51) or alglucosidase alfa 20 mg/kg (n = 49) once every 2 weeks for 49 weeks [5, 10]. After completion of the study, patients entered an open-label, long-term, follow-up phase of up to 5 years, in which patients in the alglucosidase alfa arm were switched to treatment with avalglucosidase alfa 20 mg/kg once every 2 weeks [5, 10].
Avalglucosidase alfa was non-inferior, but not superior, to alglucosidase alfa in improving respiratory function [5, 11]. At week 49, the estimated least-squares (LS) mean change from baseline in FVC (% predicted) was 2.9% in patients receiving avalglucosidase alfa compared with 0.5% in patients receiving alglucosidase alfa (between-group difference 2.43%; noninferiority p = 0.0074; noninferiority margin 1.1%). Treatment with avalglucosidase alfa also resulted in significantly greater improvement in the 6-Minute Walk Test (6MWT) distance compared with treatment with alglucosidase alfa (LS mean change from baseline 32.2 vs. 2.2 m; between-group difference 30 m; nominal p = 0.04). At all assessments over the 49-week treatment period, avalglucosidase alfa provided numerically greater improvements in FVC (% predicted) and 6MWT distance than treatment with alglucosidase alfa [5, 11].
Key clinical trials of avalglucosidase alfa conducted by Genzyme/Sanofi
Drug(s) | Indication | Phase | Status | Location(s) | Identifier |
|---|---|---|---|---|---|
Avalglucosidase alfa, alglucosidase alfa | LOPD | 3 | Ongoing | Multinational | COMET; NCT02782741; EudraCT2020-004686-39 |
Avalglucosidase alfa | LOPD | 2 | Ongoing | USA, Belgium, Denmark, France, Germany, Netherlands, UK | NEO-EXT; NCT02032524; EudraCT2013-003321-28 |
Avalglucosidase alfa | LOPD | 1 | Completed | USA, Belgium, Denmark, France, Germany, Netherlands, UK | NEO1; NCT01898364; EudraCT2012-004167-42 |
Avalglucosidase alfa | IOPD | 3 | Ongoing | Multinational | Baby-COMET; NCT04910776; EudraCT2020-004686-39 |
Avalglucosidase alfa, alglucosidase alfa | IOPD | 2 | Ongoing | USA, France, Japan, Taiwan, UK | Mini-COMET; NCT03019406; EudraCT2016-003475-21 |
2.3.1.2 NEO1 and NEO-EXT
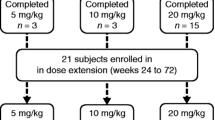
Avalglucosidase alfa generally stabilized or improved respiratory function and movement endurance in treatment-naïve and treatment-experienced (≥ 9 months treatment with alglucosidase alfa) patients with late-onset Pompe disease who were participating in the phase 1, open-label, ascending-dose NEO1 study (NCT01898364) [7]. Patients included were aged ≥18 years, GAA deficient, ambulatory for ≥ 50 m (without stopping or using an assistive device) and had an upright FVC of ≥ 50% predicted. Patients received avalglucosidase alfa 5, 10 or 20 mg/kg once every 2 weeks for 24 weeks, with 9 patients in the treatment-naïve group (n = 3 per dose group) and 12 patients in the treatment-experienced group (n = 3, 4 and 5 in the respective dose groups) completing the study. Following 24 weeks’ treatment with avalglucosidase alfa, outcomes measures including respiratory function (upright FVC % predicted, maximum expiratory pressure [MEP], and maximum inspiratory pressure [MIP]) and the 6MWT % predicted distance generally remained stable or improved across treatment groups. In treatment-naïve patients who received avalglucosidase alfa 20 mg/kg, the mean changes from baseline in FVC, MEP, MIP and 6MWT distance % predicted were 6.2%, 12.0%, 7.9% and 3.9%, respectively; corresponding values for treatment-experienced patients were 1.4%, 6.0%, − 0.2% and – 1.3%, respectively [7].
