
Abstract
Paratek Pharmaceuticals are developing omadacycline (NUZYRA™), a first-in-class orally active aminomethylcycline antibacterial, as a treatment for various bacterial infections. The drug, which is available in intravenous and oral formulations, has a broad spectrum of antibacterial activity and was recently approved in the USA as a treatment for the treatment of community acquired bacterial pneumonia (CABP) and acute bacterial skin and skin structure infections (ABSSSI) in adults. This article summarizes the milestones in the development of omadacycline leading to this first global approval for the treatment of CABP and ABSSSI.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
1 Introduction
Omadacycline (NUZYRA™), a first-in-class, orally active, aminomethylcycline antibacterial agent, is being developed by Paratek Pharmaceuticals as a treatment for various bacterial infections, and recently received approval in the USA for the treatment of community acquired bacterial pneumonia (CABP) and acute bacterial skin and skin structure infections (ABSSSI) in adults [1] Omadacycline is also under evaluation in the EU for the same indications [2]. The drug has demonstrated activity against Gram-positive, Gram-negative and atypical bacteria and is active against tetracycline-resistant bacterial pathogens expressing either ribosomal protection or efflux resistance genes, the two most common forms of tetracycline resistance [3, 4].
The recommended dosage of omadacycline, which is available in both intravenous (IV) and oral formulations, is a 200 mg IV loading dose on day 1 (either as a single 200 mg dose or two 100 mg doses) followed by a once daily 100 mg IV or 300 mg oral maintenance dose for 7 to 14 days. The option of commencing treatment with a 450 mg oral dose on day 1 is available for patients with ABSSSI [5].
1.1 Company Agreements
In February 1997 Paratek Pharmaceuticals entered into a license agreement with Tufts University, granting Paratek an exclusive licence to patent applications and other intellectual property belonging to Tufts related to drug resistance, for the purpose of developing and commercialising products for the treatment or prevention of microbial infections. In return Tufts received shares in Paratek and the right to milestone payments of up to $US0.3 million upon the achievement of specified development and regulatory approval outcomes, as well as a minimum royalty payment of $US25,000 per year. In addition, Paratek agreed to pay Tufts royalties based on gross sales of products and a percentage in the single digits of any sublicense fee received in relation to its license agreement with Tufts.
In October 2016, Paratek entered into a cooperative agreement with the U.S. Army Medical Research Institute of Infectious Diseases to investigate the use of omadacycline as a defence to potential bioweapons, including Yersinia pestis and Bacillus anthracis [6].
In April 2017 Paratek entered into a collaboration agreement with Zai Lab, with the latter obtaining exclusive rights to develop, manufacture and commercialise omadacycline in China, Hong Kong, Macau, and Taiwan. In return, Paratek received an upfront payment of $US7.5 million and is eligible to receive additional milestone payments related to development, regulatory, and commercial milestones as well as royalty payments on net sales in the territories covered by the agreement [7].
Paratek has also entered into collaboration agreements with Bayer (in 2003) [8], Merck (in 2006) [9] and Novartis (in 2009) [10] to develop omadacycline, all of which were subsequently terminated.

Key milestones in the development of omadacycline for the treatment of CABP and ABSSSI, focussing on phase III trials. ABSSSI acute bacterial skin and skin structure infections, CABP community-acquired bacterial pneumonia, MAA Marketing Authorization Application, NDA New Drug Application, UTI urinary tract infections
1.2 Patent Information
The patent portfolio of omadacycline, directed to cover composition of matter, formulations, salts and polymorphs, manufacturing methods and methods of use, are owned by Paratek Pharmaceuticals. In some corresponding foreign patents and patent applications, omadacycline is covered along with other compounds in patents and patent applications that are owned jointly by Paratek and Tufts University, subject to a license agreement. The issued composition of matter patent in the United States (US patent no. 7 553 828) is expected to expire in 2023.
