
Abstract
It is well recognised that the majority of the impact of multiple sclerosis (MS), both personal and societal, arises in the progressive phase where disability accumulates inexorably. As such, progressive MS (PMS) has been the target of pharmacological therapies for many years. However, there are no current licensed treatments for PMS. This stands in marked contrast to relapsing remitting MS (RRMS) where trials have resulted in numerous licensed therapies. PMS has proven to be a more difficult challenge compared to RRMS and this review focuses on secondary progressive MS (SPMS), where relapses occur before the onset of gradual, irreversible disability, and not primary progressive MS where disability accumulation occurs without prior relapses. Although there are similarities between the two forms, in both cases pinpointing when PMS starts is difficult in a condition in which disability can vary from day to day. There is also an overlap between the pathology of relapsing and progressive MS and this has contributed to the lack of well-defined outcomes, both surrogates and clinically relevant outcomes in PMS. In this review, we used the search term ‘randomised controlled clinical drug trials in secondary progressive MS’ in publications since 1988 together with recently completed trials where results were available. We found 34 trials involving 21 different molecules, of which 38% were successful in reaching their primary outcome. In general, the trials were well designed (e.g. double blind) with sample sizes ranging from 35 to 1949 subjects. The majority were parallel group, but there were also multi-arm and multidose trials as well as the more recent use of adaptive designs. The disability outcome most commonly used was the Expanded Disability Status Scale (EDSS) in all phases, but also magnetic resonance imaging (MRI)-measured brain atrophy has been utilised as a surrogate endpoint in phase II studies. The majority of the treatments tested in SPMS over the years were initially successful in RRMS. This has a number of implications in terms of targeting SPMS, but principally implies that the optimal strategy to target SPMS is to utilise the prodrome of relapses to initiate a therapy that will aim to both prevent progression and slow its accumulation. This approach is in agreement with the early targeting of MS but requires treatments that are both effective and safe if it is to be used before disability is a major problem. Recent successes will hopefully result in the first licensed therapy for PMS and enable us to test this approach.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Secondary progressive multiple sclerosis (MS) is differentiated from primary progressive MS by the prodrome of relapses but it shares many pathological and clinical features with the primary progressive form. This includes the difficulty in identifying its onset and measuring its impact over time. |
Secondary progressive MS is responsible for the majority of the disability and cost associated with MS but does not as yet have a licensed treatment that can modify the disease course. This is despite being targeted in 34 trials of which 34% were successful in reaching their primary outcome. One recent phase III study has successfully achieved its primary outcome and may result in licensed therapy. |
Targeting the pathology with large well conducted modern trial designs and understanding the route to licensing will, hopefully in the near future, result in the first licensed treatment for this disabling condition but also set the scene for the development of subsequent therapies. |
1 Introduction
Multiple sclerosis (MS) is a chronic autoimmune disease of the central nervous system (CNS). It is the most common cause of disability in younger adults and, aside from the enormous social cost, has an estimated total annual fiscal cost to Europe of approximately €14.6 billion [1] and US$10 billion in the USA [2], with costs increasing as disability develops [3]. MS is characterized pathologically by focal and diffuse inflammation, causing demyelination, which presents clinically as relapses. In addition, axonal damage and neuronal loss occur early in the disease course and underlie the progressive accumulation of permanent clinical disability, which exerts the most significant burden on patients and their family [4].
Over the last 20 years, there has seen unprecedented progress in the development of new treatments for relapsing remitting MS (RRMS) [5]. Unfortunately, these advances have not, as yet, translated to a successful licensed therapy for progressive MS (PMS). However, some recent clinical trials have shown promising results, demonstrating a slowing of progression that will hopefully lead to a licensed therapy in the near future [6–9]. In this review, we addressed the clinical challenge of quantifying progression, understanding the underlying pathology and the limitations of the outcome measures. In addition, we summarise evidence from previous studies of therapies for secondary progressive MS (SPMS).
2 Tackling the Challenge of Studies in SPMS
2.1 Defining Progression: A Clinical Problem
Among patients with RRMS, the early course is characterised by ‘acute attacks’ or ‘relapses’, interspersed with periods of ‘remission’, during which symptoms remain stable, although they can resolve (Fig. 1). This presentation occurs in about 85% of cases and it is followed, in the majority of cases, by a progressive course, which develops on average about 15 years after the disease onset, and is termed SPMS [10]. In roughly 15% of cases, MS presents with a progressive course from clinical onset and is termed primary progressive (PPMS). Primary and secondary progressive MS can be complicated by relapses, overlapping their course [11]. Progression is characterized by a gradual accumulation of irreversible disability, but this definition contains important unresolved questions [12].

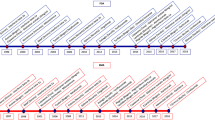
The various presentations of multiple sclerosis and the surmised relationship to the underlying disease process. a Mild RRMS where the cell loss never becomes clinically evident and no progression is evident. b Classic RRMS with relapses preceding progression—secondary progressive (SP) MS. c RRMS with accelerated onset of SPMS with frequent early relapses (highly active/rapidly evolving severe) where accelerated cell loss leads to a shorter time to progression. d PPMS lower inflammatory activity results in relapses never becoming evident. RRMS relapsing remitting multiple sclerosis, MRI magnetic resonance imaging, SPMS secondary progressive multiple sclerosis, PPMS primary progressive multiple sclerosis
Gradual implies ‘taking place by degrees’ and differs from the neurological worsening associated with a relapse. However, the current definition does not specify the minimal duration of progressive disability accumulation, making it difficult to distinguish progression from deterioration related to relapses [12]. In addition, as in clinical practice, patients are only seen at intervals, identifying a change in phenotype, and its timing is challenging and requires time. To document an objective stepwise change entails at least three visits: an initial examination, and a subsequent assessment to identify a deterioration, which has to be confirmed during a third visit. In addition, pinpointing the onset of SPMS is particularly difficult when patients experience many relapses, which often cloud the clinical occurrence of progression [13]. Indeed, it has been calculated that the process of re-classifying RRMS to SPMS in a standard clinical setting takes on average 3 years [14]. Similarly, diagnosing PPMS takes about 3 years from the onset of symptoms [6, 15].
Irreversible, in the current definition, equates with confirmed progression, e.g. an increase in neurological dysfunction, which is permanent over time. This is an essential requirement for defining progression, as recovery from relapses can occur after up to 12 months. However, the interval for confirming the irreversible accumulation of disability is not currently specified [12, 16], although in previous definitions disability worsening had to persist for at least 1 year [17]. Indeed, a recent study evaluating various criteria of disability progression and testing 11 different definitions of progression, demonstrated regression of disability occurred in 11–34% of the progression events over the five subsequent years. The most important determinant of progression stability was the length of the confirmation period. The accuracy of the progression criteria increased with duration of the confirmation period, suggesting that the disability outcomes based on 3- to 6-month confirmed disability progression overestimate irreversible disability by up to 30% [18]. In contrast, in clinical trials confirmed progression is most commonly defined as a persistent deterioration for 3 months [19]. A recent global prospective cohort study compared the accuracy of 576 data-derived onset definitions for SPMS to a consensus opinion of neurologists and then subsequently evaluated these against 5-year disease outcomes post assignment of SPMS. It was concluded the best definition included a three-strata progression magnitude in the absence of a relapse, confirmed after 3 months [20]. Notably, drug regulatory agencies place an emphasis on confirmation after at least 6 months [21].
Disability in MS is most commonly apparent as deterioration of mobility. However, MS has a plethora of effects on the CNS that can impact patients’ functionality. Although many scales have been recently developed to try to capture the widely ranging impact of MS on individuals [22], the most commonly used disability measurement remains the Extended Disability Status Score (EDSS) [23, 24]. The EDSS is an ordinal, not linear, scale ranging from 0 to 10, in 0.5-point increments; a worsening of 0.5/1 point is commonly used to define an increase in disability, often depending on the baseline value (e.g. ≤ or >5.5 points). Nevertheless, progression may occur at separate rates in the different neurological functional systems that are scored in the EDSS, complicating assessments [12]. Although the EDSS has a number of significant limitations and it is strongly biased toward ambulation impairment, it remains the favoured outcome measure for regulators [25].
2.2 Targeting the Pathology of Progression
RRMS is characterised by focal white matter inflammation, which is driven, in part, by the acute influx of lymphocytes across the blood-brain barrier, leading to demyelination [26] and massive activation of macrophage/microglial cells [27]. It is widely believed that the focal inflammatory activity underlays clinical acute attacks. White matter lesions are visualised, using magnetic resonance imaging (MRI) scanning, as T2 focal lesions, which can enhance with gadolinium (Gd+), indicating a recent breakdown of the blood-brain barrier [28]. Focal white matter inflammation and relapses become increasingly rare in PMS. In contrast, brain atrophy in the grey and white matter and diffuse axonal loss within the normal appearing white matter (NAWM) become prominent [29–31]. The neurodegenerative process is accompanied by a more diffuse inflammation, compartmentalised within the CNS, and located behind a normal or repaired blood-brain barrier [28, 31, 32]. In about 40% of SPMS patients, B cell-rich lymphocytic aggregates are present in the meninges and drive the cortical damage by releasing soluble factors and activating the microglia [33, 34]. The cortical pathology is recognised as one of the most important determinants of the long-term outcome [35]. The axonal loss is also caused by mitochondrial injury and ion channel dysfunction, which are triggered by reactive oxygen and nitric oxide species [36–39]. This cascade of events may be amplified by iron accumulation and by the hypoxic damage, secondary to the inflammation and the mitochondrial injury [40–42].
Despite relapsing and progressive MS being distinguished by different clinical features, their underlying pathophysiological mechanisms overlap. Indeed, the axonal loss begins in the early stage of the relapsing phase and increases over time, gradually leading to the clinical onset of the progressive phase, reinforced by the failure of compensatory mechanisms (Fig. 1) [43]. Therefore, treatment strategies aimed at halting or slowing progression are likely to be most effective if given early in the disease process, and in combination with drugs that target focal white matter inflammation. It is therefore plausible that aggressively targeting focal early inflammation can achieve a better disease control in the long term [43].
2.3 Measuring Progression: Biomarkers and Surrogate Markers
A biomarker is an objective quantifiable measurement that reflects the activity of a disease process [44]. Prentice defined a surrogate marker as a subset of biomarkers characterized by specific criteria [43], which means that they can be used in trials as a substitute for a clinically meaningful endpoint, which is a direct measure of how a patient feels, functions or survives. A surrogate marker, therefore, is expected to predict the effect of the therapy [45].
Unfortunately, there is no consensus on biomarkers or surrogate markers for progression. The development of such a marker is desperately required to speed up the development of effective treatments. Extensive work into potential serum and cerebrospinal fluid (CSF) biomarkers has not yet led to a marker that fulfils the criteria for surrogacy. Ocular coherence tomography (OCT) has shown potential as a biomarker for cell loss in the retina [46] and has been used for monitoring the neuroprotective effect of some therapies [47]. Although MRI focal inflammatory measures are used for monitoring the disease activity in RRMS, they have limited use for assessing progression. Spinal cord imaging offers further useful information and is especially related to mobility measures, but it is less sensitive to change with current techniques, although this is evolving with newer techniques [48]. Other radiological measures, such as increasing number and volume of T1 hypo-intense lesions, brain volume loss and changes in magnetization transfer imaging and diffusion tensor imaging, are being explored [49, 50]. Brain and spinal cord atrophy have shown the best correlation with the development of long-term disability [51] and have been used to monitor progression in some clinical studies [6, 9, 52].
3 Completed Pharmacological Studies in PMS
The following studies were identified from a search in PubMed and Cochrane Central Register of Controlled Trials in October 2015, using the terms ‘randomised controlled clinical drug trials in secondary progressive MS’ including only human and non-retracted publications from 1988 onwards. Studies specifying the inclusion of PPMS only were excluded. This resulted in 27 publications that fulfilled the criteria. Additional publications and subsequent ongoing trials were found through hand searching and discussion with leaders in the field. Thirty-four randomised studies in SPMS were identified after full text review and are described below and in Table 1. The studies have mixed populations of MS patients including RRMS, but also progressive populations that include SPMS and PPMS.
3.1 Beta Interferon
3.1.1 Mechanism of Action
Beta interferon (IFNβ) is used in RRMS [5]. IFNβ inhibits T-cell activation and proliferation by downregulating the level of expression of major histocompatibility complex (MHC) class II on antigen presenting cells (APCs) and expression of the co-stimulatory molecules CD40L and CD28 found on T cells [53–55]. IFNβ administration also promotes apoptosis due to an upregulation of cytotoxic T-lymphocyte antigen 4 (CTLA4) and Fas surface molecules on CD4+ T cells [56]. IFNβ also decreases the migratory capacity of pathogenic T cells into the CNS by downregulating expression of the integrin very late antigen 4 (VLA-4) [57] and enhancing suppressive T-cell function, possibly by upregulation of interleukin (IL)-10 and transforming growth factor (TGF) [58].
3.1.2 Clinical Trials
A systematic review of the five studies in SPMS included 1829 IFNβ subjects and 1293 placebo subjects [59]. The review found that IFNβ did not prevent the development of permanent physical disability, although there were reduced risks of relapse at 3 years (relative risk [RR], 0.91, 95% confidence interval [95% CI] 0.84–0.97) and a reduced risk of progression confirmed at 3 months (RR, 0.88, 95% CI 0.80–0.97).
