
Abstract
Idiopathic pulmonary fibrosis (IPF) is an aging-associated, recalcitrant lung disease with historically limited therapeutic options. The recent approval of two drugs, pirfenidone and nintedanib, by the US Food and Drug Administration in 2014 has heralded a new era in its management. Both drugs have demonstrated efficacy in phase III clinical trials by retarding the rate of progression of IPF; neither drug appears to be able to completely arrest disease progression. Advances in the understanding of IPF pathobiology have led to an unprecedented expansion in the number of potential therapeutic targets. Drugs targeting several of these are under investigation in various stages of clinical development. Here, we provide a brief overview of the drugs that are currently approved and others in phase II clinical trials. Future therapeutic opportunities that target novel pathways, including some that are associated with the biology of aging, are examined. A multi-targeted approach, potentially with combination therapies, and identification of individual patients (or subsets of patients) who may respond more favourably to specific agents are likely to be more effective.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Approval of pirfenidone and nintedanib has provided much-needed options to treat patients with idiopathic pulmonary fibrosis (IPF). |
Several new therapeutic agents are currently being studied in phase II trials. |
Advances in the understanding of IPF pathobiology have led to an unprecedented expansion in the number of novel therapeutic targets being investigated in pre-clinical studies. |
1 Introduction
Idiopathic pulmonary fibrosis (IPF) is a chronic, progressive lung disease with high mortality and morbidity [1–3]. The worldwide prevalence of IPF continues to rise [4, 5]. Early disappointments with several antifibrotic agents [6–9] have led to clinical guidelines strongly recommending against use of most drugs previously studied [10]. Significant advances have been made in our understanding of the pathobiology of lung fibrosis [11–13]. These insights have led to an unprecedented expansion in the number of therapeutic targets that have undergone testing in clinical trials. Two drugs—pirfenidone and nintedanib—were approved by the US Food and Drug Administration (FDA) in 2014. Here, we review data supporting the use of these drugs for the treatment of IPF and look ahead to emerging drugs and novel therapeutic targets for this recalcitrant disease.
2 Aging as a Paradigm in IPF Pathogenesis
Our understanding of the pathogenesis of IPF has evolved over the past three decades, and the role of aging in this disease process is gaining greater attention [14, 15]. The diagnosis of IPF is typically made beyond the fifth decade of life, and there is an increase in both the incidence and the prevalence of the disease with advancing age [4, 5, 16–18]. The biological hallmarks of aging [19]—namely, genomic instability, telomere attrition, epigenetic alterations, loss of proteostasis (including impaired autophagy), deregulated nutrient sensing, mitochondrial dysfunction, cellular senescence, stem cell exhaustion and altered intercellular communication—are being linked to key pathobiological processes in fibrosis [15].
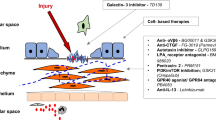
Both clinicians and scientists intuitively approach fibrosis as a pathological process; however, it can be argued that fibrosis serves an adaptive host response function [20]. Accordingly, fibrosis may be viewed as a physiological response conserved through evolution for survival after tissue injury, even at the cost of a loss in organ structure/function. This ‘trade-off’ would be predicted to select fibrotic repair over ‘perfect’ organ regeneration in environments of limited bioenergetic resources. Progressive fibrosis may occur when the normal bidirectional signalling between the epithelium and the mesenchyme (fibroblasts) that coordinates repair becomes aberrant in the context of chronic injury and aging. This aberrant signalling may result from several factors, such as elevated oxidative stress, impaired fibrinolysis, and alterations in cytokines, chemokines, growth factors and eicosanoids [21]. Ultimately, the causes of pathological fibrosis likely involve impaired ability to clear antigens, autoimmunity, impaired regeneration and aberrant activation of developmental or wound-healing genes [20]. In the context of aging, senescence of both the epithelium and the mesenchyme may be predicted to give rise to cell phenotypes and fates that are characteristic of non-resolving fibrosis (Fig. 1). Consequently, the putative therapeutic targets are myriad and provide an opportunity for a multipronged approach to intervene along a spectrum, from halting the progression of fibrosis to reversing remodelling in order to achieve normal organ structure and function.

