
Abstract
Brodalumab (Lumicef®) is a human monoclonal immunoglobulin G antibody that is being developed by Kyowa Hakko Kirin in Japan, where it has been approved for the treatment of psoriasis vulgaris, psoriatic arthritis, pustular psoriasis and psoriatic erythroderma. Brodalumab binds with high affinity to interleukin (IL)-17 receptor A, thereby inhibiting several pro-inflammatory cytokines from the IL-17 family. Regulatory applications for brodalumab in plaque psoriasis are also under review in the USA, EU and Canada. This article summarizes the milestones in the development of brodalumab leading to this first approval for the treatment of psoriasis.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
1 Introduction
The interleukin (IL)-17 family of pro-inflammatory cytokines plays an important role in the pathogenesis of several immune-mediated diseases, including psoriasis (PsO) [1]. Blockade of the IL-17 receptor A (IL-17RA) inhibits the activity of multiple ligands from the IL-17 family, and can potentially provide beneficial clinical outcomes in patients with PsO and psoriatic arthritis (PsA) [2].
Brodalumab (Lumicef®) is a monoclonal antibody against IL-17RA that is being developed for the subcutaneous treatment of psoriatic disorders. In July 2016 [3], the Ministry of Health, Labour and Welfare in Japan granted manufacturing and marketing approval for subcutaneous brodalumab for the treatment of adults with psoriasis vulgaris (PsV), PsA, pustular PsO and psoriatic erythroderma (PsE) [4]. The recommended dosage of brodalumab is 210 mg administered subcutaneously at weeks 0, 1 and 2, and every 2 weeks (q2w) thereafter [4]. Two phase III trials in Japanese patients with moderate to severe plaque PsO (i.e. PsV and PsA) [4827-003 study; NCT01782924] and generalized pustular PsO or PsE (4827-004 study; NCT01782937) have been completed.
Similar to other monoclonal antibodies, the Japanese prescribing information for brodalumab carries a boxed warning regarding the risk of serious infections, including tuberculosis reactivation [4]. The development of malignant tumours has also been reported during treatment with monoclonal antibody agents, although a causal relationship has not been demonstrated with brodalumab. Brodalumab is contraindicated in patients with a history of hypersensitivity to any of the components of the drug, and those with symptoms of serious infection or active tuberculosis [4].
Acceptance of the US FDA Biologics License Application for brodalumab was supported by the results of the phase III AMAGINE-1 (NCT01708590), AMAGINE-2 (NCT01708603) and AMAGINE-3 (NCT01708629) trials in patients with moderate to severe plaque PsO [5]. Although AMAGINE-1, -2 and -3 met their co-primary endpoints after 12 weeks’ treatment, they were subsequently terminated by the study sponsor (Amgen) after adverse events of suicidal ideation and behaviour were observed [6]. Phase III trials in patients with PsA have been

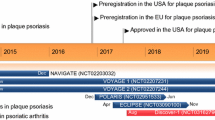
Key developmental milestones of subcutaneous brodalumab for psoriasis. BLA Biologics License Application, MAA marketing authorization application, PsE psoriatic erythroderma, PsO psoriasis, PsV psoriasis vulgaris, asterisk denotes trials terminated by Amgen
completed (AMVISION-2; NCT02024646) or terminated (AMVISION-1, NCT02029495). Recently, an FDA advisory committee recommended the approval of brodalumab in adults with moderate to severe PsO, under the condition that a suicide risk-mitigation plan for the drug was implemented, including a boxed warning and an ongoing patient registry to evaluate the risk of suicide [7].
Phase II trials in patients with inadequately controlled asthma (NCT01199289) or rheumatoid arthritis (NCT00950989) have been completed, although subsequent phase II trials in these indications (NCT01059448 and NCT01902290) were terminated; the extension trial in rheumatoid arthritis (NCT01902290) was terminated due to a lack of efficacy. A phase II trial in patients with axial spondylarthritis was withdrawn prior to enrolment (NCT02429882), and development of brodalumab in asthma, rheumatoid arthritis and spondylarthritis has since been discontinued. An intravenous formulation of brodalumab has been investigated in a Japanese phase I trial in healthy volunteers and patients with moderate to severe plaque psoriasis (NCT01488201), while phase II trials in patients with Crohn’s disease (NCT01150890, NCT01199302) were terminated. Development of the intravenous formulation of brodalumab appears to have been discontinued.
1.1 Company Agreements
In October 2010, Kyowa Hakko Kirin acquired exclusive licensing rights from Amgen for the development and marketing of brodalumab in Japan and other Asian countries [8]. In April 2012, Amgen entered into an agreement with AstraZeneca for the development and commercialization of five Amgen monoclonal antibodies, including brodalumab [9]. Under this agreement, AstraZeneca would make an upfront payment of $US50 million and fund ≈65 % of the development through to 2014, after which the costs were expected to be shared equally. After payment of single-digit royalties to Amgen, the profits were expected to be shared equally between the companies. Commercial development of brodalumab was to be split, with Amgen leading development of dermatology indications in the USA and Canada and rheumatology indications in the USA, Canada and Europe, and AstraZeneca being responsible for marketing in respiratory and dermatology indications in regions outside of the USA, Canada and markets covered under existing agreements with Amgen [9].