The European Study Group (1998) [60] found IFNβ delays sustained neurological deterioration in patients with SPMS (n = 358). This multicentre, double-masked, placebo-controlled trial, randomised patients with SPMS and EDSS scores of 3.0–6.5 receiving either 8 million IU IFNβ every other day or placebo subcutaneously for up to 3 years. The primary outcome was the time to confirmed progression of disability as measured by a 1.0-point increase on the EDSS, sustained for at least 3 months, or a 0.5-point increase if the baseline EDSS was 6.0 or 6.5. 358 patients with SPMS were allocated placebo and 360 were allocated IFNβ; 57 patients (31 placebo, 26 IFNβ) were lost to follow-up. There was a highly significant difference in time to confirmed progression of disability in favour of IFNβ (p = 0.0008). IFNβ delayed progression for 9–12 months over a study period of 2–3 years. This beneficial effect was seen both in patients with superimposed relapses and in patients who had progressive deterioration without relapses. There were also benefits seen in time to becoming wheelchair-bound, relapse rate and severity, number of steroid treatments and hospital admissions, as well as on MRI variables. The study was stopped after the interim results gave clear evidence of efficacy.
A second study (n = 436) showed IFNb-1a slowed disease progression in SPMS and demonstrated a benefit on MS functional composite (MSFC) progression, relapses, quality of life and MRI activity in SPMS [61]. Entry criteria included an EDSS score of 3.5–6.5. Subjects were randomised to receive IFNb-1a (60 µg) or placebo by weekly intramuscular injection for 2 years. The primary outcome measure, used for the first time in a large-scale MS trial, was a baseline to month 24 change in the MSFC. The MSFC comprises tests of ambulation (Timed 25-Foot Walk [T25FW]), arm function (Nine-Hole Peg Test [9HPT]) and cognition (Paced Auditory Serial Addition Test [PASAT]). Median MSFC Z-score change was reduced 40.4% in IFNb-1a subjects (p = 0.033). The effect was driven by the 9HPT and PASAT. There was no discernible benefit on the EDSS. IFNb-1a subjects had 33% fewer relapses (p = 0.008), and new or enlarging T2-hyperintense brain MRI lesions and Gd+ lesions were reduced at months 12 and 24 (both p < 0.001).
However, the Nordic Study Group and North American Study Group did not replicate these positive results. The Nordic Study Group performed a double-blind, placebo-controlled study that examined the effects of low-dose IFNb-1a. 371 patients were randomised to receive either placebo or subcutaneous IFNb-1a, 22 µg once weekly for 3 years with 6-monthly clinical assessments. The patient population was less clinically active than SPMS populations studied in other trials with less superimposed relapses. The primary outcome was time to sustained disability, as defined by time to first confirmed 1.0-point increase on the EDSS. Secondary outcomes included relapse rate. Treatment had no beneficial effect on time to confirmed progression on the EDSS (p = 0.45 for IFNb-1a vs. placebo) nor on the secondary disability outcome—Regional Functional Status Scale (p = 0.67). Annual relapse rate was 0.27 with placebo and 0.25 with IFNb-1a (p = 0.55). No significant gender differences were noted. Given some of the earlier trial results in SPMS, it was concluded that higher doses of IFNb-1a would be required in future trials [62].
The 3-year North American multicentre, double-blind, placebo-controlled, randomised trial of IFNβ included 939 subjects from the USA and Canada with SPMS and EDSS scores ranging from 3.0 to 6.5 [63]. Subjects were randomised to receive either placebo or two doses of IFNβ (250 µg or 160 µg/m2 body surface area) subcutaneously every other day. The primary outcome was time to progression of 1.0 EDSS point or 0.5 point if EDSS score was 6.0–6.5 at entry; any deterioration had to be confirmed at 6 months. No significant difference in time to confirmed progression of EDSS scores was found. However, IFNβ treatment resulted in improvement on secondary outcome measures including clinical relapses, new active MRI lesions and accumulated burden of disease on T2-weighted MRI. Effects were similar for both IFNβ treatment groups. Neutralizing antibodies to IFNβ were detected in 23–32% of recipients, but their presence did not consistently affect clinical or MRI outcomes.
The SPECTRIMS group tested two doses of IFNb-1a in patients with SPMS who could have had relapses but their main problem had to be accumulating disability. It was a multicentre, randomised, parallel-group, placebo-controlled study. 618 patients received subcutaneous placebo or IFNb-1a 22 µg or 44 µg three times weekly for 3 years [64]. The primary outcome, time to confirmed progression in disability, was not significantly affected by treatment (p = 0.146 for 44 µg vs. placebo). The relapse rate was reduced from 0.71 per year with placebo to 0.50 per year with treatment (p < 0.001 for both doses). Significant treatment effects were seen on other exacerbation-related outcomes and on a composite measure incorporating five separate clinical and MRI outcomes. Treatment did not significantly affect disability progression in this cohort, although significant benefit was observed on exacerbation-related outcomes. Exploratory post-hoc analyses suggested greater benefit in women and in patients who had reported at least one relapse in the 2 years before the study.
3.2 Glatiramer Acetate
3.2.1 Mechanism of Action
Glatiramer acetate (GA) is thought to drive preferential differentiation of CD4+ T cells into anti-inflammatory T helper-2 cells and increase T-regulatory cell numbers and optimise their function. It may also exert an immunomodulatory effect on B cells [65–67]. Some evidence suggests that it may have neuroprotective properties, via upregulation of brain-derived neurotropic factor [68].
3.2.2 Clinical Trials
A Cochrane review found two studies that have studied the effect of GA in PMS and found no benefit [69]. The first study [70] randomised 106 subjects aged 20–60 years, with a chronic-progressive course for at least 18 months, less than two exacerbations in the previous 24 months, and an EDSS of 2–6.5. These criteria were assessed in a pre-trial observation period lasting no more than 15 months and resulted in the exclusion of 47% of candidate participants. Treatment was with GA 20 mg or placebo subcutaneously twice a day. The primary outcome was confirmed progressive worsening of 1 EDSS point or 1.5 points according to baseline EDSS (≤5); any deterioration had to be confirmed at 3 months. This endpoint was observed in nine (17.6%) treated and 14 (25.5%) placebo patients. The differences between the overall survival curves were not significant. Progression rates at 12 and 24 months were higher for the placebo group (p = 0.088), with 2-year probabilities of progressing of 20.4% for GA and 29.5% for placebo. Two-year progression rates for two secondary endpoints, unconfirmed progression and progression of 0.5 EDSS points, were significant. This was followed by a large trial of 943 subjects with PPMS that did not meet its primary endpoint [71].
3.3 Mitoxantrone
3.3.1 Mechanism of Action
Mitoxantrone is an anthracenedione cytotoxic agent that inhibits DNA replication and DNA-dependent RNA synthesis and inhibits topoisomerase II activity preventing DNA repair [72]. Peripherally mitoxantrone inhibits T-cell activation, abrogates proliferation of B and T cells, diminishes antibody production, and deactivates macrophages [73]. It induces apoptosis of dendritic cells, decreases secretion of proinflammatory cytokines, such as tumour necrosis factor alfa (TNFα) and IL-2, inhibits B-cell function and increases T-cell suppressor function [74]. Mitoxantrone can cross the disrupted blood-brain barrier in MS, and in vitro evidence suggests it can induce microglial death [75, 76].
3.3.2 Clinical Trials
Mitoxantrone use in MS was part of a Cochrane review in which the data in PMS arose from a single study [77]. The review recommended that mitoxantrone should be used in SPMS when there is evidence of persistent inflammatory activity [78].
The MIMS phase III study assigned 194 patients with worsening RRMS or SPMS to receive placebo or mitoxantrone (5 g/m2 or 12 mg/m2) every 3 months intravenously [77]. The primary endpoint was a multivariate analysis of five clinical measures. Analyses of mitoxantrone 12 mg/m2 versus placebo were based on patients who received at least one dose and returned for at least one assessment of efficacy. Patients receiving 12 mg/m2 showed significantly reduced disability progression and had fewer clinical exacerbations. At 24 months, the mitoxantrone group experienced statistically significant benefits compared with the placebo group in the primary outcome (difference 0.30 [95% CI 0.17–0.44]; p < 0.0001) and the pre-planned univariate analyses: change in EDSS (0.24 [0.04–0.44]; p = 0.0194), change in ambulation index (0.21 [0.02–0.40]; p = 0.0306), adjusted total number of treated relapses (0.38 [0.18–0.59]; p = 0.0002), time to first treated relapse (0.44 [0.20–0.69]; p = 0.0004), and change in standardised neurological status (0.23 [0.03–0.43]; p = 0.0268).
3.4 Azathioprine
3.4.1 Mechanism of Action
Azathioprine (AZ) is a purine antagonist that impairs DNA replication. It impairs T-lymphocyte function and is more selective for T lymphocytes than for B lymphocytes [79]. AZ and its metabolites also impact T-lymphocyte apoptosis by modulation of Rac1 activation upon CD28 co-stimulation. As a result, the Rac1 target genes, such as mitogen-activated protein kinase, and nuclear factor-kappaB (NF-κB) and bcl-x (L) are suppressed, leading to apoptosis [80].
3.4.2 Clinical Trials
In 2007, the Cochrane Collaboration conducted a systematic review of the efficacy of AZ in the treatment of MS. Only randomised, double-blind, placebo-controlled trials that lasted at least 1 year were included, and the primary outcome was whether AZ prevented disability progression [81]. Five trials, including a total of 698 MS patients, were included; one included PMS subjects and three included mixed RRMS/PMS populations, and only one had an exclusively RRMS population [82]. Data from only 87 patients were available to calculate the number of patients who progressed during the first 2–3 years. There was a statistically significant benefit (RR 0.42, 95% CI 0.07–0.64) of AZ therapy at 3 years’ follow-up; this result was robust after sensitivity analyses and there was no heterogeneity among the trials. One further small add-on study performed in 2009 did not show any effects in SPMS [83].
The largest trial included RRMS and PMS patients, but was not included in the Cochrane review progression analysis. 354 patients were randomised to receive either AZ 2.5 mg/kg daily or placebo [84]. After 3 years, only small differences emerged between the groups. The mean increase in EDSS was 0.62 with AZ and 0.80 with placebo, a difference of 0.18. There were fewer relapses in the AZ group (average 2.2) than in the placebo group (average 2.5), but the difference was not statistically significant.
A further trial in PMS included both PPMS and SPMS patients [85]. Ninety-eight patients were recruited to a 3-year clinical trial to determine the effect of AZ alone, or AZ in combination with methylprednisolone (MP) for the first 36 weeks (group AM). Group AP received AZ and placebo instead of MP. A fourth group PP took placebos for both drugs. The AZ dose was adjusted to maintain the total white blood cell count within 3000–4000/mm3; MP was given in a fixed-dose ‘pulse’ and alternate-day regimen. The ‘intent-to-treat’ groups had no statistically significant differences in the rates of progression among the three treatments. Subgroup analyses suggest that the AM group who completed treatment exactly according to protocol did statistically significantly better than the placebo recipients using the sum of Standard Neurological Examination (SNE) scores; they were slightly better using the quantitative neuro-performance tests, but no better using Mickey’s Illness Severity Scores (ISS) or EDSS. The AZ-treated groups had half the relapse rate of the placebo-treated group. Adverse reactions to AZ accounted for most withdrawals; this was mainly due to haematological and hepatic abnormalities. The addition of MP to the AZ non-significantly improved efficacy, but also increased the adverse effects.
A further single-blind randomised parallel group trial of 18 months’ treatment with follow-up was reported [86]. 185 subjects (74 RRMS (40%); 111 SPMS (60%)) were randomised: 93 to AZ (2.5 mg/kg/daily) and 92 to placebo. Only 135 (73%) were included in the analysis (AZ 69; placebo 66). Outcomes included the mean number of relapses in the two groups at 18 months and patients defined as having worsened between the final and initial Kurtzke DSS score. Out of 37 patients with relapsing-progressive disease course in the AZ group, 19 improved or were stable in terms of DSS over 18 months (51%), while 25 out of 44 placebo patients were reported as stable or improved (57%).
A study reported in 1993 [87] randomised 40 patients in a double-blind trial. Nineteen received AZ (2 mg/kg daily) and 21 placebo over 3 years; 47.5% had a relapsing course and 52.5% had PMS. Chronic-progressive patients had to have a steady progression of disability of at least 1 point on the EDSS scale in the year prior to study entry. Outcome measures were the number of relapses per year, number of patients experiencing relapses during the study, progression on EDSS and number of patients who remained stable during the study. Twenty-one patients dropped out of the study (12 AZ and nine placebo). At the end of the study, 82% of the placebo group had deteriorated by one or more points on the EDSS compared with 38% in the AZ group, with a statistical significance of p = 0.051.
The ASPIRE study assessed the efficacy, safety and tolerability of AZ when added to IFNβ in patients with SPMS who had an incomplete response to IFNβ. The primary endpoint was change in MSFC over the 2-year double-blind and 1-year open-treatment periods. Secondary endpoints included EDSS and MSQOL-54. Eighty-five patients were randomised, 42 to AZ and IFNβ and 43 to placebo and IFNβ treatment. At the end of 36 months, 45 patients completed the study (23 and 21, respectively), with a high number of dropouts. MSQOL-54 data showed a slight worsening in the AZ group. MSFC data were compared with baseline data and showed a slight worsening at 36 months in the AZ group in comparison with placebo. MRI data showed a reduction in enhancing lesions only at 12 months of treatment in the AZ group but not at 24 and 36 months [83].