Proposed mechanisms for the predisposition to fibrosis with aging. Aging may result in ‘immunosenescence’, which results in impaired antigen clearance and autoimmunity; additionally, senescence of epithelial cells and fibroblasts results in impaired regeneration and aberrant recapitulation of developmental genes. These processes may perpetuate epithelial injury/apoptosis and fibroblast activation, which result in failed re-epithelialization, fibroblast persistence and progressive fibrosis
3 Review of Current Drugs
3.1 Pirfenidone
About 3 years after its approval in Europe in 2011, pirfenidone was approved by the US FDA for the treatment of IPF. Two decades have passed since the first report in the mid-1990s of its potential efficacy in amelioration of experimental lung fibrosis in hamsters [22]. Since then, it has been studied extensively in several animal models and was found to be effective in preventing/treating fibrosis involving the lungs, heart, liver and kidneys [23]. Recent studies have employed local delivery of pirfenidone-containing nanoparticles to treat lung fibrosis in mice [24]. Interestingly, the precise mechanisms of action of pirfenidone that mediate antifibrotic effects in vivo are ill defined, although they are purported to involve multiple pathways, including down-regulation of inflammatory cytokines [such as tumour necrosis factor-α (TNF-α)], profibrotic cytokines [such as transforming growth factor-β (TGF-β)] and oxidative stress [25].
In the large, multicentre, phase III ASCEND clinical trial, which ultimately led to the approval of pirfenidone in the USA, treatment with the drug (compared with placebo) significantly reduced the decline in lung function [as measured by an absolute decrease in forced vital capacity (FVC)] and exercise tolerance, and improved progression-free survival in patients with IPF [26]. In this study of 555 patients, 278 received pirfenidone and 277 received placebo. While this trial failed to show a significant mortality benefit, pooling of the data with those from the earlier, multicentre, phase III CAPACITY trials [27] revealed a statistically significant reduction in all-cause mortality (hazard ratio 0.52), as well as in IPF-related mortality (hazard ratio 0.32), in patients receiving pirfenidone.
The target dosing of pirfenidone is 2403 mg/day in three divided doses. Adverse effects are common with pirfenidone therapy. In the ASCEND trial, 36 % of patients receiving pirfenidone had nausea (versus 13.4 % of those receiving placebo) and 28 % had a rash (versus 8.7 %, respectively). Other known adverse effects include upper gastrointestinal (GI) symptoms, such as vomiting, dyspepsia and oesophageal reflux, in addition to elevated hepatic enzymes, asthenia and weight loss [26, 27].
3.2 Nintedanib
Nintedanib is a multiple tyrosine kinase inhibitor (TKI) known to inhibit the receptor tyrosine kinases of platelet-derived growth factor (PDGF), fibroblast growth factor (FGF) and vascular endothelial growth factor (VEGF) [28]. There have been relatively fewer studies of the efficacy of nintedanib in pre-clinical animal models [29]. Originally used in trials to treat various cancers [30], nintedanib was studied in the phase II TOMORROW trial in IPF [31]. Encouraging results from this trial formed the basis for the larger, phase III INPULSIS trials, which led to its approval for use in IPF in Europe and USA [32]. Recent studies have attempted to define the mechanisms of the antifibrotic action of nintedanib via its inhibitory effect on receptor tyrosine kinases, leading to inhibition of fibroblast proliferation and migration, myofibroblast differentiation, extracellular matrix (ECM) synthesis and inflammation [33, 34]. We have recently reported that nintedanib mediates other actions on fibroblasts, including induction of autophagy and direct inhibition of TGF-β receptor signalling [35].
In the two large, multicentre INPULSIS trials, a total of 1061 patients with mild–moderate IPF [FVC 50 % or more of predicted; diffusion capacity of the lung for carbon monoxide (DLCO) 30–79 % of predicted] were studied; 638 received nintedanib and 423 received placebo [32]. In both studies, nintedanib significantly decreased the rate of decline in FVC. Nintedanib also decreased the incidence of acute exacerbations in INPULSIS 2 but not in INPULSIS 1.
The dosing of nintedanib is at 300 mg/day in two divided doses. Adverse effects are common with nintedanib therapy. In the INPULSIS trials, >60 % patients treated with nintedanib reported diarrhoea (versus ≈18 % of those treated with placebo). Other known adverse effects include elevated hepatic enzymes and weight loss [32]. Overall, higher rates of drug discontinuation due to adverse effects were noted in the drug-treated groups than in the placebo-treated groups (21 % versus 10.8 % in INPULSIS 1, 17.6 versus 15.1 % in INPULSIS 2).
3.3 Impact of the Approved Drugs on the Treatment Paradigm in IPF
The approval of pirfenidone and nintedanib has provided much-needed options to treat patients with IPF. However, these come with a set of new questions and uncertainties. First, the criteria for selecting one drug over the other for a particular patient are unclear [36]. An informed decision-making process is necessary on the part of both physician and patient. In our experience, this decision is currently being driven primarily by the side-effect profile of each drug. Adverse effects leading to potential drug discontinuation have been considered in the ‘conditional recommendation with moderate confidence in estimates of effect’ for both drugs in the current guidelines for management of IPF [10]. Second, it is unknown if a combination of pirfenidone and nintedanib would confer added benefit over either drug alone. Third, among the other unknowns are the utility of either drug in patients with advanced IPF and/or advanced age with varied comorbidities. Fourth, there are concerns that the drugs may not be cost effective, at least in the prevalent health-care delivery systems in some countries [37]. Lastly, these drugs will alter the design of future studies of newer drugs, with the requirement for a new standard-of-care (control) arm in randomized clinical trials.