2 Scientific Summary
2.1 Pharmacodynamics
Omadacycline is semisynthetic aminomethylcycline, characterized by an aminomethyl group at the C9 position of the core tetracyclic D ring. Like other tetracyclines, the antibacterial action of omadacycline is due to binding to the bacterial ribosomal 30S subunit, thereby disrupting bacterial protein synthesis. Modification at position C9 is thought to overcome common tetracycline resistance mechanisms [5, 11]. In vitro, omadacycline is active against tetracycline-resistant Gram-positive bacteria expressing either ribosomal protection proteins (tet M) or active efflux pumps (tet K and tet L) and against Enterobacteriaceae that carry the tet B efflux gene. Like other tetracyclines, omadacycline is bacteriostatic; however, bactericidal activity against some Streptococcus pneumoniae and Haemophilus influenzae isolates has been demonstrated [5].
Omadacycline effectively inhibited bacterial protein synthesis in vitro. In contrast to tetracycline, the inhibitory effect of omadacycline was not affected by the presence of the ribosomal protection protein Tet(O) and the drug efficiently competed with tritium-labelled tetracycline for binding to 70S ribosomes in vitro. Tetracycline resistant [minimum inhibitory concentration (MIC) 32 to > 64 µg/mL] strains of Staphylococcus aureus (MRSA5 and RN4250 with ribosomal and efflux tetracycline resistance mechanisms, respectively) and S. pneumoniae (PBS382, ribosomal protection) were susceptible to omadacycline in vitro with MICs of < 0.06 to 0.25 µg/L [12].
Omadacycline has also been tested against Gram-positive and Gram-negative isolates collected in the USA and Europe as part of the 2017 SENTRY antimicrobial surveillance program (14,000 clinical isolates) [3]. The drug had potent in vitro activity against S. aureus, including methicillin-susceptible (MSSA) and methicillin-resistant (MRSA) isolates (overall MIC90 0.25 µg/mL, 2684 strains), coagulase-negative staphylococci, including MRSA isolates (overall MIC90 0.5 µg/mL, 373 strains), S. pneumoniae, including penicillin-resistant isolates (overall MIC90 0.12 µg/mL, 968 strains), β-haemolytic streptococci, including S. agalactiae (MIC90 0.25 µg/mL, 261 strains) and S. pyogenes (overall MIC90 0.12 µg/mL, 299 strains), viridans group streptococci (MIC90 0.12 µg/mL, 132 strains), Enterococcus faecalis, including vancomycin-resistant isolates (overall MIC90 0.25 µg/mL, 427 strains), E. faecium, including vancomycin-resistant isolates (overall MIC90 0.12 µg/mL, 218 strains), H. influenzae (MIC90 1 µg/mL, 556 strains) and Moraxella catarrhalis (MIC90 0.25 µg/mL, 313 strains).

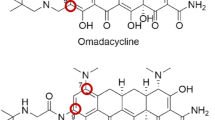
Chemical structure of omadacycline
Omadacycline has activity against most Enterobacteriaceae strains (MIC90 8 µg/mL with 87% of isolates susceptible at ≤ 4 µg/mL, 5993 strains), including Escherichia coli [overall MIC90 2 µg/mL, 2581 strains, including extended-spectrum β-lactamase (ESBL) producing isolates] and Klebsiella pneumoniae, (overall MIC90 8 µg/mL, 1269 strains including ESBL producing isolates) [3].
The US FDA identified MIC breakpoints for omadacycline for pathogens associated with ABSSSI are: S. aureus (includes MRSA isolates) susceptible ≤ 0.05 µg/mL, resistant ≥ 2 µg/mL; E. faecalis susceptible ≤ 0.25 µg/mL, resistant ≥ 1 µg/mL; S. lugdunensis, S. anginosus group and S. pyogenes susceptible ≤ 0.12 µg/mL, resistant ≥ 0.5 µg/mL; Enterobacteriaceae (K. pneumoniae and E. cloacae) susceptible ≤ 4 µg/mL, resistant ≥ 16 µg/mL. Those for pathogens associated with CABP are: S. aureus (MSSA only) susceptible ≤ 0.25 µg/mL, resistant ≥ 1 µg/mL; S. pneumoniae susceptible ≤ 0.12 µg/mL, resistant ≥ 0.5 µg/mL; Haemophilus spp. susceptible ≤ 2 µg/mL, resistant ≥ 8 µg/mL; Enterobacteriaceae (K. pneumoniae) susceptible ≤ 4 µg/mL, resistant ≥ 16 µg/mL [13].
