
Abstract
No specific antivirals are currently available for two emerging infectious diseases, Middle East respiratory syndrome (MERS) and severe acute respiratory syndrome (SARS). A literature search was performed covering pathogenesis, clinical features and therapeutics, clinically developed drugs for repurposing and novel drug targets. This review presents current knowledge on the epidemiology, pathogenesis and clinical features of the SARS and MERS coronaviruses. The rationale for and outcomes with treatments used for SARS and MERS is discussed. The main focus of the review is on drug development and the potential that drugs approved for other indications provide for repurposing. The drugs we discuss belong to a wide range of different drug classes, such as cancer therapeutics, antipsychotics, and antimalarials. In addition to their activity against MERS and SARS coronaviruses, many of these approved drugs have broad-spectrum potential and have already been in clinical use for treating other viral infections. A wealth of knowledge is available for these drugs. However, the information in this review is not meant to guide clinical decisions, and any therapeutic described here should only be used in context of a clinical trial. Potential targets for novel antivirals and antibodies are discussed as well as lessons learned from treatment development for other RNA viruses. The article concludes with a discussion of the gaps in our knowledge and areas for future research on emerging coronaviruses.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
The outbreaks of Middle East respiratory syndrome (MERS) and severe acute respiratory syndrome (SARS) were caused by emerging coronaviruses. |
A variety of approaches for developing therapeutics are discussed with emphasis on drugs that have been approved for other indications and could be repurposed for treating emerging coronaviral infections. |
The recent MERS and SARS outbreaks highlight the importance of a panel of well-characterized broad-spectrum antivirals for treating emerging viral infections |
1 Introduction
An electronic literature search for countermeasures against Middle East respiratory syndrome coronavirus (MERS-CoV) and severe acute respiratory syndrome coronavirus (SARS-CoV) was performed using PubMed and Google Scholar from 2000 through April 17, 2017. The search (key words: Middle East Respiratory Syndrome, Severe Acute Respiratory Syndrome, inhibitors, antivirals, therapeutics, FDA-approved) produced 1677 citations. References selected discussed (1) pathogenesis and history of disease, (2) clinical countermeasures used during the 2003 SARS and 2012 MERS outbreaks and outcomes, and (3) the efficacy of countermeasures targeting viral components and cellular targets of MERS-CoV and SARS-CoV. The main emphasis was on references for drug repurposing as an alternative to the costly development of novel drugs for emerging coronaviral infections.
1.1 Epidemiology of MERS and SARS
Since 2003, two human coronaviruses, SARS-CoV and MERS-CoV, emerged as global public health threats. SARS-CoV was first identified in February 2003 in Guangdong Province, Peoples Republic of China and was transmitted to humans from infected civets, likely infected from bats [1, 2]. SARS-CoV spread to 29 additional countries and was associated with high morbidity in humans (e.g. atypical pneumonia). Ultimately, SARS was contained in 2004 following a highly effective public health response but resulted in 8098 confirmed cases and 774 deaths (Fig. 1a) [3]. In 2012, MERS emerged in The Kingdom of Saudi Arabia and presented as a severe respiratory disease, with frequent gastrointestinal and renal complications. MERS-CoV, the causative agent of MERS, was later identified as a coronavirus. MERS-CoV has subsequently spread to 27 additional countries (Fig. 1B) [4]. As of September 12, 2017, 2080 confirmed cases of MERS and 722 deaths were reported [5].

Maps of the severe acute respiratory syndrome (SARS) (a) and Middle East respiratory syndrome (MERS) (b) outbreaks with confirmed case numbers
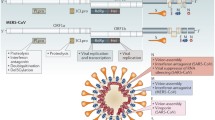
Coronaviruses are enveloped, single-stranded, positive-sense RNA viruses (Fig. 2). They are members of the Coronavirinae subfamily of viruses and together with the Torovirinae subfamily comprise the Coronaviridae virus family (order Nidovirales). Coronavirinae is divided into four genera: alpha coronavirus, beta coronavirus, gamma coronavirus, and delta coronavirus. The coronaviruses share a similar genome organization. The open reading frame 1a and 1b comprise nearly 2/3 of the genome and encode the nonstructural proteins. The multiple structural proteins, including spike, nucleocapsid, envelope, and membrane proteins are encoded by downstream open reading frames (Fig. 2) [6,7,8]. SARS-CoV and MERS-CoV belong to the beta coronavirus genus. However, SARS-CoV belongs to lineage B, and MERS-CoV belongs to lineage C along with bat coronaviruses HKU4 and HKU5. As MERS-CoV and bat coronaviruses are part of lineage C and MERS-CoV RNA was found in a bat sample in The Kingdom of Saudi Arabia, researchers hypothesize that bats may be a natural reservoir for MERS-CoV [9, 10]. Results from a recent study support that bats may be a reservoir for MERS-CoV; however, camels and goats are thought to be intermediate hosts [11]. In this study, MERS-CoV was isolated from nasal secretions of MERS-CoV-infected dromedary camels that had a short, mild disease progression.

Genomes of Middle East respiratory syndrome coronavirus (MERS-CoV) and severe acute respiratory syndrome coronavirus (SARS-CoV) indicating the open reading frames for nonstructural (1a and 1b) and structural proteins (numbered 3–9, and E, M, N, S). E envelope, M membrane, N nucleocapsid, S Spike
The suspected reservoir for SARS-CoV is the Chinese horseshoe bat [2]. However, the mechanism of emergence and adaptation to make the virus zoonotic is still not definitely understood [2]. SARS-CoV-like isolates from these bats have up to 95% sequence similarity to human and civet SARS-CoV. During the initial outbreak, SARS-CoV was originally isolated from palm civets found in a Chinese market; but, SARS-CoV was not found in the wild palm civet population [12]. Bats harbor many coronaviruses and are considered the main reservoir for later infections in an intermediate host, such as civets or camels, which spread the disease to humans [2]. Human-to-human transmission has been most commonly associated with health-care workers and those with close, unprotected contact with infected patients [13, 14].
1.2 Clinical Features
The clinical features of MERS and SARS are similar and can range from asymptomatic or mild disease to severe pneumonia with acute respiratory distress syndrome (ARDS) and multi-organ failure [15]. Although MERS and SARS are clinically similar, the MERS mortality rate is 40% and SARS’s mortality rate is 10% [16]. Approximately 75% of MERS cases were associated with underlying comorbidities with a 60% mortality rate in this subgroup (including cardiopulmonary abnormalities, obesity, and diabetes). In contrast, 10–30% of patients with SARS have comorbidities with a mortality rate of 46% within this subgroup [15, 16].
The development of symptomatic MERS and SARS mostly occurs in adults (median age of 50 years; 40 years for SARS). MERS and SARS symptoms typically follow a mean incubation time of ~ 5 days (range 2–13 and 2–14 days, respectively) and include fever, chills, cough (some associated with blood), shortness of breath, myalgia, headache, nausea, vomiting, diarrhea, sore throat, and malaise [15,16,17]. Progression from mild to severe disease is more rapid with MERS as compared to SARS with means of 7 and 11 days, respectively [15]. Secondary bacterial infections have occurred in patients with severe MERS; however, the role of these coinfections in MERS pathogenesis has yet to be determined [18,19,20]. Laboratory abnormalities associated with MERS and SARS patients include elevated lactate dehydrogenase, elevated liver enzymes; thrombocytopenia; lymphopenia and leukopenia [21,22,23].
Radiographic abnormalities consistent with viral pneumonitis and ARDS are common in MERS and SARS. Radiographic progression in the lower lobes has been reported to be more rapid for MERS than SARS [21,22,23]. For SARS, disease in the lower lobes mimics pneumonia, radiographic progression includes ground-glass opacification and lobe thickening [17]. MERS-CoV (intact virus or viral genome) is found at higher concentrations in the lower respiratory tract than in the upper respiratory tract in MERS patients and this may account for inefficient inter-human transmission [15, 24]. Currently, no approved therapeutics for patients with MERS or SARS are available, and clinical management has relied primarily on supportive care.
2 Therapeutic Agents
2.1 Clinical Usage
2.1.1 Treatment of SARS
Effectiveness of antiviral treatments used during the SARS epidemic has been mainly based on case studies and retrospective analysis of patient cohorts. Few randomized, blinded, clinical trials of anti-SARS treatments were performed, which adds complexity when interpreting the available data (Table 1). Ribavirin, a nucleoside analog that prevents RNA and DNA virus replication, was initially used in the treatment of SARS due to its broad-spectrum efficacy. For example, in a Taiwanese study, 51 SARS patients were treated daily with fluoroquinolone antibiotics, [levofloxacin (500 mg) or moxifloxacin (400 mg)] following diagnosis. Out of 51 patients, 44 SARS patients were also treated intravenously (IV) with 2000 mg of ribavirin then orally daily with 1200 mg while 7 SARS patients did not receive ribavirin. Corticosteroids, IV methylprednisolone, or oral prednisolone were administered as needed to treat worsening lung infiltrates and fever [25]. Ribavirin treatment led to hypoxia and anemia and increased risk for death in SARS patients. In a retrospective analysis, a cohort of 229 patients from Hong Kong, Singapore, and Toronto were treated with ribavirin in conjunction with corticosteroids, immunoglobulins, and/or antibiotics [26]; ribavirin did not demonstrate efficacy. Patients in Hong Kong and Singapore were treated with ribavirin at 1200 mg orally at diagnosis, followed by oral treatments with 2400 mg daily, or continual IV ribavirin therapy [8 mg/kg every 8 h (h)]. In Toronto, patients received ribavirin IV treatment with 2000 mg, followed by 1000 mg every 6 h for 4 days, and 300 mg every 8 h for 3 days. Unfortunately, fatality rates were similar between the ribavirin-treated and control groups. Later, researchers demonstrated that the ribavirin dosage required to be effective against SARS-CoV in vitro was not clinically achievable [27]. Ribavirin treatment also resulted in adverse effects including anemia, hypoxemia and decreased hemoglobin levels, and did not improve patient outcome [26]. Due to the increasing adverse effects and lack of efficacy, Health Canada stopped permitting the use of ribavirin for SARS [25].
Additional studies tested the efficacy of ribavirin in conjunction with lopinavir, an anti-retroviral agent. Lopinavir demonstrated in vitro activity against SARS-CoV [28]. In a non-randomized, open-enrollment trial of 152 suspected SARS patients [27], all patients were treated with ribavirin and corticosteroids similar to the previously described studies. In addition, 41 of the confirmed SARS patients were also treated with a combination of lopinavir (400 mg) and ritonavir (100 mg). Mean viral loads in nasopharyngeal swabs within this treatment group decreased to undetectable levels by day 10. Overall, SARS-related symptoms subsided, disease progression was milder, and no adverse effects were reported as compared to the historical control group.
In an open-label, non-randomized study of 22 SARS patients, 9 patients who received subcutaneous (SC) injections of interferon (IFN)α, alfacon-1, for 10 days at an initial dose of 9 µg/day for 2 days increasing to 15 µg/day with disease progression. All 9 patients survived with minor adverse effects [29].
2.1.2 Treatment of MERS
The evaluation of treatments in MERS patients has been hampered as high-quality clinical data from randomized clinical trials are limited. Ribavirin (with or without IFN, or corticosteroids) was the primary treatment during the MERS outbreak. In a retrospective analysis, a cohort of 20 patients was treated with oral ribavirin and SC pegylated IFN-α2a at a dose of 180 µg/week for 2 weeks (Table 2) [30]. The initial dose of ribavirin was 2000 mg, followed by a 200–1200 mg dose depending on creatinine clearance. A group of 24 patients that received supportive care and corticosteroids were considered the control group. At 14 days after confirmed diagnosis of MERS, survival was increased in the treated group (70%) compared to the control group (29%). By 28 days post-diagnosis, 30% of treated subjects survived versus 17% of the control group [30]. In an additional case study, a 69-year-old Greek patient who contracted MERS in Jeddah was treated with oral lopinavir/ritonavir (400/100 mg twice daily), pegylated IFN (180 µg SC once per week for 12 weeks), and ribavirin (2000 mg initial dose; 1200 mg every 8 h for 8 days, initiated on day 13 post-diagnosis). Two days after treatment initiation, viremia could not be detected; however, viral RNA was detected in several patient samples (feces, respiratory secretions, and serum) up to 14 weeks post-diagnosis. Despite prolonged survival, the patient succumbed from septic shock 2 months post-diagnosis [31]. An ongoing randomized clinical trial in Saudi Arabia is evaluating treatment of MERS patients with IFN-β1b in combination with lopinavir/ritonavir [32].
2.2 Drugs with Repurposing Potential for Treatment of Coronaviral Infections
Drug repurposing is an attractive alternative drug discovery strategy because it eliminates many steps usually required at the early phase of drug development. Over the past decade, interest in drug repurposing has increased as pharmaceutical companies are challenged with decreasing product pipelines, high costs associated with de novo drug discovery, and the imminent expiration of many drug patents. Some examples for successfully repurposed drugs include Viagra (Pfizer) for erectile dysfunction (original indication: angina) and raloxifene (Eli Lilly) for treatment of invasive breast cancer (original indication: osteoporosis).
The time required for traditional drug development is often discordant with the urgent need for novel therapies for emerging infectious diseases such as SARS and MERS. Outbreaks can occur anywhere in the world and frequently in resource-limited settings. Commonly, the treatment strategies that are available for emerging infectious diseases are less than adequate to improve patient outcome. Although specific antivirals for MERS-CoV and SARS-CoV are in development, drug repurposing could present an important arm in generating additional therapeutics for future coronaviruses. First, if these drugs are confirmed to have beneficial effects in vitro and in animal studies, they could be used to build a panel of approved drugs for use as a first-line of defense for newly emerging coronaviruses. Second, these drugs could be made accessible relatively quickly to patients under Emergency Use Authorization. Extending the choices of treatment by generating a panel of broad-spectrum antivirals would provide a real improvement to healthcare communities struggling to cope during an outbreak of emerging infections. A great example of how repurposing can benefit in the search of treatments for emerging infections is the drug zidovudine. Zidovudine was originally developed in 1964 as a cancer drug. In 1985, zidovudine was found to be active against human immunodeficiency virus (HIV), and 2 years later it became the first drug to be approved for the treatment of acquired immunodeficiency syndrome (AIDS) [33].
A number of research groups have identified and investigated the usefulness of approved drugs for the treatment of viral infections including coronaviruses. Below, we summarize several drug classes with antiviral activity against MERS-CoV and SARS-CoV that have repurposing potential (Fig. 3, Table 3). Some of the drugs described have activity against other virus families indicating potential broad-spectrum applications and have already been in clinical use for treating other viral infections. We would like to emphasize that none of the therapeutics described in this section are recommended for clinical use outside a clinical trial setting.

Candidate drugs for repurposing for coronaviral infections. Several drug classes (A through I) have been studied, and the steps/processes of the viral replication cycle that they most likely target are indicated. AKT serine/threonine kinase, CAD cationic amphiphilic drug, Cyps cytochrome P-450s, E envelope, ER endoplasmic reticulum, ERGIC ER–Golgi intermediate compartment, ERK extracellular signal-reduction kinase, IFN interferon, MAPK mitogen-activated protein kinase, M membrane, MPA mycophenolic acid, mTOR mechanistic target of rapamycin, N nucleocapsid, NFAT nuclear factor of activated T cells, ORF open reading frame, PI3K phosphoinositide 3-kinase, S Spike
2.2.1 Antidiarrheal Agents
Loperamide, an approved anti-diarrheal agent, is on the World Health Organization (WHO) Model List of Essential Medicines and is available in many countries. The drug acts on the opioid receptor and reduces intestinal motility [34]. Results from pharmacokinetic (PK) studies show that oral loperamide is well absorbed from the gut with less than 1% of the drug entering systemic circulation [35]. Loperamide demonstrated anti-MERS-CoV, anti-SARS, and anti-HCoV229E activity in an in vitro screen of approved drugs [36], although the mechanism of action is unknown. Interestingly, loperamide was suggested for limiting gastrointestinal fluid and electrolyte losses in patients with Ebola virus disease (EVD) [37].
2.2.2 Antimalarial Agents
The antimalarial agents, chloroquine (CQ), amodiaquine, and mefloquine have activity against SARS-CoV and MERS-CoV in vitro [36, 38, 39]. CQ is a U.S. Food and Drug Administration (FDA)-approved antimalarial agent that is also used to treat autoimmune disease such as rheumatoid arthritis due to its anti-inflammatory effects [40]. CQ has activity against a number of viruses in vitro and in vivo including flaviviruses [dengue virus (DENV)], Togaviruses [chikungunya virus (CHIKV)], paramyxoviruses (Hendra, Nipah virus), influenza viruses, HIV, and filoviruses [Ebola virus (EBOV)] [41,42,43,44,45,46,47].