3.5 Cyclosporine
3.5.1 Mechanism of Action
Cyclosporine (CsA) is a neutral lipophilic cyclic undecapeptide isolated from the fungus Hypocladium inflatum gams that has been widely used for the treatment of allograft rejection and graft-versus-host disease [88]. CsA inhibits T-cell activation by blocking the transcription of cytokine genes, including IL-2 and IL-4 [89]. CsA is an inhibitor of the calcineurin/nuclear factor of activated T-cell pathway and also the mitogen-associated protein kinase, c-Jun NH (2)-terminal protein kinases, Stress-Activated Protein Kinase and the p38 signalling pathways [90].
3.5.2 Clinical Trials
Only one trial could be identified using cyclosporine in PMS [91]. Patients were recruited with clinically definite (CD) MS, an entry EDSS score between 3.0 and 7.0, and a progressive course defined by an increase in the EDSS of between 1 and 3 points in the year prior to entry. Patients were randomised to receive either CsA (n = 273) or placebo (n = 274) in a 2-year, double-blind, multicentre trial. CsA dosage was adjusted for toxicity. The mean increase in EDSS score was 0.39 ± 1.07 with CsA and 0.65 ± 1.08 with placebo until the time of early withdrawal or completion of the study (p = 0.002). Of three primary efficacy criteria, CsA delayed the time to becoming wheelchair bound (p = 0.038), but there were no statistically significant effects on time to sustained progression or on a composite score of ‘activities of daily living’. CsA did have a favourable effect on secondary measures that included: an endpoint analysis of change from baseline EDSS for the patients completing 12 and 24 months of study, overall clinical assessments by blinded neurologists, and global assessment of change from baseline to 12 and 24 months’ completion. There was a large and differential withdrawal rate for CsA (44 vs. 32% for placebo) mostly due to nephrotoxicity and hypertension, but this did not explain the effect of CsA in delaying disease progression [91].
3.6 Methotrexate
3.6.1 Mechanism of Action
Methotrexate (MTX) inhibits dihydrofolic acid reductase, reducing dihydrofolate to tetrahydrofolate in the synthesis of purine nucleotides and thymidylate. MTX interferes with DNA synthesis, repair and cellular replication [92].
3.6.2 Clinical Trials
A Cochrane review identified one trial involving 60 participants with PMS where MTX produced a non-significant trend to reduce sustained EDSS progression and number of relapses [93]. Oral weekly low-dose MTX 7.5 mg versus placebo over 2 years was studied in a randomised, double-blind, placebo-controlled clinical trial in 60 patients with progressive CDMS aged between 21 and 60 years with a disease duration of >1 year and an EDSS between 3.0 and 6.5. Apart from the non-significant results above, the MTX group had significantly less progression of impairment as measured by validated tests of upper-extremity function in the absence of clinically significant toxicity [94].
3.7 Cyclophosphamide
3.7.1 Mechanism of Action
Cyclophosphamide (CPM) is a widely used alkylating chemotherapeutic agent that is metabolised in the liver to form active metabolites that bind DNA and interfere with mitosis and cell replication [95]. CPM depletes lymphocytes in both the peripheral blood and cerebrospinal fluid, and decreases immunoglobulin production [96]. CPM decreases secretion of interferon-gamma (IFNγ) and IL-12 from the pro-inflammatory T helper (Th) 1 cells, and increases secretion of anti-inflammatory Th2 cytokines IL-4 and IL-10 in CSF and peripheral blood [97, 98].
3.7.2 Clinical Trials
Two studies have used CPM, one of which was placebo controlled [99] and the second was without placebo [100].
CPM was assessed in a single randomised, placebo-controlled, single-blind study of 168 patients with PMS, who had a deterioration of ≥1.0 point on the EDSS in the previous year. Patients were randomised to receive intravenous CPM and oral prednisone (n = 55); daily oral CPM, alternate-day prednisone (22 weeks), and weekly plasma exchange (20 weeks) (n = 57), or placebo medications and sham plasma exchange (n = 56). All patients were followed for at least 12 months (mean 30.4 months). There were no significant differences between the groups in this primary analysis (19 [35%] treatment failures with CPM; 18 [32%] with plasma exchange; 16 [29%] with placebo). There were also no differences in the proportions improved, stabilised or worsened at each 6-month assessment or in the mean change in the EDSS at the final assessment (0.81 CPM; 0.69 plasma exchange; 0.69 placebo) [99].
The other study was of CPM versus MP in SPMS in a multicentre, randomised, double-blind, controlled phase III clinical trial. Two parallel arms compared intravenous (IV) CPM (750 mg/m2 body surface area) to IV MP every 4 weeks (1 g) during the first year and every 8 weeks during the second year on evolution of physical disability in SPMS. Inclusion criteria were age 18–65 years, EDSS between 4.0 and 6.5 inclusive, progressive deterioration >6 months and <3 years, with a documented reduction of walking distance in the last 12 months. The primary outcome was the delay to confirmed EDSS deterioration. Secondary outcomes were walking distance, proportion of patients with EDSS deterioration, relapse rate, MSFC and safety. Intention-to-treat analysis was carried out. The primary endpoint was analysed using the Kaplan-Meier method and the delay of EDSS deterioration was compared between treatment groups by a log-rank test. 138 SPMS patients were randomised and allocated in the two groups. The mean duration of treatment was 94 weeks. For the primary endpoint, 18.1% of CPM patients experienced a deterioration compared with 31.8% of MP patients (p = 0.06). Fifty-two patients stopped treatment prematurely; in 24 this was for adverse events. Patients who were able to stay on CPM had a lower risk of further disability, but their dropout rate from treatment was twice that for MP [100].
3.8 Linomide
3.8.1 Mechanism of Action
Linomide (quinoline-3-carboxamide) is a synthetic immunomodulator that increases natural killer (NK) cell activity. It was shown to have an inhibitory effect on the clinical and histological signs of acute and chronic relapsing experimental autoimmune encephalomyelitis (EAE), inducing suppression of lymphocyte response to antigens and autoantibody production, and induces activation of NK and suppressor-inducer cells [101]. Linomide treatment significantly increased the percentage of the CD4+/CD45RA+ cells and decreased CD4+/CD45RO+ cells. Linomide also induced a transient increase in NK 1.1 cells, and CD5 B-cells [102].
3.8.2 Clinical Trials
Two studies have used linomide: an initial phase II study [103] that was followed by a phase III study that was stopped due to side effects [104].
A double-blind, phase II study found linomide-treated MS patients had significantly less active lesions on serial monthly MRI scans and a tendency to stabilise clinically. Thirty patients with SPMS with an EDSS of 3–7 were treated with linomide (2.5 mg) or placebo. Twenty-four patients completed at least 6 months of treatment. At 24 weeks, the mean shift in EDSS was +0.27 ± 0.16 with placebo versus −0.17 ± 0.17 with linomide (p = 0.045). The percentage of patients with evidence of ‘activity’ on their MRI (new, enlarging or new Gd-enhancing lesions) throughout the treatment period was 75% with placebo and 33% with linomide (p = 0.021) [103].
A subsequent trial with linomide was terminated due to serious side effects. In this randomised, double-blind, placebo-controlled trial, 715 patients with active RRMS (n = 90) or SPMS (n = 625) were randomised to receive either linomide (1.0, 2.5 or 7.5mg) or placebo. Patients were evaluated at 3-month intervals, clinically and with MRI. The planned primary outcome was the time to the development of ‘confirmed’ clinical worsening defined as an increase of ≥1.0 EDSS score if entry EDSS was ≤5.0, or ≥0.5 point with an entry EDSS ≥5.5, that was not associated with an acute relapse [104]. The trial was terminated 1 month after it became fully enrolled due to unanticipated life-threatening cardiopulmonary toxicities. Significant arthralgia, myalgia, bursitis and facial and peripheral oedema were common adverse events. The trial was too brief to determine unequivocal clinical benefits.
3.9 Cladribine
3.9.1 Mechanism of Action
Cladribine (CL) (2-chlorodeoxyadenosine) is an adenosine deaminase-resistant purine nucleoside chemotherapeutic agent. CL enters the cell via the purine nucleoside transporters and is phosphorylated by deoxycytidine kinase. In cells in which the ratio of deoxycytidine kinase to deoxynucleotidase is high, such as lymphocytes and monocytes, CL is phosphorylated to the active triphosphate deoxynucleotide, 2-chlorodeoxyadenosine-ATP, the accumulation of which disrupts cellular metabolism and damages DNA, causing cell death. These processes lead to lymphocyte depletion and long-lasting lymphopenia [105].
3.9.2 Clinical Trials
Initially, CL was tested intravenously in PMS in one randomised, double-blind clinical trial [106] and subsequently orally in one trial [107]. Intravenously, it was tested in 51 subjects with PMS with a crossover to placebo after 1 year [106]. There was a reported favourable influence on both EDSS and Scripps Neurologic Rating Scale (SNRS) scores. There were also similar favourable changes on the MRI findings in patients treated with CL. In the first year the most striking finding was that while clinical deterioration continued in the placebo-treated patients, the condition of patients who received CL stabilized or even improved slightly [108].
Orally, CL was studied in 159 patients with a median baseline EDSS of 6.0 who were randomised to placebo or CL 0.07 mg/kg/day for five consecutive days every 4 weeks for either two or six cycles (total dose, 0.7 mg/kg or 2.1 mg/kg, respectively), followed by placebo, for a total of eight cycles. Thirty percent had PPMS and 70% had SPMS. EDSS and SNRS scores were assessed bi-monthly and MRI was performed every 6 months. The primary outcome measure was mean change in EDSS, which did not differ among the groups at the end of the 12-month double-blind phase. Both CL treatments were superior to placebo for the proportion of patients having Gd+ lesions (p = 0.003). Differences were statistically significant at the 6-month evaluation and were maintained at the final evaluation. The effect segregated largely with the SPMS group. The T2 burden of disease showed a modest improvement in CL-treated patients and worsened in placebo-treated patients. Most adverse events were mild or moderate in severity and were not treatment limiting. No statistically significant treatment effects were seen in terms of changes in EDSS or SNRS scores [107].
3.10 Immunoglobulin
3.10.1 Mechanism of Action
Immunoglobulin (IVIG) can protect oligodendrocytes against complement-mediated injury and potentially enhance remyelination. In addition, IVIG can modulate microglial functions in vitro creating a microenvironment permissive for remyelination [109]. In EAE, infusions of IVIG significantly reduced disease symptoms as well as the underlying CNS pathology. IVIG was only effective in EAE when administered in a prophylactic treatment protocol; IVIG in established EAE did not alter the disease course or the degree of inflammation [110].
3.10.2 Clinical Trials
Two studies have studied the effect of IVIG in PMS.
In the European study, 318 SPMS patients (mean age 44 years) were randomly assigned IVIG 1 g/kg per month (n = 159) or an equivalent volume of placebo (albumin 0.1%; n = 159) for 27 months. Clinical assessments were made 3-monthly and the MRI was repeated after 12 months and 24 months. The primary outcome was confirmed worsening of disability as defined by the time to first confirmed progression on the EDSS. Analyses were by intention to treat. Nineteen patients in the IVIG group and 39 in the placebo group terminated the study treatment prematurely but were included in the analyses. IVIG treatment had no beneficial effect on time to confirmed EDSS progression (hazard ratio 1.11 [95% CI 0.80–1.53] for IVIG vs. placebo). The annual relapse rate was 0.46 in both groups. No statistically significant differences between the treatment groups were found in any of the other clinical outcome measures nor in the change of T2-lesion load over time. The treatment was generally well tolerated, although deep venous thrombosis, pulmonary embolism or both occurred in seven patients with risk factors for thromboembolism (six who had IVIG and one who had placebo) [111].
A further trial investigated the influence of IVIG in PPMS and SPMS [112]. 231 patients stratified for PPMS (n = 34) and SPMS (n = 197) were randomly assigned to IVIG 0.4 g/kg per month or placebo for 24 months. Primary endpoints were: (i) the time to confirmed progression of disease identified as worsening of the EDSS, confirmed at 3 months, and (ii) improvement of neurological function as defined by a patient’s best EDSS score. Secondary endpoints were the proportion of patients with confirmed progression, relapse rate, assessment of fine motor skills, visual evoked potentials, contrast sensitivity, depression and quality of life. Analysis of the intention-to-treat (ITT) population of combined PPMS and SPMS patients showed that the mean time to confirmed progression was 74 weeks in the IVIG compared with 62 weeks in the placebo group (p = 0.041). When PPMS and SPMS patients were analysed separately, the time to confirmed progression was also longer in the IVIG group, but the difference was not significant. In the combined ITT population, there were fewer patients with confirmed progression in the IVIG than in the placebo group (p = 0.028). There was no IVIG-mediated improvement in neurological function. In the combined per-protocol treated patients, IVIG treatment prolonged time to confirmed progression by 13 weeks (p = 0.0396). PPMS patients, but not SPMS patients, showed a slight favourable IVIG effect on the best EDSS score. The difference was significant in PPMS (p = 0.016) but not in SPMS patients. There was a trend for a favourable IVIG effect on the proportion of patients with confirmed progression. In patients with PPMS, IVIG reached significance over placebo (p = 0.036). Other secondary endpoints did not show significant differences between treatment groups. Eighteen patients with PPMS and 102 patients with SPMS withdrew from the study. The main reasons for withdrawal were a lack of efficacy, adverse events, withdrawal of consent, protocol violation, personal problems and one death.
