
Abstract
Burosumab (Crysvita®; Kyowa Hakko Kirin Co., Ltd. and Ultragenyx Pharmaceutical Inc.) is a fully human monoclonal antibody directed at fibroblast growth factor 23 (FGF23). Excessive FGF23 production has been implicated in various hypophosphataemic diseases. Inhibition of FGF23 by burosumab results in increased renal phosphate reabsorption and increased serum levels of phosphorus and active vitamin D. In February 2018, the EMA granted subcutaneous burosumab conditional marketing authorization for the treatment of X-linked hypophosphataemia (XLH) with radiographic evidence of bone disease in children one year of age and older and adolescents with growing skeletons. In April 2018, the US FDA approved burosumab for the treatment of XLH in adults and children one year of age and older. Multinational phase III trials of burosumab are currently underway in adult and paediatric patients with XLH. Burosumab is also being evaluated in the phase II setting in adults with tumour-induced osteomalacia and epidermal nevus syndrome in the USA, as well as in Japan and Korea. This article summarizes the milestones in the development of burosumab leading to its first global approval in the EU for XLH in paediatric patients.
Similar content being viewed by others
Avoid common mistakes on your manuscript.
1 Introduction
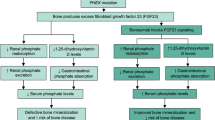
Burosumab (KRN23; Crysvita®), a fully human recombinant IgG1 monoclonal antibody directed at fibroblast growth factor 23 (FGF23) and discovered by Kyowa Hakko Kirin Co., Ltd., is being jointly developed by Kyowa Hakko Kirin Co., Ltd. and Ultragenyx Pharmaceutical Inc. for the treatment of hypophosphataemic diseases caused by excessive FGF23 production. In X-linked hypophosphataemia (XLH), the most common form of hereditary rickets (estimated prevalence 1:20,000 to 1:25,000 [1,2,3,4,5]), mutations in the phosphate-regulating endopeptidase homolog (PHEX) gene result in elevated circulating levels of FGF23 [6, 7]. Excess FGF23 activity results in impaired bone mineralization due to low serum phosphate levels caused by reduced renal tubular phosphate reabsorption. FGF23 also reduces renal 1α-hydroxylase activity, lowering serum levels of 1,25-dihydroxyvitamin D and thus reducing phosphate absorption from the gastrointestinal tract. Conventional therapy for XLH involves multiple daily doses of calcitriol and phosphate supplements, which can improve bone mineralization but do not target the underlying deficiencies in renal phosphate reabsorption and 1,25-dihydroxyvitamin D production [6, 7].

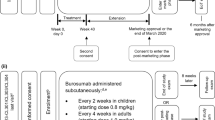
Burosumab (solution for injection) has received conditional marketing authorization in the EU and Iceland as a subcutaneous therapy for XLH with radiographic evidence of bone disease in children one year of age and older and adolescents with growing skeletons [8, 9]. This approval followed a positive opinion from the EU Committee for Medicinal Products for Human Use (CHMP) in December 2017 [9]. Conditional authorization was based on results from phase II trials in patients aged 5–12 years (NCT02163577; UX023-CL201) and 1–4 years (NCT02750618; UX023-CL205), and requires the completion of ongoing trials in paediatric patients [10]. The recommended starting dose is 0.4 mg/kg, with a maintenance dose of 0.8 mg/kg (up to a maximum dose of 90 mg) administered once every 2 weeks [8]. Fasting serum phosphate should be measured every 2 weeks during the initial month of treatment, every 4 weeks during the subsequent 2 months and thereafter as appropriate (including 4 weeks after any dose adjustment). The target fasting serum phosphate level should be in the lower end of the normal reference range for age. Burosumab is contraindicated in patients with fasting serum phosphate above the normal range for age, patients with severe renal impairment or end stage renal disease and patients receiving concomitant oral phosphate and vitamin D analogues [8].
The European Medicines Agency (EMA) listed burosumab as a medicine for children that represents an outstanding contribution to public health in their 2017 Human Medicines Highlights [11].
In the USA, burosumab was assigned priority review status and, in April 2018, was approved for the treatment of XLH in adults and children one year of age and older [12]. This followed the US FDA having previously granted burosumab a breakthrough therapy designation for the treatment of XLH in paediatric patients one year of age and older, and a rare paediatric disease designation for the treatment of XLH [13]. Burosumab has orphan drug status in both the EU and USA [14]. Ongoing multinational trials are evaluating burosumab in XLH in paediatric patients (phase II and III) and adult patients (phase III). In Japan, a phase III trial is underway in paediatric patients with XLH.