In May 2015, Amgen announced the termination of its involvement in the development of brodalumab with AstraZeneca due to Amgen’s decision to focus on other key molecules and the incidence of suicidal ideation and behaviour in clinical trials, which Amgen believed would have labelling which limited the appropriate patient population [10]. AstraZeneca confirmed the ongoing transfer of the brodalumab programme from Amgen in July 2015, and announced that potential partnering options were being investigated [11]. In September 2015, AstraZeneca entered into a collaborative agreement with Valeant Pharmaceuticals, in which Valeant was granted an exclusive
Features and properties of brodalumab
Alternative names | Lumicef®, KHK-4827, AMG-827, IL-17 receptor mAb |
Class | Anti-inflammatories, antirheumatics, monoclonal antibodies, skin disorder therapies |
Mechanism of action | IL-17 receptor antagonist |
Route of administration | Subcutaneous |
Pharmacodynamics | Binds IL-17 receptor A with high affinity (Kd = 239 pmol/L) and blocks binding of IL-17A, IL-17F, IL-17E (IL-25) and IL17A/F, which inhibits downstream inflammatory activities of IL-17 receptor A |
Pharmacokinetics | Two-compartment model with linear and non-linear elimination, estimated bioavailability of 58 % after subcutaneous administration, V of 4.62 L, CL of 0.223 L/day, Vmax of 5.40 mg/day |
Adverse events | |
Most frequent | Nasopharyngitis, URTI, headache and arthralgia |
Occasional | Neutropenia, Candida spp. infections |
Rare | Suicidal ideation and behaviour |
ATC codes | |
WHO ATC code | L04AC12 (interleukin inhibitors) |
EphMRA ATC code | Not yet assigned |
license to develop and commercialize brodalumab globally (except in Japan and other Asian countries where Kyowa Hakko Kirin hold these rights) [12]. Under this agreement, Valeant would make an upfront payment of $US100 million, and additional pre-launch (up to $US170 million) and sales-related post-launch (up to $US175 million) milestone payments. Following the approval, the profits will be shared between AstraZeneca and Valeant [12].
In July 2016, AstraZeneca entered into an agreement with LEO Pharma A/S for the exclusive licence to brodalumab in Europe, whereby LEO Pharma will acquire the rights to brodalumab in Europe under similar conditions to those agreed with Valeant, with continued payment of a low single-digit inventor royalty to Amgen [13]. Simultaneously, Valeant’s rights to develop and commercialize brodalumab in Europe were terminated [14]. Under the terms of its amended agreement with AstraZeneca, Valeant will continue to develop and commercialize brodalumab in the USA and any other territories outside of Europe. AstraZeneca will make an upfront payment and certain sales-based milestone payments to Valeant, and one of the pre-launch milestone payments due under the original agreement will be reduced [14].
1.2 Patent Information
Amgen holds a patent with an estimated expiry date of 2027 that covers the polynucleotide and polypeptide composition of brodalumab in the USA and Europe [15].
2 Scientific Summary
2.1 Pharmacodynamics
Brodalumab is a human IgG2 monoclonal antibody that binds with high affinity to human IL-17RA (dissociation constant of 239 pmol/L) and inhibits the biological activity of IL-17A, IL-17F and IL-17E (IL-25) homodimers and the IL17A/F heterodimer [16, 17]. By blocking IL-17RA ligand binding, brodalumab inhibits the downstream synergistic and pleitropic inflammatory activities of this receptor, thus reducing the severity of PsO [17]. After a single dose of subcutaneous brodalumab in patients with PsO, significant and rapid improvements were observed in lesional skin mRNA levels for several IL-17-modulated keratinocyte-derived factors, as well as reductions in skin mRNA levels of IL-17A, IL-17C, IL17F, IL-22 and both subunits of IL-23 [16]. In a study of human psoriatic skin before and after treatment with brodalumab, blockade of IL-17RA was associated with rapid transcriptomal changes in keratinocyte-expressed genes, with subsequent normalization of leukocyte abnormalities [18].
2.2 Pharmacokinetics
In population pharmacokinetic analyses in healthy volunteers and patients with PsO, the serum concentration–time profile of brodalumab was described by a two-compartment model with parallel linear and non-linear elimination pathways [17, 19]. In the final population model, brodalumab had a volume of distribution of 4.62 L, a serum clearance of 0.223 L/day and a non-linear clearance rate of 5.40 mg/day, indicating mainly serum distribution with limited tissue penetration [19]. After subcutaneous administration, brodalumab had an estimated bioavailability of 58 % [17].
Based on final model simulations for brodalumab 140 and 210 mg, the area under the serum concentration–time curve at steady state was predicted to be greater than twofold higher in patients weighing <75 kg compared with reference subjects, while age and diagnosis (healthy vs. PsO) were not expected to affect the pharmacokinetics of brodalumab to a clinically relevant extent [19]. In Japanese patients with moderate to severe plaque PsO, brodalumab exposure increased more than dose proportionally in the dose range of 70–210 mg [20]. Based on population pharmacokinetic/pharmacodynamic modelling, whereby brodalumab inhibits plaque formation indirectly via a signalling compartment, brodalumab has a half-maximal inhibitory effect concentration of 2.86 μg/mL [17].
2.3 Therapeutic Trials
2.3.1 Japanese Patients with Plaque Psoriasis, Pustular Psoriasis or Psoriatic Erythroderma
In Japanese patients with moderate to severe plaque PsO (including PsA), subcutaneous brodalumab was more effective than placebo in a 12-week, randomized, multicentre, double-blind, phase II trial (4827-002 study; NCT01748539) [20], and provided sustained clinical response over 52 weeks’ treatment in a phase III extension trial (4827-003 study; NCT01782924) [21].
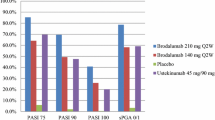
In the 4827-002 study, mean percentage improvements from baseline to week 12 in Psoriasis Area and Severity Index (PASI) scores (primary endpoint) were significantly (p < 0.001) greater with brodalumab 70, 140 and 210 mg q2w (38, 82 and 97 %, respectively) than with placebo (9 %) [20]. Furthermore, the proportion of patients with ≥75, ≥95 and 100 % improvements in PASI scores (PASI75, 90 and 100) at week 12 were significantly (p < 0.001) greater with brodalumab 140 and 210 mg q2w than placebo. Significantly (p < 0.05) more brodalumab than placebo recipients had static Physician Global Assessment (sPGA) scores of “clear” or “almost clear” (i.e. 0 or 1) at week 12, and mean percentage improvements from baseline in body surface area (BSA) involvement were significantly (p < 0.05) greater with brodalumab than placebo. In patients with PsA, 20 % improvement in American College of Rheumatology (ACR) response criteria (ACR20) was observed in one (20 %), two (40 %) and four (100 %) patients receiving brodalumab 70, 140 and 210 mg q2w, respectively, versus none of the patients receiving placebo. In addition, one patient receiving brodalumab 210 mg q2w met the criteria for 70 % ACR improvement (ACR70) at week 12. In all patients, there were significantly (p < 0.001) greater improvements from baseline in Dermatology Life Quality Index (DLQI) scores with brodalumab 140 and 210 mg q2w than placebo [20].