3.11 Dirucotide
3.11.1 Mechanism of Action
Dirucotide is a synthetic peptide of myelin basic protein (MBP) and a molecular replicate of the site of attack that is dominant in MS patients with HLA haplotypes DR-2 or DR-4. These haplotypes are present in 65–75% of patients. The apparent mechanism of action of dirucotide is the induction or restoration of immunological tolerance with respect to ongoing immune attack at this molecular site [113].
3.11.2 Clinical Trials
Based on the results of a small trial [113], a phase III trial was designed targeting a subgroup of subjects [114]. The initial small double-blind, placebo-controlled phase II study of dirucotide in PMS failed to slow the time to disease progression but post-hoc analysis suggested efficacy in a subset of patients with HLA DR2/DR4 [113]; this represented 62.5% of the MS population [110].
As a result, MAESTRO, a multicentre, randomised, 2-year, double-blind, placebo-controlled study was initiated. It included 612 subjects with a diagnosis of SPMS and an EDSS of 3.5–6.5. Subjects were stratified according to baseline EDSS score (3.5–5.0, or 5.5–6.5) and HLA haplotype (DR2+/DR4+, or DR2−/DR4−). Upon entry of 100 DR2−/DR4− subjects, further study enrolment was limited to DR2+/DR4+ subjects. Subjects were randomly assigned to either 500 mg dirucotide or placebo, given by IV injection once every 6 months for 2 years. The primary outcome measure was time to progression by ≥1.0 EDSS point or 0.5 point if baseline EDSS was 5.5 or higher, confirmed at 6 months. Secondary outcomes included: mean change in EDSS, mean change in MSFC, MRI changes, annualized relapse rate and quality of life. There were no significant differences between treatment groups in either the primary or secondary endpoints. Dirucotide was well tolerated in all treated subjects with no safety issues identified [114].
3.12 Antibodies to Interferon γ or Tumor Necrosis Factor α
3.12.1 Mechanism of Action
Cytokines upregulating cellular immunity and produced by monocytes (IL-1, TNFα) and by Th1 helpers (IL-2, IFNγ, TNF-b) are thought to drive the pathological process and tissue injury in MS, while cytokines produced by Th2 cells (IL-4, IL-10) have been shown to suppress Th1 cells and cell-mediated immunity [115]. Therefore, preventing these effects using antibody blockage could potentially be beneficial.
3.12.2 Clinical Trials
In a double-blind, placebo-controlled trial, 45 patients with active SPMS were randomised to three groups of 15 patients, each receiving a short course of antibodies to IFNγ, TNFα or placebo. After 12 months EDSS, lymphocyte subpopulations, cytokine production levels, MRI and evoked potentials were assessed. Only patients who received antibodies to IFNγ showed a significant increase in the number of patients without confirmed disability progression versus placebo. MRI also showed a decrease in the number of active lesions and a decrease in IL-1b, TNFα and IFNγ concentrations in supernatants of activated blood cells and an increase in TGF-β production [116].
3.13 Simvastatin
3.13.1 Mechanism of Action
HMG-CoA reductase inhibitors (‘statins’) have multiple potentially positive effects but the relevant mechanism of action of statins remains unknown. Simvastatin rescued Aβ-mediated cerebrovascular and cognitive deficits in a transgenic mouse model of Alzheimer’s disease [117]. Simvastatin restored baseline levels of nitric oxide (NO), NO- and KATP channel-mediated dilations and endothelin-1-induced contractions. Simvastatin significantly reduced vasculopathy with arteriogenic remodelling and string vessel pathology in TGF mice. Statins can inhibit matrix metalloproteinases (MMP) production and activity. MMPs are increased in neural cells and leukocytes that infiltrate into the CNS. Upregulated MMPs can disrupt the blood-brain barrier and potentially mediate neuroinflammation and demyelination, and cause direct toxicity to axons and neurons. Immunomodulatory effects of statins have also been noted including inhibition of CD40 and adhesion molecule expression, and blockage of lymphocyte function-associated antigen-mediated co-stimulation [118]. Simvastatin was found to inhibit IL-1β, IL-23, TGF-β, IL-21 and IL-12p70, and induces IL-27 secretion from dendritic cells providing an inhibitory cytokine milieu for Th17 and Th1-cell differentiation [119].
3.13.2 Clinical Trials
To date, only one trial has been completed in SPMS [9]. The MS STAT trial, a double-blind, controlled trial randomised 140 participants aged 18–65 years with SPMS to receive 80 mg of simvastatin or placebo. The primary outcome was annualised rate of whole-brain atrophy. Notably the last MRI was performed 1 month off treatment. Analyses were by ITT and per protocol. The mean annualised atrophy rate was significantly lower in patients in the simvastatin group (0.288% per year [SD 0.521]) than in those in the placebo group (0.584% per year [0.498]). The adjusted difference in atrophy rate between groups was −0.254% per year (95% CI −0.422 to −0.087; p = 0.003), a 43% reduction in annualised rate. Simvastatin also showed benefits in both the EDSS and in the Multiple Sclerosis Impact Scale 29.
3.14 Lamotrigine
3.14.1 Mechanism of Action
Lamotrigine (LTG) acts via sodium channel blockade and also has antiglutamatergic and neuroprotective actions [120]. Excessive accumulation of sodium in axons affected by inflammation or demyelination makes axons vulnerable to injury by favouring calcium accumulation via the sodium-calcium exchanger [121, 122]. Partial blockade of voltage-gated sodium channels is neuroprotective in several experimental models of inflammatory axonal injury [123, 124].
3.14.2 Clinical Trials
One hundred and twenty patients with SPMS were studied in a double-blind, parallel-group trial. Patients were randomly assigned to receive LTG (target dose 400 mg/day) or placebo for 2 years. The primary outcome was the rate of change of partial (central) cerebral volume over 24 months. The mean change in partial (central) cerebral volume per year was −3.18 ml (SD −1.25) in the LTG group and −2.48 ml (−0.97) in the placebo group (difference −0.71 ml, 95% CI −2.56 to 1.15; p = 0.40). However, in an exploratory modelling analysis, LTG treatment seemed to be associated with greater partial (central) cerebral volume loss than was placebo in the first year (p = 0.04), and volume increased partially after treatment stopped (p = 0.04). LTG treatment reduced the deterioration of the T25FW (p = 0.02) but did not affect other secondary clinical outcome measures. The effect of LTG on cerebral volume of patients with SPMS did not differ from that of placebo over 24 months, but LTG seemed to cause early volume loss that reversed partially on discontinuation of treatment [125].
3.15 Dronabinol
3.15.1 Mechanism of Action
Laboratory evidence has found that cannabinoids have a neuroprotective action. Glutamate toxicity was reduced by cannabidiol, a nonpsychoactive constituent of marijuana, and the psychotropic cannabinoid Δ9-tetrahydrocannabinol (THC). Cannabinoids were also protective against neurotoxicity mediated by N-methyl-d-aspartate receptors and kainate receptors [126].
3.15.2 Clinical Trials
The CAMS study used oral Δ9-tetrahydrocannabinol (Δ9-THC) as a treatment for spasticity. However, a post-hoc analysis suggested there may be effects on disability [127]. This led to CUPID, a multicentre, parallel, randomised, double-blind, placebo-controlled study, in which patients aged 18–65 years with PPMS or SPMS were randomly assigned (2:1) to receive dronabinol (Δ (9)-tetrahydrocannabinol) or placebo for 36 months. The maximum dose was 28 mg/day, titrated against bodyweight and adverse effects. Primary outcomes were EDSS progression (≥1 point from baseline EDSS of 4.0–5.0 or ≥0.5 point from a baseline EDSS of ≥5.5, confirmed at 6 months) and change from baseline in the physical impact subscale of the 29-item multiple sclerosis impact scale (MSIS-29-PHYS). Of the 498 patients randomly assigned to a treatment group, 329 received at least one dose of dronabinol and 164 received at least one dose of placebo (five did not receive the allocated intervention). 145 patients in the dronabinol group had EDSS score progression (0.24 first progression events per patient-year compared with 73 in the placebo group); the hazard ratio for prespecified primary analysis was 0.92 (p = 0.57). The mean yearly change in MSIS-29-PHYS score was 0.62 points (SD 3.29) in the dronabinol group versus 1.03 points (3.74) in the placebo group. Primary analysis with a multilevel model gave an estimated between-group difference (dronabinol vs. placebo) of −0.9 points (95% CI −2.0 to 0.2) [128].
3.16 Masitinib
3.16.1 Mechanism of Action
Brain mast cells are located perivascularly and actively participate in the pathogenesis of MS, in part because they release large amounts of mediators that sustain the inflammatory network by disrupting the blood-brain barrier [129]. Masitinib, a selective oral tyrosine kinase inhibitor, effectively inhibits the survival, migration and activity of mast cells [130].
3.16.2 Clinical Trials
In a multicentre, randomised, placebo-controlled, proof-of-concept trial in PMS, masitinib was administered orally at 3–6 mg/kg/day for at least 12 months, with dose adjustment permitted in the event of insufficient response without toxicity. The primary outcome was the change relative to baseline MSFC. Clinical response was defined as an increase in MSFC score relative to a baseline of >100%. Thirty-five patients were randomised to receive masitinib (n = 27) or placebo (n = 8). Masitinib appeared to have a positive effect on MS-related impairment for PPMS and relapse-free SPMS patients, as evidenced by an improvement in MSFC scores relative to baseline, compared with a worsening MSFC score in placebo; +103% ± 189 versus −60% ± 190 at month 12, respectively. This positive, albeit not statistically significant response, was observed as early as month 3 and sustained to month 18, with similar trends seen in the PPMS and SPMS subpopulations. A total of 7/22 (32%) assessible masitinib patients reported clinical response following 12 months of treatment (according to the modified intent-to-treat population, observed cases) compared with none in the placebo group. The EDSS remained stable in both treatment groups. These data supported a larger placebo-controlled trial [131]. The current phase III trial (NCT01433497) has recruited and was due to report in 2015, but no results are as yet posted. The trial used masitinib (6 mg/kg per day) or placebo in 450 PMS patients over 2 years; the primary outcome was MSFC.
3.17 Natalizumab
3.17.1 Mechanism of Action
Natalizumab (NTL) is a humanised monoclonal antibody targeting the a4-integrin molecule, a component of VLA-4, and is approved for highly active MS in most countries. Binding of NTL to a4b1-integrin blocks its interaction with the receptor vascular cell adhesion molecule-1 on endothelial cells at the blood-brain barrier, thus interfering with leukocyte attachment and subsequent transmigration into the CNS. It has been proposed that NTL additionally interferes with T-cell activation and alters cell survival [132].
3.17.2 Clinical Trials
NTL was tested in SPMS in a multicentre, randomised, double-blind, placebo-controlled trial (ASCEND Trial). The primary objective of the study was to investigate whether treatment with NTL slows the accumulation of disability not related to relapses in SPMS. Secondary objectives included assessment of the proportion of participants with consistent improvement in T25FW, the change in the 12-item MS Walking Scale (MSWS-12), the change in the ABILHAND Questionnaire, the impact on the Multiple Sclerosis Impact Scale-29 Physical (MSIS-29 Physical) score, the change in MRI-assessed whole brain volume between the end of study and week 24, and the proportion of participants experiencing progression of disability as measured by individual physical EDSS system scores. 889 patients were enrolled in the study but it did not achieve its primary objective (see https://clinicaltrials.gov/ct2/show/NCT01416181).
3.18 Biotin
3.18.1 Mechanism of Action
Biotin is a vitamin acting as a coenzyme for carboxylases involved in key steps of energy metabolism and fatty acid synthesis. Biotin activates acetylCoA carboxylase, a potentially rate-limiting enzyme in myelin synthesis. It is hypothesized that biotin may help to promote remyelination and reduce axonal hypoxia [133].
3.18.2 Clinical Trials
A pilot uncontrolled, non-blinded, proof-of-concept study with 23 consecutive patients with PPMS and SPMS, originating from three different French MS centres, treated patients with high doses of biotin (100–300 mg/day) from 2 to 36 months (mean 9.2 months). In four patients with prominent visual impairment related to optic nerve injury, visual acuity improved significantly. Visual evoked potentials in two patients exhibited progressive reappearance of P100 waves, with normalization of latencies in one case. Proton magnetic resonance spectroscopy (H-MRS) in one case showed a progressive normalization of the choline/creatine ratio. One patient with left homonymous hemianopia continued to improve from 2 to 16 months following treatment’s onset. Sixteen out of 18 patients (89%) with prominent spinal cord involvement were considered as improved as confirmed by blinded review of videotaped clinical examination in nine cases. In all cases, improvement was delayed from 2 to 8 months following onset of treatment [7].
A further study recruited 154 patients aged 18–75 years with PPMS/SPMS and an EDSS score of 4.5–7 (NCT02220933). During the previous 2 years, they must have had experienced EDSS progression of at least 1 point if their baseline EDSS score was 4.5–5.5, and of 0.5 point if their baseline EDSS score was 6–7. Patients were randomly assigned to placebo (n = 51) or to oral biotin (n = 103) 300 mg/day. About 41% of the treatment group and 55% of the placebo group were also taking fampridine, a drug used to manage MS symptoms. The primary outcome was an improvement at 9 months confirmed at 12 months, using either a change in EDSS score of at least 1 point (if baseline EDSS was 4.5–5.5) and 0.5 point (if baseline EDSS was 6–7), or a decrease in T25FW of 20% compared with baseline. At the end of the double-blind phase, 13 out of 103 patients (12.6%) in the biotin group and 0 out of 51 (0%) in the placebo control met the definition of the primary endpoint [8]. In the treatment group, there was a mean EDSS decrease of 0.03 at month 12, compared with a mean increase of 0.13 in the placebo group (p = 0.014) [8]. Biotin has recently been filed for a licence in the European Union.
