
Abstract
Enasidenib (Idhifa®) is an oral isocitrate dehydrogenase-2 (IDH2) inhibitor developed by Celgene Corporation under a global, exclusive license from Agios Pharmaceuticals. Enasidenib has been approved in the USA for the treatment of adults with relapsed or refractory acute myeloid leukaemia (AML) and an IDH2 mutation as detected by an FDA-approved test. It is at various stages of development in other countries for AML, myelodysplastic syndromes and solid tumours. This article summarizes the milestones in the development of enasidenib leading to this first global approval in the USA for the treatment of adults with relapsed or refractory IDH2-mutated AML.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
1 Introduction
Acute myeloid leukaemia (AML) is the most common acute leukaemia that affects adults in the USA, and it is characterized by the clonal proliferation of myeloid cells in the bone marrow that can spread quickly to the blood and other tissues [1,2,3]. Isocitrate dehydrogenase-1 (IDH1) and IDH2 mutations affect ≈20% of adults with AML, and mutant IDH1 and IDH2 enzymes have been found to increase an oncometabolite [the R-enantiomer of 2-hydroxyglutarate (2-HG)] that induces a block in cell differentiation and causes cells to lose the ability to advance from an immature to a fully differentiated state [1]. In adults with AML, IDH2 mutations are more common (8–19% of patients) than IDH1 mutations (7–14%), and mutant IDH2 enzymes (located in the mitochondria) generate more of the oncometabolite than mutant IDH1 enzymes (located in the cytoplasm) [1].
Enasidenib (Idhifa®) is an oral IDH2 inhibitor developed by Celgene Corporation under a global, exclusive license from Agios Pharmaceuticals that received approval in the USA on 1 August 2017 for the treatment of adults with relapsed or refractory AML and an IDH2 mutation as detected by an FDA-approved test [4, 5]. The Abbott RealTime™ IDH2 companion diagnostic test (CDx), which received FDA approval concurrently with enasidenib, is indicated for the identification of AML patients with IDH2 mutations who may be candidates for treatment with enasidenib [2, 6, 7].
Enasidenib is a first-in-class small molecule IDH2 inhibitor, and is the first oral targeted therapy that has become available for adults with relapsed or refractory AML and an IDH2 mutation [2, 8]. The recommended dosage of oral enasidenib for this indication is 100 mg once daily for a minimum of 6 months (to allow time for clinical response) or until disease progression or unacceptable toxicity [4]. The US prescribing information does not list contraindications to enasidenib therapy. However, it contains a boxed warning regarding differentiation syndrome (DS), which can be life-threatening or fatal if left untreated [4]. DS is a syndrome of cardiac and respiratory distress that is characterized by acute renal failure, fever, hypotension, interstitial lung infiltrates, pericardial or pleural effusions, respiratory distress, unexplained fever, and weight gain, none of which are pathognomonic [9]. If DS is suspected (there is no diagnostic test), corticosteroid therapy and haemodynamic monitoring should be initiated until symptom resolution [4].
Enasidenib is undergoing phase 3 development in Australia, Brazil, Canada, Russia, South Korea, Taiwan, Turkey and countries in the EU for AML (late-stage, second-line or greater), phase 1/2 development in Australia, Canada, South Korea, the USA and countries in the EU for AML (combination first-line therapy for newly diagnosed disease) and phase 1/2 development in France and the USA for myelodysplastic syndromes (late-stage disease) and solid tumours (first-line therapy) [8].
1.1 Company Agreements
Agios Pharmaceuticals and Celgene Corporation entered into a collaborative agreement in April 2010 with the goal of discovering, developing and delivering novel disease-altering cancer therapies based on Agios Pharmaceuticals’ innovative cancer metabolism research platform [10]. Pursuant to the terms of the agreement, Agios Pharmaceuticals received an initial payment and led the discovery and early development of all its cancer metabolism programs, and Celgene Corporation received a period of exclusivity in which it had the option to license any drug emerging from Agios’ cancer metabolism research platform (at the end of phase 1 development) and fund and lead the global development and commercialization of the drug. Celgene Corporation had the option of extending the period of exclusivity through additional funding, and Agios Pharmaceuticals was eligible to receive milestone payments and royalties on each programme [10]. In addition, Agios Pharmaceuticals retained developing and commercializing rights for certain products in the USA [10, 11]. The companies agreed in October 2011 to extend the period of exclusivity from three to four years [11], and agreed in December 2013 to extend the period by one additional year (until April 2015) [12].