Several mechanisms of action have been identified for the antiviral effect of CQ and suggest that the drug acts nonspecifically at virus entry or at the later stages of virus production. CQ accumulates within acidic organelles such as endosomes, Golgi vesicles, and lysosomes, where the drug is protonated resulting in increased pH within the vesicle [48]. Viruses depend on these acidic organelles for entry, viral replication, and maturation of virus progeny. Similarly, MERS-CoV entry into cells depends on several proteases. Dipeptidyl peptidase 4 (DPP4) acts as functional virus receptor [49], and cellular proteases [e.g. type II transmembrane serine protease (TMPRSS2) and members of the cathepsin family] activate the viral spike (S) glycoprotein [50]. CQ may have an effect on any of these proteases. CQ also affects the glycosylation step within the Golgi that directs trafficking and maturation of viral proteins [51,52,53]. For SARS-CoV, the antiviral activity of CQ has also been attributed to a deficit in glycosylation of the receptor angiotensin-converting enzyme 2 (ACE2) [54].
The broad-spectrum antiviral activity makes CQ an attractive antiviral for repurposing and treating coronaviral and other emerging viral infections. In vivo activity of CQ in MERS or SARS animal models has not yet been reported. However, the antiviral activity of the drug has been evaluated against other viruses in preclinical and clinical studies with mixed results. CQ plasma steady state concentrations in mice are similar to those reported for humans (10 µM) and are within range of the EC50 values determined for MERS-CoV (3.6 µM) and SARS-CoV (2.3 µM) [43, 55]. Preclinical studies with CQ in mice against other viruses have shown survival benefits for influenza and EBOV infections.
In clinical studies, CQ was effective at reducing viral loads in asymptomatic HIV patients [56, 57], but results of CQ treatment of CHIKV and DENV infections were mixed [58, 59].
In summary, CQ has broad-spectrum potential and the information gained from studies on other viruses can be used to plan the most appropriate strategies for evaluating its specific clinical value for treating for MERS-CoV and SARS-CoV infections. CQ has several advantages including rapid absorption from gastrointestinal tract, low cost, and very effective biodistribution. CQ may be an excellent candidate for combinatorial treatments with other antivirals. However, considerable challenges remain for the treatment of viral infections including increased understanding of the pharmacodynamics of CQ, achievement of sufficient plasma concentrations in patients, and toxicity concerns [60]. Importantly, hydroxychloroquine, a CQ derivate, may provide an alternative due to lower toxicity and similar pharmacology profile [55].
A related antimalarial drug, amodiaquine, also has activity in vitro against MERS-CoV and SARS-CoV [38]. Previous investigations have demonstrated that amodiaquine inhibits filovirus replication, and the mechanism of action is hypothesized to be similar to that of CQ [43]. Amodiaquine is well tolerated and is commonly used for malaria treatments in many countries. Further, amodiaquine in combination with artesunate was administered to EVD patients during the 2013–2016 epidemic, and the resulting decrease in fatality rates may have been associated with the use of amodiaquine as an antimalarial agent [61]. Nonhuman primate (NHP) studies are currently underway to investigate the effect of amodiaquine treatment on EVD [62].
Mefloquine, a synthetic analog of quinine, is another antimalarial drug with activity against MERS-CoV and SARS-CoV [38]. It belongs to the WHO Model List of Essential Medicines. Mefloquine is known to penetrate the blood-brain barrier and was found to inhibit JC virus infection and replication at concentrations generally achieved in the brains of patients given mefloquine for malaria [63] leading to the clinical evaluation of this drug for the treatment of progressive multifocal leukoencephalopathy [64, 65]. In 2013, the FDA added a boxed warning to the US label of mefloquine regarding the potential for neuropsychiatric side effects. Additional investigations are warranted to determine if amodiaquine or mefloquine have value for repurposing for treatment of MERS or SARS.
2.2.3 Cyclophilin Inhibitors
Cyclophilins are ubiquitous host proteins believed to have multiple roles in trafficking, protein folding and T cell activation [66]. Cyclosporine A (CysA), forms a complex with cyclophilin A, thereby blocking T cell activation. CysA is licensed for use in organ transplantation to suppress the immune response. CysA has also been shown to inhibit coronaviruses including SARS-CoV and MERS-CoV effectively in cell culture [67, 68]; however, the mechanism has yet to be determined. There is increasing evidence that cyclophilins are involved in viral replication of RNA viruses such as hepatitis C virus (HCV) and West Nile virus, and this may also apply to coronaviruses [69]. Although the immunosuppressive properties of CysA are considered a risk for treating viral infections in patients, nonimmunosuppressive analogs of CysA that bind to cyclophilins with higher affinity have been developed and some are in clinical trials as HCV therapeutics [70,71,72].
2.2.4 Interferons
Interferons (IFNs) are approved by the FDA for other indications such as hepatitis C. Although IFN-α reduced SARS-CoV replication in mice and NHPs [73, 74], efficacy of IFN-α treatment in SARS patients was mixed (see Sect. 2.1.1). From in vitro studies, another type I interferon, IFN-β1a, may be more effective than IFN-α either alone or in combination with IFN-γ [75,76,77]. Combinations of IFN-β and -γ were synergistic against SARS-CoV in vitro [77].
With regards to MERS, in vitro and in vivo preclinical studies have indicated that IFN-α2b alone or in combination with ribavirin, may have a therapeutic effect if given early in disease [78, 79]. In clinical trials, however, IFN-α2b (given in combination with other treatments) did not lead to a significant benefit to patients (see Sect. 2.1.2). IFN-β1a (EC50 = 1.37 IU/mL) was superior in activity against MERS-CoV infection in vitro compared to IFN-α2a, IFN-α2b, and IFN-γ; these IFNs had EC50 values of 160.8, 21.4, and 56.5 IU/mL, respectively [80]. IFN-β1b is currently under evaluation for MERS-CoV in a randomized clinical trial (in combination with lopinavir/ritonavir) [32]. Investigating the IFN-β subtypes (1a and 1b) in combination with other antivirals may be worthwhile as potential synergistic combinations could reduce the effective drug dosage and IFN-associated adverse effects.
2.2.5 Kinase Inhibitors
Many cellular processes are regulated independently of changes in transcription or translation through kinase-mediated cell signaling pathways. As a testament to the biological importance of kinases, there have been over 500 kinases identified along with more than 900 genes encoding proteins with kinase activity [81, 82]. As of April 2015, 28 kinase inhibitors have been granted approval by the US FDA with over half gaining approval from 2012–2015. Further, kinases are the most frequently targeted gene class in cancer therapy, second only to the G protein-coupled receptors as therapeutic targets [83, 84].
The therapeutic potential for host-targeted immunomodulatory agents in viral infections has received considerable attention [85,86,87]. Recently, Dyall et al. identified two Abelson (Abl) kinase inhibitors (imatinib and dasatinib) that inhibited MERS-CoV and SARS-CoV infection through a cell-screening assay [38]. Both compounds significantly inhibited MERS-CoV and SARS-CoV with micromolar EC50 values and low cytotoxicity. Abl2 has been identified as critical for MERS-CoV and SARS-CoV virus entry, and may be the target that imatinib inhibits to block entry of both viruses [88]. A recent systems kinome analysis investigation of in vitro MERS-CoV infection suggested that ERK/MAPK and PI3 K/Akt/mTOR signaling pathways were specifically modulated during infection [89]. Subsequent analysis of licensed kinase inhibitors targeting these pathways demonstrated that kinase inhibitors targeting the ERK/MAPK signal pathway (selumetinib and trametinib) inhibited MERS-CoV infection by ≥ 95% when added pre- or post-infection [89]. Further, trametinib demonstrated significantly stronger inhibitory activity against MERS-CoV than selumetinib suggesting that specific intermediates of the ERK/MAPK signaling pathway may represent crucial foci during early (viral entry) and late (viral replication) events in the viral life cycle. In contrast, sorafenib, an inhibitor of Raf-1 and B-Raf, components of the ERK/MAPK signaling pathway, and vascular endothelial growth factor receptor 2 (VEGFR2), inhibited MERS-CoV infection by > 90% when added to cells prior to infection; however, the inhibitory activity was reduced to < 30% when added post-infection suggesting Raf kinases were primarily involved in early viral life cycle events. In addition, the inhibitory activity dabrafenib, a Raf kinase inhibitor, was also largely ablated when added post-infection. Miltefosine, an alkyl phospholipid, considered to be an inhibitor of protein kinase B (Akt), garnered FDA approval for infectious disease-related treatments (cutaneous or mucosal leishmaniasis) [90]. In 2013, miltefosine became directly available from the US Centers for Diseases Control and Prevention for the treatment of free-living amoeba infections [91]. Pre-treatment of cells with miltefosine reduced MERS-CoV infection by 28%, but had no effect when added post-infection [89]. In contrast, inhibition of mTOR with sirolimus or everolimus reduced MERS-CoV infection by ~ 60% when added prior pre- or post-infection suggesting a critical role for mTOR in MERS-CoV infection. A recent clinical investigation by Wang et al. [92] evaluated sirolimus and corticosteroids in addition to standard antiviral treatment in a randomized controlled trial in patients with severe H1N1 pneumonia and acute respiratory failure [92]. Importantly, the addition of sirolimus was associated with improved patient outcomes including decreased hypoxia and multi-organ dysfunction, reduced mean times for liberation from mechanical ventilation, and increased clearance of virus. Thus, it may be prudent to extend the study of repurposed kinase inhibitors beyond stand-alone therapeutic investigations and also consider their potential as adjunctive therapies.
2.2.6 Neurotransmitter Inhibitors
Numerous neurotransmitter receptors inhibitors showed activity against MERS-CoV and SARS-CoV infection [38]. These drugs were initially developed as antipsychotics, antihistamines, and sedatives. Five neurotransmitter receptor antagonists belong to the chemical class of phenothiazines: chlorpromazine, triflupromazine, thiethylperazine, promethazine and fluphenazine. Phenothiazines were breakthrough medications developed in the 1950s for treating mental health patients and reduced episodes of bizarre behavior, hallucinations, and irrational thoughts [93]. Although phenothiazines primarily block dopamine receptors, they also have anticholinergic, antihistamine, and antiemetic effects.
The phenothiazines, chlorpromazine and triflupromazine, are approved antipsychotics. Chlorpromazine has been used off-label for short-term treatment of nausea and migraines. Triflupromazine is used to treat severe emesis, but the drug has more serious side effects than chlorpromazine including akathisia and tardive dyskinesia. The antiviral effect of chlorpromazine has been extensively studied, and the drug interferes with clathrin-mediated endocytosis, a process that many viruses exploit for host cell entry. Chlorpromazine inhibits entry of Junin virus [94], West Nile virus [95], EBOV [96], HCV [97], and Japanese encephalitis virus [98] suggesting broad-spectrum activity that could be exploited early during a novel virus outbreak. Chlorpromazine may have similar effects on coronaviruses as the drug effectively inhibits MERS-CoV, SARS-CoV, and human coronavirus 229E expressing green fluorescent protein [36, 38]. However, time-of-addition studies indicate that the inhibitory activity against MERS-CoV is retained whether added pre- or post-infection suggesting that there are additional effects to clathrin-mediated entry impairment [36].
Thiethylperazine is an approved antiemetic. Both chlorpromazine and thiethylperazine have been shown to inhibit alphaviruses, Semliki forest virus (SFV) and chikungunya virus (CHIKV) [45, 99]. As these drugs cross the brain-blood barrier, use of these drugs could be beneficial in the treatment of CHIKV, including common neurologic complications. Promethazine is an antihistamine used as a sedative in many countries under different brand names, but also acts as a weak anti-psychotic activity. Fluphenazine is a common antipsychotic used to treat chronic psychoses (primarily schizophrenia) and belongs to the WHO Model List of Essential Medicines. Promethazine and fluphenazine have shown in vitro activity against MERS-CoV and SARS-CoV and may have value as candidates for repurposing for coronaviral infections [38].
Benztropine mesylate, an approved anticholinergic used to treat Parkinson’s, had activity against MERS-CoV and SARS-CoV [38]. Benztropine was also identified in other screens of clinically approved drugs for antiviral activities against HCV and EBOV [100,101,102]. Although the detailed mechanism of action is unknown, HCV studies indicate that benztropine inhibits at a virus entry step, while not interfering with viral genome replication, transcription or production of viral progeny or virus production of viral progeny [102]. It has been argued that a virus entry inhibitor may have value in decreasing the incidence of relapse in chronic HCV patients that receive liver transplants [102]. However, the peak plasma concentrations of benztropine may be too low to be effective for treating an acute infection [103]. Benztropine was also independently identified in two drug screens for EBOV antivirals [100, 101]. A recent report suggests that a step after virus attachment, but prior to viral/cell membrane fusion is targeted by benztropine [104].
Clomipramine, a tricyclic antidepressant, and thiothixene, a thioxanthene antipsychotic, have also been shown to inhibit MERS-CoV and SARS-CoV infection in vitro [38]. In addition, they were found to inhibit EVD VLP entry [101]. Both are approved clinically, and clomipramine belongs to the WHO Model List of Essential Medicines.
Several other neurotransmitter inhibitors, astemizole, promethazine, chlorphenoxamine, and fluspirilene, were active against MERS-CoV and SARS-CoV in cell culture, but we were not able to find reports on activity against other viruses [38]. Astemizole is an H1-histamine receptor antagonist for treating allergic rhinitis that was withdrawn from the market. Cardiac adverse events due to drug overdose have been reported, but are extremely rare [105]. Recently, astemizole has gained renewed interest as an anticancer and antimalarial drug [106, 107].
Chlorphenoxamine is an antihistamine and anticholinergic that is currently in preclinical trials for malaria. Fluspirilene is an approved antipsychotic for treatment of schizophrenia. It is a known autophagy inducer [108]. Autophagy is a cellular degradative pathway that viruses exploit for their propagation [109]. Modulators of autophagy may perturb MERS-CoV or SARS-CoV infection, and investigation of their broad-spectrum potential for the treatment of coronaviral infections would be interesting [110].
2.2.7 Nucleic Acid Synthesis Inhibitors
Several RNA/DNA synthesis inhibitors have broad-spectrum activity against viruses including SARS-CoV and MERS-CoV [38, 80, 111,112,113,114]. Inosine monophosphate dehydrogenase (IMPDH) inhibitors such as ribavirin, mycophenolic acid, and mizoribine inhibit an important step in de novo synthesis of nucleic acids although the potency of these drugs against viruses varies. Ribavirin has been used in combination with IFN in the clinic for treatment of viral infections such as hepatitis C. Treatment regimens with ribavirin are well characterized and have been used in SARS and MERS patients with mixed results (see 2.1). Ribavirin weakly inhibits MERS-CoV in vitro, and conflicting data have been reported for the activity of ribavirin against SARS-CoV [80, 111, 115]. Many of the studies on ribavirin were performed in Vero cells that reportedly have a defect in ribonucleoside uptake, which could explain lack of activity for ribavirin in these cells [116]. Another coronavirus, mouse hepatitis virus (MHV), becomes sensitive to ribavirin when its exoribouclease activity is inactivated. In presence of exoribonuclease, ribavirin does not inhibit MHV replication [117]. The MHV exoribonuclease has been suggested to function as a ‘proofreading’ viral enzyme that is necessary for high-fidelity replication of MHV. Similarly, the exoribonuclease activity of MERS-CoV and SARS-CoV could possibly counteract inhibitory activity of ribavirin.
Mycophenolic acid (MPA), an immunosuppressant used to prevent organ rejection, has broad-spectrum antiviral activities, and antifungal, antibacterial, anticancer, and antipsoriatic properties [45, 118, 119]. Although MPA has weak inhibitory activity against SARS-CoV in vitro, it has promising activity against MERS-CoV [80, 112]. A potential alternative to MPA, the prodrug mycophenolate mofetil, has improved oral bioavailability [120]. Mycophenolate mofetil evaluated in the common marmoset model of MERS did not reduce disease manifestations compared to that observed in control subjects [121]. However, the MERS marmoset model does not recapitulate human disease due to its rapid onset and pathology associated with exposure methods [122]. Mizoribine, an approved immunosuppressant in organ transplantation with limited adverse side effects, has shown in vitro activity against HCV and bovine viral diarrhea virus (BVDV), and was considered as an alternative to ribavirin/IFN combinations for treatment of HCV infections [123]. In vivo analysis of ribavirin and other IMPDH inhibitors in SARS-CoV-infected mice have suggested that these agents would be of limited benefit [111].