The efficacy of avalglucosidase alfa was sustained during long-term (up to 6 years) treatment in an ongoing phase 2 extension of NEO1 (NEO-EXT; NCT02032524) [12]. Following 24 weeks’ treatment with avalglucosidase alfa (5–20 mg every 2 weeks) in NEO1, 8 treatment-naïve and 11 treatment-experienced patients entered the extension and continued receiving their NEO1 dose until 2016. Thereafter, all patients received avalglucosidase alfa 20 mg/kg every 2 weeks. Slope estimates of efficacy parameters after up to 6 years’ treatment with avalglucosidase alfa showed that both in treatment-naive and treatment-experienced groups, the upright FVC% predicted (− 0.473/year and − 0.648/year, respectively) remained generally stable; the MEP and MIP % predicted, although more variable among patients, were also stable overall. The slope estimates for 6MWT% predicted in treatment-naïve and -experienced patients were − 0.701/year and − 0.846/year, but remained stable for the majority of patients, with the distance walked generally improving in patients aged ≤ 50 years at enrolment [12].
Avalglucosidase alfa may also assist in muscle preservation in patients with late-onset Pompe disease. In NEO-EXT, measurement of skeletal muscle fat fraction (using quantitative magnetic resonance imaging) at baseline (week 27), year 2 and year 4 of treatment showed that overall, % fat fractions in quadriceps and hamstring were generally stable in most participants (across treatment groups, mean changes from baseline for quadriceps were 0.30–1.45 and hamstring were – 0.21 to 5.29) [13].
2.3.2 Infantile-Onset Pompe Disease
Preliminary efficacy data from the phase 2, open-label ascending-dose, 3-cohort Mini-COMET study (NCT03019406) demonstrated a positive clinical impact of avalglucosidase alfa in patients with infantile-onset Pompe disease [14]. Patients aged < 18 years who had previously received alglucosidase alfa for ≥ 6 months and demonstrated either clinical decline or sub-optimal response were included in the study. During the initial 25-week treatment period, patients (n = 22; aged 1–12 years) received avalglucosidase alfa 20 or 40 mg/kg every 2 weeks or their ongoing stable dose of alglucosidase alfa (range 20 mg/kg every 2 weeks to 40 mg/kg weekly) [14, 15]. Preliminary efficacy data for avalglucosidase alfa indicated overall positive trends (stabilization or improvement) in Gross Motor Function Measure-88, Pompe-Paediatric Evaluation of Disability Inventory functional skills scale: mobility domain, and echocardiographic left ventricular mass Z-score (quantitative data not available) [14]. Patient-level analyses also suggest that avalglucosidase alfa relative to alglucosidase alfa regimens improved or better stabilized symptoms, as assessed by motor outcomes, cardiac parameters and eyelid measures, with the avalglucosidase alfa 40 mg/kg dose providing additional benefits (no quantitative data available) [14]. Following 25 weeks of treatment, a trend for improvement in eyelid position was observed in patients receiving avalglucosidase alfa 40 mg/kg dose compared with stabilization or decline in patients receiving avalglucosidase alfa 20 mg/kg or alglucosidase alfa regimens [15]. Treatment benefits with avalglucosidase alfa 40 mg/kg were more pronounced for interpalpebral fissure distance relative to those for margin reflex distance-1 or margin pupil distance [15].
2.4 Adverse Events
Avalglucosidase alfa was generally well tolerated in patients with Pompe disease according to pooled data from four clinical studies [5]. A total of 141 patients (118 adult and 23 paediatric patients) in these studies were treated with avalglucosidase alfa for up to 85 months (mean exposure 26 months). The most frequently (incidence >5%) reported adverse reactions in the pooled safety population included headache, diarrhoea, nausea, fatigue, arthralgia, myalgia, dizziness, rash, vomiting and pyrexia, abdominal pain, pruritus, erythema, abdominal pain upper, chills, cough, urticaria, dyspnea, hypertension and hypotension. Serious adverse reactions in adult and paediatric patients were similar, with respiratory distress, chills and pyrexia occurring in at least two avalglucosidase alfa-treated patients. A total of five avalglucosidase alfa recipients permanently discontinued treatment because of adverse reactions, with two patients discontinuing treatment due to serious adverse reactions. IARs occurred in 48 (34%) patients receiving avalglucosidase alfa, with IARs such as chills, cough, diarrhoea, erythema, fatigue, headache, influenza-like illness and nausea occurring in more than one patient [5].