In June 2014, Celgene Corporation exercised its option to license enasidenib (AG-221) from Agios Pharmaceuticals’ IDH portfolio, and did so early based on emerging phase I data for the drug [13]. Celgene Corporation received exclusive global development and commercialization rights for enasidenib. Agios Pharmaceuticals continued to conduct enasidenib early clinical development and regulatory activities in collaboration with Celgene, and retained the right to conduct a part of any commercialization activities for enasidenib in the USA [13].
In December 2014, Celgene Corporation decided to extend the period of exclusivity further to April 2016, at which time the discovery phase of the 2010 agreement would expire and Agios Pharmaceuticals would wholly own all other cancer metabolism programmes [14, 15]. In May 2016, the two companies amended certain rights from the 2010 agreement and established a new metabolic immune-oncology collaboration [15].
2 Scientific Summary
2.1 Pharmacodynamics
Enasidenib is a small molecule inhibitor of IDH2, an enzyme located in the mitochondria that is critical to the Krebs cycle (i.e. citric acid cycle) [1, 4]. Enasidenib binds to the active catalytic site of the mutant IDH2 enzyme and prevents the conformational change that drives the formation of the oncometabolite (R)-2-HG [1]. Enasidenib targets IDH2 mutant variants R140Q, R172K and R172S at approximately 40-fold lower concentrations than it does the wild-type enzyme in vitro [4].
Enasidenib decreases 2-HG levels and induces cell differentiation in AML to promote a clinical response [16]. In an analysis of 125 patients with relapsed or refractory IDH2-mutated AML in a phase 1/2 study (Sect. 2.3), enasidenib decreased total 2-HG levels [indicative of (R)-2-HG levels] in patients with relapsed or refractory IDH2-mutated AML from baseline by 90.6% (maximum extent of suppression). Although the median suppression of 2-HG was significantly greater in patients with an IDH2 R140 mutation than in patients with an IDH2 R172 mutation (94.9 vs. 70.9%; p < 0.001), enasidenib was therapeutically active [with an overall response rate (ORR) of ≈40%] in these patients regardless of the type of IDH2 mutation. Of note, 2-HG suppression alone did not predict response (patients without a response also exhibited 2-HG suppression). The clinical response to enasidenib was associated with the induction of myeloid differentiation. In the same study, enasidenib treatment led to a near-normalization of the ratio of immature to mature cells in patients who achieved a complete response (CR) or a partial response (PR) [in patients with either IDH2 mutation] [16]. In general, myeloid differentiation was observed without evidence of cytotoxicity and with the retention of cytogenetic abnormalities and the IDH2 mutation (indicative of abnormal myeloblasts, rather than non-malignant cells, evolving into mature cells) [17].
Enasidenib can cause DS (associated with rapid myeloid cell proliferation and differentiation), cause non-infectious leucocytosis (through myeloid proliferation), interfere with bilirubin metabolism (through inhibition of UGT1A1) and increase the risk of tumour lysis syndrome (through myeloid proliferation causing a rapid reduction in tumour cells) [4]. Further details can be found in Sect. 2.4.
Enasidenib is not associated with large QTc interval changes [4]. In an open-label study of patients with IDH2-mutated advanced haematological malignancies, large mean changes in the QTc interval (>20 ms) were not observed following the administration of singles doses of enasidenib 30–650 mg or multiple doses of enasidenib 100 mg daily in a fasted state [4].
2.2 Pharmacokinetics
Enasidenib reaches a peak plasma concentration (Cmax) of 1.3 mcg/mL in 4 h (median time) after a single oral dose of 100 mg, and 13 mcg/mL at steady state with 100 mg daily [4]. Steady-state plasma concentrations are reached within 29 days of once-daily dosing. Following an oral dose of 100 mg, the absolute bioavailability of enasidenib is 57%. Enasidenib exposure [area under the concentration time curve (AUC)] increases in an approximately dose-proportional manner with daily doses of 50–450 mg. In vitro, enasidenib and its metabolite AGI-16903 are highly (≥96.6%) protein bound. The mean volume of distribution is 55.8 L [4].