Burosumab is also currently undergoing phase II development in tumour-induced osteomalacia (TIO) and epidermal nevus syndrome (ENS) in the USA, Japan and Korea. The US FDA granted burosumab an orphan drug designation for the treatment of TIO in June 2017. Development of an intravenous formulation of burosumab appears to have been discontinued.
1.1 Company Agreements
In August 2013, Kyowa Hakko Kirin entered into a collaboration and license agreement with Ultragenyx [15]. Under this agreement, the companies will co-develop burosumab in the USA, the EU and Canada. Developmental activities in the XLH indication will be led by Ultragenyx [15]. In the USA and Canada, for the first five years post-launch, Ultragenyx will launch burosumab and share development and commercial costs evenly with Kyowa Hakko Kirin [16]. After the first five years, Kyowa Hakko Kirin will take over the majority of commercialization efforts in the USA and Canada. Throughout these periods, Kyowa Hakko Kirin will book sales for burosumab in the USA and Canada. Kyowa Hakko Kirin will be responsible for the global manufacture and supply of burosumab, and will commercialise burosumab in the EU. Ultragenyx will develop and commercialise burosumab in Mexico, Central and South America [16].
In May 2017, Ultragenyx entered into an agreement with a subsidiary of Kyowa Hakko Kirin granting Ultragenyx the right to commercialize burosumab in Turkey for a certain period of time, after which the subsidiary will have the option to take over commercialization efforts [17].
2 Scientific Summary
2.1 Pharmacodynamics
Burosumab is a fully human recombinant IgG1 monoclonal antibody that binds to FGF23 (KD ≈ 10−11 M for human, monkey and rabbit FGF23 [18]), inhibiting FGF23 signalling and thus increasing renal tubular phosphate reabsorption and serum levels of 1,25-dihydroxyvitamin D [8].
In rabbits, a single intravenous dose of burosumab 3, 10 or 30 mg/kg increased serum phosphate and 1,25-dihydroxyvitamin D levels and decreased urinary phosphate and calcium levels [18]. Similar effects were observed in adult monkeys; a single intravenous dose of burosumab increased serum phosphate, tubular maximal reabsorption of phosphate per glomerular filtration rate (TmP/GFR) and serum 1,25-dihydroxyvitamin D levels (minimum effective dose 0.1 and 0.3 mg/kg for increases in serum phosphate and 1,25-dihydroxyvitamin D levels, respectively) [18]. In both rabbits and monkeys administered burosumab, ectopic mineralization of tissues and organs was associated with supraphysiological serum phosphate levels > 8 mg/dL (normal range for paediatric patients: 3.2–6.1 mg/dL) [18].
In a PHEX-deficient mouse (Hyp mouse) model of XLH, a single subcutaneous 4 or 16 mg/kg dose of a murine anti-FGF23 antibody increased serum phosphate and 1,25-dihydroxyvitamin D levels in a dose-dependent manner [19]. Serum phosphate levels were normalized within 3 days of the injection. The antibody also normalized renal phosphate excretion and increased the expression of type IIa sodium-phosphate co-transporter and 1α-hydroxylase in the kidney while decreasing 24-hydroxylase expression. With weekly dosing during the growth period, the murine anti-FGF23 antibody significantly improved elongation of the femur (p < 0.01 vs. control Hyp mice). Improvements in bone mineralization and cartilage development were also observed [19]. In adult Hyp mice, weekly subcutaneous 4 or 16 mg/kg doses of murine anti-FGF23 antibody improved bone mineralization but did not correct short bones [18].
In a single-dose, dose-escalation phase I trial (NCT00830674; KRN23-US-02) in adults with XLH (n = 38), burosumab produced significant increases in TmP/GFR, serum phosphate [(measured as inorganic phosphorus (Pi)] levels and serum 1,25-dihydroxyvitamin D levels (p < 0.05 vs. placebo for intravenous doses 0.03–0.3 mg/kg and subcutaneous doses 0.3–1 mg/kg) [20]. Subcutaneous burosumab had a longer duration of action than intravenous burosumab and was generally not associated with elevations in serum calcium levels, 24-h urine calcium excretion, or fasting 2-h urine calcium/creatinine ratio. With both modes of administration, the magnitude and duration of responses were dose related [20]. In patients with XLH administered repeated doses of burosumab in clinical trials, effects of burosumab were consistent with those observed in the single-dose trial (i.e. increased TmP/GFR, serum phosphorus and serum 1,25-dihydroxyvitamin D) [18]. In a phase II trial in paediatric patients with XLH (NCT02163577; UX023-CL201), burosumab increased biomarkers of bone turnover (P1NP and CTx), while decreasing biomarkers of rickets (ALP and BALP) [18]. Burosumab pharmacodynamics do not differ substantially between adult and paediatric patients [8].
