
Abstract
Microsatellite instability-high/DNA mismatch repair deficient tumors are found across the cancer spectrum and often harbor markedly increased numbers of mutations when compared to microsatellite stable/DNA mismatch repair proficient tumors. As a result of this high mutational load, tumor-infiltrating lymphocyte density is increased and more immunogenic neoepitopes are expressed, leading to upregulation of immune checkpoints in these tumors. Checkpoint inhibitors such as pembrolizumab and nivolumab, both immunoglobulin G4 (IgG4) monoclonal antibodies that block interactions between the programmed cell death receptor-1 and its ligands, have significant activity in this tumor class. This review will focus on hypermutated tumors and immuno-oncology drug development for this biologically unique tumor type, with an emphasis on FDA-approved immunotherapies for these cancers, as well as a short discussion of the many therapeutic and scientific challenges ahead in order to optimize the uses of this new class of drug.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
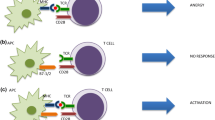
DNA repair systems normally correct DNA replication errors; however, a defect in this system can lead to microsatellite instability, and consequently, differences in tandem nucleotide repeats between tumor DNA and normal DNA result. |
Microsatellite instability-high (MSI-H)/DNA mismatch repair deficient (dMMR) tumors have high mutational loads and numbers of antigenic neoepitopes. |
Pembrolizumab and nivolumab are IgG4 monoclonal antibodies that block interactions between the programmed cell death receptor-1 (PD-1) and its ligands, PD-L1 and PD-L2. |
Checkpoint inhibitors, especially those targeting PD-1 and PD-L1, have significant activity in MSI-H/dMMR tumors. |
1 Introduction
Advances in immunotherapy have revolutionized the field of cancer medicine over the last several years, and agents such as checkpoint inhibitors and others hold promise to continue this trend. By reversing the immunosuppressive mechanisms elicited by tumor cells, checkpoint inhibitors such as pembrolizumab and nivolumab have demonstrated significant benefit in some tumors [1,2,3,4,5,6,7,8,9,10,11,12,13,14,15,16,17]. One specific family of tumors, classified either as microsatellite instability-high (MSI-H) or mismatch repair deficient (dMMR), harbors larger mutational loads and more immunogenic neoepitopes than most tumors [18]. As such, MSI-H/dMMR cancers are particularly interesting targets for immunotherapeutic agents such as checkpoint inhibitors. In this review, we will focus on the investigation of pembrolizumab and nivolumab in the treatment of MSI-H/dMMR tumors given their recent approvals by the United States Food and Drug Administration (US FDA) in this setting. Correlative scientific discoveries surrounding these agents and others will be explored. Furthermore, we will briefly discuss current and future challenges in the use of immuno-oncology agents in MSI-H/dMMR tumors.
2 The DNA Repair System, Microsatellite Instability and Neoepitopes in the Immunogenicity of Cancer
Microsatellite instability (MSI) exists when there is a difference in the length of DNA nucleotide repeats between tumor DNA and normal DNA, and tumors manifesting this phenomenon are classified as MSI-High or MSI-H tumors [19]. Clinically, somatic MSI testing is often performed using either a polymerase chain reaction (PCR)-based assay for mononucleotide and dinucleotide microsatellite markers, or analysis of mismatch repair protein expression by immunohistochemistry (IHC). MSI-H has long been an important biomarker in cancer, as it can serve as a screening test for the inherited cancer syndrome Lynch syndrome, as well as provide predictive and prognostic information for patients with MSI-H colorectal cancer, and tumors originating in other primary sites [20,21,22,23,24,25,26,27,28,29,30,31,32]. When microsatellite instability is due to a germline mutation in one of the DNA mismatch repair (MMR) genes (MLH1, MSH2, MSH6, or PMS2), the patient is diagnosed with Lynch syndrome [33]; however, microsatellite instability can also be seen sporadically, with somatic inactivation of the mismatch repair mechanism via methylation of the MLH1 gene [34], or through somatic MMR mutations [35, 36]. More broadly, defects in MMR interfere with the DNA repair system’s correction of errors that occur during DNA replication, leading to microsatellite instability and a “hypermutator” phenotype [37,38,39,40,41,42,43,44,45]. This phenotype is seen across a multitude of tumor types with varying frequencies, including endometrial, gastric, esophageal, small intestine, colorectal, cervical, neuroendocrine, sarcoma, renal, ovarian, prostate, head and neck, hepatocellular, lung, bladder, glioblastoma multiforme, low-grade glioma, breast, melanoma and thyroid [18, 46].