3.19 Rituximab
3.19.1 Mechanism of Action
Rituximab is a chimeric monoclonal antibody (mAb) of the IgG1j type that targets CD20, which is expressed by more than 95% of B cells [134]. Rituximab mediates B-cell death by antibody-dependent cell-mediated cytotoxicity and complement-dependent cytotoxicity and by inducing apoptotic mechanisms [135, 136].
3.19.2 Clinical Trials
Rituximab was used in a randomised trial in PPMS in 439 subjects and was not effective, but a subgroup analysis highlighted that responders may be of a younger age with Gd+ lesions [137]. Targeting this group may have underpinned the success of a subsequent trial using ocrelizumab, a related mAb targeting B cells, in PPMS [6].
A double-blind combination of rituximab in the Rituximab by Intravenous and Intrathecal Injection vs Placebo in patients with Low-Inflammatory SPMS (n = 80 planned, EDSS 3–7) (RIVITaLISe) trial involved a 1-year pretreatment baseline series of visits, followed by a 2-year treatment period. In the treatment arm patients received 25 mg of rituximab into the CSF and 200 mg of rituximab intravenously at month 0, followed by an additional 200 mg of rituximab intravenously at month 0.5 and another 25 mg of rituximab into the CSF at months 1.5 and 12.
Quantitative neuroimaging measures of CNS tissue destruction and clinical and electrophysiological measures of neurological disability will be collected every 6–12 months. Additionally, biomarkers focusing on analysis of CSF B cells and immunological responses to EBV will be collected at baseline and during treatment. The trial was powered for progression of brain atrophy as detected by SIENA methodology.
The trial had an adaptive design and all defined outcome measures collected in the first 30 enrolled patients were to be transformed into z-scores and compared for the robustness of longitudinal change over the coefficient of variation. As a result, the primary outcome measure of this trial was the comparison of individualized rates of brain atrophy progression between the rituximab and placebo groups after 2 years of treatment, unless the predetermined analysis established that one of the secondary outcome measures has a higher z-score.
The trial was terminated early as a planned interim analysis for the efficacy of B-cell depletion from the intrathecal compartment fitted the pre-defined stopping criteria for futility: if less than 50% of intrathecal B cells were depleted by active treatment (measured by <25% decrease in CSF CXCL13 and <50% increase in CSF BAFF). At that point, 43 subjects had entered the trial. Sixteen subjects did not proceed from year 1 into the treatment phase and nine subjects received placebo and 18 rituximab (see https://clinicaltrials.gov/ct2/show/NCT01212094).
3.20 Lipoic Acid
3.20.1 Mechanism of Action
Lipoic acid (LA) is an inexpensive and readily available oral antioxidant supplement. LA decreases inflammation and reduces optic nerve and spinal cord atrophy in EAE [138–140]. LA may exert its effects by reducing microglial activation and has beneficial effects on inflammatory cytokines in RRMS [141].
3.20.2 Clinical Trials
A 2-year, double-blind, randomised, controlled trial of 1200 mg daily LA versus placebo has recently been presented in poster form only (see https://clinicaltrials.gov/ct2/show/NCT01188811) [142]. The primary outcome was a reduction in MRI whole brain atrophy. Secondary outcomes included atrophy of brain substructures, spinal cord atrophy, retinal and macular atrophy, changes in neurological examination, walking, cognition, fatigue and quality of life. Pharmacokinetic laboratory markers were drawn at baseline and at month 12. Adverse events and safety laboratory measures were monitored.
Of the 54 randomised subjects, 51 subjects took at least one dose of study drug and were included in the analysis. Twenty-seven took LA and 24 took placebo. The average age was 58.5 ± 5.9 years; 61% were female. Average disease duration was 29.6 ± 9.5 years and median EDSS 6.0 (3.0–9.0). Four subjects terminated early (one each for glomerulonephritis, testicular cancer, renal failure and MRI intolerance). There was a baseline difference in whole brain volume with the LA having a significantly larger brain volume (p < 0.004). However, there was a significant reduction in brain volume loss over 96 weeks in the LA group (−0.4 ± 0.7) versus placebo (−1.3 ± 1.1). None of the secondary outcomes were significant. It was concluded that a larger trial was warranted to determine the clinical benefits and to confirm the safety.
3.21 Siponimod
3.21.1 Mechanism of Action
Siponimod is a selective S1P1 and S1P5 modulator that inhibits lymphocyte migration into the CNS. The non-selective S1P modulator fingolimod is effective in RRMS and brain atrophy but it was not effective in a double-blind, multicentre, parallel-group study of 969 PPMS patients with flexible treatment duration (minimum 3 years; maximum 5 years) [15]. Using fingolimod as an initial lead structure a novel chemical series was developed that focused on S1P1 potency, selectivity against S1P3, and safety and pharmacokinetics. This resulted in the development of siponimod [143]. Using MRI as an outcome, siponimod was tested in a double-blind adaptive dose-ranging phase II study in RRMS [144]; a 24-month extension confirmed effects on MRI and relapse rates [145].
3.21.2 Clinical Trials
Siponimod has been tested in the largest phase III randomised double-blind, placebo-controlled study in SPMS. Patients were aged between 18 and 60 years and had an EDSS score of 3.0–6.5. Eligible patients were randomised to receive either 2 mg of once-daily siponimod following initial dose titration starting at 0.25 mg or matching placebo. The primary outcome measure was time to 3-month confirmed disability progression as measured by an increase in EDSS. The key secondary outcomes were time to confirmed worsening of ≥20% from baseline in the T25FW and T2 lesion volume change from baseline. 1651 patients across 31 countries were enrolled (mean age 48.0 ± 7.9 years). Relapses in the 2 years prior to study start were documented in 35.1% patients (n = 578), and 55.2% had an EDSS ≥6. In patients with prior relapses, median duration of MS since first symptom/conversion to SPMS was 14.3/1.8 years and in those without prior relapses median duration of MS since first symptom/conversion to SPMS was 16.9/3.12 years [146].
The trial was composed of a core and an extension. 1363 patients completed the first core phase up to a maximum of 3 years. Eighty-seven percent were followed for at least 1 year, and the median follow-up time was 21 months. Siponimod reduced the risk of progression by 21% when looking at 3-month intervals. When analysing disease progression in 6-month intervals, the effect was even greater. This slowing of disability progression was seen in different types of patients, including those who did not have relapses. In addition, siponimod reduced the yearly relapse rate, brain volume loss and the volume of brain lesions measured by MRI. However, the drug did not improve performance on the T25FW [147].
4 Ongoing Pharmacological Studies in SPMS
There are a number of ongoing trials in SPMS that are currently running or have completed and the results are awaited (Table 2).
4.1 Ibudilast
Ibudilast is a phosphodiesterase inhibitor which showed a reduction in brain atrophy in a phase II study in RRMS [148]. On the basis of this atrophy benefit in RRMS, ibudilast is currently being studied in a phase II study in PMS (SPRINT-MS; NCT01982942). Patients can be untreated or remain on MS disease-modifying therapy (GA or IFNβ). The primary outcome is the covariate-adjusted mean rate of change in brain atrophy over 96 weeks as measured by brain parenchymal fraction. The trial was fully recruited in 2016 and is due to complete in summer 2017.
4.2 Fluoxetine, Riluzole and Amiloride
The MS-Secondary Progressive Multi-Arm Randomisation Trial (MS-SMART; NCT01910259) is a phase IIB double-blind, placebo-controlled clinical trial comparing the efficacy of three neuroprotective drugs (fluoxetine 20 mg twice daily (bd), riluzole 50 mg bd or amiloride 5 mg bd) in SPMS (440 patients, EDSS 4.0–6.5). All three drugs have shown promise in early-phase human MS clinical trials and target one or more of the pivotal neurodegenerative pathways implicated in SPMS. Patients will be followed up for 96 weeks and the primary outcome is change in brain volume. Patient recruitment was complete at the time of writing.
4.3 Imilecleucel-T Intervention
Abili-T (NCT01684761) is a phase II, double-blind, placebo controlled, multicentre study to evaluate the efficacy and safety of Tcelna (imilecleucel-T, autologous T-Cell Immunotherapy) in SPMS. Subjects whose myelin reactive T cell can be identified by epitope profiling assay will be randomised and provide blood to manufacture Tcelna. Tcelna is an autologous pool of myelin reactive T cells (MRTC) expanded ex vivo with immunodominant epitopes selected from the three myelin antigens, MBP, proteolipid protein and myelin oligodendrocyte glycoprotein on a per-subject basis then attenuated by irradiation to prevent further proliferation before releasing product for administration. Subjects will receive either Tcelna or placebo and will complete baseline assessments and will receive five doses in year 1. Subjects will be evaluated for changes in disability and cognitive function every 3 months, and radiographic changes annually.
5 Discussion
We have identified 34 randomised, controlled trials testing the potential efficacy of pharmacological treatments among patients affected by SPMS that have been published since 1988. This review did not include trials of treatments in PPMS alone, although primary and secondary progression differ little in their clinical [13] and pathological features [4].
5.1 Clinical Trial Design and Statistical Analysis in SPMS
5.1.1 The Evolution of Clinical Trial Methodology in SPMS
Although to date there does not appear to be an effective treatment for SPMS, at least 13 trials (38%) were found to have met their primary outcome; these appeared to be well conducted, with sound methodology using standard design and analysis methods. Recent studies have demonstrated clear advances in the methodology over time.
Among the 34 trials included in this review, in only one study was the randomization procedure not accurately described [113]. The majority of trials were double-blind (32/34 trials; 94%) with the exceptions of one open trial [86] and one single-blind trial [99]. In many studies, dropout was not higher than 20–25%, which is commonly observed in long running trials. However, some studies were troubled by high dropout rates. These included trials with some of the older immunosuppressive treatments, such as cyclosporine [91], AZ [83, 87] and CPM [100] as well as IVIG [112]. Total sample sizes ranged from 35 in small proof-of-concept trials [131] to 1949 in large confirmatory trials [146].
The included trials predominantly had two-arm parallel-group designs (25/34, 74%) with some exceptions: notably, one crossover trial [106] and eight multi-arm trials. This latter advance in design approach also included five multidose trials. Most trials included a placebo or add-on placebo control, with the exception of [100].
All trials except the siponimod trial [147] used a fixed follow-up period for each patient. This approach of utilising an event rate to dictate the trial length enables the trial to adapt to an uneven event rate that has troubled many of the previous studies.
A total of 24 (71%) studies used the DSS or EDSS as the primary endpoint. The EDSS changes were assessed with different statistical analyses. Early trials used mean EDSS changes, whereas later trials used proportions of patients with progressions or time-to-progression, which can be considered the current standard (see also [24]).
5.1.2 Lessons Learned
Failure to demonstrate efficacy in a number of trials might be due to the fact that the drugs were simply not effective or might be due to a number of design features including poorly defined study populations (e.g. relapsing progressive vs. purely progressive), insensitive outcomes (e.g. EDSS) and insufficient statistical power (sample sizes as well as study length). These problems, to an extent, can be addressed through advances in trial methodology, and attempts to do so are seen emerging in the trials described here.
The important lesson to be learned is that although 38%, a substantial proportion, of the trials could be considered successful and some of the drugs had been licensed for other indications previously (repurposing), this did not result in any drug being licensed for the treatment of SPMS. It remains to be seen whether the recent success of siponimod in SPMS and ocrelizumab in PPMS in phase III trials will deliver a licensed therapy, but it appears that developing a treatment requires a more strategic approach than just performing a trial.
5.1.3 Future Developments
The large number of failing trials in SPMS indicates the need for more robust and efficient study designs, endpoints and analyses. Blinded sample size re-estimation, which was proposed in relapsing MS in order to reduce uncertainty in the planning stages [149], has more recently been suggested as a possible solution for improving SPMS trial design [150]. Further improvement can be achieved with futility stopping, flexible follow-up and adaptive multi-arm trials [24, 151]. Although these new technologies can make clinical trials in SPMS more robust and efficient, at the same time the planning of these trials becomes more demanding and usually requires trial simulation [152]. The most recent results demonstrate the use of adaptive design, with the rituximab trial stopping early following a failed futility analysis, but also with siponimod, where an adaptive design was used in determining optimal dosing [144].
5.2 Relapses: A Treatable Prodrome to Prevent Progression in SPMS?
The current lack of licensed treatment options in PMS highlights the importance of optimising current approaches to treating RRMS with the aim of stopping progression by preventing its occurrence in the first place. In contrast to PPMS, this is a key potential opportunity in SPMS, as the prodromal relapsing phase offers an opportunity to target the disease process early on (Fig. 2). Focal inflammatory processes initially play a dominant role early in the disease course. By the time the disease enters the progressive phase, the inflammation gradually subsides and the axonal loss becomes the dominant pathological phenomenon [153]. However, axonal damage is also an early pathological feature of the disease, detected within actively demyelinated lesions [154] and in the normal appearing white matter [155]. The concept is further reinforced by radiological evidence of global and focal grey matter damage, occurring even before the disease onset, among subjects with a radiologically isolated syndrome (RIS) [156]. This is where radiological changes consistent with demyelination are identified on a routine MRI scan without related clinical symptoms [151].