The terminal half-life of enasidenib is 137 h, and the mean total body clearance is 0.74 L/h [4]. According to in vitro studies, enasidenib metabolism is mediated by multiple CYP enzymes (e.g. CYP1A2, 2C9, 2C19, 2D6, 3A4) as well as UGTs (e.g. UGT1A1, 2B7, 2B15). The metabolite AGI-16903 is further metabolized by enzymes such as CYP1A2, 2C19 and 3A4, and UGT1A1, 1A3 and 1A9. Following the administration of radiolabelled enasidenib, 89% of the circulating radioactivity is attributed to enasidenib, and 10% of the circulating radioactivity is attributed to AGI-16903. Enasidenib is eliminated mainly in faeces (89%), with 11% of the drug eliminated in the urine. Unchanged enasidenib accounts for 34% of radiolabelled enasidenib excreted in the faeces and 0.4% excreted in the urine [4].
Age (19–100 years), bodyweight (39–136 kg), body surface area, ethnicity, gender, mild hepatic impairment and renal impairment (with creatinine clearance ≥30 mL/min) are not expected to have clinically meaningful effects on enasidenib pharmacokinetics [4].
Features and properties of enasidenib
Alternative names | Idhifa®; AG-221; CC-90007 |
Class | Antineoplastics; propanols; pyridines; small molecules; triazines |
Mechanism of action | IDH2 inhibitor |
Route of administration | Oral |
Pharmacodynamics | Decreases 2-HG levels and induces myeloid differentiation |
Pharmacokinetics | Reaches Cmax in 4 h (median), reaches steady state plasma concentrations within 29 days of once-daily dosing |
Adverse events | |
Most frequent | Elevated bilirubin, nausea, diarrhoea, decreased appetite and vomiting |
Occasional | Differentiation syndrome, non-infectious leucocytosis, dysgeusia |
Rare | Tumour lysis syndrome |
ATC codes | |
WHO ATC code | L01 (antineoplastic agents) |
EphMRA ATC code | L1 (antineoplastics) |
Chemical name | 2-Methyl-1-[(4-[6-(trifluoromethyl)pyridin-2-yl]-6-{[2-(trifluoromethyl)pyridin-4-yl]amino}-1,3,5-triazin-2-yl)amino]propan-2-ol methanesulfonate |
2.3 Therapeutic Trials
Enasidenib treatment induced haematological responses in adults with relapsed or refractory IDH2-mutated AML in an open-label, multinational, phase 1/2 study (NCT01915498) [17]. The study enrolled patients aged ≥18 years with IDH2-mutated advanced myeloid malignancies [AML or myelodysplastic syndromes (MDS)] who had an ECOG performance status of 0–2. IDH2 mutational status was identified locally and/or confirmed with the Abbott RealTime™ IDH2 assay [4, 17]. This study is ongoing [18], and results are reported from interim analyses (three data cuts) [4, 17, 19].
In the phase 1 dose-escalation part of the study, patients received oral enasidenib 30–150 mg twice daily or 50–650 once daily under a 3 + 3 dose escalation scheme [17]. A maximum tolerated dose was not reached with these dosages, and enasidenib 100 mg once daily was selected as the dosage for the expansion phase based on demonstrated efficacy and pharmacological profiles. The phase 1 expansion part of the study consisted of four patient cohorts: (1) patients aged ≥60 years with relapsed or refractory AML (or patients of any age who had relapsed after haematopoietic cell transplantation); (2) patients aged <60 years with relapsed or refractory AML and no prior transplantation; (3) patients aged ≥60 years with untreated AML who were ineligible for induction chemotherapy; and (4) patients ineligible for other expansion cohorts [17]. The phase 2 expansion part of the study consisted of a fifth patient cohort: patients with IDH2-mutated AML in a second or later relapse, those who have relapsed after allogeneic transplantation or those refractory to second-line induction or reinduction therapy [20].
Efficacy outcomes were assessed in the largest patient subgroup, patients with relapsed or refractory AML [4, 17, 19]. The first two analyses examined patients from cohorts 1 and 2 (across all doses [17] and/or in patients who received enasidenib 100 mg/day [4, 17]), and the third analysis examined patients from cohorts 1, 2 and 5 who had received enasidenib 100 mg/day [19]. At baseline (across all data cuts), patients who were assigned to receive enasidenib 100 mg/day had a median age of 67–68 years (range 19–100 years) [4, 17, 19]. At baseline (at the second data cut), among patients who were assigned to receive enasidenib 100 mg/day, the majority (78%) of patients had an IDH2 R140 mutation (whereas 22% of patients had an IDH2 R172 mutation), 13% of patients had undergone prior stem cell transplantation and patients had a median of two prior anticancer regimens [4].