In clinical trials, anti-drug antibodies (ADAs) to burosumab were detected in a small percentage of burosumab recipients [8]. These patients tested positive for ADAs prior to receiving burosumab and the presence of ADAs did not impact the efficacy or tolerability of burosumab [8].
2.2 Pharmacokinetics
Subcutaneous burosumab has linear time-invariant pharmacokinetics, with peak serum concentration (Cmax) and area under the concentration-time curve (AUC) increasing in a dose-proportional manner (dose range 0.1–2.0 mg/kg) [8]. There is a direct pharmacokinetic-pharmacodynamic relationship between serum concentrations of burosumab and serum levels of phosphate, with these variables increasing together and peaking at approximately the same time after dose administration [8]. In adults with XLH, increases in the AUC for change from baseline in TmP/GFR, serum Pi and serum 1,25-dihydroxyvitamin D were linearly correlated with increasing burosumab AUC [6].
Following subcutaneous injection, burosumab is absorbed slowly and maximum serum concentrations are reached in 5–10 days [8]. Absolute bioavailability is almost 100% [20]. The distribution of burosumab appears to be limited; the observed volume of distribution approximates the volume of plasma in patients with XLH [8].
Burosumab is expected to be metabolized and eliminated via immunoglobulin clearance pathways, with the antibody being broken down into small peptides and amino acids; hepatic mechanisms are unlikely to be involved [8]. Burosumab clearance is low and dependent on body weight [8]. In a typical 70 kg adult and 30 kg paediatric patient with XLH, estimated clearance is 0.290 and 0.136 L/day respectively after subcutaneous administration [8, 18]. The mean terminal half-life of burosumab is ≈ 19 days [8].
Burosumab pharmacokinetics have not been studied in patients with renal or hepatic impairment [18]. The pharmacokinetic properties of burosumab do not differ substantially between adult and paediatric patients [8].
Features and properties of burosumab
Alternative names | Crysvita; KRN-23; UX 023 |
Class | Monoclonal antibodies |
Mechanism of action | FGF23 inhibitor |
Route of administration | Subcutaneous (approved), intravenous (discontinued) |
Pharmacodynamics | Increases tubular maximal reabsorption of phosphate per glomerular filtration rate (TmP/GFR) and serum phosphate and 1,25-dihydroxyvitamin D levels in vivo |
Pharmacokinetics | Linear pharmacokinetics; direct pharmacokinetic-pharmacodynamic relationship between serum concentrations of burosumab and serum levels of phosphate; no substantial differences in pharmacokinetic properties between adult and paediatric patients |
Most frequent adverse events | Headache, cough, tooth abscess, injection site reaction or erythema, nasopharyngitis, upper respiratory tract infection, vomiting, pain in extremity or back, restless legs syndrome, nausea, dizziness |
ATC codes | |
WHO ATC code | D11A-X (other dermatologicals); M05B-X05 (burosumab) |
EphMRA ATC code | D11A (other dermatological preparations); M5 (other drugs for disorders of the musculo-skeletal system) |
Chemical name | Immunoglobulin G1-kappa, anti-(human Fibroblast growth factor 23 (FGF-23, Phosphatonin, Tumor-derived hypophosphatemia-inducing factor)); human monoclonal antibody: gamma1 heavy chain (1-447) (human VH (human IGHV1-46*01 (95%) -(IGHD)-IGHJ3*02 (94%)) (8.8.10) (1-117) -human IGHG1*01 (118-447)) (220-213’)-disulfide with kappa light chain (1’-213’) (human V-KAPPA (human IGKV1D-13*01 (98%) -IGKJ3*01) (6.3.8) (1’-106’) human IGKC*01 (107’-213’)) dimer (226-226’’:229-229’’)-bisdisulfide |
2.3 Therapeutic Trials
2.3.1 In Paediatric Patients with XLH