In the 4827-003 extension study, the proportion of patients with sustained PASI75 at week 52 was 78 and 94 % with brodalumab 140 and 210 mg q2w [21]. Similarly, the rate of PASI90 was 71 and 88 % and the rate of PASI100 was 44 and 56 % with brodalumab 140 and 210 mg q2w. At week 52 in patients with PsA, brodalumab 210 mg q2w was associated with ACR20 response in six out of eight patients (75 %) and ACR50 or 70 response in five out of eight patients (63 %), while brodalumab 140 mg q2w provided ACR20 response in three out of eight patients (38 %) [21].
In the 4827-002 study (n = 151), adult patients (aged 20–70 years) with stable PsO for ≥6 months, PASI scores of ≥12 and ≥10 % BSA PsO involvement received subcutaneous brodalumab 70, 140 or 210 mg or placebo at baseline and weeks 1, 2, 4, 6, 8 and 10 [20]; those who completed the 12-week trial were eligible to enrol in the 52-week open-label extension (4827-003 study) [20, 21]. In the 4827-003 study (n = 145), patients who previously received brodalumab 140 or 210 mg q2w continued brodalumab at the same dosage, and those who previously received brodalumab 70 mg q2w or placebo were assigned to receive brodalumab 140 or 210 mg q2w [21].
Subcutaneous brodalumab 140 or 210 mg q2w was also effective in Japanese patients with generalized pustular PsO or PsE in a long-term, noncomparative, open-label, phase III trial (4827-004 study; NCT01782937) [22]. After 52 weeks’ treatment, Clinical Global Impression (CGI) scores (primary endpoint) showed improvement from baseline or remission in 83 % of patients with pustular PsO and 89 % of patients with PsE. With regard to key secondary endpoints, mean percentage improvements from baseline to week 52 in PASI scores were 93 and 95 % in patients with pustular PsO and PsE, and PASI75, 90 and 100 were achieved by 90, 90 and 60 %, respectively, of pustular PsO patients and 94, 94 and 63 %, respectively of PsE patients [22]. At week 52, patients with pustular PsO had a mean pustular symptom score of 0.5, and 94 % of patients with PsE had sPGA scores of “clear” or “almost clear” (i.e. 0 or 1). DLQI scores of 0 or 1 at week 52 were reported in 80 and 94 % of patients with pustular PsO and PsE [22].
The 4827-004 study enrolled adults (aged ≥18 years) who met the criteria for generalized pustular PsO according to guidelines for diagnosis and treatment (n = 12) and those with >80 % BSA involvement who were designated as PsE (n = 18) [22]. Patients received brodalumab 140 mg at baseline, week 1 and 2 and then q2w until week 52. Patients with inadequate response at week 4 could receive an increased dosage of 210 mg q2w (n = 8) [22].
Subcutaneous brodalumab remained effective during long-term treatment of Japanese patients with plaque PsO (including PsV and PsA), generalized pustular PsO or PsE in an interim analysis of a phase III extension trial (4827-005 study; NCT02052609) [23]. Over >80 weeks’ treatment, brodalumab q2w (140 or 210 mg) or every 4 weeks (q4w) [140 mg] was associated with mean percentage improvements from baseline in PASI of 89 and 90 %, and the rates of PASI75, 90 and 100 were 88, 73 and 33 % across all patients. Among patients with generalized pustular PsO, 100 % had pustular symptom scores of 0–2 at the final evaluation time point. In the 4827-005 study (n = 155), patients who had completed the 4827-002, 4827-003 or 4827-004 studies received brodalumab 140 mg q4w, with a switch to 140 or 210 mg q2w from weeks 2–8 depending on disease severity; patients with generalized pustular PsO or PsE who received brodalumab 210 mg q2w in the 4827-004 study could continue to receive brodalumab at the same dosage [23].
2.3.2 Patients with Plaque Psoriasis
In patients with moderate to severe plaque PsO, subcutaneous brodalumab 140 and 210 mg q2w was more effective than placebo in the randomized, multinational, double-blind, phase III AMAGINE-1 (NCT01708590) [24], AMAGINE-2 (NCT01708603) and AMAGINE-3 (NCT01708629) [25] trials. Brodalumab 210 mg q2w was also more effective than bodyweight-based ustekinumab every 12 weeks in AMAGINE-2 and -3 [25].
After a 12-week induction period, the percentage of patients with PASI75 (co-primary endpoint) was significantly (p < 0.001) greater with brodalumab 140 and 210 mg q2w than placebo in AMAGINE-1 (60 and 83 vs. 3 %) [24], -2 (67 and 86 vs. 8 %) and -3 (69 and 85 vs. 6 %) [25]. In AMAGINE-1, significant (p < 0.001) differences in PASI75 between the brodalumab and placebo groups was observed as early as week 2 [24]. The rate of sPGA success (i.e. score of 0 or 1) at week 12 (co-primary endpoint) was also significantly (p < 0.001) higher with both dosages of brodalumab than placebo in AMAGINE-1 (54 and 76 vs. 1 %) [24], -2 (58 and 79 vs. 4 %) and -3 (60 and 80 vs. 4 %) [25].
In a comparison of brodalumab 210 mg q2w with ustekinumab every 12 weeks, the proportion of patients with PASI100 at week 12 (co-primary endpoint) was significantly (p < 0.001) higher with brodalumab than ustekinumab in AMAGINE-2 (44 vs. 22 %) and -3 (37 vs. 19 %) [25]. With regard to this co-primary endpoint, patients in the bodyweight-based brodalumab group (i.e. patients with baseline bodyweight ≤100 kg who received 140 mg q2w and those with baseline bodyweight >100 kg who received 210 mg q2w) also had significantly (p < 0.001) higher rates of PASI100 at week 12 than patients in the ustekinumab group in AMAGINE-2 (34 vs. 22 %) and AMAGINE-3 (30 vs. 19 %). The median time to PASI75 response with brodalumab 140 or 210 mg q2w was significantly (p < 0.001) shorter than with ustekinumab in AMAGINE-2 (6.0 and 4.1 vs. 8.1 h) and AMAGINE-3 (5.9 and 4.1 vs. 8.1 h) [25].