4 Drug Targets/Therapeutics in Current Clinical Trials
Several newer agents are currently in phase II clinical trials for IPF, and these are summarized in Table 1. Here, we discuss the rationale and important pre-clinical data that support the testing of these agents.
4.1 IL-13
Interleukin-13 (IL-13) is known to be increased in patients with IPF [38] and in animal models of aging-associated progressive fibrosis [39]. IL-13 signalling, via multiple mechanisms—including interplay with TGF-β and macrophage chemokine (C–C motif) ligand 2 (CCL2)—drives fibrosis in vitro and in vivo [40–43]. Blocking IL-13 ameliorates experimental fibrosis in animal models [44]. The IL-13 signalling axis has been a target of active investigation for pulmonary fibrosis with several anti-IL-13 monoclonal antibodies. A phase II trial of QAX576 (ClinicalTrials.gov study ID NCT01266135) was terminated, with the cause yet unpublished. Phase II, randomized, double-blind, placebo-controlled trials of two other monoclonal antibodies against IL-13, tralokinumab (ClinicalTrials.gov study ID NCT01629667) and lebrikizumab (ClinicalTrials.gov study ID NCT01872689), are ongoing. Of note, these two monoclonal antibodies are also being evaluated in phase III trials for treatment of severe asthma. Interestingly, blocking the IL-13 receptor α2 (IL-13Rα2) appears to mediate pleiotropic effects in fibrosis. While studies support a profibrotic role for IL-13Rα2 via TGF-β1 signalling [45], IL-13Rα2 may act as a decoy to IL-13 and thus diminish its profibrotic effects [46].
4.2 CCL2
CCL2, also referred to as monocyte chemotactic protein 1 (MCP1), is purported to contribute to the pathogenesis of pulmonary fibrosis via multiple mechanisms, including effector functions downstream of IL-13 signalling [43, 47–49]. Interestingly, CCL2 has been reported to mediate receptor-independent pleiotropic effects [50]. A phase II clinical trial of the anti-CCL2 monoclonal antibody carlumab (CNTO 888) in 126 IPF patients was completed recently but found no evidence of benefit [51].
4.3 CTGF
Connective tissue growth factor (CTGF) is a matricellular protein, which modulates cellular responses to the ECM. CTGF regulates fibroblast functions and mediates TGF-β actions on fibroblasts [52, 53]. In patients with IPF, CTGF expression is increased in bronchoalveolar lavage (BAL) fluid and in lung tissue, specifically type II alveolar epithelial cells and interstitial fibroblasts [54, 55]. In animal models of lung fibrosis, targeting CTGF diminishes fibrosis [56, 57]. A phase II, randomized, double-blind, placebo-controlled trial (ClinicalTrials.gov study ID NCT01890265) of a CTGF-neutralizing antibody, FG-3019 (FibroGen), is currently recruiting IPF patients, after encouraging interim results from an earlier trial were reported at the 2014 American Thoracic Society (ATS) Conference [58]. As of 2014, out of 53 patients studied, 39 had completed the treatment period of 48 weeks; 27 of these had either no decrease or only a modest decrease (<5 %) in FVC. Of the 19 patients who accepted extended treatment, nine had demonstrated improved or stable FVC through 81 weeks of treatment.
4.4 Lysyl Oxidase-Like 2
Lysyl oxidases (LOXs) are matrix cross-linking enzymes, which catalyse formation of aldehydes from lysine residues in collagen and elastin. Their normal function helps stabilize the ECM to provide tensile strength to tissues [59, 60]. Increased activity of lysyl oxidase-like 2 (LOXL2) has been found to play a role in fibrotic disease; it is highly expressed in fibrotic regions of lungs with IPF, and its inhibition attenuates experimental lung fibrosis [61]. Serum LOXL2 has been noted to increase in patients with progressive IPF, suggesting its potential utility as a biomarker, pending validation studies [62]. A phase II trial (ClinicalTrials.gov study ID NCT01769196) in IPF patients using simtuzumab, a monoclonal antibody against LOXL2, is underway.
4.5 Integrin αvβ6
TGF-β signalling is central to fibrogenesis involving multiple organ systems [21, 63]. Global TGF-β inhibition may adversely affect its homeostatic functions, including immune suppression and tumour suppression [64]. Activation of latent TGF-β occurs locally in areas of fibrogenesis, and αvβ6 integrin is known to be an activator of such latent TGF-β complexes [65, 66]. Importantly, αvβ6 expression is minimal in normal lungs [66]. Lack of this integrin or loss of its function protects against experimental fibrosis in animal models [67, 68]. An ongoing phase II study (ClinicalTrials.gov study ID NCT01371305) of STX-100, a monoclonal antibody against αvβ6, is currently recruiting patients.