In a randomized, open-label, dose-finding phase II trial (NCT02163577; UX023-CL201), paediatric patients receiving subcutaneous burosumab once every 2 weeks (n = 26) or once every 4 weeks (n = 26) had significant improvements in Rickets Severity Score (RSS) total score from baseline to week 40 (primary efficacy endpoint) [least-squares mean (LSM) change of − 1.06 and − 0.73, respectively; p < 0.0001; intent-to-treat (ITT) analysis] [8, 18]. RSS total scores at baseline and week 40 were 1.92 and 0.75 in patients receiving burosumab every 2 weeks and 1.67 and 1.06 in patients receiving burosumab every 4 weeks [18]. Significant improvements from baseline were also observed in RSS total score at week 64, RSS wrist scores at weeks 40 and 64, and RSS knee scores at weeks 40 and 64 in patients receiving burosumab every 2 weeks (p < 0.0001) or every 4 weeks (p ≤ 0.0022) [18]. Burosumab was associated with significant improvements from baseline in Radiographic Global Impression of Change (RGI-C) global score, wrist score and knee scores at weeks 40 and 64 (p < 0.0001 in both regimen groups) [18]. Mean serum phosphate level increased from 0.77 mmol/L at baseline to 1.07 and 1.08 mmol/L at weeks 40 and 64 in patients receiving burosumab every 2 weeks [8]. In the group receiving burosumab every 2 weeks (EU-approved regimen), the average dose of burosumab was 0.73, 0.98 and 1.04 mg/kg at weeks 16, 40 and 60 respectively (maximum dose 2.0 mg/kg). Patients in this study were children aged 5–12 years (inclusive; mean age 8.5 years) with open growth plates. The majority of patients had radiographic evidence of rickets at baseline and had received prior conventional therapy for XLH [8].
In a multicentre, open-label phase II trial (NCT02750618; UX023-CL205), paediatric patients receiving subcutaneous burosumab once every 2 weeks (n = 13) had significant improvements in mean serum phosphorus level from baseline to each study visit (weeks 1, 4, 8, 12, 15, 20, 32 and 40; p < 0.0001) [primary endpoint] [18]. Mean serum phosphate level increased from 0.81 mmol/L at baseline to 1.12 mmol/L at week 40 [8, 18], while mean serum total alkaline phosphatase activity decreased from 549 U/L at baseline to 335 U/L at week 40 [8]. Burosumab also significantly improved RSS total score from baseline to week 40 (2.92 vs. 1.19; LSM change of − 1.73; p < 0.0001) and RGI-C global score from baseline to week 40 (p < 0.0001) [8]. At week 40, all patients were considered RGI-C responders (i.e. RGI-C global score ≥ + 2.0) [8, 18]. Burosumab was administered at a starting dose of 0.8 mg/kg, which could be increased to 1.2 mg/kg during the trial [18]. Patients were aged 1–4 years (inclusive; mean age 2.9 years) with radiographic evidence of rickets at baseline [8]. The majority of patients had received prior conventional therapy for XLH [8].
2.3.2 In Adult Patients with XLH
In a randomized, placebo-controlled phase III trial (NCT02526160; UX023-CL303), significantly more burosumab recipients (n = 68) than placebo recipients (n = 66) achieved serum phosphorus levels above the lower limit of normal at week 24 (94 vs. 8%; p < 0.0001; primary endpoint) [media release presentation] [21]. Burosumab was also associated with a significant improvement in stiffness (p = 0.0122) [as measured using the Western Ontario and McMaster Universities Osteoarthritis Index (WOMAC)] relative to placebo at week 24. There was no significant difference between burosumab and placebo recipients in pain at week 24 [21]. The rate of fracture healing in the 52% of patients with identified fractures or pseudofractures at baseline was 43% in the burosumab group and 8% in placebo group at week 24 (media release presentation) [22]. Benefits of burosumab were generally maintained during open-label treatment over weeks 24–48, with patients who continued treatment with burosumab showing sustained normal phosphorus levels and patients who crossed over from placebo to burosumab at the end of the double-blind period showing serum phosphorus normalization; both groups showed improvements in stiffness, pain, physical function and fracture healing during the open-label period [22]. Patients in this study were randomized to receive either subcutaneous burosumab 1 mg/kg or placebo every 4 weeks for a double-blind period of 24 weeks, followed by an open-label period in which all patients received burosumab 1 mg/kg every 4 weeks [21].