MSI-H/dMMR tumors are known to be associated with increased tumor-infiltrating lymphocyte (TIL) density, particularly CD3+CD8+ cytotoxic TILs, perhaps triggering an immune response in the host in this way [47,48,49]. MSI-H/dMMR tumors and their surrounding stroma are infiltrated by TILs because of their higher mutational load and the concomitant increased density of abnormal cell surface proteins compared to microsatellite stable (MSS) tumors. These tumors often harbor >1000 coding somatic mutations per tumor cell genome compared with 50–100 somatic mutations in their MSS counterparts [50, 51]. The increase in frameshift mutations within coding sequences seen in MSI-H/dMMR tumors leads to increased synthesis of inactive peptides, including peptides located on the cell membrane [52, 53]. These proteins can be presented through the tumor’s major histocompatibility complex (MHC) I to cytotoxic T-lymphocytes as neoantigens [54]. Furthermore, MSI-H/dMMR tumors can express genes encoding checkpoint receptors at higher levels than MSS tumors, and TILs from MSI-H tumors often manifest increased programmed cell death receptor-1 (PD-1) and programmed death-ligand 1 (PD-L1) expression when compared to their MSS counterparts [55, 56].
Due to this MSI-H/dMMR phenotype, a hypothesis emerged that checkpoint inhibitors could lead to activation of the immune system and cause blockade of immune checkpoint proteins, thereby enabling prolonged T-cell responses against cancer cells in this particular class of tumor. While checkpoint inhibitors have been studied in a variety of tumors, and are now approved by the FDA in several of these, the investigation of these agents in the treatment of MSI-H/dMMR tumors has been a particularly interesting one.
3 Immune Checkpoint Inhibitors in MSI-H/dMMR Cancers
3.1 Pembrolizumab
The first-in-human phase I dose-escalation study of the highly selective, IgG4-κ humanized monoclonal antibody pembrolizumab, KEYNOTE-001, was undertaken in patients with advanced solid tumors by Patnaik et al. [57]. Initially, ten patients received the anti-PD-1 antibody pembrolizumab at 1, 3, or 10 mg/kg intravenously every 2 weeks until disease progression or intolerable toxicity. No dose-limiting toxicities were observed, and seven additional patients received treatment with 10 mg/kg pembrolizumab every 2 weeks. Thirteen patients participated in a 3-week intrapatient dose escalation followed by 2 or 10 mg/kg every 3 weeks. Patients with melanoma and Merkel cell carcinoma experienced complete radiographic responses to therapy, and an additional three patients had partial responses. Two of the melanoma patients who had partial responses were found to have PD-L1-positive tumors, defined as PD-L1 expression in > 5% of cells. Microsatellite instability/deficient mismatch repair testing was not undertaken in this early Phase I trial.
Le et al. then conducted a multicenter phase II study of pembrolizumab in patients with advanced, treatment-refractory cancer with or without mismatch-repair deficiency [18, 58]. Early results of the first 41 patients treated on this groundbreaking study were initially published in 2015 [58]. In this study, tumor mismatch repair status was assessed through the use of the MSI Analysis System by Promega, through the evaluation of selected microsatellite sequences that are particularly prone to copying errors when mismatch repair is compromised [30, 58,59,60]. Co-primary endpoints of immune-related objective response rate (irORR) and 20-week immune-related progression-free survival (irPFS) rate were established. Pembrolizumab at a dose of 10 mg/kg was given intravenously every 14 days to patients with treatment refractory, progressive dMMR colorectal adenocarcinoma (CRC), pMMR colorectal adenocarcinoma, and dMMR non-colorectal cancer. Of the first 41 patients treated, 32 had colorectal cancer progressive on at least two lines of therapy (11 dMMR and 21 pMMR), and 9 patients had dMMR non-colorectal (ampullary, cholangiocarcinoma, endometrial, small bowel or gastric) cancers progressive on at least one treatment regimen. Interestingly, 18% of patients in the dMMR colorectal cancer cohort had evidence of germline MMR gene mutations or known Lynch syndrome, as did 44% of the dMMR non-colorectal cancer cohort; no patients in the pMMR colorectal cancer cohort had this finding, as would be expected.