At a data cut-off date of 15 April 2016, the ORR in patients with relapsed or refractory IDH2-mutated AML across all doses (n = 176) was 40.3%, and included a best response of CR in 19.3% of patients, CR with incomplete haematological recovery (CRh) or CR with incomplete platelet recovery in 6.8% of patients, PR in 6.3% of patients or morphological leukaemia-free state in 8.0% of patients [17]. The median time to first response was 1.9 months, and the median duration of response (DOR) was 5.8 months. In patients who achieved a CR, the time to CR was 3.8 months, and the DOR was 8.8 months. In terms of patients in this group who received enasidenib 100 mg/day (n = 109), the ORR was 38.5% [best response of CR (20.2% of patients), CRh or CR with incomplete platelet recovery (6.4%), PR (2.8%) or morphological leukaemia-free state (9.2%)], with a median time to first response of 1.9 months and a median DOR of 5.6 months (3.7 months and 8.8 months, respectively, in patients who achieved a CR). At a median follow-up of 7.7 months in all patients with relapsed or refractory IDH2-mutated AML, the median overall survival (OS) was 9.3 months (19.7 months in patients who achieved a CR and 8.0 months in patients who received ≥2 prior anticancer regimens), and 1-year survival was estimated at 39% [17].
At a median follow-up of 6.6 months (range 0.4–27.7 months) in patients with relapsed or refractory IDH2-mutated AML who had been assigned to receive enasidenib at a starting dosage of 100 mg/day (n = 199), 19% of patients had a CR and 4% of patients had a CRh [4]. These rates were similar regardless of IDH2 mutation variant (R140 or R172). In patients who achieved a CR or CRh, the median time to first response was 1.9 months (range 0.5–7.5 months), the median time to best response was 3.7 months (range 0.6–11.2 months) [the majority (85%) of patients achieved a best response of CR/CRh within 6 months of starting enasidenib therapy] and the median DOR was 8.2 months. Among patients who were transfusion dependent (red blood cells and/or platelets) at baseline (n = 157), 34% became transfusion independent during any 56-day post-baseline period. Of patients who were transfusion independent at baseline (n = 42), 76% continued to be transfusion independent during any 56-day post-baseline period [4].
Results from a data cut-off date of 14 October 2016 included additional patients from the phase 2 expansion cohort (n = 105) and were consistent with previously reported data [19]. The ORR was 37% (primary endpoint), and included a CR rate of 20.1%. The median time to first response was 1.9 months, and the median DOR was 5.6 months. In patients who achieved a CR, the median time to CR was 3.7 months, and the median DOR was 8.8 months. Some patients became transfusion independent, even in the absence of a CR. The median OS was 8.3 months. By this data cut-off, 345 patients had been enrolled in the study, of which 281 patients had relapsed or refractory AML, 214 of which received enasidenib 100 mg daily [19].
Key clinical trials of enasidenib
Drug(s) | Indication | Phase | Status | Location(s) | Identifier (s) | Sponsor(s) |
|---|---|---|---|---|---|---|
Enasidenib | Advanced haematological malignancies with an IDH2 mutation | 1/2 | Ongoing | Multinational | NCT01915498; AG221-C-001 | Celegene Corporation, Agios Pharmaceuticals |
Enasidenib | Advanced solid tumours with an IDH2 mutation | 1/2 | Completed | Multinational | NCT02273739; AG221-C-003 | Celgene Corporation |
Enasidenib or ivosidenib, in combination with standard induction or consolidation | Newly diagnosed AML | 1b | Recruiting | Multinational | NCT02632708; AG120-221-C-001 | Agios Pharmaceuticals, Celgene Corporation |
Enasidenib or ivosidenib, in combination with azacitadine | Newly diagnosed AML | 1b/2 | Recruiting | Multinational | NCT02677922; AG-221-AML-005 | Celgene Corporation |
Enasidenib, azacitadine (one arm of a multi-substudy trial) | Previously untreated IDH2-mutated AML | 1b/2 | Recruiting | USA | NCT03013998; BEAT AML; BAML-16-001 | BEAT AML (The Leukemia and Lymphoma Society) |
Enasidenib, conventional care | Older patients with relapsed/refractory AML with an IDH2 mutation | 3 | Ongoing | Multinational | NCT02577406; IDHENTIFY; AG-221-AML-004 | Celgene Corporation |
2.4 Adverse Events
In patients with relapsed or refractory IDH2-mutated AML who had received enasidenib 100 mg/day for a median duration of exposure of 4.3 months in the phase 1/2 study (NCT01915498) [n = 214], the most commonly reported AEs of any grade (occurring in ≥20% of patients) were elevated bilirubin (81% of patients) [37% of whom had levels ≥2 × the upper limit of normal at least once], nausea (50%), diarrhoea (43%), decreased appetite (34%) and vomiting (34%) [4]. Of note, DS of any grade occurred in 14% of patients. Other clinically significant AEs that occurred in ≤10% of patients included acute respiratory distress syndrome and pulmonary oedema. The most common grade ≥3 AEs (occurring in ≥5% of patients) were elevated bilirubin (15% of patients), decreased potassium (15%), decreased calcium (8%), decreased phosphorus (8%), diarrhoea (8%), DS (7%), tumour lysis syndrome (6%), noninfectious leucocytosis (6%) and nausea (5%) [4].