In an open-label phase I/II trial (NCT01340482; KRN23-INT-001), subcutaneous burosumab administered once every 4 weeks resulted in a maximum fasting serum Pi within the normal range (> 2.5 to ≤ 4.5 mg/dL) being achieved by 14.8, 37.0, 74.1 and 88.5% of patients on day 7 after doses 1, 2, 3 and 4 respectively (vs. 3.7% at baseline; primary efficacy outcome) [23]. Patients in this study (n = 28) were administered four doses of burosumab with stepwise dose escalation (0.05, 0.1, 0.3 and 0.6 mg/kg for doses 1, 2, 3 and 4, respectively) [23]. The majority of patients (n = 22) subsequently enrolled in a 12-month extension study (NCT01571596; KRN23-INT-002) [23].
During the open-label, phase I/II extension study (NCT01571596; KRN23-INT-002), burosumab increased serum Pi in all 22 patients, with 57.9–85.0% of patients attaining normal maximum fasting serum Pi (> 2.5 to ≤ 4.5 mg/dL) in each dose cycle [23]. At trough levels, 25.0–42.9% of patients maintained a normal serum Pi. From dose 2 of the dose-escalation trial to dose 12 of the extension study, all post-dose serum Pi values were significantly (p < 0.05) higher than dose-escalation baseline value (with the exception of the visits 5–6 weeks after the last dose of each trial). Following each dose, peak serum 1,25-dihydroxyvitamin D was significantly higher than at dose-escalation baseline (p < 0.01). Throughout the extension study, TmP/GFR levels (including troughs) were significantly greater (p < 0.05) than at dose-escalation baseline (with the exception of the visits > 4 weeks after the last dose). In this extension study, patients received burosumab 0.1–1.0 mg/kg every 4 weeks for 12 months. The first dose of burosumab was administered at a mean of 53 days after the last dose of burosumab was received in the dose-escalation trial [23].
In an open-label, phase IIb long-term extension study (NCT02312687; UX023-CL203) in patients who had participated in NCT01340482 (KRN23-INT-001) or NCT01571596 (KRN23-INT-002) [n = 20], burosumab significantly increased mean serum phosphorus, TmP/GFR and serum 1,25-dihydroxyvitamin D from baseline to week 24 (p < 0.005) [24]. Patients received subcutaneous burosumab 0.3, 0.6 or 1.0 mg/kg every 4 weeks. All patients received their first dose of burosumab > 12 months after their last dose in the earlier trial [24].
2.3.3 In Adult Patients with TIO or ENS
In an open-label phase II trial (NCT02304367; UX023T-CL201), subcutaneous burosumab (0.3–2.0 mg/kg) once every 4 weeks was associated with significant increases in mean serum Pi (p < 0.0001), TmP/GFR (p = 0.0002), serum 1,25-dihydroxyvitamin D (p = 0.003) and serum CTx (p = 0.01) from baseline to week 24 in an interim analysis of data for 8 patients who had completed ≥ 24 weeks of therapy [25]. Mean serum Pi increased from 0.55 mmol/L at baseline to 0.84 mmol/L at week 24. In one patient with bone biopsy data available for both baseline at week 48, there was an improvement from severe to mild osteomalacia. Of the patients in this interim analysis, seven had persistent TIO and one had ENS-associated osteomalacia. At baseline, osteomalacia was observed in all evaluable iliac crest bone biopsies (n = 5) [25].