In AMAGINE-1, brodalumab at both dosages was associated with significantly (p < 0.001) higher rates than placebo of PASI90, PASI100 and sPGA score of 0 at week 12 [24]. Brodalumab recipients also experienced improvements from baseline to week 12 in the Hospital Anxiety and Depression Scale (HADS) scores, with statistically significant model-based treatment differences versus placebo (nominal p < 0.001) [24]. At week 12, brodalumab was additionally associated with significantly (p < 0.001) higher rates of Psoriasis Symptom Inventory (PSI) response (i.e. total score ≤8, no item scores >1), DLQI and treatment satisfaction than placebo [26], as well as significant (p < 0.001) improvements in all health-related quality of life aspects covered by the short-form health survey and the EuroQol 5-dimension scale [27].
Similarly in AMAGINE-2 and -3, both dosages of brodalumab were associated with significantly (p < 0.001) higher rates of PASI100, sPGA score of 0 and PSI response than placebo [25]. In these trials, the percentage of patients with PASI90 at week 12 was significantly (p < 0.001) higher with both dosages of brodalumab than placebo. Compared with ustekinumab, brodalumab 210 mg q2w was associated with a significantly (p < 0.001) greater proportion of patients with PASI90 at week 12 in both AMAGINE-2 and -3. With brodalumab 140 mg q2w, the rates of PASI100 and PASI75 at week 12 were significantly higher than with ustekinumab in AMAGINE-3 (p = 0.007), but not in AMAGINE-2 [25].
Following the induction phase in AMAGINE-1, brodalumab recipients with sPGA success at week 12 were re-randomized to brodalumab (at the same dosage) or placebo for a further 40 weeks’ treatment [24]. At week 52, the rate of sPGA success was significantly (p < 0.001) higher with brodalumab at both dosages than placebo in the re-randomized groups, and the rates of PASI90 and PASI100 were maintained for up to 52 weeks among week-12 responders who continued brodalumab treatment [24]. In AMAGINE-2 and -3, where brodalumab recipients were re-randomized to one of four different brodalumab maintenance regimens at week 12, the percentage of patients with sPGA success at week 52 was significantly (p < 0.001) higher among those who were re-randomized to brodalumab 140 or 210 mg q2w than those who were re-randomized to brodalumab every 4 or 8 weeks [25].
Key clinical trials of subcutaneous brodalumab
Drug(s) | Indication | Phase | Status | Location(s) | Identifier | Company |
|---|---|---|---|---|---|---|
Brodalumab, placebo | Moderate to severe plaque psoriasis | II | Completed | Japan | NCT01748539 (4827-002 study) | Kyowa Hakko Kirin |
Brodalumab | Moderate to severe plaque psoriasis | III | Completed | Japan | NCT01782924 (4827-003 study) | Kyowa Hakko Kirin |
Brodalumab | Pustular psoriasis or psoriatic erythroderma | III | Completed | Japan | NCT01782937 (4827-004 study) | Kyowa Hakko Kirin |
Brodalumab | Pustular psoriasis, psoriatic erythroderma or plaque psoriasis | III | Ongoing | Japan | NCT02052609 (4827-005 study) | Kyowa Hakko Kirin |
Brodalumab, placebo | Moderate to severe plaque psoriasis | III | Terminated | Multinational | NCT01708590 (AMAGINE-1) | Amgen |
Brodalumab, ustekinumab, placebo | Moderate to severe plaque psoriasis | III | Terminated | Multinational | NCT01708603 (AMAGINE-2) | Amgen |
Brodalumab, ustekinumab, placebo | Moderate to severe plaque psoriasis | III | Terminated | Multinational | NCT01708629 (AMAGINE-3) | Amgen |
Brodalumab, placebo | Moderate to severe plaque psoriasis | II | Completed | Multinational | NCT00975637 | Amgen |
Brodalumab | Moderate to severe plaque psoriasis | II | Terminated | Multinational | NCT01101100 | Amgen |
Brodalumab, placebo | Psoriatic arthritis | III | Completed | Multinational | NCT02024646 (AMVISION-2) | Amgen |
Brodalumab, placebo | Psoriatic arthritis | III | Terminated | Multinational | NCT02029495 (AMVISION-1) | Amgen |
Brodalumab, placebo | Psoriatic arthritis | II | Terminated | USA, Canada | NCT01516957 | Amgen |
Brodalumab, placebo | Asthma | II | Completed | Multinational | NCT01199289 | Amgen |
Brodalumab, placebo | Asthma | II | Terminated | Multinational | NCT01902290 | Amgen |
Brodalumab | Rheumatoid arthritis | II | Completed | Multinational | NCT00950989 | Amgen |
Brodalumab | Rheumatoid arthritis | II | Terminated | Multinational | NCT01059448 | Amgen |
AMAGINE-1 (n = 661) [24], -2 (n = 1831) and -3 (n = 1881) [25] were 52-week, phase III trials in adults (aged 18–75 years) with PASI scores of ≥12, sPGA scores of ≥3 and ≥10 % BSA PsO involvement. Patients were assigned in a 1:1:1 ratio to receive subcutaneous brodalumab 140 or 210 mg q2w or placebo in AMAGINE-1 [24] and a 2:2:1:1 ratio to receive subcutaneous brodalumab 140 or 210 mg q2w, placebo or ustekinumab (45 or 90 mg in patients weighing ≤100 or >100 kg) every 12 weeks in AMAGINE-2 and -3 [25]. Patients assigned to brodalumab received doses on day 1 and weeks 1, 2, 4, 6, 8 and 10 [24, 25], and those assigned to ustekinumab received doses on day 1, week 4 and every 12 weeks thereafter [25]. All trials included a 12-week induction phase, followed by a withdrawal/retreatment [24] or maintenance [25] phase of 40 weeks. At week 12 in AMAGINE-1, brodalumab recipients with sPGA success were re-randomized in a 1:1 ratio to receive brodalumab (at the same dosage) or placebo, while those with sPGA scores of ≥2 were assigned to receive brodalumab 210 mg q2w [24]. At week 12 in AMAGINE-2 and -3, brodalumab recipients were re-randomized in a 2:2:2:1 ratio to receive brodalumab 210 mg q2w or 140 mg every 2, 4 or 8 weeks [25]. Placebo recipients were switched to brodalumab 210 mg q2w at week 12 in all three trials [24, 25], while ustekinumab recipients continued to receive ustekinumab until week 52 in AMAGINE-2 and -3 [25].