The chemotherapeutic gemcitabine, has shown in vitro activity against MERS-CoV and SARS-CoV [38]. The drug’s anti-cancer mechanism is attributed to its ability to inhibit ribonucleotide reductase essential for de novo pyrimidine biosynthesis. Gemcitabine has been shown to suppress influenza virus RNA transcription and replication by targeting ribonucleotide reductase and showed anti-retroviral activity in vivo in the mouse model for murine leukemia virus [124, 125].
2.2.8 Protease Inhibitors
MERS-CoV and SARS-CoV require activation of their envelope glycoproteins by host proteases for cell entry by the endosomal or the non-endosomal pathways. Inhibitors of host cell proteases are being investigated as possible antivirals [126]. The serine protease TMPRRSS2 mediates entry via the non-endosomal pathway for both MERS-CoV and SARS-CoV [50, 127,128,129]. Camostat mesylate, which has been used in the treatment of chronic pancreatitis, inhibits TMPSSR2-mediated glycoprotein activation of MERS-CoV, SARS-CoV, and influenza virus [126, 130,131,132]. K11777, a cysteine protease inhibitor, is in clinical development for treating parasitic infections. K11777 has broad-spectrum activity against coronaviruses (MERS-CoV, SARS-CoV, HCoV-229E), filoviruses (EBOV, Marburg virus), and paramyxoviruses (Nipah virus) [133]. Interestingly, Zhou et al. [133] demonstrated that Camostat and K11777 had inhibitory activity against SARS-CoV whereas EBOV was only inhibited by K11777, suggesting differential host protease requirements for these viruses [133]. E-64-D, an inhibitor of an endosomal cysteine protease currently in Phase III trials for the treatment of muscular dystrophy, inhibits both MERS-CoV and SARS-CoV in vitro [38]. E-64-D also inhibits filovirus cell entry [134, 135]. The dependency of viruses for specific serine or cysteine host proteases must be considered in the selection of protease inhibitors for antiviral therapeutic applications. Therefore, an increased understanding of the relationship between host proteases and viral pathogenesis will determine the most effective treatment options for viral infections.
Lopinavir was identified as an inhibitor of MERS-CoV and SARS-CoV in vitro, and time-of-addition experiments indicate that the drug acts at an early stage of viral entry [36]. Lopinavir, an inhibitor of the HIV protease, is used clinically for the treatment of HIV infections [136]. It is given in combination with ritonavir, an inhibitor of cytochrome P450 3A4, to increase blood concentrations because of the low bioavailability of lopinavir [136]. Lopinavir also inhibits human papilloma virus and is currently under development for the topical treatment of cervical cancer [137]. Treatment with lopinavir/ritonavir resulted in reduced mortality in a NHP model of MERS [121]. Lopinavir has been shown to target the main protease (Mpro) of SARS-CoV [138]. However, lopinavir has also been shown to act on other intracellular processes that are involved in coronavirus replication. Additional studies are needed to fully understand the mechanism of action of lopinavir involving cellular proteases. During the 2003 SARS outbreak, patients in open clinical trials were treated with lopinavir/ritonavir in combination with ribavirin had a milder disease course and reduction in fatality rate compared to that observed with historical controls [27, 28].
2.2.9 Protein Synthesis Inhibitors
Three protein synthesis inhibitors with activity against coronaviruses were identified, emetine, anisomycin and omacetaxine mepesuccinate [38, 139]. Emetine, a natural plant alkaloid, and anisomycin, an antibiotic, both inhibit protein elongation and were identified as anti-protozoals [140, 141]. While emetine is approved for amoebiasis treatment, anisomycin did not move beyond clinical trials [141, 142]. Dehydroemetine, a synthetic emetine derivative, has fewer side effects and is available as an investigational new drug [143, 144]. Anisomycin was originally discovered as a peptidyl transferase inhibitor, but also activates the MAP kinase signaling pathway [145]. In addition to activity against MERS-CoV and SARS-CoV, emetine and anisomycin inhibit the animal picornavirus encephalomyocarditis virus [146]. Anisomycin has in vitro activity against poliovirus [147]. Omacetaxine mepesuccinate, a plant-derived alkaloid, is an anticancer therapeutic that received FDA approval in 2012 for the treatment of chronic myeloid leukemia. Omacetaxine inhibits MERS-CoV, bovine coronavirus, human enteric coronavirus and hepatitis B virus [139, 148]. In spite of omacetaxine broad spectrum anti-coronavirus activity, the drug had no activity against SARS-CoV [38]. Drugs that inhibit coronaviruses by targeting protein synthesis may have potential in the development of combination therapies with drugs that target other antiviral pathways.
2.2.10 Selective Estrogen Receptor Modulators
Recent investigations have demonstrated the potential of estrogen receptor (ER) antagonists for repurposing as anti-coronavirus compounds [38]. For example, toremifene citrate and tamoxifen citrate with activity against SARS-CoV and MERS-CoV were developed and approved as anticancer therapeutics. Both drugs have shown activity against HCV replication in vitro [149, 150]. Mechanistic studies revealed that the ER is functionally associated with HCV replication [150]. ER promotes the interaction between the HCV replication complex and the HCV polymerase NS5B. ER–mediated regulation of HCV genome replication is abrogated by tamoxifen.
Toremifene and tamoxifen also effectively inhibit EBOV infection in vitro [151]. However, in contrast to HCV, mechanistic studies have shown that toremifene-mediated EBOV inhibition is independent of the ER pathway as toremifene was still active against EBOV in cells that did not express ER [151, 152]. Toremifene acts at a late step of virus entry after internalization of EBOV and may prevent fusion between the viral and endosomal membranes [151,152,153]. Based on the chemical structure, toremifene is a cationic amphiphilic drug (CAD) that is known to be lysosomotropic and could affect endosomal processes during virus entry [151, 154]. Treatment with toremifene led to 50% survival of EBOV–infected mice confirming that this drug has an effect in vivo as well [151].
In terms of clinical application, toremifene and tamoxifen have good bioavailability, safety and tolerability profiles combined with a long history of use in the clinic. However, prolongation of the QT interval has been noted for toremifene and should not be prescribed to patients with congenital or acquired long QT syndrome, uncorrected hypokalemia or uncorrected hypomagnesemia [155]. Tamoxifen can increase uterine malignancies, stroke and pulmonary embolism in women with ductal carcinoma in situ or at high risk for breast cancer [156]. Despite these side effects, the drugs may have substantial value for short-term treatment of acute coronaviral infections. Advanced patient studies and careful evaluation of the pharmacokinetic profiles may facilitate dosing strategies that limit the risk of adverse events.
2.2.11 Sterol Metabolism Inhibitors
Two sterol synthesis inhibitors, terconazole and triparanol, have shown activity against MERS-CoV and SARS-CoV [38]. Studies with virus-like particles (VLPs) have demonstrated that terconazole inhibits coronavirus cell entry, including MERS-CoV and SARS-CoV. The sterol synthesis pathway has been shown to be required for infection by several viruses including HCV [157, 158]. Terconazole, approved for vaginal yeast infections, can be administered orally, topically or by suppository. Triparanol was approved for lowering plasma cholesterol, but was withdrawn due to numerous side effects. Both are CADs that induce accumulation of cholesterol in late endosomes and have been shown to inhibit EBOV entry [152].
2.3 Drugs in Development
2.3.1 Potential Targets for Inhibition of MERS-CoV and SARS-CoV
In addition to drug repurposing, development of novel antiviral countermeasures is needed for emerging coronaviruses. To this end, design or development strategies have targeted the viral replication cycle and host pathways essential for viral replication (Table 4). Two nucleoside inhibitors of viral RNA-dependent RNA polymerases, GS-5734 and BCX4430, have potential as broad-spectrum antivirals [159, 160]. Both drugs are active against MERS-CoV and SARS-CoV in cell culture, but in vivo efficacy remains to be investigated. In addition, a new class of nucleosides with a flexible purine base has anti-coronaviral activity, and further optimization could generate potent inhibitors of the coronaviral polymerase [161]. The surface glycoprotein (S) of SARS-CoV and MERS-CoV and other coronaviruses consists of two domains: S1, containing the receptor-binding domain (RBD) needed for extracellular binding; and S2, containing the fusion peptide needed for membrane fusion and release. Endocytosis of SARS-CoV is facilitated by the binding of RBD with the angiotensin converting enzyme 2 (ACE2) receptor on host cells. Membrane-bound cathepsin L cleaves the S protein revealing the S2 fusion protein, which fuses with the membrane and releases the viral RNA. Inhibitors of cathepsin L, the ACE2–SARS–S1 complex, or the S2 fusion peptide could be suitable targets to inhibit SARS-CoV entry [162]. Results from recent studies have identified inhibitors of viral entry, viral proteases, and helicases that potently inhibit both MERS-CoV and SARS-CoV [162]. Proteases, such as papain-like protease and 3C-like protease, could also be useful as antiviral targets for drug development as they are required for cleaving non-structural proteins for viral maturation. Most protease inhibitors are “suicide” protease inhibitors that bind to the target irreversibly. However, reversible protease inhibitors may have greater potential as they are less toxic and better tolerated [113]. Recent studies with helicase inhibitors show that three domain targets, N-terminal metal binding domain, a hinge domain, and a NTP/helicase domain, have potential for the development of new drugs [163].
2.3.2 RNA Interference
Directed RNA interference (RNAi) presents a powerful approach for the development of novel virus-specific therapeutics based on gene silencing [164]. Recent studies have shown that small interfering RNAs (siRNAs) or short hairpin RNAs can inhibit expression of viral genes and thereby block the replication of SARS-CoV in cultured cells [165,166,167,168,169,170,171,172]. Intranasal delivery of a combination of small interfering RNA (siSC2-5) targeting SARS-CoV open reading frame 1 and S protein decreased SARS pathogenesis in NHPs [173]. Potential RNAi candidates identified through computational modeling for MERS-CoV require further in vitro and preclinical investigation [174]. Several RNAi therapeutics for the treatment of viral infections have entered clinical trials including TKM-Ebola, a siRNA/lipid nanoparticle platform targeting EBOV [164, 175, 176]. This technology has great potential for therapeutics for emerging viruses as viral genome sequencing can now be completed in a very short time frame that is crucial in situations when an epidemic of a novel emerging viral infection unfolds. The main obstacle for RNAi strategies lies in the identification of suitable viral targets and in the delivery efficiency of nucleic acids to target cells in vivo.
2.3.3 Peptide Entry Inhibitors
Peptides share common physicochemical properties with CADs that facilitate interaction with cell membranes and interference with the fusion of cellular and viral membranes during virus entry. Researchers are making progress in defining the mechanism of action of peptide entry inhibitors of enveloped viruses such as enfuvirtide, an approved HIV inhibitor [177]. Enfuvirtide is a 36-residue peptide derived from the amphipathic loop/C-helix heptad repeat domain of HIV gp41. A rational approach based on scanning fusion protein sequences for amphipathic sequences has led to the discovery of additional peptide inhibitors for other viruses including MERS-CoV and SARS-CoV [178,179,180]. Chemical modifications of peptides have increased their in vivo stability and bioavailability, improving their potential for clinical applications as novel broad-spectrum viral entry inhibitors [181,182,183].
2.4 Antibody Therapy
The success of palivizumab for treatment of respiratory syncytial virus infection has reinvigorated efforts to develop monoclonal antibody-based therapeutics for infectious diseases [184]. ZMapp, a monoclonal antibody cocktail targeting EBOV, has been tested in NHPs with success and was moved to Phase I and II clinical trials in humans during the EVD epidemic [185, 186]. Similarly, a monoclonal antibody against Hendra virus has been administered to humans on a “compassionate use” basis [187,188,189]. These examples demonstrate the potential for antibody therapy to combat emerging/re-emerging viruses, and similar strategies have been pursued by multiple groups for development of antibodies to MERS-CoV [190,191,192,193,194,195,196,197]. Monoclonal antibodies to MERS-CoV have been sourced from humanized mice libraries or human antibody libraries [192, 194, 197, 198]. Antibodies target the S RBD, S1, or S2 subunits, and have demonstrated efficacy in animal models as reviewed in Ying et al. [199]. Monoclonal antibody therapy can impart a selective pressure for generation of resistant viruses. Although mutants escaping monoclonal antibody pressure tend to be less fit, analysis of the emergence of mutations that confer resistance to the monoclonal antibody will need to be performed.
An alternative to monoclonal antibody therapy is polyclonal antibody therapy using convalescent sera (sera sourced from a nonhuman or humanized animal). Polyclonal antibodies provide an advantage over monoclonal antibodies in that escape mutants are less likely to emerge [200, 201]. Convalescent sera have been recommended for MERS, and a Phase II interventional clinical trial is ongoing to determine efficacy [198]. However, the availability of a suitable donor subject presents a significant complication for this approach. Nonhuman animal sera has been considered, but safety concerns limit this option. Fractionation of nonhuman sera is an alternative; however, antibody-mediated clearance can be limited due to failure of the human Fc receptors to recognize the antibody heavy chain.
An alternative is de-speciating the antibodies by using only the Fab antibody fragment; however, cost and sufficient material may make mass production of Fab fragments difficult. The use of sera from humanized mice or other small laboratory animals is complicated by sample acquisition/volume restraints. Larger laboratory animals may provide a potential alternative. SAB Biotherapeutics has developed a trans-chromosomic bovine platform for the generation of human IgG antibodies [201]. Vaccination of trans-chromosomic cattle with S protein nanoparticles or inactivated, whole virus generated fully humanized polyclonal antibodies that demonstrated efficacy in the Ad-DPP4 murine model of MERS.
3 Lessons Learned
One of the most important lessons regarding antiviral drug development is that both highly specific and broad-spectrum antivirals bring unique advantages to the table. Antiviral agents can fall anywhere in the spectrum between “broad-spectrum” and “highly specific” [202]. A drug that targets a specific virus or virus family will have narrow activity, high potency, and low toxicity; however, such a drug may also promote resistance [202]. In contrast, broad-spectrum antivirals typically target a host factor or pathway, and often these agents have higher toxicities, lower potencies, and delayed treatment effects. However, the selective pressure for resistance is often lower with broad-spectrum agents.
A large part of our knowledge on antiviral development stems from the studies of chronic viral infections. Antiviral development strategies for DNA viruses have been successful in identifying a single drug that specifically targets a viral protein. This strategy has been less successful for RNA viruses. RNA viruses mutate at a higher rate than DNA viruses resulting in enhanced development of drug resistance.
3.1 AIDS
Despite extensive efforts over the past 30 years, a therapeutic or prophylactic HIV vaccine has remained elusive. Antiviral agents are the only available treatments for AIDS. Over 25 antivirals belonging to 6 different drug classes targeting different stages of viral replication are available (e.g. reverse transcriptase, protease, fusion, entry, integrase) [203]. Combination treatments with 2 to 3 drugs are effective and result in a sustained virologic response [204]. Two aspects have been found to be important for avoiding resistance: (1) selecting drugs with at least two different targets, and (2) selecting drugs that belong to different chemical classes. These considerations may also apply for drug combinations with synergistic effects against MERS- and SARS-CoV.
3.2 Hepatitis C
Broad-spectrum antiviral therapies can be of great value for treating emerging infections when it takes time to develop direct-acting antivirals. For treatment of chronic hepatitis C, clinicians have depended on IFN and ribavirin for a number of years [205]. Eventually, IFN and ribavirin combination was replaced by very effective fixed-combination therapies using direct-acting antivirals that target multiple steps of the HCV life cycle [206]. IFN and ribavirin contribute significantly to the treatment of viral infections for which no direct-acting antivirals exist. However, they have major side effects [207]. More options for broad-spectrum antivirals with improved safety profiles would be beneficial for use for emerging coronavirus infections.
3.3 Influenza
Influenza viruses are characterized by a high mutation rate of the RNA genome. As available vaccines may not be protective against a novel pandemic strain, antiviral agents are considered an essential component for preparedness. Combinations of direct-acting antivirals are under evaluation for additive or synergistic effects and prevention of resistance [208, 209]. One triple combination (oseltamivir, amantadine and ribavirin) is synergistic and prevents resistance in vivo [210, 211], highlighting the potential of combinatorial therapy.
3.4 Ebola Virus Disease
The recent epidemic of EVD in Western Africa has renewed the urgency for development of treatments against emerging viruses. Although vaccines and direct-acting antiviral treatment are under investigation, none are approved for clinical use [212,213,214,215,216]. The WHO prioritized a panel of drugs approved for other indications that were considered for repurposing under FDA’s Emergency Use Authorization [62]. Two of these drugs also have activity against MERS-CoV and SARS-CoV, amodiaquine (antimalarial agent) and toremifene citrate (breast cancer treatment).