As with all therapeutic proteins, there is potential for immunogenicity with avalglucosidase alfa [5]. Pooled safety data showed that after treatment with avalglucosidase alfa, ADAs were reported in 95% (21% had neutralizing antibodies) of treatment naïve patients and 55% (5% had neutralizing antibodies) of treatment-experienced patients. The incidence of IARs in patients with ADA peak titre ≥ 12,800 was 62% (8/13), in those with peak ADA titre < 12,800 was 19% (8/43) and in ADA-negative patients was 33% (1/3). The incidence of hypersensitivity reactions was approximately twice as high in patients with higher ADA titres than in those with lower titres (31% vs 14%) [5].
2.4.1 Late-Onset Pompe Disease
Avalglucosidase alfa 20 mg/kg every two weeks was generally well tolerated in patients with late-onset Pompe disease participating in the phase 3 COMET study (NCT02782741) [5, 11]. Treatment-emergent adverse events occurred in 86.3% of avalglucosidase alfa-treated and 91.8% of alglucosidase alfa-treated patients [11]. The most common (incidence > 10%) adverse reactions with avalglucosidase alfa during 49 weeks of treatment were headache (22% vs 33% with alglucosidase alfa), fatigue (18% vs 14%), diarrhoea (12% vs 16%) and nausea (12% vs 14%) [5]. Serious adverse reactions occurred in one (2%) avalglucosidase alfa and three (6%) alglucosidase alfa recipients. IARs occurred in 25% of avalglucosidase alfa and 33% of alglucosidase alfa recipients. IARs (mild to moderate) occurring in more than one avalglucosidase alfa recipient included headache, diarrhoea, pruritus, urticaria and rash. No severe IARs were reported in patients receiving avalglucosidase alfa, while severe IARs occurred in two patients receiving alglucosidase alfa [5].
Avalglucosidase alfa was also generally well tolerated in the phase 1 study (NEO1; NCT01898364) and its extension (NEO-EXT; NCT02032524) [7, 12]. The most common treatment-related adverse events with avalglucosidase alfa during long-term therapy (up to 6 years) in NEO-EXT were fatigue, headache, nausea and rash [12].
2.4.2 Infantile-Onset Pompe Disease
Avalglucosidase alfa was generally well tolerated in patients with infantile-onset Pompe disease participating in the phase 2 Mini-COMET study (NCT03019406) [14]. There were no serious or severe treatment-related adverse events with avalglucosidase alfa therapy. No treatment-emergent adverse event in avalglucosidase alfa recipients resulted in permanent discontinuation of treatment and no death was reported in the study [14].
2.5 Ongoing Clinical Trials
In addition to the ongoing phase 3 COMET, phase 2 NEO-EXT and phase 2 Mini-COMET studies discussed in Sect. 2.3, recruitment is underway for the phase 3, open-label, multinational Baby-COMET study (NCT04910776) in treatment-naïve paediatric patients aged ≤ 6 years with infantile-onset Pompe disease. The study aims to assess the efficacy, safety, pharmacodynamics and pharmacokinetics of avalglucosidase alfa in ≈ 16 patients; the primary endpoint of the study is the proportion of patients alive and free of invasive ventilation at week 52.
3 Current Status
Avalglucosidase alfa received its first approval on 6 August 2021 [4] in the USA for the treatment of patients 1 year of age and older with late-onset Pompe disease (GAA deficiency) [5]. Avalglucosidase alfa received a positive opinion in the EU on 23 July 2021 for long-term enzyme replacement therapy for the treatment of patients with Pompe disease [6].
References
Do HV, Khanna R, Gotschall R. Challenges in treating Pompe disease: an industry perspective. Ann Transl Med. 2019;7(13):291.
Meena NK, Raben N. Pompe disease: new developments in an old lysosomal storage disorder. Biomolecules. 2020;10(9):1339.
Zhu Y, Li X, Kyazike J, et al. Conjugation of mannose 6-phosphate-containing oligosaccharides to acid alpha-glucosidase improves the clearance of glycogen in pompe mice. J Biol Chem. 2004;279(48):50336–41.
US FDA. FDA approves new treatment for Pompe Disease [media release]. 6 Aug 2021. https://www.fda.gov/.
Genzyme Corp. Nexviazyme™ (avalglucosidase alfa): US prescribing information. 2021. https://products.sanofi.us/nexviazyme/nexviazyme.pdf. Accessed 16 Aug 2021.