Of 35 response-evaluable patients on this study, irORR was 40% (4 of 10 patients; 95% CI 12–74%) for dMMR metastatic CRC (mCRC) patients, and 0% (0 of 18 patients; 95% CI 0–20%) for pMMR mCRC patients. For patients with dMMR non-colorectal cancer, irORR was 71% (5 of 7 patients; 95% CI 29–96%). The co-primary endpoint of 20-week irPFS rate was found to be 78% (7 of 9 patients; 95% CI 40–97%) for dMMR mCRC patients, 11% (2 of 18 patients; 95% CI 1–35%) for pMMR mCRC patients, and 67% (4 of 6 patients; 95% CI 22–96%) for dMMR non-colorectal cancer patients. Impressively, the rate of disease control, or percentage of patients with objective response or stable disease, was 90% in mCRC patients whose tumors manifested dMMR (9 of 10 patients; 95% CI 55–100%) and 71% (5 of 7 evaluable patients; 95% CI 29–96%) in non-colorectal cancer patients whose tumors manifested dMMR. In contrast, the rate of disease control in mCRC patients whose tumors manifested pMMR was only 11% (2 of 18 patients; 95% CI 1–35%). In general, non-colorectal cancer patients whose tumors had dMMR, had faster responses to pembrolizumab than dMMR mCRC patients, with median times to response by RECIST of 12 vs. 28 weeks, respectively (p = 0.03).
At the time of data analysis and subsequent publication of this study cohort, median progression-free survival (PFS) and overall survival (OS) for dMMR mCRC patients had not been reached. For patients with pMMR mCRC, median PFS was 2.2 months (95% CI 1.4–2.8 months) and median OS was 5.0 months (95% CI, 3 not estimable). For non-colorectal cancer patients whose tumors manifested dMMR, median PFS was 5.4 months (95% CI, 3 not estimable) and median OS was not reached. No significant differences were seen with respect to the median amount of time that patients had known metastatic disease (p = 0.77 by log-rank test) or median PFS while receiving prior regimens (p = 0.60) between the dMMR mCRC and pMMR mCRC groups.
Overall, pembrolizumab was fairly well tolerated, with grade 3–4 adverse events of anemia (17%), lymphopenia (20%), diarrhea (5%), bowel obstruction (7%), elevated ALT (5%), hypoalbuminemia (10%) and hyponatremia (7%) seen. Of note, grade 1 or 2 rash or pruritus occurred in 24% of patients, grade 1 or 2 pancreatitis in 15% of patients, and grade 1 or 2 thyroiditis, hypothyroidism or hypophysitis in 10% of patients.
Whole-exome sequencing was performed on these pembrolizumab-treated patients, with a mean of 1782 somatic mutations per tumor seen in the dMMR cancer cohorts (n = 9) vs. a mean of 73 mutations per tumor in the pMMR CRC cohort (n = 6; p = 0.007 by nonparametric Wilcoxon test), verifying earlier data [50, 51]. Importantly, patients whose tumors had high numbers of somatic mutations, as well as high numbers of abnormal cell surface proteins with the potential to become mutation-associated neoantigens (MANAs), were associated with longer PFS and a trend toward objective response. Immunohistochemistry (IHC) of MSI-H tumors showed greater densities of CD8+ TILs and membranous PD-L1 expression (SP263) than were observed in the stroma surrounding the tumors among the patients with MSS tumors, with a trend toward higher objective response and stable disease rates, but not survival, in the MSI-H cohorts [58].
Due to the impressive results seen in this study in pembrolizumab-treated patients with dMMR tumors, accrual of patients with dMMR tumors on this study continued, adding gastroesophageal, neuroendocrine, osteosarcoma, pancreas, prostate, thyroid and unknown primary cancers to the original tumor types [18]. In this trial, a total of 86 patients with dMMR tumors were enrolled between September 2013 and September 2016, and their longitudinal data were pooled with those from 11 dMMR mCRC and 7 dMMR non-colorectal cancer patients from the prior portion of the study. All patients had previously received and progressed on at least one line of cancer therapy, and patients had evidence of mismatch repair deficiency as assessed by either polymerase chain reaction (PCR) or immunohistochemistry (IHC). In this study, no new safety signals for pembrolizumab administered at 10 mg/kg intravenously every 14 days were seen. A total of 74% of patients experienced an adverse event, but most of these were low grade.
In this study published by Le et al., objective responses were seen in 53% (46 of 86 patients; 95% CI 42–64%), with 8 patients (21%) achieving a complete radiographic response (CR); similar ORR were seen between the colorectal and non-colorectal dMMR cancer groups [18]. Furthermore, 66 of 86 patients (77%; 95% CI 66–85%) achieved disease control, as defined by objective response or stable disease as best response. Average time to any response was 21 weeks, with average time to complete response being 42 weeks. No difference in ORR was seen between Lynch syndrome-associated and non-Lynch syndrome-associated tumors (46 vs. 59%, p = 0.27). At the time of publication and with a median follow-up time of 12.5 months, neither median PFS nor OS had been reached for these patients. Estimated PFS at 1 and 2 years was 64 and 53%, respectively; estimated OS at 1 and 2 years was 76 and 64%, respectively. In 11 patients who achieved a CR and stopped pembrolizumab after two years of treatment per protocol, no evidence of cancer recurrence was observed, with average time off therapy at the time of publication being 8.3 months. An additional seven patients without disease progression stopped pembrolizumab at the 2-year mark per protocol or earlier due to intolerance to therapy and have not had disease progression, with an average time off therapy of 7.6 months.