4.6 LPA1
Lysophosphatidic acid (LPA) is a phospholipid derivative, which signals through multiple cell-surface G-protein coupled receptors participating in profibrotic wound-repair responses, such as fibroblast activation and resistance to apoptosis, epithelial cell apoptosis and increased vascular permeability [69]. Inhibiting the LPA1 receptor has been shown to prevent fibrosis in various pre-clinical models [70–73]. Importantly, LPA levels are increased in BAL fluid from IPF patients; LPA1 signalling has potent fibroblast chemoattractant activity [71]. An ongoing phase II study (ClinicalTrials.gov study ID NCT01766817) in IPF patients using BMS-986020, an LPA1 receptor antagonist, is currently recruiting patients.
4.7 Autoantibodies
It has long been recognized that aging is associated with immunosenescence, accompanied by an increase in autoantibodies [74, 75]. Recent pilot data indicate that targeting autoantibodies during acute IPF exacerbations might improve outcomes; strategies to reduce autoantibodies include treatment with rituximab, a monoclonal antibody against CD20 [76]. Studies have shown favourable effects on lung function in patients with scleroderma-induced interstitial lung disease with long-term use of rituximab [77], despite concerns that rituximab treatment itself can induce lung fibrosis [78–80]. A phase II study, ART-IPF (Autoantibody Reduction Therapy in Patients with Idiopathic Pulmonary Fibrosis) (ClinicalTrials.gov study ID NCT01969409), is currently recruiting patients.
4.8 Carbon Monoxide
Carbon monoxide is a gaseous molecule with multifunctional actions and typifies contextual duplicity. While it is clearly toxic at high concentrations, therapeutic beneficial effects of exposure to lower concentrations are emerging in various conditions, including inflammation, sepsis and acute lung injury [81–85]. Carbon monoxide suppresses in vitro fibroblast proliferation and bleomycin-induced lung injury in mice [86]. A phase II, multicentre trial (ClinicalTrials.gov study ID NCT01214187), studying inhaled carbon monoxide in IPF, has recently stopped enrolling, and the results are yet to be published.
4.9 Antimicrobials
Recent reports suggest that infections may contribute to IPF pathogenesis and lead to its progression, as well as acute exacerbations; however, precise cause–effect relationships have not been established [87–89]. There is also emerging interest in the role of the microbiome in IPF [87]. Use of antimicrobials to clear infection/colonization, thereby potentially altering the microbiome and the immune response, is being studied in IPF. In a study using the bleomycin-injury model in mice, azithromycin ameliorated lung fibrosis [90]. A randomized study of azithromycin in IPF (ClinicalTrials.gov study ID NCT02173145), with the primary goal of assessing its immunomodulatory functions in suppressing cough, is currently recruiting patients. In a multicentre, randomized, controlled trial of cotrimoxazole in 181 IPF patients, in those who adhered to the regimen until study completion, drug therapy was noted to confer an all-cause mortality benefit (hazard ratio 0.21) and a reduction in the need for an increase in oxygen therapy (odds ratio 0.05), despite no benefit in terms of the primary outcome of retarding the decline in lung function (FVC) and exercise capacity (6-minute walk test) [91]. In the cotrimoxazole group, while a significantly larger number of patients discontinued treatment because of adverse effects (chiefly, nausea and skin rash), respiratory infections were significantly less common in this group. A larger, phase III trial (ClinicalTrials.gov study ID NCT01777737), testing the validity of treatment of IPF with cotrimoxazole, is currently recruiting participants.
5 Emerging Drug Targets/Therapeutic Strategies
5.1 ROCKs
First discovered in 1995 [92], the Rho kinase (ROCK) family members, ROCK1 and ROCK2, are serine/threonine kinases, which regulate multiple cellular functions, including fibroblast apoptosis/survival and mechanotransduction [93, 94]. Many of their downstream targets are associated with regulation of cytoskeletal stability, stress fibre formation, focal adhesion assembly and cell contractility. Biomechanical stress-induced signal transduction by ROCKs may function as a feed-forward mechanism in the microenvironment of an already stiffened ECM with ongoing fibrosis. Recent studies have shown that inhibition of this pathway can ameliorate experimental fibrosis; more importantly, ROCK activity is known to be increased in areas of active fibrosis in human IPF; thus, its inhibition may be an effective therapeutic strategy [95]. A phase II study of an oral selective ROCK2 inhibitor, KD025, to treat IPF is being planned. It is noteworthy that this drug is being evaluated in multiple therapeutic areas, including autoimmune, fibrotic, neurological and metabolic diseases.