Additional broad-spectrum antiviral agents (including repurposed drugs) should be a top priority for future emerging infections including coronavirus infections. A panel of broad-spectrum drugs that have been carefully validated for efficacy and safety and that could be used in combination would supply a minimum of protection for patients and healthcare workers at outbreak locations. This panel of drugs could be used in situations of a known re-emerging pathogen for which specific antiviral agents and vaccines has not been approved or of an unknown novel pathogen that could arise.
4 Gaps in Knowledge and Future Outlook
4.1 Animal Models of MERS and SARS
Effective development of countermeasures depends on developing appropriate animal models that uniformly recapitulate human disease progression and severity of pathological manifestations. As with most animal models of human disease, no one animal model fully reflects SARS or MERS, therefore researchers are faced with exploring several small animal models or choosing the “best-fit” model. To date, animal models do not fully recapitulate human disease, thus animal models of MERS and SARS need further refinement. Many small animal models have been evaluated as potential MERS and SARS models including mice, hamsters, and ferrets for MERS and Syrian hamsters, and guinea pigs for SARS [217,218,219]. Four murine models have been reported for MERS. The first model that demonstrated promise involved transduction of the respiratory tract with the putative MERS-CoV receptor, human DPP4 (or CD26) [220]. The major indicator of disease in this model is viral load in the lung at 4 days post-inoculation. Although clinical signs of disease, including weight loss, were limited in this model, it has been used for pathogenesis countermeasure studies [221, 222]. Lethal, disseminated MERS infection has been demonstrated in transgenic mice expressing human DPP4 [223]. Inflammatory processes were observed in the brains of these mice in contrast to human disease in which CNS involvement has not been reported. A transgenic mouse MERS model was developed in which the mouse DPP4 gene was replaced by the human DPP4 gene under control of the endogenous mouse DPP4 promoter. Using this model, MERS-CoV-infected mice developed lung pathology [194]. In addition, administration of human monoclonal antibodies against the spike protein in these transgenic mice provided protection against MERS-CoV infection [194]. Clustered regularly interspaced short palindromic repeat-CRISPR-associated protein 9 (CRISPR-CAS9) gene editing technology was used to modify the mouse DPP4 to match human DPP4 by altering amino acids at positions 288 and 330. Interestingly, wild type virus infection of these mice did not result in an improved model of MERS. However, serial passage of MERS-CoV resulted in MERS-CoV-15. Intranasal exposure of MERS-CoV-15 in 288/330, mice led to weight loss and a severe respiratory disease that included ARDS-like signs and reduced pulmonary function [224].
MERS-CoV infection of rabbits has also been evaluated as a model for MERS. Haagmans et al. [225] demonstrated that MERS-CoV infected rabbits did not develop obvious clinical signs, but infectious virus could be detected in the upper respiratory tract [225]. Furthermore, epithelial cells of the bronchioles and terminal bronchioles respiratory tract were positive for MERS-CoV by immunohistochemistry and in-situ hybridization, which reflects tissue tropism in human disease [225]. Using the rabbit model, Houser et al. [191] demonstrated that human monoclonal antibody 336 given pre-exposure reduced viral RNA lung titer at 3 days post-exposure, but not when given post-exposure [191].
Due to phylogenetic similarities with humans, NHP models of disease have long been considered as necessary for evaluation of countermeasures to infectious diseases. Rhesus monkey and common marmoset have been evaluated as potential models for MERS. Following intratracheal instillation of MERS-CoV in rhesus monkey models, lung pathology was observed [122, 226,227,228]. Experiments using rhesus monkeys have indicated that they develop limited systemic disease and a transient respiratory disease. Radiologic evaluations have indicated inflammatory infiltrates that develop shortly after exposure. Analysis of lung tissues by reverse transcriptase- quantitative polymerase chain reaction indicated virus replication in the lung.
Similar to MERS, African green monkeys (AGMs), rhesus monkeys, cynomolgus monkeys, and common marmosets have been identified as potential models for SARS [229]. Smits et al. [230] compared SARS CoV infection in young AGMs to cynomolgus monkeys, they observed that neither species developed clinical signs during a 4-day experiment [230]. Gross pathology indicated multi-focal pulmonary consolidation with consolidated grey-red firm lungs. These lesions affected 30% of the lungs in one subject. By comparison, the cynomolgus monkeys developed small patchy macroscopic lesions. Similar to MERS-CoV infection of NHPs, viral load decreases from exposure day to day 4. A comparison between AGMs, rhesus monkeys, and cynomolgus monkeys further support AGMs as the best available NHP model for SARS [231]. AGMs developed the highest viral load and most disease when compared to cynomolgus and rhesus monkeys. Lethal disease was not observed in any of these species; therefore, further development of the SARS model is warranted since lethal respiratory tract disease was the hallmark of SARS.
As an alternative to Old World NHPs, many groups have employed marmosets as models of human infectious disease [232,233,234]. Common marmosets have been evaluated as a MERS model [121, 122, 235]. These studies have demonstrated that common marmosets develop disease following exposure to MERS-CoV as shown by histopathological analysis, radiological analysis, and RT-qPCR. However, variable results have been reported, and exposure methodology can impact disease progression. Therefore, mock-infected groups must be included to account for pathological artifacts. The virus-specific pathology could be quantified using computed tomography, and future experiments using large group sizes could be used for countermeasure evaluation. Greenough et al. [236] performed a serial euthanasia study of SARS-CoV infected marmosets [236]. Subjects were intratracheally exposed with SARS-CoV. They observed mild inconsistent clinical signs of disease. Viral loads peaked at day 4 post-infection. Histopathology indicated interstitial pneumonitis with multinucleated syncytia that were described as mild and not observed in all late time-point subjects. Overall, further research is needed to develop animal models of SARS that reflect human disease presentation.
4.2 Combinations with Synergy
Drugs with repurposing potential discussed here (Table 3) have the advantage of easy access, availability and decreased cost of development and provide a wide array of options for combination studies. The pharmacological knowledge available for such compounds may also reduce concerns regarding adverse effects in patients. The generation of a translational database encompassing pharmacodynamics data and infectious disease biology data has been proposed and would greatly facilitate decision making to pursue new drug combinations [237]. Many of the drugs have potential for broad-spectrum antiviral activity and have already been in clinical use for treating other viral infections. As novel drugs in development move from the pre-clinical to clinical phase, they also become available for combination therapy. Care should be taken with the pharmacological evaluation of each combination to avoid possible contraindications of the drugs with regards to disease or adverse effects. Novel broad-spectrum replication inhibitors, such as GS-5734 (Gilead Sciences, in Phase I clinical trial), immunomodulators (nitazoxanide; steroids; statins) along with direct-acting antiviral agents for coronaviruses that are in development represent interesting partners for combinations. Combinations can involve broad-spectrum versus specific antiviral agents; drugs with different mechanism of action; or drugs that target different steps of the viral life cycle. Identifying one or more potent combinations with activity in an animal model would greatly increase preparedness for the next coronavirus outbreak.
4.3 Structure-Based Drug Design
Elucidation of the crystal structure of viral proteins has led to novel approaches for rational drug design. Rational design investigations using protein structure information and in silico screening for affinity to active sites of viral proteins holds promise. HIV-1 protease inhibitors have been one of the big successes of rational drug design. Only 6 years after the publication of the HIV-1 protease structure, saquinavir was developed in record time from bench to bedside and was licensed for use against AIDS in 1995 [238,239,240]. In total, six antivirals against the HIV-1 protease were designed and approved between 1995 and 2000. Similarly, for HCV, computer–aided approaches based on the known crystal structure of a viral protein have successfully guided the design and synthesis of inhibitors for the HCV NS3/NS4A proteases such as the peptidomimetics, telaprevir and boceprivir [241,242,243].
Due to the power of computational modeling using crystal structures from known coronaviruses, the crystal structures for the viral proteases, Mpro and PLpro, of SARS-CoV and MERS-CoV were determined relatively quickly [244]. These structures have already been used for the discovery of inhibitors with high binding affinity to the active site of the proteases. The structures of additional MERS-CoV or SARS-CoV viral proteins have yet to be determined and would offer additional viral targets for drug discovery.
Structural design can help with development of inhibitors in preparing for future outbreaks of yet unknown emerging coronaviruses. Based on the potential of zoonotic transmission of coronaviruses from bats to humans, crystal structures for the main protease (Mpro) of different bat coronavirus families have been proposed for screening and identifying broad-spectrum antiviral agents [244]. Proof-of-principle was shown for the novel protease inhibitor SG85, which inhibits the bat coronavirus HKU4 [244]. In 2012, SG85 was quickly identified as an inhibitor of MERS-CoV through in silico docking studies with the Mpro of MERS-CoV and the bat coronavirus HKU4.
4.4 Cationic Amphiphilic Drugs: A Novel Class of Antiviral Agents?
Several drugs (e.g. toremifene citrate, terconazole, Fig. 3) belong to a group of compounds termed cationic amphiphilic drugs (CADs) [100, 151, 152, 154, 245]. Phenothiazines (e.g. fluphenazine chlorpromazine, Fig. 3) are CADs that have been shown to inhibit HCV entry at virus-host cell fusion by intercalating into the cholesterol-rich domains of the host cell membrane and increasing membrane fluidity [97]. Drugs that act through this mechanism may present an interesting new class of broad-spectrum antivirals. CADs are known to be lysomotropic and accumulate in acidic compartments where their tertiary amine groups are protonated. The compounds act as mild bases and can neutralize the low pH of the acidic environment of endo/lysosomes. CADs can intercalate into membranes, alter the biophysical properties of membranes and thereby could potentially interfere with fusion of virus with the endo/lysosomal membrane. The concept of interfering with virus entry and budding through physicochemical properties of drugs is intriguing. Many viruses would be susceptible to this type of inhibition and CADs could be used as broad-spectrum antiviral agents. Detailed structure-activity relationship studies on CADs will be required to determine the chemical core structures and physicochemical properties important for this type of antiviral agent. Future investigations regarding the conservation of this mechanism of action to coronaviruses, as well as other emerging viruses, are warranted.
4.5 Analogs of Developed Drugs
Approved drugs with activity against MERS-CoV or SARS-CoV could be used as lead compounds for further antiviral drug development. Pharmaceuticals usually have undergone multiple rounds of structure-activity relationship studies generating analogs to improve drug activity against the original indication or target. The analogs of drugs with activity against MERS-CoV and SARS-CoV may still be available and could be screened to identify other analogs with increased antiviral activity. Although drug analogs would have to go through the full licensure process, there would be little to no added initial cost associated with producing the structure-activity relationship compounds. Recycling old analogs is one approach that may have value for developing novel drugs for viral infections.
5 Conclusion
A more streamlined process is needed for development of effective treatment measures for emerging and re-emerging pathogens. The availability of a panel of approved broad-spectrum drugs would clearly be beneficial as they could be used for treating disease symptoms and reducing morbidity until more specific acting antivirals and vaccines are developed.
A large number of potential drugs and therapeutics for the treatment of MERS and SARS have been discussed. The greatest challenge will be how best to down-select and evaluate the different approaches. As we have learned from drug development for AIDS and hepatitis, alleviating disease symptoms and increasing life span may be a more achievable goal rather than looking for a treatment that will provide complete recovery. Broad-spectrum antivirals, specific antivirals, and immune modulators each have an important role in treating viral infections either individually or in combination. Effective communication between the different institute partners (government, industry, academic; national, and international partners) is essential. Combining these drug discovery efforts will increase the chance of having one or more potential therapeutic agents at an advanced development stage by the time another outbreak of an emerging coronavirus occurs.
References
Li W, Shi Z, Yu M, Ren W, Smith C, Epstein JH, et al. Bats are natural reservoirs of SARS-like coronaviruses. Science. 2005;310(5748):676–9.
Lau SK, Woo PC, Li KS, Huang Y, Tsoi HW, Wong BH, et al. Severe acute respiratory syndrome coronavirus-like virus in Chinese horseshoe bats. Proc Natl Acad Sci USA. 2005;102(39):14040–5.
Revised US. surveillance case definition for severe acute respiratory syndrome (SARS) and update on SARS cases—United States and worldwide, December 2003. MMWR Morb Mortal Wkly Rep. 2003;52(49):1202–6.
World Health Organization. Middle East respiratory syndrome coronavirus (MERS-CoV)—Saudi Arabia. Disease outbreak news. 2017. http://www.who.int/csr/don/17-january-2017-mers-saudi-arabia/en/. Accessed 24 Jan 2017.
World Health Organization. Middle East respiratory syndrome coronavirus (MERS-CoV). Fact sheet. 2017.
Fehr AR, Perlman S. Coronaviruses: an overview of their replication and pathogenesis. Methods Mol Biol. 2015;1282:1–23.
Pasternak AO, Spaan WJ, Snijder EJ. Nidovirus transcription: how to make sense…? J Gen Virol. 2006;87(Pt 6):1403–21.
Perlman S, Netland J. Coronaviruses post-SARS: update on replication and pathogenesis. Nat Rev Microbiol. 2009;7(6):439–50.
Eckerle I, Corman VM, Muller MA, Lenk M, Ulrich RG, Drosten C. Replicative capacity of MERS coronavirus in livestock cell lines. Emerg Infect Dis. 2014;20(2):276–9.
Memish ZA, Mishra N, Olival KJ, Fagbo SF, Kapoor V, Epstein JH, et al. Middle East respiratory syndrome coronavirus in bats, Saudi Arabia. Emerg Infect Dis. 2013;19(11):1819–23.
Adney DR, van Doremalen N, Brown VR, Bushmaker T, Scott D, de Wit E, et al. Replication and shedding of MERS-CoV in upper respiratory tract of inoculated dromedary camels. Emerg Infect Dis. 2014;20(12):1999–2005.
Wang LF, Shi Z, Zhang S, Field H, Daszak P, Eaton BT. Review of bats and SARS. Emerg Infect Dis. 2006;12(12):1834–40.
Chowell G, Abdirizak F, Lee S, Lee J, Jung E, Nishiura H, et al. Transmission characteristics of MERS and SARS in the healthcare setting: a comparative study. BMC Med. 2015;13:210.
Hunter JC, Nguyen D, Aden B, Al Bandar Z, Al Dhaheri W, Abu Elkheir K, et al. Transmission of Middle East respiratory syndrome coronavirus infections in healthcare settings, Abu Dhabi. Emerg Infect Dis. 2016;22(4):647–56.
Zumla A, Hui DS, Perlman S. Middle East respiratory syndrome. Lancet. 2015;386(9997):995–1007.
Rasmussen SA, Watson AK, Swerdlow DL. Middle East respiratory syndrome (MERS). Microbiol Spectr. 2016;4(3):1–23.
Chan PK, Tang JW, Hui DS. SARS: clinical presentation, transmission, pathogenesis and treatment options. Clin Sci (Lond). 2006;110(2):193–204.
Zaki AM, van Boheemen S, Bestebroer TM, Osterhaus AD, Fouchier RA. Isolation of a novel coronavirus from a man with pneumonia in Saudi Arabia. N Engl J Med. 2012;367(19):1814–20.
Guery B, Poissy J, el Mansouf L, Sejourne C, Ettahar N, Lemaire X, et al. Clinical features and viral diagnosis of two cases of infection with Middle East Respiratory Syndrome coronavirus: a report of nosocomial transmission. Lancet. 2013;381(9885):2265–72.
WHO Mers-Cov Research Group. State of knowledge and data gaps of Middle East respiratory syndrome coronavirus (MERS-CoV) in humans. PLoS Curr. 2013;5.
Arabi YM, Arifi AA, Balkhy HH, Najm H, Aldawood AS, Ghabashi A, et al. Clinical course and outcomes of critically ill patients with Middle East respiratory syndrome coronavirus infection. Ann Intern Med. 2014;160(6):389–97.
Assiri A, Al-Tawfiq JA, Al-Rabeeah AA, Al-Rabiah FA, Al-Hajjar S, Al-Barrak A, et al. Epidemiological, demographic, and clinical characteristics of 47 cases of Middle East respiratory syndrome coronavirus disease from Saudi Arabia: a descriptive study. Lancet Infect Dis. 2013;13(9):752–61.
Ajlan AM, Ahyad RA, Jamjoom LG, Alharthy A, Madani TA. Middle East respiratory syndrome coronavirus (MERS-CoV) infection: chest CT findings. AJR Am J Roentgenol. 2014;203(4):782–7.