The in vitro activity of omadacycline has also been tested against clinical isolates of various anaerobic bacteria, including Bacteroides fragilis (MIC90 4 µg/mL, 21 isolates), B. thetaiotaomicron (MIC90 4 µg/mL, 21 isolates), B. vulgatus (MIC90 1 µg/mL, 21 isolates), B. ovatus (MIC90 8 µg/mL, 15 isolates), Prevotella spp. (MIC90 2 µg/mL, 22 isolates) and P. asaccharolytica (MIC90 0.5 µg/mL, 21 isolates) [4]. Omadacycline has potent in vitro activity against the biothreat pathogens Y. pestis (MIC90 1 µg/mL, 30 strains) and B. anthracis (MIC90 0.06 µg/mL, 30 strains) [14].
As well as showing in vitro activity against tetracycline-resistant Gram-positive bacteria and Enterobacteriaceae, omadacycline was active against certain S. aureus, S. pneumoniae and H. influenzae strains carrying macrolide resistance genes (erm A, B and/or C) or ciprofloxacin resistance genes (gyrA and parC) and against β-lactamase positive H. influenzae [5]. Omadacycline showed activity against tetracycline-nonsusceptible isolates identified in patients in the phase III OPTIC, OASIS-1 and OASIS-2 trials, including S. aureus exhibiting a doxycycline nonsusceptible phenotype (MIC 0.25–0.5 µg/mL, 8 isolates) and S. pneumoniae with tetracycline and doxycycline and/or MLSB nonsusceptible phenotypes (MIC 0.03–0.06 µg/mL, 16 isolates). The drug also had activity against Gram-negative isolates carrying tetracycline efflux-pump genes, including E. coli (MIC 0.5–2 µg/mL, 8 isolates), E. cloacae (MIC 2 µg/mL, 2 isolates) and K. pneumoniae (MIC 2–16 µg/mL, 6 isolates) [15].
The area under the plasma concentration-time curve (AUC) divided by the MIC is considered the best pharmacodynamic/pharmacokinetic (PD/PK) predictor of tetracycline activity [16]. In vivo studies in murine models of thigh infection and pneumonia have confirmed that the AUC/MIC ratio is the best predictor of omadacycline efficacy (correlation coefficient 0.74 in the pneumonia model) [16, 17]. In an S. aureus thigh infection model (10 strains including MRSA), the mean 24 h free-drug AUC (fAUC)/MIC values for stasis and 1-log kill (from start of therapy) were 23.7 and 78.1 [17]. In a S. pneumoniae pneumonia model of strains with phenotypic resistance to other antibacterials, including tetracyclines, the mean 24 h plasma fAUC/MIC values for stasis and 1-log kill were 16–20 and 6.1–180, while the mean 24 h epithelial lining fluid (ELF) fAUC/MIC values for stasis and 1-log kill were 14–18 and 6.0–200. The mean 24 h plasma and ELF fAUC/MIC values for 2-log kill were 19–56 and 17–47 [16]. In PD/PK target attainment analyses of various omadacycline treatment regimens for patients with CABP, the probabilities of target attainment were ≥ 90% at an MIC of 0.5 µg/mL for S. pneumoniae (i.e. one dilution higher than MIC100) and 82.7–99.5% at an MIC of 1 µg/mL for H. influenzae (i.e. MIC90) [18].
In vitro, omadacycline inhibits binding of H-scopolamine to the M2 subtype of the muscarinic acetylcholine receptor. However, in phase III ABSSSI and CABP studies, omadacycline had no clinically meaningful effect on blood pressure, heart rate, ECG parameters and cardiac safety, and in a thorough QT study, there was no clinically meaningful prolongation of the QTc interval [5, 19].
2.2 Pharmacokinetics
Omadacycline demonstrated dose-proportional pharmacokinetics (over a 300–450 mg dose range) after administration of a single oral dose of in healthy adult volunteers [5]. The bioavailability of omadacycline is 34.5% after a single oral 300 mg dose. Exposure to omadacycline is similar between a 100 mg IV dose and a 300 mg oral dose administered in healthy, fasted volunteers. Steady-state peak plasma concentrations (Cmaxss) were achieved 0.5 h after administration of intravenous (IV) omadacycline 100 mg once daily and 2.5 h after administration of oral omadacycline 300 mg once daily. Mean Cmaxss values for the respective dosages were 2120 and 952 ng/mL and mean AUC values at steady state were 12,140 and 11,156 ng · h/mL [5].