Given the natural history of the disease (a) the use of early effective treatment in RRMS has the potential to delay the onset of SPMS (b). Treatment can act by both delaying the onset of clinical progression and reducing the impact of the disease in terms of disability by reducing the rate of progression (b). RRMS relapsing remitting multiple sclerosis, MRI magnetic resonance imaging, SPMS secondary progressive multiple sclerosis
Taken together, evidence indicates that mechanisms underlying the accumulation of permanent disability are active well in advance of the clinical onset of PMS [157]. The question of whether the suppression of the focal inflammation can prevent axonal loss is still unresolved and has a crucial importance for preventing progression [158]. Natural history studies demonstrate that patients with a larger number of relapses during the first 2–5 years have an increased probability of SPMS and accumulate disability more rapidly [159–161]. Therefore, early inflammatory changes probably contribute to set up a cascade of pathological events, leading to the development of the PMS. In contrast, late relapses do not influence the long-term outcome [161, 162], supporting the notion that the disease evolution, after the early stage, becomes largely independent of focal inflammatory mechanisms. This is in keeping with lessons from alemtuzumab [163], where effective immunosuppression in SPMS did not prevent the accumulation of disability. In contrast, early intervention, during RRMS, led to stabilization of disease activity.
These observations raised the concept of an early window of opportunity for treatments to exert their maximum efficacy. Since current therapies address mainly the inflammatory component of disease, introducing treatments at a time when inflammation is most prominent would appear the most sensible strategy. Positive results from clinically isolated syndrome (CIS) trials, demonstrating that early therapeutic intervention significantly reduces the probability of converting to CDMS [164–168] and prevents long-term disability accumulation [169, 170], further support the concept of early treatment initiation as a way of delaying the onset and evolution of SPMS (Fig. 2). Potentially treating RIS offers an extension of this approach with yet earlier intervention possible. So far we lack the evidence to support such an approach, although trials are ongoing using drugs for RRMS [NCT02739542].
5.3 Conclusions
Treating SPMS remains a challenge but after many years we are beginning to see progress in reducing its impact on the lives of people with MS. This has arisen from advances at many levels, knowledge of the natural history and the underlying pathology of the disease, clearer understanding of outcome performance and their limitations, together with advancing trial design and new drug development. Newer trials have had the benefit of these advances and this has likely contributed to recent successes in phase II and III SPMS and in phase III PPMS studies [6–9, 147] and there is hope that a licensed pharmacological treatment for SPMS will emerge.
A major issue of relevance for trials, but also in real life, is separating disability accumulation from progressive damage as opposed to the residual impact of a relapse. This will have an impact in practice on how a treatment licensed for PMS will be used by clinicians. Initial studies assumed treatments having an effect on RRMS would translate into an effect in SPMS; although this has not resulted in a licensed therapy, this review has found that 13 of the 34 trials in SPMS/PMS have been successful. This does, together with evidence from the underlying pathology, to an extent justify the early use of a potential treatment for progression in the relapsing phase. Though PPMS is not the focus of this review, the similarity in the pathological features and clinical course of SPMS and PPMS would argue for a similar approach to both conditions. This is supported now by the success in phase III trials of ocrelizumab in both RRMS and PPMS, and means that possibly a unified approach to MS will be justified in the future.
References
Olesen J, Gustavsson A, Svensson M, Wittchen HU, et al. The economic cost of brain disorders in Europe. Eur J Neurol. 2012;19(1):155–62.
Adelman G, Rane SG, Villa KF. The cost burden of multiple sclerosis in the United States: a systematic review of the literature. J Med Econ. 2013;16(5):639–47.
Jones E, Pike J, Marshall T, Ye X. Quantifying the relationship between increased disability and health care resource utilization, quality of life, work productivity, health care costs in patients with multiple sclerosis in the US. BMC Health Serv Res. 2016;22(16):294.
Reynolds R, Roncaroli F, Nicholas R, Radotra B, et al. The neuropathological basis of clinical progression in multiple sclerosis. Acta Neuropathol. 2011;122(2):155–70.
Tramacere I, Del Giovane C, Salanti G, D’Amico R, et al. Immunomodulators and immunosuppressants for relapsing-remitting multiple sclerosis: a network meta-analysis. Cochrane Database Syst Rev. 2015:CD011381.
Montalban X HB, Rammohan K, Giovannoni G, On behalf of the ORATORIO Clinical Investigators, et al. Ocrelizumab versus placebo in in primary progressive multiple sclerosis. N Engl J Med. 2017;376(3):209–220.
Sedel F, Papeix C, Bellanger A, Touitou V, et al. High doses of biotin in chronic progressive multiple sclerosis: a pilot study. Mult Scler Relat Disord. 2015;4(2):159–69.
Tourbah AL-FC, Edan G, Clanet M, et al. MD1003 (high doses of biotin) in progressive multiple sclerosis: subgroup analyses of the MS-SPI trial. ECTRIMS Online Library; 2015. p. 116698.
Chataway J, Schuerer N, Alsanousi A, Chan D, et al. Effect of high-dose simvastatin on brain atrophy and disability in secondary progressive multiple sclerosis (MS-STAT): a randomised, placebo-controlled, phase 2 trial. Lancet. 2014;383(9936):2213–21.
Scalfari A, Neuhaus A, Daumer M, Muraro PA, et al. Onset of secondary progressive phase and long-term evolution of multiple sclerosis. J Neurol Neurosurg Psychiatry. 2014;85(1):67–75.
Novotna M, Paz Soldan MM, Abou Zeid N, Kale N, et al. Poor early relapse recovery affects onset of progressive disease course in multiple sclerosis. Neurology. 2015;85(8):722–9.
Lublin FD, Reingold SC, Cohen JA, Cutter GR, et al. Defining the clinical course of multiple sclerosis: the 2013 revisions. Neurology. 2014;83(3):278–86.
Kremenchutzky M, Rice GP, Baskerville J, Wingerchuk DM, et al. The natural history of multiple sclerosis: a geographically based study 9: observations on the progressive phase of the disease. Brain. 2006;129(pt 3):584–94.
Katz Sand I, Krieger S, Farrell C, Miller AE. Diagnostic uncertainty during the transition to secondary progressive multiple sclerosis. Mult Scler. 2014;20(12):1654–7.
Lublin F, Miller DH, Freedman MS, Cree BA, et al. Oral fingolimod in primary progressive multiple sclerosis (INFORMS): a phase III, randomised, double-blind, placebo-controlled trial. Lancet. 2016;387(10023):1075–84.
Rovaris M, Confavreux C, Furlan R, Kappos L, et al. Secondary progressive multiple sclerosis: current knowledge and future challenges. Lancet Neurol. 2006;5(4):343–54.
Polman CH, Reingold SC, Banwell B, Clanet M, et al. Diagnostic criteria for multiple sclerosis: 2010 revisions to the McDonald criteria. Ann Neurol. 2011;69(2):292–302.
Kalincik T, Cutter G, Spelman T, Jokubaitis V, et al. Defining reliable disability outcomes in multiple sclerosis. Brain. 2015;138(Pt 11):3287–98.
Rover C, Nicholas R, Straube S, Friede T. Changing EDSS progression in placebo cohorts in relapsing MS: a systematic review and meta-regression. PLoS One. 2015;10(9):e0137052.
Lorscheider J, Buzzard K, Jokubaitis V, Spelman T, et al. Defining secondary progressive multiple sclerosis. Brain. 2016;139(pt 9):2395–405
European Medicines agency: Guideline on clinical investigation of medicinal products for the treatment of Multiple Sclerosis Committee for Medicinal Products for Human Use. EMA/CHMP/771815/2011, Rev. 2; 2015.
Schaffler N, Schonberg P, Stephan J, Stellmann JP, et al. Comparison of patient-reported outcome measures in multiple sclerosis. Acta Neurol Scand. 2013;128(2):114–21.
Kurtzke JF. Rating neurologic impairment in multiple sclerosis: an expanded disability status scale (EDSS). Neurology. 1983;33(11):1444–52.
Chataway J, Nicholas R, Todd S, Miller DH, et al. A novel adaptive design strategy increases the efficiency of clinical trials in secondary progressive multiple sclerosis. Mult Scler. 2011;17(1):81–8.
Meyer-Moock S, Feng YS, Maeurer M, Dippel FW, et al. Systematic literature review and validity evaluation of the Expanded Disability Status Scale (EDSS) and the Multiple Sclerosis Functional Composite (MSFC) in patients with multiple sclerosis. BMC Neurol. 2014;14:58.
Korn T. Pathophysiology of multiple sclerosis. J Neurol. 2008;255(Suppl 6):2–6.
van der Valk P, De Groot CJ. Staging of multiple sclerosis (MS) lesions: pathology of the time frame of MS. Neuropathol Appl Neurobiol. 2000;26(1):2–10.
Lassmann H. Multiple sclerosis: is there neurodegeneration independent from inflammation? J Neurol Sci. 2007;259(1–2):3–6.
Fu L, Matthews PM, De Stefano N, Worsley KJ, et al. Imaging axonal damage of normal-appearing white matter in multiple sclerosis. Brain. 1998;121(pt 1):103–13.
Rovaris M, Bozzali M, Santuccio G, Ghezzi A, et al. In vivo assessment of the brain and cervical cord pathology of patients with primary progressive multiple sclerosis. Brain. 2001;124(pt 12):2540–9.
De Stefano N, Matthews PM, Filippi M, Agosta F, et al. Evidence of early cortical atrophy in MS: relevance to white matter changes and disability. Neurology. 2003;60(7):1157–62.
Meinl E, Krumbholz M, Derfuss T, Junker A, et al. Compartmentalization of inflammation in the CNS: a major mechanism driving progressive multiple sclerosis. J Neurol Sci. 2008;274(1–2):42–4.
Howell OW, Reeves CA, Nicholas R, Carassiti D, et al. Meningeal inflammation is widespread and linked to cortical pathology in multiple sclerosis. Brain. 2011;134(pt 9):2755–71.
Magliozzi R, Howell O, Vora A, Serafini B, et al. Meningeal B-cell follicles in secondary progressive multiple sclerosis associate with early onset of disease and severe cortical pathology. Brain. 2007;130(pt 4):1089–104.
Calabrese M, Rocca MA, Atzori M, Mattisi I, et al. A 3-year magnetic resonance imaging study of cortical lesions in relapse-onset multiple sclerosis. Ann Neurol. 2010;67(3):376–83.
Waxman SG. Ion channels and neuronal dysfunction in multiple sclerosis. Arch Neurol. 2002;59(9):1377–80.
Campbell GR, Ziabreva I, Reeve AK, Krishnan KJ, et al. Mitochondrial DNA deletions and neurodegeneration in multiple sclerosis. Ann Neurol. 2011;69(3):481–92.
Fischer MT, Sharma R, Lim JL, Haider L, et al. NADPH oxidase expression in active multiple sclerosis lesions in relation to oxidative tissue damage and mitochondrial injury. Brain. 2012;135(pt 3):886–99.
Fischer MT, Wimmer I, Hoftberger R, Gerlach S, et al. Disease-specific molecular events in cortical multiple sclerosis lesions. Brain. 2013;136(Pt 6):1799–815.
Holland CM, Charil A, Csapo I, Liptak Z, et al. The relationship between normal cerebral perfusion patterns and white matter lesion distribution in 1,249 patients with multiple sclerosis. J Neuroimaging. 2012;22(2):129–36.
Davies AL, Desai RA, Bloomfield PS, McIntosh PR, et al. Neurological deficits caused by tissue hypoxia in neuroinflammatory disease. Ann Neurol. 2013;74(6):815–25.
Lansley J, Mataix-Cols D, Grau M, Radua J, et al. Localized grey matter atrophy in multiple sclerosis: a meta-analysis of voxel-based morphometry studies and associations with functional disability. Neurosci Biobehav Rev. 2013;37(5):819–30.
Hauser SL, Oksenberg JR. The neurobiology of multiple sclerosis: genes, inflammation, and neurodegeneration. Neuron. 2006;52(1):61–76.
Strimbu K, Tavel JA. What are biomarkers? Curr Opin HIV AIDS. 2010;5(6):463–6.
Prentice RL. Surrogate endpoints in clinical trials: definition and operational criteria. Stat Med. 1989;8(4):431–40.
Narayanan D, Cheng H, Bonem KN, Saenz R, et al. Tracking changes over time in retinal nerve fiber layer and ganglion cell-inner plexiform layer thickness in multiple sclerosis. Mult Scler. 2014;20(10):1331–41.
Raftopoulos R, Hickman SJ, Toosy A, Sharrack B, et al. Phenytoin for neuroprotection in patients with acute optic neuritis: a randomised, placebo-controlled, phase 2 trial. Lancet Neurol. 2016;15(3):259–69.
Kearney H, Miller DH, Ciccarelli O. Spinal cord MRI in multiple sclerosis–diagnostic, prognostic and clinical value. Nat Rev Neurol. 2015;11(6):327–38.
Ciccarelli O, Barkhof F, Bodini B, De Stefano N, et al. Pathogenesis of multiple sclerosis: insights from molecular and metabolic imaging. Lancet Neurol. 2014;13(8):807–22.
Barkhof F, Calabresi PA, Miller DH, Reingold SC. Imaging outcomes for neuroprotection and repair in multiple sclerosis trials. Nat Rev Neurol. 2009;5(5):256–66.
Fisniku LK, Brex PA, Altmann DR, Miszkiel KA, et al. Disability and T2 MRI lesions: a 20-year follow-up of patients with relapse onset of multiple sclerosis. Brain. 2008;131(Pt 3):808–17.
Ontaneda D, Fox RJ, Chataway J. Clinical trials in progressive multiple sclerosis: lessons learned and future perspectives. Lancet Neurol. 2015;14(2):208–23.