Memish ZA, Al-Tawfiq JA, Makhdoom HQ, Assiri A, Alhakeem RF, Albarrak A, et al. Respiratory tract samples, viral load, and genome fraction yield in patients with Middle East respiratory syndrome. J Infect Dis. 2014;210(10):1590–4.
Chiou HE, Liu CL, Buttrey MJ, Kuo HP, Liu HW, Kuo HT, et al. Adverse effects of ribavirin and outcome in severe acute respiratory syndrome: experience in two medical centers. Chest. 2005;128(1):263–72.
Leong HN, Ang B, Earnest A, Teoh C, Xu W, Leo YS. Investigational use of ribavirin in the treatment of severe acute respiratory syndrome, Singapore, 2003. Trop Med Int Health. 2004;9(8):923–7.
Chu CM, Cheng VC, Hung IF, Wong MM, Chan KH, Chan KS, et al. Role of lopinavir/ritonavir in the treatment of SARS: initial virological and clinical findings. Thorax. 2004;59(3):252–6.
Chan KS, Lai ST, Chu CM, Tsui E, Tam CY, Wong MM, et al. Treatment of severe acute respiratory syndrome with lopinavir/ritonavir: a multicentre retrospective matched cohort study. Hong Kong Med J. 2003;9(6):399–406.
Loutfy MR, Blatt LM, Siminovitch KA, Ward S, Wolff B, Lho H, et al. Interferon alfacon-1 plus corticosteroids in severe acute respiratory syndrome: a preliminary study. JAMA. 2003;290(24):3222–8.
Omrani AS, Saad MM, Baig K, Bahloul A, Abdul-Matin M, Alaidaroos AY, et al. Ribavirin and interferon alfa-2a for severe Middle East respiratory syndrome coronavirus infection: a retrospective cohort study. Lancet Infect Dis. 2014;14(11):1090–5.
Spanakis N, Tsiodras S, Haagmans BL, Raj VS, Pontikis K, Koutsoukou A, et al. Virological and serological analysis of a recent Middle East respiratory syndrome coronavirus infection case on a triple combination antiviral regimen. Int J Antimicrob Agents. 2014;44(6):528–32.
King Abdullah International Medical Research Center. MERS-CoV infection treated with a combination of lopinavir /ritonavir and interferon beta-1b (MIRACLE). Bethesda: National Institutes of Health; 2017. https://clinicaltrials.gov/ct2/show/NCT02845843. Accessed 12 July 2017.
Ashburn TT, Thor KB. Drug repositioning: identifying and developing new uses for existing drugs. Nat Rev Drug Discov. 2004;3(8):673–83.
Regnard C, Twycross R, Mihalyo M, Wilcock A. Loperamide. J Pain Symptom Manag. 2011;42(2):319–23.
Awouters F, Niemegeers CJ, Janssen PA. Pharmacology of antidiarrheal drugs. Annu Rev Pharmacol Toxicol. 1983;23:279–301.
de Wilde AH, Jochmans D, Posthuma CC, Zevenhoven-Dobbe JC, van Nieuwkoop S, Bestebroer TM, et al. Screening of an FDA-approved compound library identifies four small-molecule inhibitors of Middle East respiratory syndrome coronavirus replication in cell culture. Antimicrob Agents Chemother. 2014;58(8):4875–84.
Chertow DS, Uyeki TM, DuPont HL. Loperamide therapy for voluminous diarrhea in Ebola virus disease. J Infect Dis. 2015;211(7):1036–7.
Dyall J, Coleman CM, Hart BJ, Venkataraman T, Holbrook MR, Kindrachuk J, et al. Repurposing of clinically developed drugs for treatment of Middle East respiratory syndrome coronavirus infection. Antimicrob Agents Chemother. 2014;58(8):4885–93.
Keyaerts E, Vijgen L, Maes P, Neyts J, Van Ranst M. In vitro inhibition of severe acute respiratory syndrome coronavirus by chloroquine. Biochem Biophys Res Commun. 2004;323(1):264–8.
Thome R, Lopes SC, Costa FT, Verinaud L. Chloroquine: modes of action of an undervalued drug. Immunol Lett. 2013;153(1–2):50–7.
Brouwers J, Vermeire K, Schols D, Augustijns P. Development and in vitro evaluation of chloroquine gels as microbicides against HIV-1 infection. Virology. 2008;378(2):306–10.
Farias KJ, Machado PR, da Fonseca BA. Chloroquine inhibits dengue virus type 2 replication in Vero cells but not in C6/36 cells. Sci World J. 2013;2013:282734.
Madrid PB, Chopra S, Manger ID, Gilfillan L, Keepers TR, Shurtleff AC, et al. A systematic screen of FDA-approved drugs for inhibitors of biological threat agents. PLoS One. 2013;8(4):e60579.
Ooi EE, Chew JS, Loh JP, Chua RC. In vitro inhibition of human influenza A virus replication by chloroquine. Virol J. 2006;3:39.
Pohjala L, Utt A, Varjak M, Lulla A, Merits A, Ahola T, et al. Inhibitors of alphavirus entry and replication identified with a stable Chikungunya replicon cell line and virus-based assays. PLoS One. 2011;6(12):e28923.
Porotto M, Orefice G, Yokoyama CC, Mungall BA, Realubit R, Sganga ML, et al. Simulating henipavirus multicycle replication in a screening assay leads to identification of a promising candidate for therapy. J Virol. 2009;83(10):5148–55.
Randolph VB, Winkler G, Stollar V. Acidotropic amines inhibit proteolytic processing of flavivirus prM protein. Virology. 1990;174(2):450–8.
Savarino A, Boelaert JR, Cassone A, Majori G, Cauda R. Effects of chloroquine on viral infections: an old drug against today’s diseases? Lancet Infect Dis. 2003;3(11):722–7.
Raj VS, Mou H, Smits SL, Dekkers DH, Muller MA, Dijkman R, et al. Dipeptidyl peptidase 4 is a functional receptor for the emerging human coronavirus-EMC. Nature. 2013;495(7440):251–4.
Gierer S, Bertram S, Kaup F, Wrensch F, Heurich A, Kramer-Kuhl A, et al. The spike protein of the emerging betacoronavirus EMC uses a novel coronavirus receptor for entry, can be activated by TMPRSS2, and is targeted by neutralizing antibodies. J Virol. 2013;87(10):5502–11.
Di Trani L, Savarino A, Campitelli L, Norelli S, Puzelli S, D’Ostilio D, et al. Different pH requirements are associated with divergent inhibitory effects of chloroquine on human and avian influenza A viruses. Virol J. 2007;4:39.
Marzi A, Moller P, Hanna SL, Harrer T, Eisemann J, Steinkasserer A, et al. Analysis of the interaction of Ebola virus glycoprotein with DC-SIGN (dendritic cell-specific intercellular adhesion molecule 3-grabbing nonintegrin) and its homologue DC-SIGNR. J Infect Dis. 2007;15(196 Suppl 2):S237–46.
Savarino A, Lucia MB, Rastrelli E, Rutella S, Golotta C, Morra E, et al. Anti-HIV effects of chloroquine: inhibition of viral particle glycosylation and synergism with protease inhibitors. J Acquir Immune Defic Syndr. 2004;35(3):223–32.
Vincent MJ, Bergeron E, Benjannet S, Erickson BR, Rollin PE, Ksiazek TG, et al. Chloroquine is a potent inhibitor of SARS coronavirus infection and spread. Virol J. 2005;2:69.
Browning DJ. Toxicology of hydroxychloroquine and chloroquine and the pathology of the retinopathy they cause. In: Browning DJ, editor. Hydroxychloroquine and chloroquine retinopathy. New York: Springer Science + Business Media; 2014. p. 65–83.
Sperber K, Chiang G, Chen H, Ross W, Chusid E, Gonchar M, et al. Comparison of hydroxychloroquine with zidovudine in asymptomatic patients infected with human immunodeficiency virus type 1. Clin Ther. 1997;19(5):913–23.
Sperber K, Louie M, Kraus T, Proner J, Sapira E, Lin S, et al. Hydroxychloroquine treatment of patients with human immunodeficiency virus type 1. Clin Ther. 1995;17(4):622–36.
De Lamballerie X, Boisson V, Reynier JC, Enault S, Charrel RN, Flahault A, et al. On chikungunya acute infection and chloroquine treatment. Vector Borne Zoonotic Dis. 2008;8(6):837–9.
Tricou V, Minh NN, Van TP, Lee SJ, Farrar J, Wills B, et al. A randomized controlled trial of chloroquine for the treatment of dengue in Vietnamese adults. PLoS Negl Trop Dis. 2010;4(8):e785.
Falzarano D, Safronetz D, Prescott J, Marzi A, Feldmann F, Feldmann H. Lack of protection against ebola virus from chloroquine in mice and hamsters. Emerg Infect Dis. 2015;21(6):1065–7.
Gignoux E, Azman AS, de Smet M, Azuma P, Massaquoi M, Job D, et al. Effect of artesunate-amodiaquine on mortality related to Ebola virus disease. N Engl J Med. 2016;374(1):23–32.
World Health Organization. Categorization and prioritization of drugs for consideration for testing or use in patients infected with Ebola. 2015. http://www.who.int/medicines/ebola-treatment/2015_0703TablesofEbolaDrugs.pdf?ua=1. Accessed 13 Oct 2016.
Brickelmaier M, Lugovskoy A, Kartikeyan R, Reviriego-Mendoza MM, Allaire N, Simon K, et al. Identification and characterization of mefloquine efficacy against JC virus in vitro. Antimicrob Agents Chemother. 2009;53(5):1840–9.
Clifford DB, Nath A, Cinque P, Brew BJ, Zivadinov R, Gorelik L, et al. A study of mefloquine treatment for progressive multifocal leukoencephalopathy: results and exploration of predictors of PML outcomes. J Neurovirol. 2013;19(4):351–8.
Gofton TE, Al-Khotani A, O’Farrell B, Ang LC, McLachlan RS. Mefloquine in the treatment of progressive multifocal leukoencephalopathy. J Neurol Neurosurg Psychiatry. 2011;82(4):452–5.
Stamnes MA, Rutherford SL, Zuker CS. Cyclophilins: a new family of proteins involved in intracellular folding. Trends Cell Biol. 1992;2(9):272–6.
de Wilde AH, Raj VS, Oudshoorn D, Bestebroer TM, van Nieuwkoop S, Limpens RW, et al. MERS-coronavirus replication induces severe in vitro cytopathology and is strongly inhibited by cyclosporin A or interferon-alpha treatment. J Gen Virol. 2013;94(Pt 8):1749–60.
de Wilde AH, Zevenhoven-Dobbe JC, van der Meer Y, Thiel V, Narayanan K, Makino S, et al. Cyclosporin A inhibits the replication of diverse coronaviruses. J Gen Virol. 2011;92(Pt 11):2542–8.
Nagy PD, Wang RY, Pogany J, Hafren A, Makinen K. Emerging picture of host chaperone and cyclophilin roles in RNA virus replication. Virology. 2011;411(2):374–82.
Flisiak R, Horban A, Gallay P, Bobardt M, Selvarajah S, Wiercinska-Drapalo A, et al. The cyclophilin inhibitor Debio-025 shows potent anti-hepatitis C effect in patients coinfected with hepatitis C and human immunodeficiency virus. Hepatology. 2008;47(3):817–26.
Hopkins S, DiMassimo B, Rusnak P, Heuman D, Lalezari J, Sluder A, et al. The cyclophilin inhibitor SCY-635 suppresses viral replication and induces endogenous interferons in patients with chronic HCV genotype 1 infection. J Hepatol. 2012;57(1):47–54.
Lawitz E, Godofsky E, Rouzier R, Marbury T, Nguyen T, Ke J, et al. Safety, pharmacokinetics, and antiviral activity of the cyclophilin inhibitor NIM811 alone or in combination with pegylated interferon in HCV-infected patients receiving 14 days of therapy. Antivir Res. 2011;89(3):238–45.
Barnard DL, Day CW, Bailey K, Heiner M, Montgomery R, Lauridsen L, et al. Evaluation of immunomodulators, interferons and known in vitro SARS-coV inhibitors for inhibition of SARS-coV replication in BALB/c mice. Antivir Chem Chemother. 2006;17(5):275–84.
Haagmans BL, Kuiken T, Martina BE, Fouchier RA, Rimmelzwaan GF, van Amerongen G, et al. Pegylated interferon-alpha protects type 1 pneumocytes against SARS coronavirus infection in macaques. Nat Med. 2004;10(3):290–3.
Cinatl J, Morgenstern B, Bauer G, Chandra P, Rabenau H, Doerr HW. Treatment of SARS with human interferons. Lancet. 2003;362(9380):293–4.
Hensley LE, Fritz LE, Jahrling PB, Karp CL, Huggins JW, Geisbert TW. Interferon-beta 1a and SARS coronavirus replication. Emerg Infect Dis. 2004;10(2):317–9.
Sainz B Jr, Mossel EC, Peters CJ, Garry RF. Interferon-beta and interferon-gamma synergistically inhibit the replication of severe acute respiratory syndrome-associated coronavirus (SARS-CoV). Virology. 2004;329(1):11–7.
Falzarano D, de Wit E, Martellaro C, Callison J, Munster VJ, Feldmann H. Inhibition of novel beta coronavirus replication by a combination of interferon-alpha2b and ribavirin. Sci Rep. 2013;3:1686.
Falzarano D, de Wit E, Rasmussen AL, Feldmann F, Okumura A, Scott DP, et al. Treatment with interferon-alpha2b and ribavirin improves outcome in MERS-CoV-infected rhesus macaques. Nat Med. 2013;19(10):1313–7.
Hart BJ, Dyall J, Postnikova E, Zhou H, Kindrachuk J, Johnson RF, et al. Interferon-beta and mycophenolic acid are potent inhibitors of Middle East respiratory syndrome coronavirus in cell-based assays. J Gen Virol. 2014;95(Pt 3):571–7.
Wu P, Nielsen TE, Clausen MH. FDA-approved small-molecule kinase inhibitors. Trends Pharmacol Sci. 2015;36(7):422–39.
Hunter T. Protein kinases and phosphatases: the yin and yang of protein phosphorylation and signaling. Cell. 1995;80(2):225–36.
Cohen P. Protein kinases—the major drug targets of the twenty-first century? Nat Rev Drug Discov. 2002;1(4):309–15.
Hopkins AL, Groom CR. The druggable genome. Nat Rev Drug Discov. 2002;1(9):727–30.
Josset L, Menachery VD, Gralinski LE, Agnihothram S, Sova P, Carter VS, et al. Cell host response to infection with novel human coronavirus EMC predicts potential antivirals and important differences with SARS coronavirus. MBio. 2013;4(3):e00165-13.
Ludwig S. Disruption of virus-host cell interactions and cell signaling pathways as an anti-viral approach against influenza virus infections. Biol Chem. 2011;392(10):837–47.
Tisoncik JR, Korth MJ, Simmons CP, Farrar J, Martin TR, Katze MG. Into the eye of the cytokine storm. Microbiol Mol Biol Rev. 2012;76(1):16–32.
Coleman CM, Sisk JM, Mingo RM, Nelson EA, White JM, Frieman MB. Abelson kinase inhibitors are potent inhibitors of severe acute respiratory syndrome coronavirus and Middle East respiratory syndrome coronavirus fusion. J Virol. 2016;90(19):8924–33.
Kindrachuk J, Ork B, Hart BJ, Mazur S, Holbrook MR, Frieman MB, et al. Antiviral potential of ERK/MAPK and PI3 K/AKT/mTOR signaling modulation for Middle East respiratory syndrome coronavirus infection as identified by temporal kinome analysis. Antimicrob Agents Chemother. 2015;59(2):1088–99.
Paladin Therapeutics. Impavido (Miltefosine) capsules, for oral use prescribing information. Wilmington: Paladin Therapeutics; 2014. https://dailymed.nlm.nih.gov/dailymed/fda/fdaDrugXsl.cfm?setid=d6658aeb-7bc1-4eef-ad0d-0a873ddbecf5&type=display. Accessed 13 July 2017.
Centers for Disease Control and Prevention. Investigational drug available directly from CDC for the treatment of infections with free-living amebae. MMWR Morb Mortal Wkly Rep. 2013;62(33):666.
Wang CH, Chung FT, Lin SM, Huang SY, Chou CL, Lee KY, et al. Adjuvant treatment with a mammalian target of rapamycin inhibitor, sirolimus, and steroids improves outcomes in patients with severe H1N1 pneumonia and acute respiratory failure. Crit Care Med. 2014;42(2):313–21.
Ohlow MJ, Moosmann B. Phenothiazine: the seven lives of pharmacology’s first lead structure. Drug Discov Today. 2011;16(3–4):119–31.