From a correlative science standpoint, several interesting associations were seen in post-treatment tissue samples obtained by consent from patients enrolled in this study. Twelve of twenty patients who underwent tumor tissue biopsy a few months after initiation of pembrolizumab therapy were found to have no tumor cells in the biopsy specimen, but instead were found to have inflammation, fibrosis and mucin associated with an immune response [18]. This finding was a strong predictor of PFS when compared to biopsies with evidence of residual tumor (25.9 vs. 2.9 months). Through deep sequencing of T-cell receptor CDR3 regions (TCR-seq), Le and colleagues showed that checkpoint blockade induced peripheral expansion of tumor-specific T cells. Frequencies of mutation-associated neoantigen-specific T-cell clones peaked after pembrolizumab treatment and corresponded with normalization of the systemic tumor marker, often predating radiographic response by several weeks [18]. On the other hand, mutations in the gene encoding β2-microglobulin (B2M), a protein required for antigen presentation [61], were often seen in patients who developed resistance to pembrolizumab therapy over time.
As a result of these remarkable clinical findings and additional data from other clinical trials (KEYNOTE-016, KEYNOTE-164, KEYNOTE-012, KEYNOTE-028, and KEYNOTE-158), pembrolizumab was granted accelerated approval by the US Food and Drug Administration (FDA) on May 23, 2017 for adult and pediatric patients with unresectable or metastatic, microsatellite instability-high (MSI-H) or mismatch repair deficient (dMMR) solid tumors that have progressed following prior treatment and who have no satisfactory alternative treatment options, or with MSI-H or dMMR colorectal cancer that has progressed following treatment with a fluoropyrimidine, oxaliplatin and irinotecan (Table 1) [62]. The recommended pembrolizumab dose for this indication is 200 mg for adults or 2 mg/kg (up to a maximum of 200 mg) for children, administered as an intravenous infusion over 30 min every 3 weeks for up to 24 months in patients without disease progression. Interestingly, this marked the first time the FDA had granted regulatory drug approval based on a tumor’s biomarker rather than its original site, or tissue/site-agnostic approval.
3.2 Nivolumab
In the last several years, another immune checkpoint inhibitor has undergone testing and validation in the MSI-H/dMMR population after an initial efficacy signal was seen in metastatic colorectal cancer. The first-in-human, Phase I dose-escalation study of PD-1 blockade with the fully human immunoglobulin G4 (IgG4) monoclonal antibody MDX-1106, later named nivolumab, was undertaken by Brahmer et al. in 39 patients with advanced treatment-refractory solid tumors [63]. These tumors included colorectal cancer (n = 14), metastatic melanoma (n = 10), castrate-resistant prostate cancer (n = 8), non-small cell lung cancer (n = 6) and renal cell carcinoma (n = 1). Patients received a single intravenous infusion of either 0.3, 1, 3, or 10 mg/kg of MDX-1106. Fifteen additional patients were enrolled in an expansion cohort at 10 mg/kg. If patients had clinical benefit with MDX-1106, they could receive further doses of drug. MDX-1106 was well tolerated at these doses, with no maximum tolerated dose (MTD) reached. Of several responses seen in this trial, a patient with MSI-H colorectal cancer treated with a total of five doses of 3 mg/kg MDX-1106 achieved a complete radiographic response after 6 months of therapy, with partial response seen 8 weeks after a single dose of drug. Subsequent follow-up demonstrated this disease response to be durable, with no disease recurrence for almost four years at the time of follow-up publication [64]. At that time, the patient had been off anti-neoplastic therapy for 3 years. As one of the early signs of efficacy of checkpoint inhibitors in MSI-H/dMMR disease, this signal led to further study of nivolumab in patients with dMMR/MSI-H mCRC.
A recently reported Phase II study by Overman et al. further investigated the use of nivolumab in patients with metastatic dMMR/MSI-H colorectal cancer [65]. CheckMate 142 was an open-label, multicenter study of patients with metastatic pMMR and dMMR/MSI-H CRC who had progressed on or were intolerant to at least one line of standard chemotherapy and who were treated with either nivolumab monotherapy or nivolumab plus ipilimumab combination therapy. In the monotherapy arm, nivolumab was given intravenously at a dose of 3 mg/kg every 2 weeks.