5.2 NOX4
Nicotinamide adenine dinucleotide phosphate [reduced form] (NADPH) oxidase 4 (NOX4) is an oxidant-generating enzyme, which mediates myofibroblast activation and fibrogenic responses in multiple organ systems [96–101]. Its biochemical activity was first discovered as a TGF-β-responsive H2O2-generating flavoenzyme in lung fibroblasts [102], several years prior to the identification and cloning of non-phagocytic NOX family enzymes [103]. NOX4 may demonstrate antagonistic pleiotropy in aging, contributing to excessive oxidative stress, thereby increasing the predisposition to fibrosis as a response to lung injury [104, 105]. Newer insights in epigenetics have helped researchers to better understand the mechanisms involved in NOX4 up-regulation in senescence [106]. Targeting NOX4 with intranasal small interfering RNA (siRNA), as well as the small-molecule inhibitor GKT137831, reverses the otherwise persistent fibrosis seen in aged mice [107]. Specific targeting of NOX4 awaits study in human IPF.
5.3 AMPK
Adenosine monophosphate (AMP)—activated protein kinase (AMPK) is a master metabolic sensor and homeostat at the cellular level. AMPK activation is among the few interventions that control the aging process and extend lifespan in animal models [108]. Interestingly, AMPK activation has been reported to mediate antifibrotic effects in experimental models, both in vitro and in vivo, although the mechanisms are unclear [109]. AMPK activation leads to enhanced autophagy and metabolic reprogramming, both of which have potential therapeutic value in the context of aging-associated fibrosis [110, 111]. The antidiabetic drug metformin, used by millions of people worldwide, is a potent activator of AMPK and, being cost effective with a favourable safety profile, is an attractive candidate for repurposing in IPF.
5.4 Other Kinases
The concept of targeting protein kinases with TKIs in IPF has been considered for several years [112–115]. The recent results with nintedanib suggest that other TKIs may be as effective (or even more effective), and this warrants further testing. In addition, while much has been studied and proposed about the role of receptor tyrosine kinases and their therapeutic targeting in pulmonary fibrosis [116, 117], the roles of many non-receptor tyrosine and serine/threonine kinases are unclear. Recent reports suggest that inhibition of the Src family of non-receptor protein kinases can ameliorate experimental fibrosis in vitro and in vivo [118]. Future studies are likely to establish if several kinase inhibitors, which are either already approved or being tested for treatment of various cancers, may be repurposed for IPF.
5.5 RNA Inhibition
MicroRNAs (miRNAs) and siRNAs are tiny, regulatory, non-coding RNAs, which have similar functions but different sources of origin [119]. They decrease levels of target gene transcripts (messenger RNA [mRNA]), predominantly by mRNA destabilization, although they also modestly reduce translation efficiency [120]. Depending on the target(s) of the siRNA or miRNA, their activity can lead to a profibrotic or antifibrotic milieu [121]. For example, miR-29 is known to have antifibrotic effects; its levels are suppressed by TGF-β stimulation in vitro and in the lungs of bleomycin-injured mice [122, 123]. On the other hand, miR-21 is known to promote fibrosis [124]. Strategies to increase levels of antifibrotic miRNAs/siRNAs and decrease levels of profibrotic miRNAs would open up a multitude of potent and highly targeted options to treat IPF. Numerous miRNA-based therapeutics are already in development for various diseases, such as hepatitis C virus (HCV) infection, various cancers, heart failure and fibrosis [125]. Novel technologies for optimal delivery of siRNAs, miRNA mimics and antimiR oligonucleotides are being explored [126].
6 Future Directions
The year 2014 marked a watershed moment in the history of the care of IPF patients in the USA, with FDA approval of the first drugs ever for this disease. However, several questions regarding the use of pirfenidone and nintedanib remain. Can they benefit patients with more advanced disease than were enrolled in the phase III studies? Can they be given in combination to achieve potentially greater efficacy? How long should either be given to patients who continue to progress despite treatment before they are deemed treatment failures? Most importantly, given the inherent heterogeneity of IPF, how can we identify patients most likely to benefit from one or both of these drugs?
While the approval of pirfenidone and nintedanib should be considered an important landmark, much work needs to be done. With renewed interest and commitment from both academia and industry, several more drugs and drug targets are likely to follow. We have reviewed some of the emerging candidate drugs that are currently in phase II trials and several others that are in earlier stages of clinical or pre-clinical development. Deeper insights into the biology of aging in IPF pathogenesis are likely to uncover an exciting new set of drug targets. The repurposing of drugs has the potential to allow candidate drugs to gain quicker access to the clinic. At the same time, drug discovery efforts based on emerging understanding of IPF pathobiology must continue. Coupling of drug discovery with biomarker discovery has the greatest potential to realize the promise of personalized medicine. Discovery of biomarkers that serve as surrogates for clinically relevant end points—ideally mortality—would facilitate clinical trials over shorter periods and potentially with fewer subjects. Ultimately, such advancements will result in bringing drugs with greater efficacy, safety and tolerability to patients with IPF—a disease once thought to be a death sentence.