Subcutaneous brodalumab 70, 140 and 210 q2w and 280 mg q4w was more effective than placebo in patients with moderate to severe plaque PsO in a randomized, multinational, double-blind phase II trial (NCT00975637) [28]. At week 12, there were significantly (p < 0.001) greater mean improvements from baseline in PASI scores (primary endpoint) with subcutaneous brodalumab 70, 140 and 210 mg q2w and 280 mg q4w (45, 86, 86 and 76 %, respectively) than with placebo (16 %). Patients receiving brodalumab 140 or 210 mg q2w or 280 mg q4w had significantly (p < 0.001) greater mean improvements from baseline in PASI scores than those receiving brodalumab 70 mg q2w. Compared with placebo, all dosages of brodalumab were associated with significantly (p < 0.05) higher rates of PASI50, 75, 90 or 100 and significantly (p < 0.01) greater mean percentage improvements from baseline in BSA affected by PsO. This trial enrolled 198 adults (aged 18–70 years) with stable plaque PsO for ≥6 months, PASI scores of ≥12 and ≥10 % BSA psoriatic involvement [28].
In an open-label extension of this phase II trial (NCT01101100), brodalumab 140 or 210 mg q2w was associated with sPGA scores of 0 in 51 % of patients and sPGA scores of 0 or 1 in 72 % of patients after 144 weeks’ treatment [29]. In the extension trial (n = 181), patients from all treatment groups in the parent phase II study received brodalumab 210 mg q2w, with a protocol amendment after ≈1 year to reduce the dosage to 140 mg q2w in patients weighing ≤100 kg (dosage kept at 210 mg q2w in those weighing >100 kg) and a further amendment to enable patients with inadequate response to brodalumab 140 mg q2w to increase their dosage to 210 mg q2w [29].
2.3.3 Patients with Psoriatic Arthritis
Subcutaneous brodalumab 140 or 280 mg q2w was more effective than placebo in patients with PsA in a randomized, multicentre, double-blind, phase II trial (NCT01516957) [30]. At week 12, the percentage of patients with ACR20 response (primary endpoint) was significantly (p ≤ 0.03) greater with brodalumab 140 and 280 mg q2w than with placebo (37 and 39 vs. 18 %), and the rate of ACR50 response was 14 % in both brodalumab dosage groups versus 4 % in the placebo group (p = 0.05 for both comparisons). At week 12, both dosages of brodalumab were associated with significant (p < 0.01) improvements compared with placebo in Clinical Disease Activity Index (CDAI) scores, Disease Activity Score for 28-joint counts (DAS28) and individual ACR components (including tender-joint counts, swollen-joint counts and patient’s and physicians’ global assessment of disease activity). However, there were no significant differences at week 12 between either dosage of brodalumab and placebo with regard to improvements in dactylitis or enthesitis, or changes in the patient’s global assessment of joint pain or the Health Assessment Questionnaire-Disability Index response [30]. In an analysis of patients with a baseline BSA involvement of ≥3 %, brodalumab at both dosages was associated with significantly (p < 0.0001) greater mean improvements in PSI total scores (−7.8 and −11.2 vs. −1.5) and rates of PSI response (75 and 82 vs. 17 %) at week 12 than placebo, as well as significantly (p < 0.01) more patients with PSI total scores of 0 at week 12 than placebo (25 and 36 vs. 3 %) [31].
In the open-label extension phase of this trial, during which all patients received brodalumab 280 mg q2w, the rate of ACR20 response at week 24 was 51 and 64 % among patients who initially received brodalumab 140 and 280 mg q2w, and 44 % among those who switched from placebo to brodalumab 280 mg q2w [30]. The rates of ACR50 and ACR70 response also improved during the extension phase, with additional improvements over 52 weeks’ treatment among patients who initially received brodalumab 140 mg q2w [30]. Prior treatment with a biological agent did not appear to affect the efficacy of brodalumab over 52 week’s treatment in these patients [32]. Over 108 weeks’ treatment, brodalumab was associated with sustained clinically meaningful benefits with regard to ACR20 and ACR50 response, as well as individual ACR components and PSI scores [33].
In the phase II trial (n = 168), adults (aged 18–75 years) with active PsA (based on the Classification Criteria for PsA) received subcutaneous brodalumab 140 or 280 mg or placebo at day 1 and weeks 1, 2, 4, 6, 8 and 10 [30]. At week 12, all patients who had not discontinued treatment were eligible to receive open-label brodalumab 280 mg q2w for up to 5 years in the extension phase [30]. In November 2013, the protocol was amended to reduce the brodalumab dosage to 210 mg q2w for all patients [33].
Randomized, double-blind, placebo-controlled phase III trials investigating the use of subcutaneous brodalumab 140 and 210 mg in patients with PsA have been completed (NCT02024646; AMVISION-2; n = 484) or terminated (NCT02029495; AMVISION-1; n = 478); data from these trials are not available.
2.4 Adverse Events
Subcutaneous brodalumab is generally well tolerated in adults with plaque PsO, generalized pustular PsO, PsE and PsA [20–25, 30].