Teleshova N, Bao W, Kivisakk P, Ozenci V, et al. Elevated CD40 ligand expressing blood T-cell levels in multiple sclerosis are reversed by interferon-beta treatment. Scand J Immunol. 2000;51(3):312–20.
Genc K, Dona DL, Reder AT. Increased CD80(+) B cells in active multiple sclerosis and reversal by interferon beta-1b therapy. J Clin Invest. 1997;99(11):2664–71.
Jiang H, Milo R, Swoveland P, Johnson KP, et al. Interferon beta-1b reduces interferon gamma-induced antigen-presenting capacity of human glial and B cells. J Neuroimmunol. 1995;61(1):17–25.
Hallal-Longo DE, Mirandola SR, Oliveira EC, Farias AS, et al. Diminished myelin-specific T cell activation associated with increase in CTLA4 and Fas molecules in multiple sclerosis patients treated with IFN-beta. J Interferon Cytokine Res. 2007;27(10):865–73.
Muraro PA, Leist T, Bielekova B, McFarland HF. VLA-4/CD49d downregulated on primed T lymphocytes during interferon-beta therapy in multiple sclerosis. J Neuroimmunol. 2000;111(1–2):186–94.
Dhib-Jalbut S, Marks S. Interferon-beta mechanisms of action in multiple sclerosis. Neurology. 2010;5(74 Suppl 1):S17–24.
La Mantia L, Vacchi L, Di Pietrantonj C, Ebers G, et al. Interferon beta for secondary progressive multiple sclerosis. Cochrane Database Syst Rev. 2012;1:CD005181.
Placebo-controlled multicentre randomised trial of interferon beta-1b in treatment of secondary progressive multiple sclerosis. European Study Group on interferon beta-1b in secondary progressive MS. Lancet. 1998;352(9139):1491–7.
Cohen JA, Cutter GR, Fischer JS, Goodman AD, et al. Benefit of interferon beta-1a on MSFC progression in secondary progressive MS. Neurology. 2002;59(5):679–87.
Andersen O, Elovaara I, Farkkila M, Hansen HJ, et al. Multicentre, randomised, double blind, placebo controlled, phase III study of weekly, low dose, subcutaneous interferon beta-1a in secondary progressive multiple sclerosis. J Neurol Neurosurg Psychiatry. 2004;75(5):706–10.
Panitch H, Miller A, Paty D, Weinshenker B, et al. Interferon beta-1b in secondary progressive MS: results from a 3-year controlled study. Neurology. 2004;63(10):1788–95.
Secondary Progressive Efficacy Clinical Trial of Recombinant Interferon-Beta-1a in MSSG. Randomized controlled trial of interferon-beta-1a in secondary progressive MS: clinical results. Neurology. 2001;56(11):1496–504.
Kala M, Miravalle A, Vollmer T. Recent insights into the mechanism of action of glatiramer acetate. J Neuroimmunol. 2011;235(1–2):9–17.
Dhib-Jalbut S. Glatiramer acetate (Copaxone) therapy for multiple sclerosis. Pharmacol Ther. 2003;98(2):245–55.
Neuhaus O, Farina C, Wekerle H, Hohlfeld R. Mechanisms of action of glatiramer acetate in multiple sclerosis. Neurology. 2001;56(6):702–8.
Chen C, Liu X, Wan B, Zhang JZ. Regulatory properties of copolymer I in Th17 differentiation by altering STAT3 phosphorylation. J Immunol. 2009;183(1):246–53.
La Mantia L, Munari LM, Lovati R. Glatiramer acetate for multiple sclerosis. Cochrane Database Syst Rev. 2010;(5):CD004678.
Bornstein MB, Miller A, Slagle S, Weitzman M, et al. A placebo-controlled, double-blind, randomized, two-center, pilot trial of Cop 1 in chronic progressive multiple sclerosis. Neurology. 1991;41(4):533–9.
Wolinsky JS, Narayana PA, O’Connor P, Coyle PK, et al. Glatiramer acetate in primary progressive multiple sclerosis: results of a multinational, multicenter, double-blind, placebo-controlled trial. Ann Neurol. 2007;61(1):14–24.
Martinelli V, Radaelli M, Straffi L, Rodegher M, et al. Mitoxantrone: benefits and risks in multiple sclerosis patients. Neurol Sci. 2009;30(Suppl 2):S167–70.
Watson CM, Davison AN, Baker D, O’Neill JK, et al. Suppression of demyelination by mitoxantrone. Int J Immunopharmacol. 1991;13(7):923–30.
Vollmer T, Stewart T, Baxter N. Mitoxantrone and cytotoxic drugs’ mechanisms of action. Neurology. 2010;5(74 Suppl 1):S41–6.
Millefiorini E, Gasperini C, Pozzilli C, D’Andrea F, et al. Randomized placebo-controlled trial of mitoxantrone in relapsing-remitting multiple sclerosis: 24-month clinical and MRI outcome. J Neurol. 1997;244(3):153–9.
Li JM, Yang Y, Zhu P, Zheng F, et al. Mitoxantrone exerts both cytotoxic and immunoregulatory effects on activated microglial cells. Immunopharmacol Immunotoxicol. 2012;34(1):36–41.
Hartung HP, Gonsette R, Konig N, Kwiecinski H, et al. Mitoxantrone in progressive multiple sclerosis: a placebo-controlled, double-blind, randomised, multicentre trial. Lancet. 2002;360(9350):2018–25.
Martinelli Boneschi F, Vacchi L, Rovaris M, Capra R, et al. Mitoxantrone for multiple sclerosis. Cochrane Database Syst Rev. 2013;(5):CD002127.
Patel AA, Swerlick RA, McCall CO. Azathioprine in dermatology: the past, the present, and the future. J Am Acad Dermatol. 2006;55(3):369–89.
Tiede I, Fritz G, Strand S, Poppe D, et al. CD28-dependent Rac1 activation is the molecular target of azathioprine in primary human CD4+ T lymphocytes. J Clin Invest. 2003;111(8):1133–45.
Casetta I, Iuliano G, Filippini G. Azathioprine for multiple sclerosis. Cochrane Database Syst Rev. 2007;(4):CD003982.
Goodkin DE, Bailly RC, Teetzen ML, Hertsgaard D, et al. The efficacy of azathioprine in relapsing-remitting multiple sclerosis. Neurology. 1991;41(1):20–5.
Montanari E ML, Pesci I, et al. ASPIRE (azathioprine secondary progressive interferon treated patients randomised evaluation) study: 2-year double-blind and 1-year open, randomised, multicentre, pilot study. Multiple sclerosis functional composite (MSFC) and magnetic resonance data. Mult Scler. 2009;250(suppl):822.
Double-masked trial of azathioprine in multiple sclerosis. British and Dutch Multiple Sclerosis Azathioprine Trial Group. Lancet. 1988;2(8604):179–83.
Ellison GW, Myers LW, Mickey MR, Graves MC, et al. A placebo-controlled, randomized, double-masked, variable dosage, clinical trial of azathioprine with and without methylprednisolone in multiple sclerosis. Neurology. 1989;39(8):1018–26.
Ghezzi A, Di Falco M, Locatelli C, et al. Clinical controlled randomized trial of azathioprine in multiple sclerosis. In: Consette RE, Delmotte P, editors. Recent advances in multiple sclerosis therapy. Elsevier; 1989.
Milanese CLML, Salmaggi A, Eoli M. A double blind study on azathioprine efficacy in multiple sclerosis: final report. J Neurol. 1993;240:295–8.
Borel JF, Feurer C, Magnee C, Stahelin H. Effects of the new anti-lymphocytic peptide cyclosporin A in animals. Immunology. 1977;32(6):1017–25.
Granelli-Piperno A. In situ hybridization for interleukin 2 and interleukin 2 receptor mRNA in T cells activated in the presence or absence of cyclosporin A. J Exp Med. 1988;168(5):1649–58.
Matsuda S, Koyasu S. Mechanisms of action of cyclosporine. Immunopharmacology. 2000;47(2–3):119–25.
Efficacy and toxicity of cyclosporine in chronic progressive multiple sclerosis: a randomized, double-blinded, placebo-controlled clinical trial. The Multiple Sclerosis Study Group. Ann Neurol. 1990;27(6):591–605.
Calabresi P, Chabner BA. Chemotheraphy of neoplastic diseases. In: S GL, Gilman A, Rall T, Nies AS, Taylor P, editors. The Pharmacological Basis of Therapeutics. New York: Pergamon Press; 1990. p. 1202–1208.
Gray O, McDonnell GV, Forbes RB. Methotrexate for multiple sclerosis. Cochrane Database Syst Rev. 2004;(2):CD003208.
Goodkin DE, Rudick RA, VanderBrug Medendorp S, Daughtry MM, et al. Low-dose (7.5 mg) oral methotrexate reduces the rate of progression in chronic progressive multiple sclerosis. Ann Neurol. 1995;37(1):30–40.
Kovarsky J. Clinical pharmacology and toxicology of cyclophosphamide: emphasis on use in rheumatic diseases. Semin Arthritis Rheum. 1983;12(4):359–72.
Brinkman CJ, Nillesen WM, Hommes OR. T-cell subpopulations in blood and cerebrospinal fluid of multiple sclerosis patients: effect of cyclophosphamide. Clin Immunol Immunopathol. 1983;29(3):341–8.
Comabella M, Balashov K, Issazadeh S, Smith D, et al. Elevated interleukin-12 in progressive multiple sclerosis correlates with disease activity and is normalized by pulse cyclophosphamide therapy. J Clin Invest. 1998;102(4):671–8.
Smith DR, Balashov KE, Hafler DA, Khoury SJ, et al. Immune deviation following pulse cyclophosphamide/methylprednisolone treatment of multiple sclerosis: increased interleukin-4 production and associated eosinophilia. Ann Neurol. 1997;42(3):313–8.
The Canadian cooperative trial of cyclophosphamide and plasma exchange in progressive multiple sclerosis. The Canadian Cooperative Multiple Sclerosis Study Group. Lancet. 1991;337(8739):441–6.
Brochet B, Deloire MS, Perez P, et al. Double-blind, randomized, controlled study of cyclophosphamide versus methylprednisolone in secondary progressive multiple sclerosis. PLoS One. 2017;12(e0168834):2017.
Abramsky O, Lehmann D, Karussis D. Immunomodulation with linomide: possible novel therapy for multiple sclerosis. Mult Scler. 1996;2(4):206–10.
Lehmann D, Karussis DM, Fluresco D, Mizrachi-Koll R, et al. Immunomodulation of autoimmunity by linomide: inhibition of antigen presentation through down regulation of macrophage activity in the model of experimental autoimmune encephalomyelitis. J Neuroimmunol. 1997;74(1–2):102–10.
Karussis DM, Meiner Z, Lehmann D, Gomori JM, et al. Treatment of secondary progressive multiple sclerosis with the immunomodulator linomide: a double-blind, placebo-controlled pilot study with monthly magnetic resonance imaging evaluation. Neurology. 1996;47(2):341–6.
Noseworthy JH, Wolinsky JS, Lublin FD, Whitaker JN, et al. Linomide in relapsing and secondary progressive MS: part I: trial design and clinical results. North American Linomide Investigators. Neurology. 2000;54(9):1726–33.
Comi G, Hartung HP, Kurukulasuriya NC, Greenberg SJ, et al. Cladribine tablets for the treatment of relapsing-remitting multiple sclerosis. Expert Opin Pharmacother. 2013;14(1):123–36.
Beutler E, Sipe JC, Romine JS, Koziol JA, et al. The treatment of chronic progressive multiple sclerosis with cladribine. Proc Natl Acad Sci USA. 1996;93(4):1716–20.
Rice GP, Filippi M, Comi G. Cladribine and progressive MS: clinical and MRI outcomes of a multicenter controlled trial. Cladribine MRI Study Group. Neurology. 2000;54(5):1145–55.
Sipe JC, Romine JS, Koziol JA, McMillan R, et al. Cladribine in treatment of chronic progressive multiple sclerosis. Lancet. 1994;344(8914):9–13.
Stangel M, Hartung HP. Intravenous immunoglobulins in multiple sclerosis. Studies and mechanisms of action–an update. Nervenarzt. 2002;73(2):119–24.
Humle Jorgensen S, Sorensen PS. Intravenous immunoglobulin treatment of multiple sclerosis and its animal model, experimental autoimmune encephalomyelitis. J Neurol Sci. 2005;233(1–2):61–5.
Hommes OR, Sorensen PS, Fazekas F, Enriquez MM, et al. Intravenous immunoglobulin in secondary progressive multiple sclerosis: randomised placebo-controlled trial. Lancet. 2004;364(9440):1149–56.
Pohlau D, Przuntek H, Sailer M, Bethke F, et al. Intravenous immunoglobulin in primary and secondary chronic progressive multiple sclerosis: a randomized placebo controlled multicentre study. Mult Scler. 2007;13(9):1107–17.
Warren KG, Catz I, Ferenczi LZ, Krantz MJ. Intravenous synthetic peptide MBP8298 delayed disease progression in an HLA Class II-defined cohort of patients with progressive multiple sclerosis: results of a 24-month double-blind placebo-controlled clinical trial and 5 years of follow-up treatment. Eur J Neurol. 2006;13(8):887–95.
Freedman MS, Bar-Or A, Oger J, Traboulsee A, et al. A phase III study evaluating the efficacy and safety of MBP8298 in secondary progressive MS. Neurology. 2011;77(16):1551–60.