A total of 74 patients with dMMR/MSI-H mCRC [as determined by local guidelines with immunohistochemistry or polymerase chain reaction (PCR) testing] were enrolled in this study and treated with nivolumab monotherapy. However, only 53 of these patients, or 72%, were centrally confirmed to have MSI-H tumors. Fourteen patients (19%) were found by centralized and standardized testing to have non-MSI-H tumors, and seven (9%) had no central results due to insufficient tumor tissue or DNA. Interestingly, five patients with a clinical history of Lynch syndrome and dMMR results by local testing were found on PCR testing centrally to be microsatellite stable (MSS). This highlights the difficulty of dMMR/MSI-H determination by current methods of IHC, PCR, and next-generation sequencing (NGS) given the imperfect sensitivity and specificity of these tests.
At the time of data cutoff, 27 patients (36%) had discontinued nivolumab due to disease progression and six (8%) had discontinued the drug due to treatment-related toxic effects. Overall, 23 patients (31.1%, 95% CI 20.8–42.9%) had achieved a partial response, and 51 patients (69%; 95 CI 57–79%) had achieved disease control for 12 weeks or longer. Responding patients had a median time to response of 2.8 months (IQR 1.4–3.2), and only three responders ultimately had disease progression. Median duration of response and median overall survival have not yet been reached. Median progression-free survival was 14.3 months (95% CI not estimable), and 12-month overall survival was 73% (95% CI 62–82%). In the overall study population, which included patients with pMMR/MSS mCRC, there were no responses seen in pMMR tumor-bearing patients. Interestingly, 3 of 14 patients (21%) not centrally confirmed as MSI-H had responses to nivolumab.
A total of 15 patients (20%) had grade 3 or 4 drug-related adverse events, with increased amylase and lipase constituting the most common grade 3 or 4 events. Patients who discontinued treatment due to drug-related adverse events experienced increased ALT, colitis, duodenal ulcer, acute kidney injury and stomatitis. Patient-reported outcome analyses showed clinically meaningful improvements in multiple domains, including functioning, symptoms and global quality of life, among others.
Of interest in this study, tumor PD-L1 expression was not an effective predictive biomarker of response to nivolumab, as responses were seen across all patient subgroups, including those with (≥ 1%) and without (< 1%) tumor PD-L1 expression. Responses were also seen in patients with and without a history of Lynch syndrome, and with and without KRAS or BRAF mutations.
On the basis of results from CheckMate 142, the largest single cohort of dMMR/MSI-H mCRC patients given an immune checkpoint inhibitor, the FDA granted accelerated approval to nivolumab on July 31, 2017 for the treatment of patients aged 12 years and older with dMMR or MSI-H metastatic colorectal cancer that has progressed following treatment with a fluoropyrimidine, oxaliplatin and irinotecan (Table 1) [66]. The recommended nivolumab dose for this indication is 240 mg every 2 weeks.
4 Combinations Utilizing Checkpoint Inhibitors and Other Immuno-Oncology Agents
While monotherapy with pembrolizumab or nivolumab has demonstrated efficacy in dMMR/MSI-H tumors, it is currently unclear whether combinations of checkpoint inhibitors or other immune-oncology agents will improve upon these successes. One ongoing example of combinatorial therapy utilizing checkpoint inhibitors is CheckMate 142, in which a cohort of mCRC patients received both nivolumab and ipilimumab, the latter being a fully human monoclonal antibody directed against the cell surface antigen CTLA-4 [67]. Patients with previously treated dMMR/MSI-H mCRC received nivolumab 3 mg/kg and ipilimumab 1 mg/kg every 3 weeks for four doses, followed by nivolumab 3 mg/kg every two weeks until discontinuation due to disease progression or toxicity. Preliminary results showed an overall response rate of 55% and disease control rate (DCR) of 79% in a total population of 84 patients. The 9-month rates of PFS and OS were 77 and 88%, respectively, and median PFS and OS have not yet been reached. The combination of nivolumab and ipilimumab was more toxic than nivolumab monotherapy, however, with grade 3–4 treatment-related adverse events occurring in 29% of patients.
In addition to anti-CTLA-4 and anti-PD-1/PD-L1 combinations, other combinatorial immuno-oncology strategies are currently under investigation or are being planned. These include the use of other key immune regulators such as LAG-3, OX-40, TIM-3, KIR, VISTA, GITR, IDO-1,2 and toll-like receptors, among others. Moreover, additional immune checkpoint inhibitors such as atezolizumab, durvalumab, avelumab, and others, are under development and investigation in this space. While efficacy and toxicities of these combinations are currently unknown, rational immunotherapy combinations hold promise for significant therapeutic advances not only in the dMMR/MSI-H setting, but in patients with pMMR tumors as well.