Key clinical trials of burosumab
Drug(s) | Indication | Phase | Status | Location(s) | Identifier | Sponsor(s) |
|---|---|---|---|---|---|---|
Burosumab | XLH in adults | I/II | Completed | USA, Canada | NCT01340482; KRN23-INT-001 | Kyowa Hakko Kirin |
Burosumab | XLH in adults | I/II | Completed | USA, Canada | NCT01571596; KRN23-INT-002 | Kyowa Hakko Kirin |
Burosumab | XLH in pts aged 5–12 years | II | Active, not recruiting | Multinational | NCT02163577; UX023-CL201 | Ultragenyx; Kyowa Hakko Kirin |
Burosumab | XLH in adults | IIb | Active, not recruiting | USA | NCT02312687; UX023-CL203 | Ultragenyx; Kyowa Hakko Kirin |
Burosumab | TIO or ENS in adults | II | Active, not recruiting | USA | NCT02304367; UX023T-CL201 | Ultragenyx |
Burosumab | TIO or ENS in adults | II | Active, not recruiting | Japan, Korea | NCT02722798; JapicCTI-163191; KRN23-002 | Kyowa Hakko Kirin |
Burosumab | XLH in pts aged 1–4 years | II | Active, not recruiting | USA | NCT02750618; UX023-CL205 | Ultragenyx |
Burosumab, placebo | XLH in adults | III | Active, not recruiting | Multinational | NCT02526160; UX023-CL303 | Ultragenyx; Kyowa Hakko Kirin |
Burosumab | XLH in adults | III | Active, not recruiting | Multinational | NCT02537431; UX023-CL304 | Ultragenyx; Kyowa Hakko Kirin |
Burosumab, oral phosphate/active vitamin D therapy | XLH in pts aged 1–12 years | III | Active, not recruiting | Multinational | NCT02915705; UX023-CL301 | Ultragenyx; Kyowa Hakko Kirin |
Burosumab | XLH in pts aged 1–12 years | III | Active, not recruiting | Japan | NCT03233126; JapicCTI-173614; KRN23-003 | Kyowa Hakko Kirin |
2.4 Adverse Events
Burosumab was generally well tolerated by adult and paediatric patients with XLH in clinical trials [18, 23, 24]. In the safety analysis set of the double-blind phase III NCT02526160 (UX023-CL303) trial, which included adults with XLH exposed to either burosumab (n = 68) or placebo (n = 66) for up to 367 days (≈ 48 total subject-years of burosumab exposure), treatment-emergent adverse events (TEAEs) were reported in 94.1% of burosumab recipients and 92.4% of placebo recipients [18]. The most common TEAEs (incidence ≥ 10% and higher than with placebo) were back pain (14.7% of burosumab recipients vs. 9.1% of placebo recipients), nasopharyngitis (13.2 vs. 9.1%), tooth abscess (13.2 vs. 7.6%), headache (11.8 vs. 7.6%), restless legs syndrome (11.8 vs. 6.1%), nausea (10.3 vs. 9.1%) and dizziness (10.3 vs. 6.1%). TEAEs that were considered to be related to the study drug occurred in 44.1% of burosumab recipients and 39.4% of placebo recipients. Grade 3 TEAEs were reported in 11.8% of burosumab recipients and 13.6% of placebo recipients, the majority of which were not considered to be related to the study drug. Serious TEAEs were reported in 2.9% of burosumab recipients and 3.0% of placebo recipients; none of these were considered to be related to the study drug. No TEAE led to treatment discontinuation or death in either study arm [18].
With respect to TEAEs of specific interest in NCT02526160 (UX023-CL303), injection site reactions were reported in 11.8% of burosumab recipients and 12.1% of placebo recipients, hypersensitivity (immunogenicity) was reported in 5.9 and 6.1%, and hyperphosphataemia was reported in 5.9 and 0% [18]. All cases of restless legs syndrome (11.8% of burosumab recipients vs. 7.6% of placebo recipients) were of mild or moderate severity; none were considered serious. There were no cases of ectopic mineralization in either study arm [18].
In the phase II NCT02163577 (UX023-CL201) trial, 52 paediatric patients with XLH received burosumab every 2 weeks or every 4 weeks for ≥ 64 weeks (data cut-off date 1st December 2016; after week 64, all patients received burosumab every 2 weeks) [18]. TEAEs were reported in all patients. Those most commonly reported (incidence ≥ 30%) in patients who were randomized to receive burosumab every 2 weeks (EU-approved regimen) were headache (69.2%), cough (65.4%), injection site erythema (46.2%), nasopharyngitis (42.3%), upper respiratory tract infection (38.5%), vomiting (38.5%), injection site reaction (38.5%), pain in extremity (38.5%), pyrexia (34.6%), oropharyngeal pain (30.8%) and arthralgia (30.8%). TEAEs considered to be related to the study drug were reported in 65.4% of patients receiving burosumab every 2 weeks and 73.1% of patients receiving burosumab every 4 weeks. Grade 3 TEAEs were reported by one patient in each group (3.8%) and were not considered to be related to the study drug. A patient in the 4-weekly burosumab group experienced two serious TEAEs (concurrent pyrexia and myalgia) which were of moderate severity; these may have been related to the study drug. No TEAE led to treatment discontinuation or death in either study arm [18].