Candurra NA, Maskin L, Damonte EB. Inhibition of arenavirus multiplication in vitro by phenotiazines. Antivir Res. 1996;31(3):149–58.
Chu JJ, Ng ML. Infectious entry of West Nile virus occurs through a clathrin-mediated endocytic pathway. J Virol. 2004;78(19):10543–55.
Bhattacharyya S, Warfield KL, Ruthel G, Bavari S, Aman MJ, Hope TJ. Ebola virus uses clathrin-mediated endocytosis as an entry pathway. Virology. 2010;401(1):18–28.
Chamoun-Emanuelli AM, Pecheur EI, Simeon RL, Huang D, Cremer PS, Chen Z. Phenothiazines inhibit hepatitis C virus entry, likely by increasing the fluidity of cholesterol-rich membranes. Antimicrob Agents Chemother. 2013;57(6):2571–81.
Nawa M, Takasaki T, Yamada K, Kurane I, Akatsuka T. Interference in Japanese encephalitis virus infection of Vero cells by a cationic amphiphilic drug, chlorpromazine. J Gen Virol. 2003;84(Pt 7):1737–41.
Kaur P, Chu JJ. Chikungunya virus: an update on antiviral development and challenges. Drug Discov Today. 2013;18(19–20):969–83.
Johansen LM, DeWald LE, Shoemaker CJ, Hoffstrom BG, Lear-Rooney CM, Stossel A, et al. A screen of approved drugs and molecular probes identifies therapeutics with anti-Ebola virus activity. Sci Transl Med. 2015;7(290):290ra89.
Kouznetsova J, Sun W, Martinez-Romero C, Tawa G, Shinn P, Chen CZ, et al. Identification of 53 compounds that block Ebola virus-like particle entry via a repurposing screen of approved drugs. Emerg Microbes Infect. 2014;3(12):e84.
Mingorance L, Friesland M, Coto-Llerena M, Perez-del-Pulgar S, Boix L, Lopez-Oliva JM, et al. Selective inhibition of hepatitis C virus infection by hydroxyzine and benztropine. Antimicrob Agents Chemother. 2014;58(6):3451–60.
Carranza M, Snyder MR, Shaw JD, Zesiewicz TA. Parkinson’s disease. A guide to medical treatment. Turin: SEEd Medical Publishers; 2013.
Cheng H, Lear-Rooney CM, Johansen L, Varhegyi E, Chen ZW, Olinger GG, et al. Inhibition of Ebola and Marburg virus entry by G protein-coupled receptor antagonists. J Virol. 2015;89(19):9932–8.
Lindquist M, Edwards IR. Risks of non-sedating antihistamines. Lancet. 1997;349(9061):1322.
Chong CR, Chen X, Shi L, Liu JO, Sullivan DJ Jr. A clinical drug library screen identifies astemizole as an antimalarial agent. Nat Chem Biol. 2006;2(8):415–6.
Garcia-Quiroz J, Camacho J. Astemizole: an old anti-histamine as a new promising anti-cancer drug. Anticancer Agents Med Chem. 2011;11(3):307–14.
Zhang L, Yu J, Pan H, Hu P, Hao Y, Cai W, et al. Small molecule regulators of autophagy identified by an image-based high-throughput screen. Proc Natl Acad Sci USA. 2007;104(48):19023–8.
Chiramel AI, Brady NR, Bartenschlager R. Divergent roles of autophagy in virus infection. Cells. 2013;2(1):83–104.
Shoji-Kawata S, Sumpter R, Leveno M, Campbell GR, Zou Z, Kinch L, et al. Identification of a candidate therapeutic autophagy-inducing peptide. Nature. 2013;494(7436):201–6.
Barnard DL, Day CW, Bailey K, Heiner M, Montgomery R, Lauridsen L, et al. Enhancement of the infectivity of SARS-CoV in BALB/c mice by IMP dehydrogenase inhibitors, including ribavirin. Antivir Res. 2006;71(1):53–63.
Chan JF, Chan KH, Kao RY, To KK, Zheng BJ, Li CP, et al. Broad-spectrum antivirals for the emerging Middle East respiratory syndrome coronavirus. J Infect. 2013;67(6):606–16.
Cheng KW, Cheng SC, Chen WY, Lin MH, Chuang SJ, Cheng IH, et al. Thiopurine analogs and mycophenolic acid synergistically inhibit the papain-like protease of Middle East respiratory syndrome coronavirus. Antivir Res. 2015;115:9–16.
Saijo M, Morikawa S, Fukushi S, Mizutani T, Hasegawa H, Nagata N, et al. Inhibitory effect of mizoribine and ribavirin on the replication of severe acute respiratory syndrome (SARS)-associated coronavirus. Antivir Res. 2005;66(2–3):159–63.
Cinatl J Jr, Michaelis M, Hoever G, Preiser W, Doerr HW. Development of antiviral therapy for severe acute respiratory syndrome. Antivir Res. 2005;66(2–3):81–97.
Ibarra KD, Pfeiffer JK. Reduced ribavirin antiviral efficacy via nucleoside transporter-mediated drug resistance. J Virol. 2009;83(9):4538–47.
Smith EC, Denison MR. Coronaviruses as DNA wannabes: a new model for the regulation of RNA virus replication fidelity. PLoS Pathog. 2013;9(12):e1003760.
Diamond MS, Zachariah M, Harris E. Mycophenolic acid inhibits dengue virus infection by preventing replication of viral RNA. Virology. 2002;304(2):211–21.
Kitchin JE, Pomeranz MK, Pak G, Washenik K, Shupack JL. Rediscovering mycophenolic acid: a review of its mechanism, side effects, and potential uses. J Am Acad Dermatol. 1997;37(3 Pt 1):445–9.
Armstrong VW, Tenderich G, Shipkova M, Parsa A, Koerfer R, Schroder H, et al. Pharmacokinetics and bioavailability of mycophenolic acid after intravenous administration and oral administration of mycophenolate mofetil to heart transplant recipients. Ther Drug Monit. 2005;27(3):315–21.
Chan JF, Yao Y, Yeung ML, Deng W, Bao L, Jia L, et al. Treatment with lopinavir/ritonavir or interferon-beta1b improves outcome of MERS-CoV Infection in a nonhuman primate model of common marmoset. J Infect Dis. 2015;212(12):1904–13.
Johnson RF, Via LE, Kumar MR, Cornish JP, Yellayi S, Huzella L, et al. Intratracheal exposure of common marmosets to MERS-CoV Jordan-n3/2012 or MERS-CoV EMC/2012 isolates does not result in lethal disease. Virology. 2015;485:422–30.
Naka K, Ikeda M, Abe K, Dansako H, Kato N. Mizoribine inhibits hepatitis C virus RNA replication: effect of combination with interferon-alpha. Biochem Biophys Res Commun. 2005;330(3):871–9.
Clouser CL, Holtz CM, Mullett M, Crankshaw DL, Briggs JE, Chauhan J, et al. Analysis of the ex vivo and in vivo antiretroviral activity of gemcitabine. PLoS One. 2011;6(1):e15840.
Denisova OV, Kakkola L, Feng L, Stenman J, Nagaraj A, Lampe J, et al. Obatoclax, saliphenylhalamide, and gemcitabine inhibit influenza a virus infection. J Biol Chem. 2012;287(42):35324–32.
Simmons G, Zmora P, Gierer S, Heurich A, Pohlmann S. Proteolytic activation of the SARS-coronavirus spike protein: cutting enzymes at the cutting edge of antiviral research. Antivir Res. 2013;100(3):605–14.
Glowacka I, Bertram S, Muller MA, Allen P, Soilleux E, Pfefferle S, et al. Evidence that TMPRSS2 activates the severe acute respiratory syndrome coronavirus spike protein for membrane fusion and reduces viral control by the humoral immune response. J Virol. 2011;85(9):4122–34.
Matsuyama S, Nagata N, Shirato K, Kawase M, Takeda M, Taguchi F. Efficient activation of the severe acute respiratory syndrome coronavirus spike protein by the transmembrane protease TMPRSS2. J Virol. 2010;84(24):12658–64.
Shulla A, Heald-Sargent T, Subramanya G, Zhao J, Perlman S, Gallagher T. A transmembrane serine protease is linked to the severe acute respiratory syndrome coronavirus receptor and activates virus entry. J Virol. 2011;85(2):873–82.
Hosoya M, Matsuyama S, Baba M, Suzuki H, Shigeta S. Effects of protease inhibitors on replication of various myxoviruses. Antimicrob Agents Chemother. 1992;36(7):1432–6.
Kawase M, Shirato K, van der Hoek L, Taguchi F, Matsuyama S. Simultaneous treatment of human bronchial epithelial cells with serine and cysteine protease inhibitors prevents severe acute respiratory syndrome coronavirus entry. J Virol. 2012;86(12):6537–45.
Shirato K, Kawase M, Matsuyama S. Middle East respiratory syndrome coronavirus infection mediated by the transmembrane serine protease TMPRSS2. J Virol. 2013;87(23):12552–61.
Zhou Y, Vedantham P, Lu K, Agudelo J, Carrion R Jr, Nunneley JW, et al. Protease inhibitors targeting coronavirus and filovirus entry. Antivir Res. 2015;116:76–84.
Chandran K, Sullivan NJ, Felbor U, Whelan SP, Cunningham JM. Endosomal proteolysis of the Ebola virus glycoprotein is necessary for infection. Science. 2005;308(5728):1643–5.
Schornberg K, Matsuyama S, Kabsch K, Delos S, Bouton A, White J. Role of endosomal cathepsins in entry mediated by the Ebola virus glycoprotein. J Virol. 2006;80(8):4174–8.
Sham HL, Kempf DJ, Molla A, Marsh KC, Kumar GN, Chen CM, et al. ABT-378, a highly potent inhibitor of the human immunodeficiency virus protease. Antimicrob Agents Chemother. 1998;42(12):3218–24.
Hampson L, Maranga IO, Masinde MS, Oliver AW, Batman G, He X, et al. A single-arm, proof-of-concept trial of lopimune (lopinavir/ritonavir) as a treatment for HPV-related pre-invasive cervical disease. PLoS One. 2016;11(1):e0147917.
Wu CY, Jan JT, Ma SH, Kuo CJ, Juan HF, Cheng YS, et al. Small molecules targeting severe acute respiratory syndrome human coronavirus. Proc Natl Acad Sci USA. 2004;101(27):10012–7.
Cao J, Forrest JC, Zhang X. A screen of the NIH Clinical Collection small molecule library identifies potential anti-coronavirus drugs. Antivir Res. 2015;114:1–10.
Sobin BA, Tanner Jr. FW. Anisomycin, a new anti-protozoan antibiotic. J Am Chem Soc. 1954;76:4053.
Akinboye ES, Brennen WN, Rosen DM, Bakare O, Denmeade SR. Iterative design of emetine-based prodrug targeting fibroblast activation protein (FAP) and dipeptidyl peptidase IV DPPIV using a tandem enzymatic activation strategy. Prostate. 2016;76(8):703–14.
Gonzalez Constandse R. Anisomycin in intestinal amebiasis; study of 30 clinical cases. Prensa Med Mex. 1956;21(7–10):114–5.
Conte JE. Manual of antibiotics and infectious diseases: Treatment and prevention. 9th ed. Philadelphia: Lippincott Williams & Wilkins; 2001.
Gupta RS, Krepinsky JJ, Siminovitch L. Structural determinants responsible for the biological activity of (−)-emetine, (−)-cryptopleurine, and (−)-tylocrebrine: structure-activity relationship among related compounds. Mol Pharmacol. 1980;18(1):136–43.
Zinck R, Cahill MA, Kracht M, Sachsenmaier C, Hipskind RA, Nordheim A. Protein synthesis inhibitors reveal differential regulation of mitogen-activated protein kinase and stress-activated protein kinase pathways that converge on Elk-1. Mol Cell Biol. 1995;15(9):4930–8.
Ramabhadran TV, Thach RE. Specificity of protein synthesis inhibitors in the inhibition of encephalomyocarditis virus replication. J Virol. 1980;34(1):293–6.
Hwang YC, Chu JJ, Yang PL, Chen W, Yates MV. Rapid identification of inhibitors that interfere with poliovirus replication using a cell-based assay. Antivir Res. 2008;77(3):232–6.
Romero MR, Serrano MA, Efferth T, Alvarez M, Marin JJ. Effect of cantharidin, cephalotaxine and homoharringtonine on “in vitro” models of hepatitis B virus (HBV) and bovine viral diarrhoea virus (BVDV) replication. Planta Med. 2007;73(6):552–8.
Gastaminza P, Whitten-Bauer C, Chisari FV. Unbiased probing of the entire hepatitis C virus life cycle identifies clinical compounds that target multiple aspects of the infection. Proc Natl Acad Sci USA. 2010;107(1):291–6.
Watashi K, Inoue D, Hijikata M, Goto K, Aly HH, Shimotohno K. Anti-hepatitis C virus activity of tamoxifen reveals the functional association of estrogen receptor with viral RNA polymerase NS5B. J Biol Chem. 2007;282(45):32765–72.
Johansen LM, Brannan JM, Delos SE, Shoemaker CJ, Stossel A, Lear C, et al. FDA-approved selective estrogen receptor modulators inhibit Ebola virus infection. Sci Transl Med. 2013;5(190):190ra79.
Shoemaker CJ, Schornberg KL, Delos SE, Scully C, Pajouhesh H, Olinger GG, et al. Multiple cationic amphiphiles induce a Niemann-Pick C phenotype and inhibit Ebola virus entry and infection. PLoS One. 2013;8(2):e56265.
Zhao Y, Ren J, Harlos K, Jones DM, Zeltina A, Bowden TA, et al. Toremifene interacts with and destabilizes the Ebola virus glycoprotein. Nature. 2016;535(7610):169–72.
Kaufmann AM, Krise JP. Lysosomal sequestration of amine-containing drugs: analysis and therapeutic implications. J Pharm Sci. 2007;96(4):729–46.
Pharma Orion. FARESTON® (toremifene citrate) tablets prescribing information. Memphis: GTx, Distributor; 2011.
AstraZeneca. NOLVADEX® tamoxifen citrate tablets prescribing information. Wilmington: AstraZeneca; 2006.
Blanc M, Hsieh WY, Robertson KA, Watterson S, Shui G, Lacaze P, et al. Host defense against viral infection involves interferon mediated down-regulation of sterol biosynthesis. PLoS Biol. 2011;9(3):e1000598.
Owens CM, Mawhinney C, Grenier JM, Altmeyer R, Lee MS, Borisy AA, et al. Chemical combinations elucidate pathway interactions and regulation relevant to Hepatitis C replication. Mol Syst Biol. 2010;8(6):375.
Warren TK, Jordan R, Lo MK, Ray AS, Mackman RL, Soloveva V, et al. Therapeutic efficacy of the small molecule GS-5734 against Ebola virus in rhesus monkeys. Nature. 2016;531(7594):381–5.
Warren TK, Wells J, Panchal RG, Stuthman KS, Garza NL, Van Tongeren SA, et al. Protection against filovirus diseases by a novel broad-spectrum nucleoside analogue BCX4430. Nature. 2014;508(7496):402–5.
Peters HL, Jochmans D, de Wilde AH, Posthuma CC, Snijder EJ, Neyts J, et al. Design, synthesis and evaluation of a series of acyclic fleximer nucleoside analogues with anti-coronavirus activity. Bioorg Med Chem Lett. 2015;25(15):2923–6.
Adedeji AO, Sarafianos SG. Antiviral drugs specific for coronaviruses in preclinical development. Curr Opin Virol. 2014;8:45–53.
Tanner JA, Zheng BJ, Zhou J, Watt RM, Jiang JQ, Wong KL, et al. The adamantane-derived bananins are potent inhibitors of the helicase activities and replication of SARS coronavirus. Chem Biol. 2005;12(3):303–11.
Leonard JN, Schaffer DV. Antiviral RNAi therapy: emerging approaches for hitting a moving target. Gene Ther. 2006;13(6):532–40.
He ML, Zheng B, Peng Y, Peiris JS, Poon LL, Yuen KY, et al. Inhibition of SARS-associated coronavirus infection and replication by RNA interference. JAMA. 2003;290(20):2665–6.
He ML, Zheng BJ, Chen Y, Wong KL, Huang JD, Lin MC, et al. Development of interfering RNA agents to inhibit SARS-associated coronavirus infection and replication. Hong Kong Med J. 2009;15(3 Suppl 4):28–31.