5 Conclusions
While objective responses, survival benefit, and durability of responses seen of dMMR/MSI-H tumors to immune checkpoint inhibitor monotherapy have been impressive, there are still many additional unanswered questions and ongoing areas of investigation in this field. First, why do some patients with dMMR/MSI-H tumors not respond to checkpoint inhibition? Are there additional predictive biomarkers that may be helpful to identify these patients early, perhaps sparing them from potential toxicities of therapy? Of those dMMR/MSI-H patients who initially respond to checkpoint inhibition, what are the mechanisms for acquired resistance in the subset of patients who develop disease progression despite ongoing treatment? How long will responding patients continue to benefit from therapy? Does resistance to one PD-1 inhibitor confer resistance to the class of agents or just the agent to which the tumor has been exposed?
For patients who have either complete radiographic responses to therapy or extremely durable responses, is there an optimal length of checkpoint inhibitor treatment to maximize disease response and durability and minimize toxicities? Are there other checkpoint inhibitors under development that may work better in this setting and/or have fewer toxicities? Furthermore, could combinations with other immunomodulatory agents enhance response in the dMMR/MSI-H population, or cause disease regression in the microsatellite stable (MSS) population? These questions are worthy of further exploration and research. In the meantime, given the responses of dMMR/MSI-H tumors to checkpoint inhibition in the treatment-refractory setting, there are now ongoing studies evaluating the potential role for these agents in combination with and without cytotoxic chemotherapy or other immunomodulatory agents, in earlier lines of therapy for metastatic disease, and in the neoadjuvant and adjuvant settings in some tumors.
The success of checkpoint inhibition in dMMR/MSI-H tumors is strongly welcomed in the oncology field. However, given that this genotype is seen in a minority of patients [18, 46], we need to learn from this success and build upon our understanding of its biological underpinnings so that more patients can potentially benefit from the utility of checkpoint inhibition and immuno-oncology as a whole.
References
Bellmunt J, et al. Pembrolizumab as second-line therapy for advanced urothelial carcinoma. N Engl J Med. 2017;376(11):1015–26.
Garon EB, et al. Pembrolizumab for the treatment of non-small-cell lung cancer. N Engl J Med. 2015;372(21):2018–28.
Chen R, et al. Phase II study of the efficacy and safety of pembrolizumab for relapsed/refractory classic Hodgkin lymphoma. J Clin Oncol. 2017;35(19):2125–32.
Herbst RS, et al. Pembrolizumab versus docetaxel for previously treated, PD-L1-positive, advanced non-small-cell lung cancer (KEYNOTE-010): a randomised controlled trial. Lancet. 2016;387(10027):1540–50.
Nghiem PT, et al. PD-1 blockade with pembrolizumab in advanced Merkel-cell carcinoma. N Engl J Med. 2016;374(26):2542–52.
Reck M, et al. Pembrolizumab versus chemotherapy for PD-L1-positive non-small-cell lung cancer. N Engl J Med. 2016;375(19):1823–33.
Ribas A, et al. Pembrolizumab versus investigator-choice chemotherapy for ipilimumab-refractory melanoma (KEYNOTE-002): a randomised, controlled, phase 2 trial. Lancet Oncol. 2015;16(8):908–18.
Ansell SM, et al. PD-1 blockade with nivolumab in relapsed or refractory Hodgkin’s lymphoma. N Engl J Med. 2015;372(4):311–9.
Antonia SJ, et al. Nivolumab alone and nivolumab plus ipilimumab in recurrent small-cell lung cancer (CheckMate 032): a multicentre, open-label, phase 1/2 trial. Lancet Oncol. 2016;17(7):883–95.
Borghaei H, et al. Nivolumab versus docetaxel in advanced nonsquamous non-small-cell lung cancer. N Engl J Med. 2015;373(17):1627–39.
Brahmer J, et al. Nivolumab versus docetaxel in advanced squamous-cell non-small-cell lung cancer. N Engl J Med. 2015;373(2):123–35.
Ferris RL, et al. Nivolumab for recurrent squamous-cell carcinoma of the head and neck. N Engl J Med. 2016;375(19):1856–67.
Larkin J, et al. Combined nivolumab and ipilimumab or monotherapy in untreated melanoma. N Engl J Med. 2015;373(1):23–34.
Motzer RJ, et al. Nivolumab versus everolimus in advanced renal-cell carcinoma. N Engl J Med. 2015;373(19):1803–13.
Postow MA, et al. Nivolumab and ipilimumab versus ipilimumab in untreated melanoma. N Engl J Med. 2015;372(21):2006–17.
Robert C, et al. Nivolumab in previously untreated melanoma without BRAF mutation. N Engl J Med. 2015;372(4):320–30.