In Japanese patients with moderate to severe plaque PsO, the most common adverse events (AEs) over 12 weeks’ treatment with brodalumab in the 4827-002 study were nasopharyngitis (12.4 vs. 7.9 % with placebo), diarrhoea (5.3 vs. 0 %) upper respiratory tract inflammation (3.5 vs. 0 %) and folliculitis (3.5 vs. 0 %) [20]. Similarly, the most common AEs over 52 weeks’ treatment with brodalumab in the 4827-003 extension study were nasopharyngitis (35.2 %), upper respiratory tract inflammation (10.3 %) and contact dermatitis (9.7 %) [21]. Serious AEs occurred in four patients over 12 weeks’ treatment [two (5.1 %) with brodalumab 70 mg q2w (myocardial infarction in one and worsening of PsO in the other), one (2.7 %) with brodalumab 210 mg q2w (perforated appendicitis) and one (2.6 %) with placebo (tibial fracture)] [20] and eight patients over 52 weeks [four (5.5 %) with brodalumab 140 mg q2w (arthropod sting allergy, cellulitis, osteoarthritis and varicose vein in one each) and four (5.6 %) with brodalumab 210 mg q2w (myocardial ischaemia, cellulitis, infection and contact dermatitis in one each) [21]. None of the serious AEs were considered treatment-related, and there were no deaths. Over 52 weeks, one patient (1.4 %) in the brodalumab 140 mg q2w group and seven (9.6 %) in the brodalumab 210 mg q2w group developed grade 1 or 2 candidiasis, and grade 1 neutropenia was reported in one (1.4 %) patient in the brodalumab 210 mg q2w group [21]. There were no cases of suicidal ideation or behaviour in the 4827-002 or -003 studies [20, 21].
In Japanese patients with generalized pustular PsO or PsE in the 4827-004 study [22] and in patients with plaque PsO, generalized pustular PsO or PsE in the ongoing 4827-005 study [23], nasopharyngitis was the most common AE with brodalumab over 52 and >80 weeks’ treatment, occurring in 33.3 and 43.5 % of patients, respectively [22, 23]. Serious AEs occurred in five patients (16.7 %) in the 4827-004 study, although none of these were considered treatment-related and there were no deaths [22]. In the 4827-005 study, serious AEs were reported after the first brodalumab injection in 22 patients (12.4 %) [23].
In patients with moderate to severe plaque PsO in AMAGINE-1, the most common AEs (≥5 % incidence) during the 12-week induction phase with brodalumab and placebo were nasopharyngitis (9.3 vs. 10 %), upper respiratory tract infection (URTI) [8.2 vs. 6.4 %] and headache (5.2 vs. 3.2 %); the most common AEs [≥10 per 100 patient-years of exposure (PYE)] through to week 52 were nasopharyngitis, URTI, headache and arthralgia [24]. Similarly in AMAGINE-2 and -3, the most common AEs with brodalumab were nasopharyngitis, URTI, headache and arthralgia during the induction phase, with these AEs generally occurring with numerically higher incidence with brodalumab than ustekinumab or placebo (with the exception of URTIs) [25].
In AMAGINE-1, serious AEs occurred in 2.3 and 1.4 % of patients in the brodalumab and placebo groups during the induction phase, and the exposure-adjusted rate for serious AEs with brodalumab was 9.5 per 100 PYE over 52 weeks’ treatment [24]. Through to week 52, the exposure-adjusted rate of serious AEs with brodalumab and ustekinumab was 8.3 and 13.0 per 100 PYE in AMAGINE-2 and 7.9 and 4.0 per 100 PYE in AMAGINE-3 [25].
During the induction phase, neutropenia occurred in one patient in AMAGINE-1 (in the brodalumab 140 mg q2w group) [24], four patients in AMAGINE-2 (one each with brodalumab 140 and 210 mg q2w and two with ustekinumab) and 13 patients in AMAGINE-3 (five and seven with brodalumab 140 and 210 mg q2w and one with ustekinumab) [25]. Where reported [25], neutropenia was not associated with serious infections, and was mostly mild, transient and reversible. Through to week 52, the exposure-adjusted rate of neutropenia with brodalumab was 0.4, 0.2 and 1.5 per 100 PYE in AMAGINE-1 [24], -2 and -3, respectively (vs. 0.8 and 0.8 per 100 PYE with ustekinumab) [25].
Suspected Candida spp. infections were reported during the induction phase in nine patients in AMAGINE-1 (three with placebo, one with brodalumab 140 mg q2w and five with brodalumab 210 mg q2w) [24]. Over 12 weeks’ treatment in AMAGINE-2 and -3, Candida spp. infections occurred in eight and three patients receiving brodalumab 140 mg q2w and ten and eight patients receiving brodalumab 210 mg q2w (vs. two and one receiving placebo and two and one receiving ustekinumab) [25]. The exposure-adjusted event rate for Candida spp. infections with brodalumab through to week 52 was 3.5, 5.2 and 5.7 per 100 PYE in AMAGINE-1 [24], -2 and -3, respectively (vs. 4.1 and 1.6 with ustekinumab) [25].
During the induction phase, depression was reported in three patients in AMAGINE-1 (one in each treatment group) [24], nine patients in AMAGINE-2 (four, two, two and one with brodalumab 140 and 210 mg q2w, ustekinumab and placebo, respectively) and nine patients in AMAGINE-3 (four, two, one and two with brodalumab 140 and 210 mg q2w, ustekinumab and placebo, respectively) [25]. Through to week 52, the exposure-adjusted rate of depression with brodalumab was 1.2, 1.7 and 1.8 per 100 PYE in AMAGINE-1 [24], -2 and -3, respectively (vs. 3.3 and 0.8 per 100 PYE with ustekinumab) [25].
In AMAGINE-1, two patients died from suicide while receiving brodalumab 210 mg q2w, one during the withdrawal phase and the other during an open-label extension; neither of these suicide events were considered to be related to brodalumab by the investigator [24]. In AMAGINE-2, one brodalumab 210 mg q2w recipient attempted suicide during the induction phase, and two patients died from suicide after exposure to brodalumab 210 mg q2w post-induction in the long-term extension phase [25]. Where reported [25], the exposure-adjusted event rates for suicide ideation and suicide attempt with brodalumab through to week 52 were 0.1 and 0.2 per 100 PYE in AMAGINE-2 (vs. 0.0 and 0.4 per 100 PYE with ustekinumab), and 0.1 and 0.0 per 100 PYE in AMAGINE-3 (vs. 0.4 and 0.0 per 100 PYE with ustekinumab).