References
Thannickal VJ, Flaherty KR, Martinez FJ, Lynch JP 3rd. Idiopathic pulmonary fibrosis: emerging concepts on pharmacotherapy. Expert Opin Pharmacother. 2004;5(8):1671–86.
American Thoracic Society. Idiopathic pulmonary fibrosis: diagnosis and treatment. International consensus statement. American Thoracic Society (ATS), and the European Respiratory Society (ERS). Am J Respir Crit Care Med. 2000;161(2 Pt 1):646–64.
American Thoracic Society. European Respiratory Society. American Thoracic Society/European Respiratory Society international multidisciplinary consensus classification of the idiopathic interstitial pneumonias. This joint statement of the American Thoracic Society (ATS), and the European Respiratory Society (ERS) was adopted by the ATS Board of Directors, June 2001 and by the ERS Executive Committee, June 2001. Am J Respir Crit Care Med. 2002;165(2):277–304.
Nalysnyk L, Cid-Ruzafa J, Rotella P, Esser D. Incidence and prevalence of idiopathic pulmonary fibrosis: review of the literature. Eur Respir Rev. 2012;21(126):355–61.
Raghu G, Weycker D, Edelsberg J, Bradford WZ, Oster G. Incidence and prevalence of idiopathic pulmonary fibrosis. Am J Respir Crit Care Med. 2006;174(7):810–6.
Idiopathic Pulmonary Fibrosis Clinical Research Network, Raghu G, Anstrom KJ, King TE Jr, Lasky JA, Martinez FJ. Prednisone, azathioprine, and N-acetylcysteine for pulmonary fibrosis. N Engl J Med. 2012;366(21):1968–77.
King TE Jr, Brown KK, Raghu G, du Bois RM, Lynch DA, Martinez F, et al. BUILD-3: a randomized, controlled trial of bosentan in idiopathic pulmonary fibrosis. Am J Respir Crit Care Med. 2011;184(1):92–9.
King TE Jr, Albera C, Bradford WZ, Costabel U, Hormel P, Lancaster L, et al. Effect of interferon gamma-1b on survival in patients with idiopathic pulmonary fibrosis (INSPIRE): a multicentre, randomised, placebo-controlled trial. Lancet. 2009;374(9685):222–8.
Network Idiopathic Pulmonary Fibrosis Clinical Research, Zisman DA, Schwarz M, Anstrom KJ, Collard HR, Flaherty KR, et al. A controlled trial of sildenafil in advanced idiopathic pulmonary fibrosis. N Engl J Med. 2010;363(7):620–8.
Raghu G, Rochwerg B, Zhang Y, Garcia CA, Azuma A, Behr J, et al. An official ATS/ERS/JRS/ALAT clinical practice guideline: treatment of idiopathic pulmonary fibrosis. An update of the 2011 clinical practice guideline. Am J Respir Crit Care Med. 2015;192(2):e3–19.
Ding Q, Luckhardt T, Hecker L, Zhou Y, Liu G, Antony VB, et al. New insights into the pathogenesis and treatment of idiopathic pulmonary fibrosis. Drugs. 2011;71(8):981–1001.
Ahluwalia N, Shea BS, Tager AM. New therapeutic targets in idiopathic pulmonary fibrosis: aiming to rein in runaway wound-healing responses. Am J Respir Crit Care Med. 2014;190(8):867–78.
Woodcock HV, Maher TM. The treatment of idiopathic pulmonary fibrosis. F1000Prime Rep. 2014;6:16.
Thannickal VJ, Murthy M, Balch WE, Chandel NS, Meiners S, Eickelberg O, et al. Blue Journal Conference. Aging and susceptibility to lung disease. Am J Respir Crit Care Med. 2015;191(3):261–9.
Thannickal VJ. Mechanistic links between aging and lung fibrosis. Biogerontology. 2013;14(6):609–15.
Raghu G, Chen SY, Yeh WS, Maroni B, Li Q, Lee YC, et al. Idiopathic pulmonary fibrosis in US Medicare beneficiaries aged 65 years and older: incidence, prevalence, and survival, 2001–11. Lancet Respir Med. 2014;2(7):566–72.
Fell CD, Martinez FJ, Liu LX, Murray S, Han MK, Kazerooni EA, et al. Clinical predictors of a diagnosis of idiopathic pulmonary fibrosis. Am J Respir Crit Care Med. 2010;181(8):832–7.
Collard HR. The age of idiopathic pulmonary fibrosis. Am J Respir Crit Care Med. 2010;181(8):771–2.