The rate and extent of oral omadacycline absorption was reduced (Cmax by ≈ 40%; AUC by ≈ 60%) when a high-fat meal was ingested 2 h prior to drug administration. However, oral administration of the drug 4 h after a high-fat meal did not affect omadacycline pharmacokinetics. Likewise, ingesting food 2 h after administration of an oral dose of the drug had no significant effect on omadacycline pharmacokinetics. Omadacycline must be taken with water, and the patient must fast for at least 4 h prior to administration and must not have food or drink, apart from water, for 2 h and no dairy products, antacids or multivitamins for 4 h after administration [5].
The accumulation ratio of omadacycline is 1.5. Plasma protein binding is low (20%) and is concentration independent. The volume of distribution at steady state is 190 L after IV administration of omadacycline 100 mg, and the apparent volume of distribution after a single oral 300 mg dose is 794 L [5].
Omadacycline is not metabolized in the liver and is primarily excreted as the unchanged drug in urine (27% of a 100 mg IV dose and 14.4% of a radiolabelled 300 mg oral dose) and faeces (81.1% after oral administration). Systemic clearance at steady state is 8.8 L/h after IV administration of omadacycline 100 mg and apparent systemic clearance after a single oral 300 mg dose is 34.6 h. Renal clearance of omadacycline is 2.4–3.3 L/h and the terminal elimination half-life is 15.5–16 h [5].
Omadacycline has good lung penetration. At steady state, after IV administration of multiple doses of omadacycline in healthy volunteers, AUC24 in alveolar cells was 25.8-fold higher than plasma AUC24, and that in lung epithelial lining fluid was 1.5-fold higher [5, 20].
Age and gender had no clinically relevant effect on the pharmacokinetic profile of omadacycline in studies in volunteers [21]. The pharmacokinetic profile of omadacycline was similar in patients with end-stage renal disease on haemodialysis and volunteers, indicating dosage adjustment is not required in the presence of any degree of renal impairment [22]. The pharmacokinetics of omadacycline are not affected by hepatic impairment and dosage adjustments are not required [5].
2.3 Therapeutic Trials
2.3.1 Community-Acquired Bacterial Pneumonia
Omadacycline was non-inferior to moxifloxacin for the treatment of CABP in the phase III OPTIC study (NCT02531438). Patients with CABP were randomized to 7–14 days’ treatment with IV omadacycline 100 mg once every 12 h for two doses then once daily (n = 386) or IV moxifloxacin 400 mg once daily (n = 388). Patients had the option to transition to oral treatment (omadacycline 300 mg once daily, moxifloxacin 400 mg once daily) after ≥ 3 days of IV treatment. In the intent-to-treat (ITT) population, 81.1% of omadacycline and 82.7% of moxifloxacin recipients achieved early clinical response [assessed 72–120 h after the first dose and defined as survival, no rescue antibacterial therapy and improvement in at least two of four subject symptoms (cough, sputum production, pleuritic chest pain, dyspnoea) without deterioration in any of these four symptoms], and 87.6 and 85.1% achieved clinical success at post-therapy evaluation (assessed at 5–10 days after the last dose and defined as survival with resolution of signs and symptoms of the infection to the extent that further antibacterial therapy was not necessary). Clinical success at post therapy evaluation in patients with ≥ 1 causative bacterial pathogen identified (from respiratory or blood cultures or a culture-independent method) was 85.2 and 87.5% in omadacycline and moxifloxacin recipients with causative Gram-positive bacteria identified, 84.8 and 81.2% in patients with causative Gram-negative bacteria identified and 92.4 and 91.5% in patients with atypical pathogens identified. In the clinically evaluable population (n = 340 in the omadacycline group and 345 in the moxifloxacin group), clinical success at post-therapy evaluation was achieved in 92.9 and 90.4% of patients [23].