Cavallo MG, Pozzilli P, Thorpe R. Cytokines and autoimmunity. Clin Exp Immunol. 1994;96(1):1–7.
Skurkovich S, Boiko A, Beliaeva I, Buglak A, et al. Randomized study of antibodies to IFN-gamma and TNF-alpha in secondary progressive multiple sclerosis. Mult Scler. 2001;7(5):277–84.
Tong XK, Hamel E. Simvastatin restored vascular reactivity, endothelial function and reduced string vessel pathology in a mouse model of cerebrovascular disease. J Cereb Blood Flow Metab. 2015;35(3):512–20.
Malfitano AM, Marasco G, Proto MC, Laezza C, et al. Statins in neurological disorders: an overview and update. Pharmacol Res. 2014;88:74–83.
Zhang X, Tao Y, Wang J, Garcia-Mata R, et al. Simvastatin inhibits secretion of Th17-polarizing cytokines and antigen presentation by DCs in patients with relapsing remitting multiple sclerosis. Eur J Immunol. 2013;43(1):281–9.
Ketter TA, Manji HK, Post RM. Potential mechanisms of action of lamotrigine in the treatment of bipolar disorders. J Clin Psychopharmacol. 2003;23(5):484–95.
Stys PK. General mechanisms of axonal damage and its prevention. J Neurol Sci. 2005;233(1–2):3–13.
Frohman EM, Filippi M, Stuve O, Waxman SG, et al. Characterizing the mechanisms of progression in multiple sclerosis: evidence and new hypotheses for future directions. Arch Neurol. 2005;62(9):1345–56.
Bechtold DA, Miller SJ, Dawson AC, Sun Y, et al. Axonal protection achieved in a model of multiple sclerosis using lamotrigine. J Neurol. 2006;253(12):1542–51.
Kapoor R, Davies M, Blaker PA, Hall SM, et al. Blockers of sodium and calcium entry protect axons from nitric oxide-mediated degeneration. Ann Neurol. 2003;53(2):174–80.
Kapoor R, Furby J, Hayton T, Smith KJ, et al. Lamotrigine for neuroprotection in secondary progressive multiple sclerosis: a randomised, double-blind, placebo-controlled, parallel-group trial. Lancet Neurol. 2010;9(7):681–8.
Hampson AJ, Grimaldi M, Axelrod J, Wink D. Cannabidiol and (−)Delta9-tetrahydrocannabinol are neuroprotective antioxidants. Proc Natl Acad Sci USA. 1998;95(14):8268–73.
Zajicek JP, Sanders HP, Wright DE, Vickery PJ, et al. Cannabinoids in multiple sclerosis (CAMS) study: safety and efficacy data for 12 months follow up. J Neurol Neurosurg Psychiatry. 2005;76(12):1664–9.
Zajicek J, Ball S, Wright D, Vickery J, et al. Effect of dronabinol on progression in progressive multiple sclerosis (CUPID): a randomised, placebo-controlled trial. Lancet Neurol. 2013;12(9):857–65.
Theoharides TC, Kempuraj D, Kourelis T, Manola A. Human mast cells stimulate activated T cells: implications for multiple sclerosis. Ann N Y Acad Sci. 2008;1144:74–82.
Dubreuil P, Letard S, Ciufolini M, Gros L, et al. Masitinib (AB1010), a potent and selective tyrosine kinase inhibitor targeting KIT. PLoS One. 2009;4(9):e7258.
Vermersch P, Benrabah R, Schmidt N, Zephir H, et al. Masitinib treatment in patients with progressive multiple sclerosis: a randomized pilot study. BMC Neurol. 2012;12:36.
Klotz L, Wiendl H. Monoclonal antibodies in neuroinflammatory diseases. Expert Opin Biol Ther. 2013;13(6):831–46.
Sedel F, Bernard D, Mock DM, Tourbah A. Targeting demyelination and virtual hypoxia with high-dose biotin as a treatment for progressive multiple sclerosis. Neuropharmacol. 2016;110(pt B):644–53.
Maloney DG, Grillo-Lopez AJ, White CA, Bodkin D, et al. IDEC-C2B8 (Rituximab) anti-CD20 monoclonal antibody therapy in patients with relapsed low-grade non-Hodgkin’s lymphoma. Blood. 1997;90(6):2188–95.
Browning JL. B cells move to centre stage: novel opportunities for autoimmune disease treatment. Nat Rev Drug Discov. 2006;5(7):564–76.
Dalakas MC. B cells as therapeutic targets in autoimmune neurological disorders. Nat Clin Pract Neurol. 2008;4(10):557–67.
Hawker K, O’Connor P, Freedman MS, Calabresi PA, et al. Rituximab in patients with primary progressive multiple sclerosis: results of a randomized double-blind placebo-controlled multicenter trial. Ann Neurol. 2009;66(4):460–71.
Marracci GH, Jones RE, McKeon GP, Bourdette DN. Alpha lipoic acid inhibits T cell migration into the spinal cord and suppresses and treats experimental autoimmune encephalomyelitis. J Neuroimmunol. 2002;131(1–2):104–14.
Morini M, Roccatagliata L, Dell’Eva R, Pedemonte E, et al. Alpha-lipoic acid is effective in prevention and treatment of experimental autoimmune encephalomyelitis. J Neuroimmunol. 2004;148(1–2):146–53.
Chaudhary P, Marracci G, Yu X, Galipeau D, et al. Lipoic acid decreases inflammation and confers neuroprotection in experimental autoimmune optic neuritis. J Neuroimmunol. 2011;233(1–2):90–6.
Khalili M, Azimi A, Izadi V, Eghtesadi S, et al. Does lipoic acid consumption affect the cytokine profile in multiple sclerosis patients: a double-blind, placebo-controlled, randomized clinical trial. NeuroImmunoModulation. 2014;21(6):291–6.
Spain RI MC, Horak F, Simon J, et al. P1.373—lipoic acid for neuroprotection in secondary progressive multiple sclerosis. In: 68th Annual meeting of the American academy of neurology, Vancouver; 2016.
Pan S, Gray NS, Gao W, Mi Y, et al. Discovery of BAF312 (Siponimod), a potent and selective S1P Receptor modulator. ACS Med Chem Lett. 2013;4(3):333–7.
Selmaj K, Li DK, Hartung HP, Hemmer B, et al. Siponimod for patients with relapsing-remitting multiple sclerosis (BOLD): an adaptive, dose-ranging, randomised, phase 2 study. Lancet Neurol. 2013;12(8):756–67.
Kappos L, Li DK, Stuve O, Hartung HP, et al. Safety and efficacy of siponimod (BAF312) in patients with relapsing-remitting multiple sclerosis: dose-blinded, randomized extension of the phase 2 BOLD study. JAMA Neurol. 2016;73(9):1089-98.
Kappos L B-OA, Cree B, Fox R, et al. Baseline subgroup characteristics of EXPAND: a phase 3 study of siponimod (BAF312) for the Treatment of Secondary progressive multiple sclerosis (P3.084). Neurology. 2016;86(16):suppl. P3.084.
Kappos L B-OA, Cree B, et al. Efficacy and safety of siponimod in secondary progressive multiple sclerosis—results of the placebo controlled, double-blind, phase III EXPAND study. ECTRIMS 2016. London: ECTRIMS Online Library; 2016.
Barkhof F, Hulst HE, Drulovic J, Uitdehaag BM, et al. Ibudilast in relapsing-remitting multiple sclerosis: a neuroprotectant? Neurology. 2010;74(13):1033–40.
Nicholas R Straube S, Schmidli H, Schneider S, et al. Trends in annualized relapse rates in relapsing remitting multiple sclerosis and consequences for clinical trial design. Mult Scler. 2011;17(10):1211–7.
Friede T, Pohlmann H, Schmidli H. Blinded sample size reestimation in event driven clinical trials: methods and an application in multiple sclerosis. 2017 (in preparation).
Friede T, Parsons N, Stallard N, Todd S, et al. Designing a seamless phase II/III clinical trial using early outcomes for treatment selection: an application in multiple sclerosis. Stat Med. 2011;30(13):1528-40.
Friede T, Nicholas R, Stallard N, Todd S, et al. Refinement of the Clinical Scenario Evaluation Framework for Assessment of Competing Development Strategies with an Application to Multiple Sclerosis. Drug Inf J. 2010;44(6):713–718.
Mahad DH, Trapp BD, Lassmann H. Pathological mechanisms in progressive multiple sclerosis. Lancet Neurol. 2015;14(2):183–93.
Trapp BD, Peterson J, Ransohoff RM, Rudick R, et al. Axonal transection in the lesions of multiple sclerosis. N Engl J Med. 1998;338(5):278–85.
Evangelou N, DeLuca GC, Owens T, Esiri MM. Pathological study of spinal cord atrophy in multiple sclerosis suggests limited role of local lesions. Brain. 2005;128(pt 1):29–34.
Giorgio A, Stromillo ML, Rossi F, Battaglini M, et al. Cortical lesions in radiologically isolated syndrome. Neurology. 2011;77(21):1896–9.
Bjartmar C, Trapp BD. Axonal degeneration and progressive neurologic disability in multiple sclerosis. Neurotox Res. 2003;5(1–2):157–64.
Fox RJ, Thompson A, Baker D, Baneke P, et al. Setting a research agenda for progressive multiple sclerosis: the International Collaborative on Progressive MS. Mult Scler. 2012;18(11):1534–40.
Confavreux C, Vukusic S, Adeleine P. Early clinical predictors and progression of irreversible disability in multiple sclerosis: an amnesic process. Brain. 2003;126(pt 4):770–82.
Leray E, Yaouanq J, Le Page E, Coustans M, et al. Evidence for a two-stage disability progression in multiple sclerosis. Brain. 2010;133(pt 7):1900–13.
Scalfari A, Neuhaus A, Degenhardt A, Rice GP, et al. The natural history of multiple sclerosis: a geographically based study 10: relapses and long-term disability. Brain. 2010;133(pt 7):1914–29.
Tremlett H, Yousefi M, Devonshire V, Rieckmann P, et al. Impact of multiple sclerosis relapses on progression diminishes with time. Neurology. 2009;73(20):1616–23.
Coles AJ, Cox A, Le Page E, Jones J, et al. The window of therapeutic opportunity in multiple sclerosis: evidence from monoclonal antibody therapy. J Neurol. 2006;253(1):98–108.
Jacobs LD, Beck RW, Simon JH, Kinkel RP, et al. Intramuscular interferon beta-1a therapy initiated during a first demyelinating event in multiple sclerosis. CHAMPS Study Group. N Engl J Med. 2000;343(13):898–904.
Miller AE, Wolinsky JS, Kappos L, Comi G, et al. Oral teriflunomide for patients with a first clinical episode suggestive of multiple sclerosis (TOPIC): a randomised, double-blind, placebo-controlled, phase 3 trial. Lancet Neurol. 2014;13(10):977–86.
Comi G, Martinelli V, Rodegher M, Moiola L, et al. Effect of glatiramer acetate on conversion to clinically definite multiple sclerosis in patients with clinically isolated syndrome (PreCISe study): a randomised, double-blind, placebo-controlled trial. Lancet. 2009;374(9700):1503–11.
Kappos L, Freedman MS, Polman CH, Edan G, et al. Effect of early versus delayed interferon beta-1b treatment on disability after a first clinical event suggestive of multiple sclerosis: a 3-year follow-up analysis of the BENEFIT study. Lancet. 2007;370(9585):389–97.
Comi G, Filippi M, Barkhof F, Durelli L, et al. Effect of early interferon treatment on conversion to definite multiple sclerosis: a randomised study. Lancet. 2001;357(9268):1576–82.
Kinkel RP, Kollman C, O’Connor P, Murray TJ, et al. IM interferon beta-1a delays definite multiple sclerosis 5 years after a first demyelinating event. Neurology. 2006;66(5):678–84.
Kappos L, Freedman MS, Polman CH, Edan G, et al. Long-term effect of early treatment with interferon beta-1b after a first clinical event suggestive of multiple sclerosis: 5-year active treatment extension of the phase 3 BENEFIT trial. Lancet Neurol. 2009;8(11):987–97.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Funding
No funding was received for the publication of this article
Conflict of interest
Dr. Ashwini Nandoskar has no conflicts of interest.
Dr. Joel Raffel has no conflicts of interest.
Dr. Antonio Scalfari has received honoraria for speaking (Teva and Genzyme) and support for attending conferences (Biogen, Teva, Genzyme, Novartis).
Professor Tim Friede has received personal fees for consultancies (including data monitoring committees) from Novartis, Bayer, Biogen, AstraZeneca, Janssen, Grünenthal, Pharmalog, SGS and Roche, all outside the submitted work.
Richard Nicholas has received an honorarium for advisory boards (Biogen Idec, Roche, Novartis, Genzyme), speaking (Biogen Idec, Novartis, Genzyme), funds for organising education (Biogen Idec, Genzyme), and for staff/research (Biogen). He has been a principal investigator in RRMS (Biogen, Novartis) and progressive MS (dirucotide, simvastatin, dronabinol, natalizumab, siponimod, MS-SMART) trials.
Rights and permissions
About this article

Cite this article
Nandoskar, A., Raffel, J., Scalfari, A.S. et al. Pharmacological Approaches to the Management of Secondary Progressive Multiple Sclerosis. Drugs 77, 885–910 (2017). https://doi.org/10.1007/s40265-017-0726-0
Published:
Issue Date:
DOI: https://doi.org/10.1007/s40265-017-0726-0