Lopez-Otin C, Blasco MA, Partridge L, Serrano M, Kroemer G. The hallmarks of aging. Cell. 2013;153(6):1194–217.
Thannickal VJ, Zhou Y, Gaggar A, Duncan SR. Fibrosis: ultimate and proximate causes. J Clin Invest. 2014;124(11):4673–7.
Thannickal VJ, Toews GB, White ES, Lynch JP 3rd, Martinez FJ. Mechanisms of pulmonary fibrosis. Annu Rev Med. 2004;55:395–417.
Iyer SN, Wild JS, Schiedt MJ, Hyde DM, Margolin SB, Giri SN. Dietary intake of pirfenidone ameliorates bleomycin-induced lung fibrosis in hamsters. J Lab Clin Med. 1995;125(6):779–85.
Schaefer CJ, Ruhrmund DW, Pan L, Seiwert SD, Kossen K. Antifibrotic activities of pirfenidone in animal models. Eur Respir Rev. 2011;20(120):85–97.
Trivedi R, Redente EF, Thakur A, Riches DW, Kompella UB. Local delivery of biodegradable pirfenidone nanoparticles ameliorates bleomycin-induced pulmonary fibrosis in mice. Nanotechnology. 2012;23(50):505101.
Maher TM. Pirfenidone in idiopathic pulmonary fibrosis. Drugs Today (Barc). 2010;46(7):473–82.
King TE Jr, Bradford WZ, Castro-Bernardini S, Fagan EA, Glaspole I, Glassberg MK, et al. A phase 3 trial of pirfenidone in patients with idiopathic pulmonary fibrosis. N Engl J Med. 2014;370(22):2083–92.
Noble PW, Albera C, Bradford WZ, Costabel U, Glassberg MK, Kardatzke D, et al. Pirfenidone in patients with idiopathic pulmonary fibrosis (CAPACITY): two randomised trials. Lancet. 2011;377(9779):1760–9.
Hilberg F, Roth GJ, Krssak M, Kautschitsch S, Sommergruber W, Tontsch-Grunt U, et al. BIBF 1120: triple angiokinase inhibitor with sustained receptor blockade and good antitumor efficacy. Cancer Res. 2008;68(12):4774–82.
Chaudhary NI, Roth GJ, Hilberg F, Muller-Quernheim J, Prasse A, Zissel G, et al. Inhibition of PDGF, VEGF and FGF signalling attenuates fibrosis. Eur Respir J. 2007;29(5):976–85.
McCormack PL. Nintedanib: first global approval. Drugs. 2015;75(1):129–39.
Richeldi L, Costabel U, Selman M, Kim DS, Hansell DM, Nicholson AG, et al. Efficacy of a tyrosine kinase inhibitor in idiopathic pulmonary fibrosis. N Engl J Med. 2011;365(12):1079–87.
Richeldi L, du Bois RM, Raghu G, Azuma A, Brown KK, Costabel U, et al. Efficacy and safety of nintedanib in idiopathic pulmonary fibrosis. N Engl J Med. 2014;370(22):2071–82.
Wollin L, Maillet I, Quesniaux V, Holweg A, Ryffel B. Antifibrotic and anti-inflammatory activity of the tyrosine kinase inhibitor nintedanib in experimental models of lung fibrosis. J Pharmacol Exp Ther. 2014;349(2):209–20.
Wollin L, Wex E, Pautsch A, Schnapp G, Hostettler KE, Stowasser S, et al. Mode of action of nintedanib in the treatment of idiopathic pulmonary fibrosis. Eur Respir J. 2015;45(5):1434–45.
Rangarajan S, Kurundkar A, Kurundkar D, Bernard K, Sanders YY, Ding Q, et al. Novel mechanisms for the anti-fibrotic action of nintedanib. Am J Respir Cell Mol Biol (Epub 2015 Jun 13).
Loveman E, Copley VR, Scott DA, Colquitt JL, Clegg AJ, O’Reilly KM. Comparing new treatments for idiopathic pulmonary fibrosis—a network meta-analysis. BMC Pulm Med. 2015;15:37.
Loveman E, Copley VR, Colquitt JL, Scott DA, Clegg AJ, Jones J, et al. The effectiveness and cost-effectiveness of treatments for idiopathic pulmonary fibrosis: systematic review, network meta-analysis and health economic evaluation. BMC Pharmacol Toxicol. 2014;15:63.
Hancock A, Armstrong L, Gama R, Millar A. Production of interleukin 13 by alveolar macrophages from normal and fibrotic lung. Am J Respir Cell Mol Biol. 1998;18(1):60–5.
Cieslik KA, Taffet GE, Carlson S, Hermosillo J, Trial J, Entman ML. Immune-inflammatory dysregulation modulates the incidence of progressive fibrosis and diastolic stiffness in the aging heart. J Mol Cell Cardiol. 2011;50(1):248–56.