There were no cases of serum phosphorus exceeding the upper limit of normal in the either of the phase II studies in paediatric patients, nor were there any cases of restless legs syndrome [18]. Injection site reactions (generally of mild severity) occurred in ≈ 57% of paediatric patients receiving burosumab in phase II trials [8]. The potential hypersensitivity reactions that occurred most frequently were rash (22%), injection site rash (6%) and urticaria (4%). These were of mild or moderate severity [8].
2.5 Ongoing Clinical Trials
There are a number of ongoing phase II and III trials of burosumab (not recruiting). A multinational, randomized, open-label, active-controlled phase III trial (NCT02915705; UX023-CL301) is currently comparing burosumab to conventional therapy (oral phosphate and active vitamin D) in paediatric patients aged 1–12 years with XLH (a specific obligation for the EU conditional approval [18]). The phase II trials in paediatric patients with XLH aged 5–12 years (NCT02163577; UX023-CL201) and 1–4 years (NCT02750618; UX023-CL205) are still underway. In Japan, an open-label phase III trial (NCT03233126; JapicCTI-173614; KRN23-003) evaluating burosumab in patients aged 1–12 years with X-linked hypophosphataemic rickets/osteomalacia has been initiated.
Ongoing multinational phase III trials that are evaluating burosumab in adults with XLH include the randomized, double-blind, placebo-controlled NCT02526160 (UX023-CL303) trial and the open-label NCT02537431 (UX023-CL304) trial, the latter of which aims to establish effects on bone quality and osteomalacia. In the USA, the open-label, long-term extension study (NCT02312687; UX023-CL203) of burosumab in adults with XLH who participated in NCT01340482 (KRN23-INT-001) or NCT01571596 (KRN23-INT-002) is still underway.
Burosumab is currently under investigation in TIO and ENS in open-label phase II trials in the USA (NCT02304367; UX023T-CL201) and Japan/Korea (NCT02722798; JapicCTI-163191; KRN23-002).
3 Current Status
Burosumab received its first global approval on 23rd February 2018 for the treatment of X-linked hypophosphataemia (XLH) with radiographic evidence of bone disease in children one year of age and older and adolescents with growing skeletons in the EU.
References
Ruppe MD. GeneReviews®: X-linked hypophosphatemia. 2017. https://www.ncbi.nlm.nih.gov/. Accessed 6 Apr 2018.
Beck-Nielsen SS, Brock-Jacobsen B, Gram J, et al. Incidence and prevalence of nutritional and hereditary rickets in southern Denmark. Eur J Endocrinol. 2009;160:491–7.
Shore RM, Chesney RW. Rickets: part II. Pediatr Radiol. 2013;43(2):152–72.
Endo I, Fukumoto S, Ozono K, et al. Nationwide survey of fibroblast growth factor 23 (FGF23)-related hypophosphatemic diseases in Japan: prevalence, biochemical data and treatment. Endocr J. 2015;62(9):811–6.
Carpenter TO. New perspectives on the biology and treatment of X-linked hypophosphatemic rickets. Pediatr Clin N Am. 1997;44(2):443–66.
Zhang X, Imel EA, Ruppe MD, et al. Pharmacokinetics and pharmacodynamics of a human monoclonal anti-FGF23 antibody (KRN23) in the first multiple ascending-dose trial treating adults with X-linked hypophosphatemia. J Clin Pharmacol. 2016;56(2):176–85.
Zhang X, Peyret T, Gosselin NH, et al. Population pharmacokinetic and pharmacodynamic analyses from a 4-month intradose escalation and its subsequent 12-month dose titration studies for a human monoclonal anti-FGF23 antibody (KRN23) in adults with X-linked hypophosphatemia. J Clin Pharmacol. 2016;56(4):429–38.
European Medicines Agency. CRYSVITA (Burosumab): summary of product characteristics. 2018. http://ec.europa.eu/. Accessed 4 Apr 2018.
Ultragenyx Pharmaceutical Inc. Kyowa Kirin and Ultragenyx announce Crysvita® (burosumab) receives conditional marketing authorization in Europe for the treatment of X-linked hypophosphatemia in children. [media release]. 23 Feb 2018. http://ir.ultragenyx.com/.
European Medicines Agency. New medicine for rare bone disease: Crysvita, a medicine for the treatment of X-linked hypophosphataemia, recommended for conditional approval. 2017. http://www.ema.europa.eu/. Accessed 4 Apr 2018.