Major adverse cardiovascular events (MACEs) occurred in five patients (1.0 per 100 PYE) during the withdrawal phase of AMAGINE-1, including two patients who had been re-randomized to placebo and re-treated with brodalumab 210 mg q2w and three who had received constant brodalumab 210 mg q2w; none of these MACEs were considered related to brodalumab [24]. In AMAGINE-2 and -3, adjudicated MACEs were reported in one and two patients during the induction phase (all receiving brodalumab 140 mg q2w), and had an exposure-adjusted rate of 0.4 and 0.7 per 100 PYE through to week 52 (vs. 0.8 and 0.0 per 100 PYE with ustekinumab) [25].
In patients with PsA, the most common AEs over 12 weeks’ treatment with brodalumab (≥5 % incidence) were URTI (12 vs. 7 % with placebo), fatigue (7 vs. 4 %), diarrhoea (6 vs. 4 %) and headache (6 vs. 7 %) [30]. Over 52 weeks, the most common AEs with brodalumab were nasopharyngitis (14 %), URTI (12 %), psoriatic arthropathy (10 %), arthralgia (9 %), bronchitis (7 %), nausea (7 %), oropharyngeal pain (7 %), sinusitis (7 %), cough (6 %), diarrhoea (6 %), urinary tract infection (6 %), influenza (5 %), muscle spasms (5 %), peripheral oedema (5 %), depression (5 %), dizziness (5 %), fatigue (5 %) and rash (5 %). Serious AEs occurred in four patients during the first 12 weeks’ treatment [three (3 %) with brodalumab and one (2 %) with placebo] and ten patients (6 %) through to week 52. There were no reported AEs of neutropenia over 52 weeks’ treatment; however, laboratory tests revealed five cases of neutropenia (all grade 1) during the first 12 weeks’ treatment [four (4 %) with brodalumab and one (2 %) with placebo] and eight cases of neutropenia (six grade 1 and two grade 2) through to week 52. There were no deaths in this trial [30].
With regard to immunogenicity, anti-brodalumab antibodies were not detected during the 4827-002 study [20], but were detected in two brodalumab recipients in the 4827-003 study [21] and one brodalumab recipient in the 4827-004 study [22]. Neutralizing antibodies were not detected in the 4827-003 or -004 studies [21, 22]. Where reported in the AMAGINE trials [25], anti-brodalumab antibodies emerged during 52 weeks’ treatment with brodalumab in 28 patients (1.8 %) in AMAGINE-2 and 37 patients (2.3 %) in AMAGINE-3. None of these patients developed neutralizing antibodies and the emergence of anti-brodalumab antibodies was not associated with any AEs or loss of efficacy [25]. In patients with PsA, non-neutralizing anti-brodalumab antibodies were detected in two patients during the extension phase (at weeks 16 and 24) [30].
2.5 Ongoing Clinical Trials
The open-label, phase III 4827-005 extension study (NCT02052609) in Japanese patients with plaque PsO (PsV or PsA), generalized pustular PsO or PsE is evaluating the safety and efficacy of long-term exposure to brodalumab in patients who completed the 4827-002, 4827-003 or 4827-004 studies. This trial is expected to be completed in December 2016.
3 Current Status
Brodalumab received its first global approval on 4 July 2016 for the treatment of PsV, PsA, pustular PsO and PsE in Japan [3].
References
Campa M, Mansouri B, Warren R, et al. A review of biologic therapies targeting IL-23 and IL-17 for use in moderate-to-severe plaque psoriasis. Dermatol Ther (Heidelb). 2016;6(1):1–12.
Kivelevitch DN, Menter A. Use of brodalumab for the treatment of psoriasis and psoriatic arthritis. Immunotherapy. 2015;7(4):323–33.
Kyowa Hakko Kirin. Lumicef® approved in Japan [media release]. 4 July 2016. http://www.kyowa-kirin.com.
Kyowa Hakko Kirin Co. Ltd. Lumicef® subcutaneous injection 210 mg syringe: brodalumab (gene recombination) formulation: Japanese prescribing information. 2016. http://www.kksmile.com. Accessed 5 Aug 2016.
Valeant Pharmaceuticals International. Valeant announces FDA acceptance of BLA submission for brodalumab in moderate-to-severe plaque psoriasis [media release]. 25 Jan 2016. http://www.valeant.com.
Farahnik B, Beroukhim K, Abrouk M, et al. Brodalumab for the treatment of psoriasis: a review of phase III trials. Dermatol Ther (Heidelb). 2016;6(2):111–24.
FirstWord Pharma. FDA advisory panel backs Valeant’s psoriasis drug brodalumab, with suicide risk-mitigation plan [media release]. 19 July 2016. http://www.firstwordpharma.com.
Kyowa Hakko Kirin Co Ltd. Kyowa Hakko Kirin announces results of the phase 3 studies of KHK4827 in subjects with psoriasis in Japan [media release]. 23 July 2015. http://www.kyowa-kirin.com.
Amgen, AstraZeneca. Amgen and AstraZeneca announce collaboration to jointly develop and commercialize clinical-stage inflammation portfolio [media release]. 2 Apr 2012. http://www.astrazeneca.com.
Amgen. Amgen to terminate participation in co-development and commercialization of brodalumab [media release]. 22 May 2015. http://www.amgen.com.
AstraZeneca. AstraZeneca PLC H1 2015 results [media release]. 30 July 2015. http://www.astrazeneca.com.
Valeant Pharmaceuticals International. Valeant and AstraZeneca to partner on brodalumab [media release]. 1 Sep 2015. http://www.valeant.com.
AstraZeneca. AstraZeneca enters licensing agreements with LEO Pharma in skin diseases [media release]. 1 July 2016. http://www.astrazeneca.com.