Lu A, Zhang H, Zhang X, Wang H, Hu Q, Shen L, et al. Attenuation of SARS coronavirus by a short hairpin RNA expression plasmid targeting RNA-dependent RNA polymerase. Virology. 2004;324(1):84–9.
Wang Z, Ren L, Zhao X, Hung T, Meng A, Wang J, et al. Inhibition of severe acute respiratory syndrome virus replication by small interfering RNAs in mammalian cells. J Virol. 2004;78(14):7523–7.
Zheng BJ, Guan Y, Tang Q, Du C, Xie FY, He ML, et al. Prophylactic and therapeutic effects of small interfering RNA targeting SARS-coronavirus. Antivir Ther. 2004;9(3):365–74.
Zhang Y, Li T, Fu L, Yu C, Li Y, Xu X, et al. Silencing SARS-CoV spike protein expression in cultured cells by RNA interference. FEBS Lett. 2004;560(1–3):141–6.
Wu CJ, Huang HW, Liu CY, Hong CF, Chan YL. Inhibition of SARS-CoV replication by siRNA. Antivir Res. 2005;65(1):45–8.
Akerstrom S, Mirazimi A, Tan YJ. Inhibition of SARS-CoV replication cycle by small interference RNAs silencing specific SARS proteins, 7a/7b, 3a/3b and S. Antivir Res. 2007;73(3):219–27.
Li BJ, Tang Q, Cheng D, Qin C, Xie FY, Wei Q, et al. Using siRNA in prophylactic and therapeutic regimens against SARS coronavirus in rhesus macaque. Nat Med. 2005;11(9):944–51.
Hasan MM, Akter R, Ullah MS, Abedin MJ, Ullah GM, Hossain MZ. A computational approach for predicting role of human microRNAs in MERS-CoV genome. Adv Bioinform. 2014;2014:967946.
Thi EP, Mire CE, Lee AC, Geisbert JB, Zhou JZ, Agans KN, et al. Lipid nanoparticle siRNA treatment of Ebola-virus-Makona-infected nonhuman primates. Nature. 2015;521(7552):362–5.
Dunning J, Sahr F, Rojek A, Gannon F, Carson G, Idriss B, et al. Experimental treatment of Ebola virus disease with TKM-130803: a single-arm phase 2 clinical trial. PLoS Med. 2016;13(4):e1001997.
Badani H, Garry RF, Wimley WC. Peptide entry inhibitors of enveloped viruses: the importance of interfacial hydrophobicity. Biochim Biophys Acta. 2014;1838(9):2180–97.
Lu L, Liu Q, Zhu Y, Chan KH, Qin L, Li Y, et al. Structure-based discovery of Middle East respiratory syndrome coronavirus fusion inhibitor. Nat Commun. 2014;5:3067.
Sainz B Jr, Mossel EC, Gallaher WR, Wimley WC, Peters CJ, Wilson RB, et al. Inhibition of severe acute respiratory syndrome-associated coronavirus (SARS-CoV) infectivity by peptides analogous to the viral spike protein. Virus Res. 2006;120(1–2):146–55.
Sainz B Jr, Rausch JM, Gallaher WR, Garry RF, Wimley WC. Identification and characterization of the putative fusion peptide of the severe acute respiratory syndrome-associated coronavirus spike protein. J Virol. 2005;79(11):7195–206.
Bird GH, Madani N, Perry AF, Princiotto AM, Supko JG, He X, et al. Hydrocarbon double-stapling remedies the proteolytic instability of a lengthy peptide therapeutic. Proc Natl Acad Sci USA. 2010;107(32):14093–8.
Kindrachuk J, Scruten E, Attah-Poku S, Bell K, Potter A, Babiuk LA, et al. Stability, toxicity, and biological activity of host defense peptide BMAP28 and its inversed and retro-inversed isomers. Biopolymers. 2011;96(1):14–24.
Walensky LD, Bird GH. Hydrocarbon-stapled peptides: principles, practice, and progress. J Med Chem. 2014;57(15):6275–88.
Shadman KA, Wald ER. A review of palivizumab and emerging therapies for respiratory syncytial virus. Expert Opin Biol Ther. 2011;11(11):1455–67.
Qiu X, Wong G, Audet J, Bello A, Fernando L, Alimonti JB, et al. Reversion of advanced Ebola virus disease in nonhuman primates with ZMapp. Nature. 2014;514(7520):47–53.
PREVAIL II Writing Group for the Multinational PREVAIL Study Team. A randomized, controlled trial of ZMapp for Ebola virus fnfection. N Engl J Med. 2016;375(15):1448–56.
Bossart KN, Zhu Z, Middleton D, Klippel J, Crameri G, Bingham J, et al. A neutralizing human monoclonal antibody protects against lethal disease in a new ferret model of acute nipah virus infection. PLoS Pathog. 2009;5(10):e1000642.
Zhu Z, Dimitrov AS, Bossart KN, Crameri G, Bishop KA, Choudhry V, et al. Potent neutralization of Hendra and Nipah viruses by human monoclonal antibodies. J Virol. 2006;80(2):891–9.
Bossart KN, Geisbert TW, Feldmann H, Zhu Z, Feldmann F, Geisbert JB, et al. A neutralizing human monoclonal antibody protects African green monkeys from hendra virus challenge. Sci Transl Med. 2011;3(105):105ra3.
Corti D, Zhao J, Pedotti M, Simonelli L, Agnihothram S, Fett C, et al. Prophylactic and postexposure efficacy of a potent human monoclonal antibody against MERS coronavirus. Proc Natl Acad Sci USA. 2015;112(33):10473–8.
Houser KV, Gretebeck L, Ying T, Wang Y, Vogel L, Lamirande EW, et al. Prophylaxis with a Middle East respiratory syndrome coronavirus (MERS-CoV)-specific human monoclonal antibody protects rabbits from MERS-CoV infection. J Infect Dis. 2016;213(10):1557–61.
Jiang L, Wang N, Zuo T, Shi X, Poon KM, Wu Y, et al. Potent neutralization of MERS-CoV by human neutralizing monoclonal antibodies to the viral spike glycoprotein. Sci Transl Med. 2014;6(234):234ra59.
Johnson RF, Bagci U, Keith L, Tang X, Mollura DJ, Zeitlin L, et al. 3B11-N, a monoclonal antibody against MERS-CoV, reduces lung pathology in rhesus monkeys following intratracheal inoculation of MERS-CoV Jordan-n3/2012. Virology. 2016;490:49–58.
Pascal KE, Coleman CM, Mujica AO, Kamat V, Badithe A, Fairhurst J, et al. Pre- and postexposure efficacy of fully human antibodies against Spike protein in a novel humanized mouse model of MERS-CoV infection. Proc Natl Acad Sci USA. 2015;112(28):8738–43.
Qiu H, Sun S, Xiao H, Feng J, Guo Y, Tai W, et al. Single-dose treatment with a humanized neutralizing antibody affords full protection of a human transgenic mouse model from lethal Middle East respiratory syndrome (MERS)-coronavirus infection. Antivir Res. 2016;14(132):141–8.
Tang XC, Agnihothram SS, Jiao Y, Stanhope J, Graham RL, Peterson EC, et al. Identification of human neutralizing antibodies against MERS-CoV and their role in virus adaptive evolution. Proc Natl Acad Sci USA. 2014;111(19):E2018–26.
Ying T, Du L, Ju TW, Prabakaran P, Lau CC, Lu L, et al. Exceptionally potent neutralization of Middle East respiratory syndrome coronavirus by human monoclonal antibodies. J Virol. 2014;88(14):7796–805.
Mair-Jenkins J, Saavedra-Campos M, Baillie JK, Cleary P, Khaw FM, Lim WS, et al. The effectiveness of convalescent plasma and hyperimmune immunoglobulin for the treatment of severe acute respiratory infections of viral etiology: a systematic review and exploratory meta-analysis. J Infect Dis. 2015;211(1):80–90.
Ying T, Li H, Lu L, Dimitrov DS, Jiang S. Development of human neutralizing monoclonal antibodies for prevention and therapy of MERS-CoV infections. Microbes Infect. 2015;17(2):142–8.
Berry JD, Gaudet RG. Antibodies in infectious diseases: polyclonals, monoclonals and niche biotechnology. New Biotechnol. 2011;28(5):489–501.
Luke T, Wu H, Zhao J, Channappanavar R, Coleman CM, Jiao JA, et al. Human polyclonal immunoglobulin G from transchromosomic bovines inhibits MERS-CoV in vivo. Sci Transl Med. 2016;8(326):326ra21.
De Clercq E. Strategies in the design of antiviral drugs. Nat Rev Drug Discov. 2002;1(1):13–25.
AIDSinfo. HIV treatment. FDA-approved HIV medicines. 2016. https://aidsinfo.nih.gov/education-materials/fact-sheets/21/58/fda-approved-hiv-medicines. Accessed 13 Oct 2016.
World Health Organization. Consolidated guidelines for the use of antiretroviral drugs for treating and preventing HIV infection. Recommendations for a public health approach, 2nd edn. 2016. http://www.who.int/hiv/pub/arv/arv-2016/en/. Accessed 13 Oct 2016.
American Association for the Study of Liver Diseases, Infectious Diseases Society of America. HCV guidance: recommendations for testing, managing, and treating hepatitis C. 2017. http://www.hcvguidelines.org./sites/default/files/full-guidance-pdf/HCVGuidance_April_12_2017_b.pdf. Accessed 28 July 2017.
Kohli A, Shaffer A, Sherman A, Kottilil S. Treatment of hepatitis C: a systematic review. JAMA. 2014;312(6):631–40.
Ogawa E, Furusyo N, Kajiwara E, Takahashi K, Nomura H, Tanabe Y, et al. Evaluation of the adverse effect of premature discontinuation of pegylated interferon alpha-2b and ribavirin treatment for chronic hepatitis C virus infection: results from Kyushu University liver disease study. J Gastroenterol Hepatol. 2012;27(7):1233–40.
Govorkova EA, McCullers JA. Therapeutics against influenza. Curr Top Microbiol Immunol. 2013;370:273–300.
Dunning J, Baillie JK, Cao B, Hayden FG. International Severe Acute Respiratory Emerging Infection Consortium. Antiviral combinations for severe influenza. Lancet Infect Dis. 2014;14(12):1259–70.
Hayden FG. Advances in antivirals for non-influenza respiratory virus infections. Influenza Other Respir Viruses. 2013;7(Suppl 3):36–43.
Seo S, Englund JA, Nguyen JT, Pukrittayakamee S, Lindegardh N, Tarning J, et al. Combination therapy with amantadine, oseltamivir and ribavirin for influenza A infection: safety and pharmacokinetics. Antivir Ther. 2013;18(3):377–86.
Agnandji ST, Huttner A, Zinser ME, Njuguna P, Dahlke C, Fernandes JF, et al. Phase 1 Trials of rVSV Ebola Vaccine in Africa and Europe. N Engl J Med. 2016;374(17):1647–60.
Ewer K, Rampling T, Venkatraman N, Bowyer G, Wright D, Lambe T, et al. A monovalent chimpanzee adenovirus Ebola vaccine boosted with MVA. N Engl J Med. 2016;374(17):1635–46.
Ledgerwood JE, DeZure AD, Stanley DA, Coates EE, Novik L, Enama ME, et al. Chimpanzee adenovirus vector Ebola vaccine. N Engl J Med. 2017; 376(10):928–38.
Regules JA, Beigel JH, Paolino KM, Voell J, Castellano AR, Hu Z, et al. A recombinant vesicular stomatitis virus Ebola vaccine. N Engl J Med. 2017;376(4):330–41.
Henao-Restrepo AM, Longini IM, Egger M, Dean NE, Edmunds WJ, Camacho A, et al. Efficacy and effectiveness of an rVSV-vectored vaccine expressing Ebola surface glycoprotein: interim results from the Guinea ring vaccination cluster-randomised trial. Lancet. 2015;386(9996):857–66.
Baseler L, de Wit E, Feldmann H. A comparative review of animal models of Middle East respiratory syndrome coronavirus infection. Vet Pathol. 2016;53(3):521–31.
Sutton TC, Subbarao K. Development of animal models against emerging coronaviruses: from SARS to MERS coronavirus. Virology. 2015;479–480:247–58.
van Doremalen N, Munster VJ. Animal models of Middle East respiratory syndrome coronavirus infection. Antivir Res. 2015;122:28–38.
Zhao J, Li K, Wohlford-Lenane C, Agnihothram SS, Fett C, Zhao J, et al. Rapid generation of a mouse model for Middle East respiratory syndrome. Proc Natl Acad Sci USA. 2014;111(13):4970–5.
Channappanavar R, Fett C, Zhao J, Meyerholz DK, Perlman S. Virus-specific memory CD8 T cells provide substantial protection from lethal severe acute respiratory syndrome coronavirus infection. J Virol. 2014;88(19):11034–44.
Channappanavar R, Zhao J, Perlman S. T cell-mediated immune response to respiratory coronaviruses. Immunol Res. 2014;59(1–3):118–28.
Agrawal AS, Garron T, Tao X, Peng BH, Wakamiya M, Chan TS, et al. Generation of a transgenic mouse model of Middle East respiratory syndrome coronavirus infection and disease. J Virol. 2015;89(7):3659–70.
Cockrell AS, Yount BL, Scobey T, Jensen K, Douglas M, Beall A, et al. A mouse model for MERS coronavirus-induced acute respiratory distress syndrome. Nat Microbiol. 2016;28(2):16226.
Haagmans BL, van den Brand JM, Provacia LB, Raj VS, Stittelaar KJ, Getu S, et al. Asymptomatic Middle East respiratory syndrome coronavirus infection in rabbits. J Virol. 2015;89(11):6131–5.
de Wit E, Rasmussen AL, Falzarano D, Bushmaker T, Feldmann F, Brining DL, et al. Middle East respiratory syndrome coronavirus (MERS-CoV) causes transient lower respiratory tract infection in rhesus macaques. Proc Natl Acad Sci USA. 2013;110(41):16598–603.
Munster VJ, de Wit E, Feldmann H. Pneumonia from human coronavirus in a macaque model. N Engl J Med. 2013;368(16):1560–2.
Yao Y, Bao L, Deng W, Xu L, Li F, Lv Q, et al. An animal model of MERS produced by infection of rhesus macaques with MERS coronavirus. J Infect Dis. 2014;209(2):236–42.
Clay CC, Donart N, Fomukong N, Knight JB, Overheim K, Tipper J, et al. Severe acute respiratory syndrome-coronavirus infection in aged nonhuman primates is associated with modulated pulmonary and systemic immune responses. Immun Ageing. 2014;11(1):4.
Smits SL, van den Brand JM, de Lang A, Leijten LM, van Ijcken WF, van Amerongen G, et al. Distinct severe acute respiratory syndrome coronavirus-induced acute lung injury pathways in two different nonhuman primate species. J Virol. 2011;85(9):4234–45.
McAuliffe J, Vogel L, Roberts A, Fahle G, Fischer S, Shieh WJ, et al. Replication of SARS coronavirus administered into the respiratory tract of African green, rhesus and cynomolgus monkeys. Virology. 2004;330(1):8–15.
Hartman AL, Powell DS, Bethel LM, Caroline AL, Schmid RJ, Oury T, et al. Aerosolized Rift Valley fever virus causes fatal encephalitis in African green monkeys and common marmosets. J Virol. 2014;88(4):2235–45.
Kramski M, Matz-Rensing K, Stahl-Hennig C, Kaup FJ, Nitsche A, Pauli G, et al. A novel highly reproducible and lethal nonhuman primate model for orthopox virus infection. PLoS One. 2010;5(4):e10412.
Mucker EM, Chapman J, Huzella LM, Huggins JW, Shamblin J, Robinson CG, et al. Susceptibility of marmosets (Callithrix jacchus) to monkeypox virus: a low dose prospective model for monkeypox and smallpox disease. PLoS One. 2015;10(7):e0131742.
Falzarano D, de Wit E, Feldmann F, Rasmussen AL, Okumura A, Peng X, et al. Infection with MERS-CoV causes lethal pneumonia in the common marmoset. PLoS Pathog. 2014;10(8):e1004250.
Greenough TC, Carville A, Coderre J, Somasundaran M, Sullivan JL, Luzuriaga K, et al. Pneumonitis and multi-organ system disease in common marmosets (Callithrix jacchus) infected with the severe acute respiratory syndrome-associated coronavirus. Am J Pathol. 2005;167(2):455–63.
Bai JP. Pharmacodynamics and systems pharmacology approaches to repurposing drugs in the wake of global health burden. J Pharm Sci. 2016;105(10):3007–12.
Brik A, Wong CH. HIV-1 protease: mechanism and drug discovery. Org Biomol Chem. 2003;1(1):5–14.