The most common serious AEs (occurring in ≥2% of patients) were leucocytosis (10% of patients), DS (8%) [where symptoms included acute renal failure, hypoxia, multi-organ failure, pyrexia, or respiratory failure], diarrhoea (6%), nausea (5%), tumour lysis syndrome (5%), vomiting (3%) and decreased appetite (3%) [4]. AEs led to dose interruption in 43% of patients [most commonly because of DS (4% of patients) and leucocytosis (3%)], dose reduction in 5% of patients, and treatment discontinuation in 17% of patients [most commonly because of leucocytosis (1% of patients)]. Of note, the 30-day mortality rate was 4.2% of patients, and the 60-day mortality rate was 11.7% [4].
The AE profile of enasidenib in a subsequent analysis of a group of patients including patients from the phase 2 expansion cohort was consistent with that of previously reported data [19]. The most common treatment-emergent AEs included nausea (48% of patients), fatigue (41%), diarrhoea (41%), decreased appetite (34%) and elevated bilirubin (33%). Treatment-related serious AEs were reported in 26.1% of all patients in the study, and included DS (7% of patients), leucocytosis (4%), tumour lysis syndrome (3%) and hyperbilirubinaemia (2%) [19].
2.5 Companion Diagnostic
In October 2016, Celgene Corporation entered into a collaborative agreement with Abbott to develop and commercialize a CDx for enasidenib that would help identify AML patients with IDH mutations [21]. This led to the development of the Abbott RealTime™ IDH2, an in vitro polymerase chain reaction assay capable of qualitatively detecting single nucleotide variants coding nine IDH2 mutations (R140G, R140L, R140Q, R140W, R172G, R172K, R172M, R172S, R172W) in DNA extracted from human blood or bone marrow [6]. The Abbott RealTime™ IDH2 CDx received approval from the FDA in August 2017 to be used as an aid in the identification of AML patients with IDH2 mutations to be treated with enasidenib [2].
2.6 Ongoing Clinical Trials
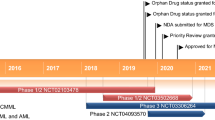
In August 2013, Celgene Corporation and Agios Pharmaceuticals initiated the first-in-human phase 1/2 trial (NCT01915498) on which approval of enasidenib in AML in the USA is based [5, 17]. It is ongoing and has an estimated completion date of December 2018 [18].
In October 2014, Celgene Corporation initiated the second clinical trial of the enasidenib development program [22]. The open-label, multinational, phase 1/2 study (NCT02273739) examined the clinical activity, safety and tolerability of enasidenib in patients with IDH2-mutated advanced solid tumours (including angioimmunoblastic T-cell lymphoma as well as gliomas) [18, 22]. In phase 1 (the dose-escalation phase) of the trial, patients received increasing doses of enasidenib in order to determine the maximum tolerated dose [18]. In phase 2 (the planned dose expansion phase) of the trial, three cohorts of patients received enasidenib 100 mg once daily until disease progression or unacceptable toxicity [18, 22]. Although the trial is complete [18], results are not yet available.