European Medicines Agency. Nexviadyme (avalglucosidase alfa): summary of opinion (initial authorisation). 2021. https://www.ema.europa.eu/en/documents/smop-initial/chmp-summary-positive-opinion-nexviadyme_en.pdf. Accessed 16 Aug 2021.
Pena LDM, Barohn RJ, Byrne BJ, et al. Safety, tolerability, pharmacokinetics, pharmacodynamics, and exploratory efficacy of the novel enzyme replacement therapy avalglucosidase alfa (neoGAA) in treatment-naive and alglucosidase alfa-treated patients with late-onset Pompe disease: a phase 1, open-label, multicenter, multinational, ascending dose study. Neuromuscul Disord. 2019;29(3):167–86.
Zhu Y, Jiang JL, Gumlaw NK, et al. Glycoengineered acid alpha-glucosidase with improved efficacy at correcting the metabolic aberrations and motor function deficits in a mouse model of Pompe disease. Mol Ther. 2009;17(6):954–63.
Schoser B, Barohn R, Dimachkie M, et al. NEO1 and NEO-EXT studies: pharmacodynamic and exploratory biomarker assessments following repeat avalglucosidase alfa dosing for up to 4.5 years in patients with late-onset Pompe disease [abstract no. EPR3085]. Eur J Neurol. 2019;26(Suppl 1):326.
Straub V, Choi YC, Manera JD, et al. Comet methodology: comparison of the efficacy and safety of the enzyme replacement therapies, neogaa and alglucosidase alfa, in treatment-naive patients with late-onset Pompe disease [abstract no. 792]. J Inborn Errors Metab Screen. 2017;5:355–6.
Kishnani PS, Attarian S, Borges JL, et al. Efficacy and safety results of the avalglucosidase alfa phase 3 COMET trial in late-onset Pompe disease patients [abstract no. 121]. Mol Genet Metab. 2021;132(2):S57.
Dimachkie MM, Barohn RJ, Byrne B, et al. NEO1/NEO-EXT studies: Safety and exploratory efficacy of repeat avalglucosidase alfa dosing after up to 6 years in participants with late-onset Pompe disease (LOPD) [abstract no. 58]. Mol Genet Metab. 2021;132(2):S34.
Carlier P, Vissing J, Diaz Manera J, et al. NEO-EXT: skeletal muscle % fat in late-onset Pompe disease after long-term avalglucosidase alfa (3-point Dixon MRI) [abstract no. EPR-189]. Eur J Neurol. 2021;28(Suppl 1):333.
Kishnani PS, Brassier A, Broomfield A, et al. Mini-COMET study: individual participant-level responses to treatment in patients with infantile-onset Pompe disease receiving repeated dose regimens of avalglucosidase alfa or alglucosidase alfa who were previously treated with alglucosidase alfa [abstract no. 122]. Mol Genet Metab. 2021;132(2):S57–8.
Davison J, Brassier A, Broomfield A, et al. Mini-COMET study: effects of repeat avalglucosidase alfa dosing on ptosis in participants with infantile-onset Pompe disease (IOPD) who were previously treated with alglucosidase alfa [abstract no. 52]. Mol Genet Metab. 2021;132(2):S31–2.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Funding
The preparation of this review was not supported by any external funding.
Authorship and Conflict of interest
During the peer review process the manufacturer of the agent under review was offered an opportunity to comment on the article. Changes resulting from any comments received were made by the authors on the basis of scientific completeness and accuracy. Sohita Dhillon is a contracted employee of Adis International Ltd/Springer Nature and declares no relevant conflicts of interest. All authors contributed to the review and are responsible for the article content.
Ethics approval, Consent to participate, Consent to publish, Availability of data and material, Code availability
Not applicable.
Additional information
This profile has been extracted and modified from the AdisInsight database. AdisInsight tracks drug development worldwide through the entire development process, from discovery, through pre-clinical and clinical studies to market launch and beyond.
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
About this article

Cite this article
Dhillon, S. Avalglucosidase alfa: First Approval. Drugs 81, 1803–1809 (2021). https://doi.org/10.1007/s40265-021-01600-3
Published:
Issue Date:
DOI: https://doi.org/10.1007/s40265-021-01600-3