2.3.2 Skin and Skin Structure Infections
2.3.2.1 Phase III
Monotherapy with once-daily IV/oral omadacycline was non-inferior to twice-daily IV/oral linezolid as treatment for ABSSSI in the double-blind phase III OASIS-1 study (NCT02378480). Patients with ABSSSI known or suspected to be caused by a Gram-positive pathogen were randomized to treatment with IV omadacycline 100 mg once every 12 h for two doses then once daily, or IV linezolid 600 mg once every 12 h. After ≥ 3 days on IV therapy, patients on omadacycline could be switched to a 300 mg once daily oral dose and patients on linezolid could be switched to a 600 mg twice daily oral dose. In the modified ITT population (patients without a sole Gram-negative pathogen at baseline) early clinical response (defined as a reduction in lesion size of ≥ 20%) was observed in 84.8% of omadacycline (n = 316) and 85.5% of linezolid (n = 311) recipients. Clinical success at post therapy evaluation was achieved in 86.1 and 83.6% of omadacycline and linezolid recipients. In the clinically evaluable population (n = 269 in the omadacycline group and 260 in the linezolid group) clinical success at post-therapy evaluation was achieved in 96.3 and 93.5% of patients [24].
Monotherapy with once-daily oral omadacycline was non-inferior to twice-daily oral linezolid as treatment for ABSSSI in the double-blind phase III OASIS-2 study (NCT02877927). Patients were randomized to omadacycline 450 mg once daily for two doses then 300 mg once daily (n = 360) or linezolid 600 mg (n = 360) twice daily for a total treatment duration of 7–14 days (median duration was 8.2 and 8.0 days). In the modified ITT population (as per OASIS-1) early clinical response was observed in 87.5 and 82.5% of omadacycline and linezolid recipients, with 84.2 and 80.8% achieving clinical success at post-therapy evaluation. In the clinically evaluable population (n = 284 in the omadacycline group and 292 in the linezolid group) clinical success at post-therapy evaluation was achieved in 97.9 and 95.5% of patients [25].
2.3.2.2 Phase II
IV/oral omadacycline was noninferior to IV/oral linezolid as treatment for complicated skin and skin structure infections in the efficacy analyses of an investigator-blind, phase II safety and tolerability trial. In the ITT population, successful clinical response was achieved by 88.3 and 75.9% of omadacycline and linezolid recipients. In the clinically evaluable population, 98.0 and 93.2% of patients in the respective treatment groups had a successful clinical response. The clinical success rate was 97.2% (70 of 72 patients) and 92.7% (51 of 55 patients), respectively, in microbiologically evaluable patients with S. aureus infections. In this study, patients were randomized to omadacycline 100 mg IV once daily (with an option to transition to 200 mg orally once daily) or linezolid 600 mg IV twice daily (with an option to transition to 600 mg orally twice daily). Patients with suspected or documented Gram-negative infections received aztreonam (2 g IV once every 12 h) or matched placebo in the linezolid and omadacycline groups, respectively. A comparison of the efficacy of omadacycline and linezolid was a key secondary outcome [26].
2.4 Adverse Events
Adverse reactions occurring in ≥ 2% of patients with CABP treated with omadacycline in the OPTIC trial (n = 382) included increased alanine aminotransferase levels [3.7% (vs. 4.6% with moxifloxacin)], hypertension (3.4 vs. 2.8%), increased gamma-glutamyl transferase levels (2.6 vs. 2.1%), insomnia (2.6 vs. 2.1%), vomiting (2.6 vs. 1.5%), constipation (2.4 vs. 1.5%), nausea (2.4 vs. 5.4%), increased aspartate aminotransferase levels (2.1 vs. 3.6%) and headache (2.1 vs. 1.3%) [5]. Diarrhoea was reported in 1% of omadacycline recipients compared with 8% of moxifloxacin recipients. No omadacycline recipients and eight moxifloxacin recipients developed Clostridium difficile infection [23]. Discontinuation of treatment due to adverse events occurred in 5.5% of omadacycline recipients and 7.0% of moxifloxacin recipients. Eight omadacycline recipients (2%) and four moxifloxacin recipients (1%) died during the OPTIC trial; all were aged > 65 years and most had multiple comorbidities [5].
Adverse reactions occurring in ≥ 2% of patients with ABSSSI treated with omadacycline in the OASIS-1 and OASIS-2 trials (n = 691) included nausea [21.9% (vs. 8.7% with linezolid)], vomiting (11.4 vs. 3.9%), infusion site reactions (5.2 vs. 3.6%), increased alanine aminotransferase levels (4.1 vs. 3.6%), increased aspartate aminotransferase levels (3.6 vs. 3.5%), headache (3.3 vs. 3.0%) and diarrhoea (3.2 vs. 2.9%). Across both trials, treatment discontinuation due to adverse events occurred in 12 (1.7%) omadacycline and 10 (1.5%) linezolid recipients. Only one omadacycline recipient in each trial discontinued treatment because of nausea and vomiting [5].