Navia MA, Fitzgerald PM, McKeever BM, Leu CT, Heimbach JC, Herber WK, et al. Three-dimensional structure of aspartyl protease from human immunodeficiency virus HIV-1. Nature. 1989;337(6208):615–20.
Wlodawer A, Miller M, Jaskolski M, Sathyanarayana BK, Baldwin E, Weber IT, et al. Conserved folding in retroviral proteases: crystal structure of a synthetic HIV-1 protease. Science. 1989;245(4918):616–21.
Gotte M, Feld JJ. Direct-acting antiviral agents for hepatitis C: structural and mechanistic insights. Nat Rev Gastroenterol Hepatol. 2016;13(6):338–51.
Hazuda DJ, Burroughs M, Howe AY, Wahl J, Venkatraman S. Development of boceprevir: a first-in-class direct antiviral treatment for chronic hepatitis C infection. Ann N Y Acad Sci. 2013;1291:69–76.
Kwong AD, Kauffman RS, Hurter P, Mueller P. Discovery and development of telaprevir: an NS3-4A protease inhibitor for treating genotype 1 chronic hepatitis C virus. Nat Biotechnol. 2011;29(11):993–1003.
Hilgenfeld R. From SARS to MERS: crystallographic studies on coronaviral proteases enable antiviral drug design. FEBS J. 2014;281(18):4085–96.
Miller ME, Adhikary S, Kolokoltsov AA, Davey RA. Ebolavirus requires acid sphingomyelinase activity and plasma membrane sphingomyelin for infection. J Virol. 2012;86(14):7473–83.
Muller MP, Dresser L, Raboud J, McGeer A, Rea E, Richardson SE, et al. Adverse events associated with high-dose ribavirin: evidence from the Toronto outbreak of severe acute respiratory syndrome. Pharmacotherapy. 2007;27(4):494–503.
Ward SE, Loutfy MR, Blatt LM, Siminovitch KA, Chen J, Hinek A, et al. Dynamic changes in clinical features and cytokine/chemokine responses in SARS patients treated with interferon alfacon-1 plus corticosteroids. Antivir Ther. 2005;10(2):263–75.
Zhao Z, Zhang F, Xu M, Huang K, Zhong W, Cai W, et al. Description and clinical treatment of an early outbreak of severe acute respiratory syndrome (SARS) in Guangzhou, PR China. J Med Microbiol. 2003;52(Pt 8):715–20.
Al-Tawfiq JA, Momattin H, Dib J, Memish ZA. Ribavirin and interferon therapy in patients infected with the Middle East respiratory syndrome coronavirus: an observational study. Int J Infect Dis. 2014;20:42–6.
Boonyasuppayakorn S, Reichert ED, Manzano M, Nagarajan K, Padmanabhan R. Amodiaquine, an antimalarial drug, inhibits dengue virus type 2 replication and infectivity. Antivir Res. 2014;106:125–34.
Bassetto M, De Burghgraeve T, Delang L, Massarotti A, Coluccia A, Zonta N, et al. Computer-aided identification, design and synthesis of a novel series of compounds with selective antiviral activity against chikungunya virus. Antivir Res. 2013;98(1):12–8.
Cruz DJ, Bonotto RM, Gomes RG, da Silva CT, Taniguchi JB, No JH, et al. Identification of novel compounds inhibiting chikungunya virus-induced cell death by high throughput screening of a kinase inhibitor library. PLoS Negl Trop Dis. 2013;7(10):e2471.
Yan Y, Zou Z, Sun Y, Li X, Xu KF, Wei Y, et al. Anti-malaria drug chloroquine is highly effective in treating avian influenza A H5N1 virus infection in an animal model. Cell Res. 2013;23(2):300–2.
Paton NI, Lee L, Xu Y, Ooi EE, Cheung YB, Archuleta S, et al. Chloroquine for influenza prevention: a randomised, double-blind, placebo controlled trial. Lancet Infect Dis. 2011;11(9):677–83.
Romanelli F, Smith KM, Hoven AD. Chloroquine and hydroxychloroquine as inhibitors of human immunodeficiency virus (HIV-1) activity. Curr Pharm Des. 2004;10(21):2643–8.
Nakagawa M, Sakamoto N, Tanabe Y, Koyama T, Itsui Y, Takeda Y, et al. Suppression of hepatitis C virus replication by cyclosporin A is mediated by blockade of cyclophilins. Gastroenterology. 2005;129(3):1031–41.
Qing M, Yang F, Zhang B, Zou G, Robida JM, Yuan Z, et al. Cyclosporine inhibits flavivirus replication through blocking the interaction between host cyclophilins and viral NS5 protein. Antimicrob Agents Chemother. 2009;53(8):3226–35.
Kambara H, Tani H, Mori Y, Abe T, Katoh H, Fukuhara T, et al. Involvement of cyclophilin B in the replication of Japanese encephalitis virus. Virology. 2011;412(1):211–9.
Bose S, Mathur M, Bates P, Joshi N, Banerjee AK. Requirement for cyclophilin A for the replication of vesicular stomatitis virus New Jersey serotype. J Gen Virol. 2003;84(Pt 7):1687–99.
Briggs CJ, Ott DE, Coren LV, Oroszlan S, Tozser J. Comparison of the effect of FK506 and cyclosporin A on virus production in H9 cells chronically and newly infected by HIV-1. Arch Virol. 1999;144(11):2151–60.
Randhawa PS, Farasati NA, Huang Y, Mapara MY, Shapiro R. Viral drug sensitivity testing using quantitative PCR: effect of tyrosine kinase inhibitors on polyomavirus BK replication. Am J Clin Pathol. 2010;134(6):916–20.
Pogliaghi M, Papagno L, Lambert S, Calin R, Calvez V, Katlama C, et al. The tyrosine kinase inhibitor Dasatinib blocks in-vitro HIV-1 production by primary CD4+ T cells from HIV-1 infected patients. AIDS. 2014;28(2):278–81.
de Wispelaere M, LaCroix AJ, Yang PL. The small molecules AZD0530 and dasatinib inhibit dengue virus RNA replication via Fyn kinase. J Virol. 2013;87(13):7367–81.
Reeves PM, Smith SK, Olson VA, Thorne SH, Bornmann W, Damon IK, et al. Variola and monkeypox viruses utilize conserved mechanisms of virion motility and release that depend on ABL and SRC family tyrosine kinases. J Virol. 2011;85(1):21–31.
Murray JL, McDonald NJ, Sheng J, Shaw MW, Hodge TW, Rubin DH, et al. Inhibition of influenza A virus replication by antagonism of a PI3 K-AKT-mTOR pathway member identified by gene-trap insertional mutagenesis. Antivir Chem Chemother. 2012;22(5):205–15.
Brennan DC, Legendre C, Patel D, Mange K, Wiland A, McCague K, et al. Cytomegalovirus incidence between everolimus versus mycophenolate in de novo renal transplants: pooled analysis of three clinical trials. Am J Transplant. 2011;11(11):2453–62.
Kobashigawa J, Ross H, Bara C, Delgado JF, Dengler T, Lehmkuhl HB, et al. Everolimus is associated with a reduced incidence of cytomegalovirus infection following de novo cardiac transplantation. Transpl Infect Dis. 2013;15(2):150–62.
Hutterer C, Wandinger SK, Wagner S, Muller R, Stamminger T, Zeittrager I, et al. Profiling of the kinome of cytomegalovirus-infected cells reveals the functional importance of host kinases Aurora A, ABL and AMPK. Antivir Res. 2013;99(2):139–48.
Cheshenko N, Trepanier JB, Stefanidou M, Buckley N, Gonzalez P, Jacobs W, et al. HSV activates Akt to trigger calcium release and promote viral entry: novel candidate target for treatment and suppression. FASEB J. 2013;27(7):2584–99.
Heredia A, Gilliam B, Latinovic O, Le N, Bamba D, Devico A, et al. Rapamycin reduces CCR5 density levels on CD4 T cells, and this effect results in potentiation of enfuvirtide (T-20) against R5 strains of human immunodeficiency virus type 1 in vitro. Antimicrob Agents Chemother. 2007;51(7):2489–96.
Kudchodkar SB, Yu Y, Maguire TG, Alwine JC. Human cytomegalovirus infection alters the substrate specificities and rapamycin sensitivities of raptor- and rictor-containing complexes. Proc Natl Acad Sci USA. 2006;103(38):14182–7.
Di Benedetto F, Di Sandro S, De Ruvo N, Montalti R, Ballarin R, Guerrini GP, et al. First report on a series of HIV patients undergoing rapamycin monotherapy after liver transplantation. Transplantation. 2010;89(6):733–8.
Ghassemieh B, Ahya VN, Baz MA, Valentine VG, Arcasoy SM, Love RB, et al. Decreased incidence of cytomegalovirus infection with sirolimus in a post hoc randomized, multicenter study in lung transplantation. J Heart Lung Transplant. 2013;32(7):701–6.
Soliman A, Fathy A, Khashab S, Shaheen N, Soliman M. Sirolimus conversion may suppress viral replication in hepatitis C virus-positive renal transplant candidates. Exp Clin Transplant. 2013;11(5):408–11.
Michaelis M, Paulus C, Loschmann N, Dauth S, Stange E, Doerr HW, et al. The multi-targeted kinase inhibitor sorafenib inhibits human cytomegalovirus replication. Cell Mol Life Sci. 2011;68(6):1079–90.
Gao M, Duan H, Liu J, Zhang H, Wang X, Zhu M, et al. The multi-targeted kinase inhibitor sorafenib inhibits enterovirus 71 replication by regulating IRES-dependent translation of viral proteins. Antivir Res. 2014;106:80–5.
Benedict A, Bansal N, Senina S, Hooper I, Lundberg L, de la Fuente C, et al. Repurposing FDA-approved drugs as therapeutics to treat Rift Valley fever virus infection. Front Microbiol. 2015;6:676.
Sasaki H, Nakamura M, Ohno T, Matsuda Y, Yuda Y, Nonomura Y. Myosin-actin interaction plays an important role in human immunodeficiency virus type 1 release from host cells. Proc Natl Acad Sci USA. 1995;92(6):2026–30.
Hirai H, Takeda S, Natori S, Sekimizu K. Inhibition of SV40 DNA replication in vitro by chlorpromazine. Biol Pharm Bull. 1993;16(6):565–7.
Yanagida K, Baba C, Baba M. Inhibition of bovine viral diarrhea virus (BVDV) by mizoribine: synergistic effect of combination with interferon-alpha. Antivir Res. 2004;64(3):195–201.
Batman G, Oliver AW, Zehbe I, Richard C, Hampson L, Hampson IN. Lopinavir up-regulates expression of the antiviral protein ribonuclease L in human papillomavirus-positive cervical carcinoma cells. Antivir Ther. 2011;16(4):515–25.
Murakami Y, Fukasawa M, Kaneko Y, Suzuki T, Wakita T, Fukazawa H. Selective estrogen receptor modulators inhibit hepatitis C virus infection at multiple steps of the virus life cycle. Microbes Infect. 2013;15(1):45–55.
Zheng K, Chen M, Xiang Y, Ma K, Jin F, Wang X, et al. Inhibition of herpes simplex virus type 1 entry by chloride channel inhibitors tamoxifen and NPPB. Biochem Biophys Res Commun. 2014;446(4):990–6.
Kilianski A, Mielech AM, Deng X, Baker SC. Assessing activity and inhibition of Middle East respiratory syndrome coronavirus papain-like and 3C-like proteases using luciferase-based biosensors. J Virol. 2013;87(21):11955–62.
Lundin A, Dijkman R, Bergstrom T, Kann N, Adamiak B, Hannoun C, et al. Targeting membrane-bound viral RNA synthesis reveals potent inhibition of diverse coronaviruses including the Middle East respiratory syndrome virus. PLoS Pathog. 2014;10(5):e1004166.
Millet JK, Whittaker GR. Host cell entry of Middle East respiratory syndrome coronavirus after two-step, furin-mediated activation of the spike protein. Proc Natl Acad Sci USA. 2014;111(42):15214–9.
Shie JJ, Fang JM, Kuo CJ, Kuo TH, Liang PH, Huang HJ, et al. Discovery of potent anilide inhibitors against the severe acute respiratory syndrome 3CL protease. J Med Chem. 2005;48(13):4469–73.
Shao YM, Yang WB, Peng HP, Hsu MF, Tsai KC, Kuo TH, et al. Structure-based design and synthesis of highly potent SARS-CoV 3CL protease inhibitors. ChemBioChem. 2007;8(14):1654–7.
Ramajayam R, Tan KP, Liu HG, Liang PH. Synthesis and evaluation of pyrazolone compounds as SARS-coronavirus 3C-like protease inhibitors. Bioorg Med Chem. 2010;18(22):7849–54.
Shao YM, Yang WB, Kuo TH, Tsai KC, Lin CH, Yang AS, et al. Design, synthesis, and evaluation of trifluoromethyl ketones as inhibitors of SARS-CoV 3CL protease. Bioorg Med Chem. 2008;16(8):4652–60.
Chen L, Gui C, Luo X, Yang Q, Gunther S, Scandella E, et al. Cinanserin is an inhibitor of the 3C-like proteinase of severe acute respiratory syndrome coronavirus and strongly reduces virus replication in vitro. J Virol. 2005;79(11):7095–103.
Lee CC, Kuo CJ, Hsu MF, Liang PH, Fang JM, Shie JJ, et al. Structural basis of mercury- and zinc-conjugated complexes as SARS-CoV 3C-like protease inhibitors. FEBS Lett. 2007;581(28):5454–8.
Lee C, Lee JM, Lee NR, Kim DE, Jeong YJ, Chong Y. Investigation of the pharmacophore space of severe acute respiratory syndrome coronavirus (SARS-CoV) NTPase/helicase by dihydroxychromone derivatives. Bioorg Med Chem Lett. 2009;19(16):4538–41.
Kim MK, Yu MS, Park HR, Kim KB, Lee C, Cho SY, et al. 2,6-Bis-arylmethyloxy-5-hydroxychromones with antiviral activity against both hepatitis C virus (HCV) and SARS-associated coronavirus (SCV). Eur J Med Chem. 2011;46(11):5698–704.
Cho A, Saunders OL, Butler T, Zhang L, Xu J, Vela JE, et al. Synthesis and antiviral activity of a series of 1’-substituted 4-aza-7,9-dideazaadenosine C-nucleosides. Bioorg Med Chem Lett. 2012;22(8):2705–7.
Shah PP, Wang T, Kaletsky RL, Myers MC, Purvis JE, Jing H, et al. A small-molecule oxocarbazate inhibitor of human cathepsin L blocks severe acute respiratory syndrome and Ebola pseudotype virus infection into human embryonic kidney 293T cells. Mol Pharmacol. 2010;78(2):319–24.
Adedeji AO, Severson W, Jonsson C, Singh K, Weiss SR, Sarafianos SG. Novel inhibitors of severe acute respiratory syndrome coronavirus entry that act by three distinct mechanisms. J Virol. 2013;87(14):8017–28.
Huentelman MJ, Zubcevic J, Hernandez Prada JA, Xiao X, Dimitrov DS, Raizada MK, et al. Structure-based discovery of a novel angiotensin-converting enzyme 2 inhibitor. Hypertension. 2004;44(6):903–6.
Acknowledgements
The authors thank Laura Bollinger and Jiro Wada for providing technical writing services and graphical support, respectively.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
The content of this publication does not necessarily reflect the views or policies of the US Department of Health and Human Services (DHHS) or of the institutions and companies affiliated with the authors.
Funding
This work was supported in part by the Division of Intramural Research and Division of Clinical Research, National Institute of Allergy and Infectious Diseases. This work was funded in part through Battelle Memorial Institute’s prime contract with the US National Institute of Allergy and Infectious Diseases (NIAID) under Contract No. HHSN272200700016I. J.D. performed this work as employee of Tunnell Government Services, Inc., subcontractor to Battelle Memorial Institute (BMI). R.G. performed this work as employee of BMI. G.G.O. performed this work as employee of MRI Global, subcontractor to BMI.
Conflict of interest
The authors have declared that they have no conflict of interest.
Rights and permissions
About this article

Cite this article
Dyall, J., Gross, R., Kindrachuk, J. et al. Middle East Respiratory Syndrome and Severe Acute Respiratory Syndrome: Current Therapeutic Options and Potential Targets for Novel Therapies. Drugs 77, 1935–1966 (2017). https://doi.org/10.1007/s40265-017-0830-1
Published:
Issue Date:
DOI: https://doi.org/10.1007/s40265-017-0830-1