Sharma P, et al. Nivolumab in metastatic urothelial carcinoma after platinum therapy (CheckMate 275): a multicentre, single-arm, phase 2 trial. Lancet Oncol. 2017;18(3):312–22.
Le DT, et al. Mismatch repair deficiency predicts response of solid tumors to PD-1 blockade. Science. 2017;357(6349):409–13.
Aaltonen LA, et al. Clues to the pathogenesis of familial colorectal cancer. Science. 1993;260(5109):812–6.
Goldstein J, et al. Multicenter retrospective analysis of metastatic colorectal cancer (CRC) with high-level microsatellite instability (MSI-H). Ann Oncol. 2014;25(5):1032–8.
Popat S, Hubner R, Houlston RS. Systematic review of microsatellite instability and colorectal cancer prognosis. J Clin Oncol. 2005;23(3):609–18.
Lochhead P, et al. Microsatellite instability and BRAF mutation testing in colorectal cancer prognostication. J Natl Cancer Inst. 2013;105(15):1151–6.
Phipps AI, et al. Association between molecular subtypes of colorectal cancer and patient survival. Gastroenterology. 2015;148(1):77–87 e2.
French AJ, et al. Prognostic significance of defective mismatch repair and BRAF V600E in patients with colon cancer. Clin Cancer Res. 2008;14(11):3408–15.
Ribic CM, et al. Tumor microsatellite-instability status as a predictor of benefit from fluorouracil-based adjuvant chemotherapy for colon cancer. N Engl J Med. 2003;349(3):247–57.
Carethers JM, et al. Use of 5-fluorouracil and survival in patients with microsatellite-unstable colorectal cancer. Gastroenterology. 2004;126(2):394–401.
Sargent DJ, et al. Defective mismatch repair as a predictive marker for lack of efficacy of fluorouracil-based adjuvant therapy in colon cancer. J Clin Oncol. 2010;28(20):3219–26.
Tajima A, et al. The mismatch repair complex hMutS alpha recognizes 5-fluorouracil-modified DNA: implications for chemosensitivity and resistance. Gastroenterology. 2004;127(6):1678–84.
Tajima A, et al. Both hMutSalpha and hMutSss DNA mismatch repair complexes participate in 5-fluorouracil cytotoxicity. PLoS One. 2011;6(12):e28117.
Boland CR, Goel A. Microsatellite instability in colorectal cancer. Gastroenterology. 2010;138(6):2073–2087 e3.
Sinicrope FA, et al. DNA mismatch repair status and colon cancer recurrence and survival in clinical trials of 5-fluorouracil-based adjuvant therapy. J Natl Cancer Inst. 2011;103(11):863–75.
Webber EM, et al. Systematic review of the predictive effect of MSI status in colorectal cancer patients undergoing 5FU-based chemotherapy. BMC Cancer. 2015;15:156.
Lynch HT, et al. Milestones of Lynch syndrome: 1895–2015. Nat Rev Cancer. 2015;15(3):181–94.
Kane MF, et al. Methylation of the hMLH1 promoter correlates with lack of expression of hMLH1 in sporadic colon tumors and mismatch repair-defective human tumor cell lines. Cancer Res. 1997;57(5):808–11.
Geurts-Giele WR, et al. Somatic aberrations of mismatch repair genes as a cause of microsatellite-unstable cancers. J Pathol. 2014;234(4):548–59.
Haraldsdottir S, et al. Colon and endometrial cancers with mismatch repair deficiency can arise from somatic, rather than germline, mutations. Gastroenterology. 2014;147(6):1308–1316 e1.
Parsons R, et al. Hypermutability and mismatch repair deficiency in RER+ tumor cells. Cell. 1993;75(6):1227–36.
Bhattacharyya NP, et al. Mutator phenotypes in human colorectal carcinoma cell lines. Proc Natl Acad Sci USA. 1994;91(14):6319–23.
Eshleman JR, et al. Increased mutation rate at the hprt locus accompanies microsatellite instability in colon cancer. Oncogene. 1995;10(1):33–7.
Modrich P. DNA mismatch correction. Annu Rev Biochem. 1987;56:435–66.
Fishel R, et al. The human mutator gene homolog MSH2 and its association with hereditary nonpolyposis colon cancer. Cell. 1993;75(5):1027–38.
Liu B, et al. hMSH2 mutations in hereditary nonpolyposis colorectal cancer kindreds. Cancer Res. 1994;54(17):4590–4.
Papadopoulos N, et al. Mutation of a mutL homolog in hereditary colon cancer. Science. 1994;263(5153):1625–9.
Bronner CE, et al. Mutation in the DNA mismatch repair gene homologue hMLH1 is associated with hereditary non-polyposis colon cancer. Nature. 1994;368(6468):258–61.