In October 2015, Celgene Corporation started enrolling patients into a randomized, open-label, multinational, phase 3 trial (IDHENTIFY; NCT02577406) to compare the efficacy and safety of enasidenib versus conventional care regimens (CCRs) in older patients (aged ≥60 years) with relapsed or refractory IDH2-mutated AML after two or three prior treatment regimens [23]. Patients will receive enasidenib or one of four CCRs (azacitadine, low-dose cytarabine, intermediate-dose cytarabine or best supportive care) in 28-day cycles. The primary endpoint is OS. Enrolment is planned to continue through 2019 [23].
Studies are also underway to evaluate the use of enasidenib combination therapy in the frontline setting in patients with newly diagnosed AML with an IDH2 mutation [24, 25].
In December 2015, Agios Pharmaceuticals and Celgene Corporation initiated an open-label, multinational, phase 1b study (NCT02632708) to evaluate the safety of enasidenib versus ivosidenib, in combination with standard induction and consolidation therapy, in adults with newly diagnosed, IDH-mutated AML who are eligible for intensive chemotherapy [25]. Patients receive enasidenib 100 mg once daily (those with an IDH2 mutation) or ivosidenib 500 mg once daily (those with an IDH1 mutation) with two types of induction therapy (cytarabine with either daunorubicin or idarubicin) and two types of consolidation therapy (mitoxantrone plus etoposide, or cytarabine). Patients achieving CR, CRh or CR with incomplete platelet recovery during induction therapy proceed to consolidation therapy, after which they may proceed to maintenance therapy and receive enasidenib or ivosidenib for up to 1 year or until relapse, haematopoietic stem cell transplant or the development of unacceptable toxicity. The primary endpoint is the determination of the safety and tolerability [25]. The study has an estimated completion date of September 2020 [18].
In March 2016, Celgene Corporation initiated a randomized, open-label, multinational, phase 1b/2 study (NCT02677922) to evaluate the efficacy and safety of enasidenib plus azacitadine and ivosidenib plus azacitadine in adults with newly diagnosed, IDH-mutated AML who are not candidates for intensive induction chemotherapy [24]. Patients receive enasidenib 100 mg once daily (those with an IDH2 mutation) or ivosidenib 500 mg once daily (those with an IDH1 mutation) continuously in 28-day cycles with a standard dose of azacitadine (75 mg/m2) daily for 7 days of each 28-day cycle. The primary endpoint of the phase 1b dose-escalation stage is determination of the safety and tolerability as well as the recommended dose of enasidenib plus azacitadine and ivosidenib plus azacitadine, and the primary endpoint in the phase 2 randomized stage is determination of the efficacy of enasidenib or ivosidenib in combination with azacitadine compared with azacitadine alone [24]. The study has an estimated completion date of December 2018 [18].
References
Medeiros BC, Fathi AT, DiNardo CD, et al. Isocitrate dehydrogenase mutations in myeloid malignancies. Leukemia. 2017;31(2):272–81.
Celgene Corporation. FDA grants approval of IDHIFA®, the first oral targeted therapy for adult patients with relapsed/refractory acute myeloid leukemia and an IDH2 mutation [media release]. 1 Aug 2017. http://www.celgene.com/.
Döhner H, Weisdorf DJ, Bloomfield CD. Acute myeloid leukemia. N Engl J Med. 2015;373(12):1136–52.
Celgene Corporation. IDHIFA® (enasidenib) tablets: US prescribing information. 2017. https://www.fda.gov. Accessed 30 Aug 2017.
US FDA. FDA approves new targeted treatment for relapsed or refractory acute myeloid leukemia [media release]. 1 Aug 2017. https://www.fda.gov.
US FDA. List of cleared or approved companion diagnostic devices (in vitro and imaging tools). 2017. https://www.fda.gov. Accessed 22 Aug 2017.
Adis Insight. Drug profile: enasidenib companion diagnostic. 2017. http://adisinsight.springer.com. Accessed 22 Aug 2017.
Adis Insight. Drug profile: enasidenib. 2017. http://adisinsight.springer.com. Accessed 22 Aug 2017.
Cardinale L, Asteggiano F, Moretti F, et al. Pathophysiology, clinical features and radiological findings of differentiation syndrome/all-trans-retinoic acid syndrome. World J Radiol. 2014;6(8):583–8.
Agios Pharmaceuticals. Celgene Corporation and Agios Pharmaceuticals announce global strategic collaboration to advance unique science of cancer metabolism [media release]. 15 Apr 2010. http://www.agios.com/.