Valeant Pharmaceuticals International. Valeant Pharmaceuticals announces new licensing arrangement for brodalumab in Europe [media release]. 1 July 2016. http://www.valeant.com.
Amgen. Form 10-K [media release]. 19 Feb 2016. http://investors.amgen.com.
Papp KA, Reid C, Foley P, et al. Anti-IL-17 receptor antibody AMG 827 leads to rapid clinical response in subjects with moderate to severe psoriasis: results from a phase I, randomized, placebo-controlled trial. J Invest Dermatol. 2012;132(10):2466–9.
Salinger DH, Endres CJ, Martin DA, et al. A semi-mechanistic model to characterize the pharmacokinetics and pharmacodynamics of brodalumab in healthy volunteers and subjects with psoriasis in a first-in-human single ascending dose study. Clin Pharmacol Drug Dev. 2014;3(4):276–83.
Russell CB, Rand H, Bigler J, et al. Gene expression profiles normalized in psoriatic skin by treatment with brodalumab, a human anti-IL-17 receptor monoclonal antibody. J Immunol. 2014;192(8):3828–36.
Endres CJ, Salinger DH, Kock K, et al. Population pharmacokinetics of brodalumab in healthy adults and adults with psoriasis from single and multiple dose studies. J Clin Pharmacol. 2014;54(11):1230–8.
Nakagawa H, Niiro H, Ootaki K. Brodalumab, a human anti-interleukin-17-receptor antibody in the treatment of Japanese patients with moderate-to-severe plaque psoriasis: efficacy and safety results from a phase II randomized controlled study. J Dermatol Sci. 2016;81(1):44–52.
Umezawa Y, Nakagawa H, Niiro H, et al. Long-term clinical safety and efficacy of brodalumab in the treatment of Japanese patients with moderate-to-severe plaque psoriasis. J Eur Acad Dermatol Venereol. 2016. doi:10.1111/jdv.13785.
Yamasaki K, Nakagawa H. Clinical efficacy and safety of brodalumab (KHK4827), antiinterleukin-17-receptor a monoclonal antibody, in Japanese patients with pustular psoriasis (generalized) and psoriatic erythroderma: a phase 3, open-label, long-term study (4827-004 study) [abstract no. 365 plus poster]. J Am Acad Dermatol. 2015;72(5 Suppl 1):AB228.
Nakagawa H, Ootaki K. Long-term efficacy and safety of brodalumab in Japanese patients with plaque psoriasis (psoriasis vulgaris, psoriatic arthritis), pustular psoriasis (generalized) and psoriatic erythroderma: an open-label extension study [abstract no. P142]. J Eur Acad Dermatol Venereol. 2016;30(S6):72.
Papp KA, Reich K, Paul C, et al. A prospective phase III, randomized, double-blind, placebo-controlled study of brodalumab in patients with moderate-to-severe plaque psoriasis. Br J Dermatol. 2016. doi:10.1111/bjd.14493.
Lebwohl M, Strober B, Menter A, et al. Phase 3 studies comparing brodalumab with ustekinumab in psoriasis. N Engl J Med. 2015;373(14):1318–28.
Strober B, Gordon K, Augustin M, et al. Improvements in patient-reported outcomes (PROs) among moderate to severe plaque psoriasis patients treated with brodalumab: results from AMAGINE-1 [abstract no. 3169]. J Am Acad Dermatol. 2016;74(5 Suppl 1):AB256.
Paul C, Barker J, Klekotka P, et al. Improvements in health-related quality of life measured by SF-36 and EQ-5D in patients with moderate to severe plaque psoriasis treated with brodalumab [abstract no. 3172]. J Am Acad Dermatol. 2016;74(5 Suppl 1):AB256.
Papp KA, Leonardi C, Menter A, et al. Brodalumab, an anti-interleukin-17-receptor antibody for psoriasis. N Engl J Med. 2012;366(13):1181–9.
Papp K, Leonardi C, Menter A, et al. Maintenance of clinical response with long-term brodalumab (AMG 827) therapy for psoriasis: week 144 results from an open-label extension study [abstract no. 956]. J Am Acad Dermatol. 2015;72(5 Suppl 1):AB240.
Mease PJ, Genovese MC, Greenwald MW, et al. Brodalumab, an anti-IL17RA monoclonal antibody, in psoriatic arthritis. N Engl J Med. 2014;370(24):2295–306.
Mease PJ, Genovese MC, Mutebi A, et al. Improvement in psoriasis signs and symptoms assessed by the psoriasis symptom inventory with brodalumab treatment in patients with psoriatic arthritis. J Rheumatol. 2016;43(2):343–9.
Genovese MC, Mease PJ, Greenwald MW, et al. Efficacy and safety of brodalumab over one year in patients with psoriatic arthritis with and without prior exposure to a biologic [abstract no. AB0752]. Ann Rheum Dis. 2014;73(Suppl 2):1052–3.
Mease P, Genovese MC, Greenwald MW, et al. Two-year clinical response to brodalumab, an anti-IL-17 receptor antibody, in patients with psoriatic arthritis [abstract no. OP0175]. Ann Rheum Dis. 2015;74(Supplement 2):136–7.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Disclosure
The preparation of this review was not supported by any external funding. During the peer review process the manufacturer of the agent under review was offered an opportunity to comment on the article. Changes resulting from any comments received were made by the author on the basis of scientific completeness and accuracy. Sarah Greig is a salaried employee of Adis, Springer SBM.
Additional information
This profile has been extracted and modified from the AdisInsight database. AdisInsight tracks drug development worldwide through the entire development process, from discovery, through pre-clinical and clinical studies to market launch and beyond.
An erratum to this article can be found at http://dx.doi.org/10.1007/s40265-016-0647-3.
Rights and permissions
About this article

Cite this article
Greig, S.L. Brodalumab: First Global Approval. Drugs 76, 1403–1412 (2016). https://doi.org/10.1007/s40265-016-0634-8
Published:
Issue Date:
DOI: https://doi.org/10.1007/s40265-016-0634-8