Eshleman JR, Markowitz SD. Mismatch repair defects in human carcinogenesis. Hum Mol Genet. 1996;5:1489–94.
Hause RJ, et al. Classification and characterization of microsatellite instability across 18 cancer types. Nat Med. 2016;22(11):1342–50.
Kim H, et al. Clinical and pathological characteristics of sporadic colorectal carcinomas with DNA replication errors in microsatellite sequences. Am J Pathol. 1994;145(1):148–56.
Alexander J, et al. Histopathological identification of colon cancer with microsatellite instability. Am J Pathol. 2001;158(2):527–35.
Dolcetti R, et al. High prevalence of activated intraepithelial cytotoxic T lymphocytes and increased neoplastic cell apoptosis in colorectal carcinomas with microsatellite instability. Am J Pathol. 1999;154(6):1805–13.
Ogino S, et al. Lymphocytic reaction to colorectal cancer is associated with longer survival, independent of lymph node count, microsatellite instability, and CpG island methylator phenotype. Clin Cancer Res. 2009;15(20):6412–20.
Vogelstein B, et al. Cancer genome landscapes. Science. 2013;339(6127):1546–58.
Jung B, et al. Loss of activin receptor type 2 protein expression in microsatellite unstable colon cancers. Gastroenterology. 2004;126(3):654–9.
Markowitz S, et al. Inactivation of the type II TGF-beta receptor in colon cancer cells with microsatellite instability. Science. 1995;268(5215):1336–8.
Lee V, et al. Mismatch repair deficiency and response to immune checkpoint blockade. Oncologist. 2016;21(10):1200–11.
Llosa NJ, et al. The vigorous immune microenvironment of microsatellite instable colon cancer is balanced by multiple counter-inhibitory checkpoints. Cancer Discov. 2015;5(1):43–51.
Gatalica Z, et al. Programmed cell death 1 (PD-1) and its ligand (PD-L1) in common cancers and their correlation with molecular cancer type. Cancer Epidemiol Biomarkers Prev. 2014;23(12):2965–70.
Patnaik A, et al. Phase I study of pembrolizumab (MK-3475; anti-PD-1 monoclonal antibody) in patients with advanced solid tumors. Clin Cancer Res. 2015;21(19):4286–93.
Le DT, et al. PD-1 blockade in tumors with mismatch-repair deficiency. N Engl J Med. 2015;372(26):2509–20.
Lynch HT, de la Chapelle A. Hereditary colorectal cancer. N Engl J Med. 2003;348(10):919–32.
Yamamoto H, Imai K, Perucho M. Gastrointestinal cancer of the microsatellite mutator phenotype pathway. J Gastroenterol. 2002;37(3):153–63.
Zaretsky JM, et al. Mutations associated with acquired resistance to PD-1 blockade in melanoma. N Engl J Med. 2016;375(9):819–29.
https://www.fda.gov/Drugs/InformationOnDrugs/ApprovedDrugs/ucm560040.htm. Cited 20 Oct 2017.
Brahmer JR, et al. Phase I study of single-agent anti-programmed death-1 (MDX-1106) in refractory solid tumors: safety, clinical activity, pharmacodynamics, and immunologic correlates. J Clin Oncol. 2010;28(19):3167–75.
Lipson EJ, et al. Durable cancer regression off-treatment and effective reinduction therapy with an anti-PD-1 antibody. Clin Cancer Res. 2013;19(2):462–8.
Overman MJ, et al. Nivolumab in patients with metastatic DNA mismatch repair-deficient or microsatellite instability-high colorectal cancer (CheckMate 142): an open-label, multicentre, phase 2 study. Lancet Oncol. 2017;18(9):1182–91.
https://www.fda.gov/drugs/informationondrugs/approveddrugs/ucm569366.htm. Cited 20 Oct 2017.
Andre T, Lonardi S, Wong KYM, et al. Combination of nivolumab + ipilimumab in the treatment of patients with deficient DNA mismatch repair/microsatellite instability metastatic colorectal cancer: CheckMate 142 study. Proc Am Soc Clin Oncol. 2017;2017:35.
Funding
No funding was received for the preparation of this article.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
Both authors (KKC, RMG) have received research funding from Merck. KKC has also received research funding from Bristol-Myers Squibb. RMG has served as a paid consultant to Merck and Merck KGA.
Rights and permissions
About this article

Cite this article
Ciombor, K.K., Goldberg, R.M. Hypermutated Tumors and Immune Checkpoint Inhibition. Drugs 78, 155–162 (2018). https://doi.org/10.1007/s40265-018-0863-0
Published:
Issue Date:
DOI: https://doi.org/10.1007/s40265-018-0863-0