Agios Pharmaceuticals. Celgene and Agios extend cancer metabolism collaboration [media release]. 5 Oct 2011. http://www.agios.com/.
Agios Pharmaceuticals. Agios advances cancer metabolism collaboration with Celgene [media release]. 11 Dec 2013. http://www.agios.com/.
Agios Pharmaceuticals. Agios Pharmaceuticals announces that Celgene exercised its option to license AG-221 under global strategic collaboration [media release]. 13 Jun 2014. http://www.agios.com/.
Agios Pharmaceuticals. Agios announces Celgene decision to extend discovery phase of global strategic collaboration to April 2016 [media release]. 8 Dec 2014. http://www.agios.com/.
Agios Pharmaceuticals. Agios and Celgene establish new collaboration in metabolic immuno-oncology and amend certain rights from 2010 agreement [media release]. 17 May 2016. http://www.agios.com/.
Amatangelo MD, Quek L, Shih A, et al. Enasidenib induces acute myeloid leukemia cell differentiation to promote clinical response. Blood. 2017;130(6):732–41.
Stein EM, DiNardo CD, Pollyea DA, et al. Enasidenib in mutant-IDH2 relapsed or refractory acute myeloid leukemia. Blood. 2017;130(6):722–31.
National Library of Medicine. Clinicaltrials.gov. 2017. https://clinicaltrials.gov. Accessed 17 Aug 2017.
Agios Pharmaceuticals. New data from phase 1/2 trial of oral IDHIFA® (enasidenib) demonstrate durable complete responses in patients with IDH2 mutant relapsed or refractory AML [media release]. 24 Jun 2017. http://www.agios.com/.
Agios Pharmaceuticals. Agios announces data from ongoing phase 1/2 trial of AG-221 showing durable responses in patients with advanced hematologic malignancies [media release]. 6 Dec 2015. http://www.agios.com/.
Celgene Corporation. Celgene and Agios announce collaborations with Abbott for diagnostic identification of IDH mutations in AML [media release]. 12 Oct 2016. http://www.celgene.com.
Agios Pharmaceuticals. Agios Pharmaceuticals announces initiation of a phase 1/2 clinical trial of AG-221 in patients with advanced solid tumors with an IDH2 mutation [media release]. 21 Oct 2014. http://www.agios.com/.
Tallman MS, Knight RD, Glasmacher AG, et al. Phase III randomized, open-label study comparing the efficacy and safety of AG-221 vs conventional care regimens (CCR) in older patients with advanced acute myeloid leukemia (AML) with isocitrate dehydrogenase (IDH)-2 mutations in relapse or refractory to multiple prior treatments: the IDHENTIFY trial [abstract no. TPS7074]. J Clin Oncol. 2016;34(15 Suppl).
Agios Pharmaceuticals. Agios announces initiation of phase 1/2 frontline combination study of AG-221 or AG-120 with VIDAZA® (azacitadine for injection) in newly diagnosed acute myeloid leukemia (AML) patients not eligible for intensive chemotherapy [media release]. 30 Mar 2016. http://www.agios.com/.
Agios Pharmaceuticals. Agios announces initiation of phase 1b frontline trial of AG-221 or AG-120 in combination with intensive chemotherapy in newly diagnosed acute myeloid leukemia (AML) patients [media release]. 18 Dec 2015. http://www.agios.com/.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Funding
The preparation of this review was not supported by any external funding.
Conflict of interest
During the peer review process the manufacturer of the agent under review was offered an opportunity to comment on the article. Changes resulting from any comments received were made by the author on the basis of scientific completeness and accuracy. Esther Kim is a salaried employee of Adis/Springer, is responsible for the article content and declares no relevant conflicts of interest.
Additional information about this Adis Drug Review can be found at http://www.medengine.com/Redeem/E7FBF06072D6F08B.
Additional information
This profile has been extracted and modified from the AdisInsight database. AdisInsight tracks drug development worldwide through the entire development process, from discovery, through pre-clinical and clinical studies to market launch and beyond.
Rights and permissions
About this article

Cite this article
Kim, E.S. Enasidenib: First Global Approval. Drugs 77, 1705–1711 (2017). https://doi.org/10.1007/s40265-017-0813-2
Published:
Issue Date:
DOI: https://doi.org/10.1007/s40265-017-0813-2