European Medicines Agency. Human medicines highlights 2017. 2018. http://www.ema.europa.eu/. Accessed 6 Apr 2018.
US FDA. FDA approves first therapy for rare inherited form of rickets, x-linked hypophosphatemia. [media release]. 17 Apr 2018. http://www.fda.gov/
Ultragenyx Pharmaceutical, Kyowa Hakko Kirin. Ultragenyx and Kyowa Hakko Kirin announce FDA acceptance and priority review designation of burosumabs biologics license application. [media release]. 10 Oct 2017. http://www.ultragenyx.com.
Ultragenyx Pharmaceutical. Ultragenyx granted EU orphan drug designation for KRN23 for the treatment of X-linked hypophosphatemia. [media release]. 30 Oct 2014. http://www.ultragenyx.com.
Ultragenyx Pharmaceutical, Inc. Ultragenyx announces collaboration with Kyowa Hakko Kirin to develop and commercialize phase 2-stage KRN23 for X-linked hypophosphatemia. [media release]. 3 Sep 2013. http://www.ultragenyx.com.
Ultragenyx Pharmaceutical Inc. Form 10-Q. 2017. http://ir.ultragenyx.com/. Accessed 4 Apr 2018.
Ultragenyx Pharmaceutical. Ultragenyx reports second quarter 2017 financial results and corporate update 2017. http://ir.ultragenyx.com/. Accessed 4 Apr 2018.
European Medicines Agency. CHMP assessment report: Crysvita (burosumab). 2017. http://www.ema.europa.eu/. Accessed 4 Apr 2018.
Aono Y, Yamazaki Y, Yasutake J, et al. Therapeutic effects of anti-FGF23 antibodies in hypophosphatemic rickets/osteomalacia. J Bone Miner Res. 2009;24:1879–88.
Carpenter TO, Imel EA, Ruppe MD, et al. Randomized trial of the anti-FGF23 antibody KRN23 in X-linked hypophosphatemia. J Clin Invest. 2014;124(4):1587–97.
Ultragenyx Pharmaceutical. Ultragenyx, Kyowa Hakko Kirin and Kyowa Kirin International announce positive 24-week data from adult phase 3 study of burosumab (KRN23) in X-linked hypophosphatemia. [media release]. 18 Apr 2017. http://www.ultragenyx.com.
Ultragenyx Pharmaceutical, Kyowa Kirin International, Kyowa Hakko Kirin. Ultragenyx and Kyowa Kirin announce positive 48-week data from adult phase 3 study of burosumab (KRN23) in X-linked hypophosphatemia. [media release]. 4 Dec 2017. http://www.ultragenyx.com.
Imel EA, Zhang X, Ruppe MD, et al. Prolonged correction of serum phosphorus in adults with X-linked hypophosphatemia using monthly doses of KRN23. J Clin Endocrinol Metab. 2015;100(7):2565–73.
Ruppe M, Peacock M, Weber T, et al. Clinical and radiographic characteristics of adult X-linked hypophosphatemia (XLH) in a cohort of patients treated with KRN23, an antibody to FGF23 [abstract no. MO0319]. In: 38th Annual Meeting of the American Society for Bone and Mineral Research. 2016.
Carpenter T, Miller PD, Weber T, et al. Effects of KRN23, an anti-FGF23 antibody, in patients with tumor-induced osteomalacia or epidermal nevus syndrome-associated osteomalacia: Interim results from a phase 2 study [abstract no. P531]. Osteoporos Int. 2017;28(Suppl 1):S337–8.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Funding
The preparation of this review was not supported by any external funding.
Conflict of interest
During the peer review process the manufacturer of the agent under review was offered an opportunity to comment on the article. Changes resulting from any comments received were made by the author on the basis of scientific completeness and accuracy. Yvette Lamb is a salaried employee of Adis/Springer, is responsible for the article content and declares no relevant conflicts of interest.
Additional information
This profile has been extracted and modified from the AdisInsight database. AdisInsight tracks drug development worldwide through the entire development process, from discovery, through pre-clinical and clinical studies to market launch and beyond.
Rights and permissions
About this article

Cite this article
Lamb, Y.N. Burosumab: First Global Approval. Drugs 78, 707–714 (2018). https://doi.org/10.1007/s40265-018-0905-7
Published:
Issue Date:
DOI: https://doi.org/10.1007/s40265-018-0905-7