Adverse events occurring in < 2% of patients treated with omadacycline in OPTIC, OASIS-1 and OASIS-2 included tachycardia, atrial fibrillation, anaemia, thrombocytosis, vertigo, abdominal pain, dyspepsia, fatigue, hypersensitivity, oral candidiasis, vulvovaginal mycotic infection, increased creatinine phosphokinase levels, increased bilirubin levels, increased alkaline phosphatase levels, dysgeusia, lethargy, oropharyngeal pain, pruritus, erythema and urticaria [5].
2.5 Ongoing Clinical Trials
A study comparing the safety and efficacy of oral omadacycline to that of oral nitrofurantoin as treatment for cystitis (NCT03425396) is currently recruiting patients.
References
Paratek Pharmaceuticals. Paratek announces FDA approval of NUZYRA™ (omadacycline) (media release). 2 Oct 2018.
Paratek Pharmaceuticals. Paratek announces acceptance of European Marketing Authorization application for oral and intravenous omadacycline (media release). 4 Oct 2018.
Huband MD, Pfaller MA, Streit JM, et al. Omadacycline in vitro activity against skin and skin structure, respiratory, and urinary tract pathogens collected from the U.S. and Europe during the SENTRY Surveillance Program (2017). (abstract no. 627 plus poster). In: ASM Microbe 2018. 2018.
Stapert L, Wolfe C, Shinabarger D, et al. In vitro activities of omadacycline and comparators against anaerobic bacteria. Antimicrob Agents Chemother. 2018;62(4):e00047–118.
Paratek Pharmaceuticals. Nuzyra (omadacycline): US prescribing information. 2018. https://www.accessdata.fda.gov/drugsatfda_docs/label/2018/209816_209817lbl.pdf. Accessed 23 Oct 2018.
Paratek Pharmaceuticals. Paratek, U.S. Department of Defense enter research agreement to study omadacycline against biodefense pathogens (media release). 11 Oct 2016.
Paratek Pharmaceuticals. Paratek Pharmaceuticals and Zai Lab announce collaboration, development and license agreement for omadacycline in China (media release). 24 Apr 2017.
Bayer AG, Bayer Pharmaceuticals Corporation, Paratek Pharmaceuticals Inc. Bayer and Paratek Pharmaceuticals sign collaboration agreement for the novel antibiotic BAY 73-6944/PTK 0796 (media release). 15 Sep 2003.
Merck & Co. Inc., Paratek Pharmaceuticals Inc. Merck and Paratek Pharmaceuticals sign collaboration agreement for novel antibiotic PTK 0796 currently in phase 1 testing (media release). 9 Mar 2006.
Novartis. Novartis gains exclusive worldwide rights to PTK 0796, in phase III study as potential first-in-class IV and oral broad-spectrum antibiotic (media release). 8 Oct 2009.
Tanaka SK, Steenbergen J, Villano S. Discovery, pharmacology, and clinical profile of omadacycline, a novel aminomethylcycline antibiotic. Bioorg Med Chem. 2016;24(24):6409–19.
Draper MP, Weir S, Macone A, et al. Mechanism of action of the novel aminomethylcycline antibiotic omadacycline. Antimicrob Agents Chemother. 2014;58(3):1279–83.
US Food and Drug Administration. Omadacycline injection and oral products: FDA identified breakpoints. 2018. https://www.fda.gov/Drugs/DevelopmentApprovalProcess/DevelopmentResources/ucm622612.htm. Accessed 9 Nov 2018.
Steenbergen J, Tanaka SK, Miller LL, et al. in vitro and in vivo activity of omadacycline against two biothreat pathogens, Bacillus anthracis and Yersinia pestis. Antimicrob Agents Chemother. 2017;61(5):e02434.
Mendes R.E., Castangiera M., Armstrong E.S., et al. Efficacy of omadacycline against molecularly characterized Gram-positive and Gram-negative pathogens causing infections in the phase 3 CABP and ABSSSI clinical trials [abstract no. 1364 plus poster]. In: ID Week 2018. 2018.
Lepak AJ, Zhao M, Marchillo K, et al. In vivo pharmacodynamic evaluation of omadacycline (PTK 0796) against Streptococcus pneumoniae in the murine pneumonia model. Antimicrob Agents Chemother. 2017;61(5):05.
Lepak AJ, Zhao M, Marchillo K, et al. In vivo pharmacodynamic evaluation of Omadacycline (PTK 0796) against Staphylococcus aureus (SA) in the murine thigh infection model [abstract no. 1531]. OFID. 2017;4(Suppl. 1):S478–9.
Bhavnani SM, Hammel JP, Lakota EA, et al. Pharmacokinetic-pharmacodynamic (PK-PD) target attainment analyses evaluating omadacycline dosing regimens for the treatment of patients with community-acquired bacterial pneumonia (CABP) for Streptococcus pneumoniae (Sp) and Haemophilus Influenzae (Hi) [abstract no. 3793]. In: ASM Microbe 2018. 2018.
Darpo B, Tzanis E, Garrity-Ryan L, et al. Cardiac safety of omadacycline in the IV/oral phase 3 acute bacterial skin and skin structure infection (ABSSSI) and in the IV/oral phase 3 community-acquired bacterial pneumonia (CABP) studies [abstract no. 1886]. OFID. 2017;4(Suppl.1):S544-S5.
Gotfried MH, Horn K, Garrity-Ryan L, et al. Comparison of omadacycline and tigecycline pharmacokinetics in the plasma, epithelial lining fluid, and alveolar cells of healthy adult subjects. Antimicrob Agents Chemother. 2017;61(9):e01135–217.
Tanaka K, Tzanis E, Villano S. Effect of age and gender on the pharmacokinetics of the oral and IV omadacycline, a new class of aminomethylcyclines [abstract no. P1318 and poster]. In: 26th ECCMID. 2016.
Berg JK, Tzanis E, Garrity-Ryan L, et al. Pharmacokinetics and safety of omadacycline in subjects with impaired renal function. Antimicrob Agents Chemother. 2018;62(2):e02057–117.
Stets R, Popescu M, Gonong J, et al. A phase 3 randomized, double-blind, multi-center study to compare the safety and efficacy of IV-to-oral omadacycline to moxifloxacin for the treatment of adult subjects with CABP (The OPTIC Study) [abstract no. 1883 plus poster]. OFID. 2017;4(Suppl. 1):S543-S4.
O’Riordan WA, Green S, Overcash JS, et al. A phase 3 randomized, double-blind, multi-centre study to compare the safety and efficacy of oral and IV omadacycline to linezolid for treating adult subjects with ABSSSI (the OASIS study) [abstract no. OS0606 and oral presentation]. In: 27th ECCMID. 2017.
O’Riordan W, Cardenas C, Sirbu A, et al. A phase-3 randomized, double-blind, multi-centre study to compare the safety and efficacy of oral omadacycline to oral linezolid for treating adult subjects with ABSSSI (OASIS-2 study) [abstract no. O0425]. In: 28th ECCMID. 2018.
Noel GJ, Draper MP, Hait H, et al. A randomized, evaluator-blind, phase 2 study comparing the safety and efficacy of omadacycline to those of linezolid for treatment of complicated skin and skin structure infections. Antimicrob Agents Chemother. 2012;56(11):5650–4.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Funding
The preparation of this review was not supported by any external funding.
Conflicts of interest
During the peer review process the manufacturer of the agent under review was offered an opportunity to comment on the article. Changes resulting from any comments received were made by the authors on the basis of scientific completeness and accuracy. A. Markham, a contracted employee of Adis/Springer, and Susan Keam, an employee of Adis/Springer, are responsible for the article content and declare no relevant conflicts of interest.
Additional information
This profile has been extracted and modified from the AdisInsight database. AdisInsight tracks drug development worldwide through the entire development process, from discovery, through pre-clinical and clinical studies to market launch and beyond.
Rights and permissions
About this article

Cite this article
Markham, A., Keam, S.J. Omadacycline: First Global Approval. Drugs 78, 1931–1937 (2018). https://doi.org/10.1007/s40265-018-1015-2
Published:
Issue Date:
DOI: https://doi.org/10.1007/s40265-018-1015-2