Lee CG, Homer RJ, Zhu Z, Lanone S, Wang X, Koteliansky V, et al. Interleukin-13 induces tissue fibrosis by selectively stimulating and activating transforming growth factor beta(1). J Exp Med. 2001;194(6):809–21.
Lee JH, Kaminski N, Dolganov G, Grunig G, Koth L, Solomon C, et al. Interleukin-13 induces dramatically different transcriptional programs in three human airway cell types. Am J Respir Cell Mol Biol. 2001;25(4):474–85.
Zhu Z, Ma B, Zheng T, Homer RJ, Lee CG, Charo IF, et al. IL-13-induced chemokine responses in the lung: role of CCR2 in the pathogenesis of IL-13-induced inflammation and remodeling. J Immunol. 2002;168(6):2953–62.
Murray LA, Argentieri RL, Farrell FX, Bracht M, Sheng H, Whitaker B, et al. Hyper-responsiveness of IPF/UIP fibroblasts: interplay between TGFbeta1, IL-13 and CCL2. Int J Biochem Cell Biol. 2008;40(10):2174–82.
Murray LA, Zhang H, Oak SR, Coelho AL, Herath A, Flaherty KR, et al. Targeting interleukin-13 with tralokinumab attenuates lung fibrosis and epithelial damage in a humanized SCID idiopathic pulmonary fibrosis model. Am J Respir Cell Mol Biol. 2014;50(5):985–94.
Fichtner-Feigl S, Strober W, Kawakami K, Puri RK, Kitani A. IL-13 signaling through the IL-13alpha2 receptor is involved in induction of TGF-beta1 production and fibrosis. Nat Med. 2006;12(1):99–106.
Lumsden RV, Worrell JC, Boylan D, Walsh SM, Cramton J, Counihan I, et al. Modulation of pulmonary fibrosis by IL-13Ralpha2. Am J Physiol Lung Cell Mol Physiol. 2015;308(7):L710–8.
Baran CP, Opalek JM, McMaken S, Newland CA, O’Brien JM Jr, Hunter MG, et al. Important roles for macrophage colony-stimulating factor, CC chemokine ligand 2, and mononuclear phagocytes in the pathogenesis of pulmonary fibrosis. Am J Respir Crit Care Med. 2007;176(1):78–89.
Liu X, Das AM, Seideman J, Griswold D, Afuh CN, Kobayashi T, et al. The CC chemokine ligand 2 (CCL2) mediates fibroblast survival through IL-6. Am J Respir Cell Mol Biol. 2007;37(1):121–8.
Moore BB, Murray L, Das A, Wilke CA, Herrygers AB, Toews GB. The role of CCL12 in the recruitment of fibrocytes and lung fibrosis. Am J Respir Cell Mol Biol. 2006;35(2):175–81.
Kalderen C, Stadler C, Forsgren M, Kvastad L, Johansson E, Sydow-Backman M, et al. CCL2 mediates anti-fibrotic effects in human fibroblasts independently of CCR2. Int Immunopharmacol. 2014;20(1):66–73.
Raghu G, Martinez FJ, Brown KK, Costabel U, Cottin V, Wells AU, et al. A phase II, randomized, double-blind, placebo-controlled, parallel-group, dose-ranging study of the safety and efficacy of CNTO 888 (carlumab) in patients with idiopathic pulmonary fibrosis [abstract]. Am J Respir Crit Care Med. 2013;187:A3376.
Duncan MR, Frazier KS, Abramson S, Williams S, Klapper H, Huang X, et al. Connective tissue growth factor mediates transforming growth factor beta-induced collagen synthesis: down-regulation by cAMP. FASEB J. 1999;13(13):1774–86.
Grotendorst GR. Connective tissue growth factor: a mediator of TGF-beta action on fibroblasts. Cytokine Growth Factor Rev. 1997;8(3):171–9.
Allen JT, Knight RA, Bloor CA, Spiteri MA. Enhanced insulin-like growth factor binding protein-related protein 2 (connective tissue growth factor) expression in patients with idiopathic pulmonary fibrosis and pulmonary sarcoidosis. Am J Respir Cell Mol Biol. 1999;21(6):693–700.
Pan LH, Yamauchi K, Uzuki M, Nakanishi T, Takigawa M, Inoue H, et al. Type II alveolar epithelial cells and interstitial fibroblasts express connective tissue growth factor in IPF. Eur Respir J. 2001;17(6):1220–7.
Lasky JA, Ortiz LA, Tonthat B, Hoyle GW, Corti M, Athas G, et al. Connective tissue growth factor mRNA expression is upregulated in bleomycin-induced lung fibrosis. Am J Physiol. 1998;275(2 Pt 1):L365–71.
Lipson KE, Wong C, Teng Y, Spong S. CTGF is a central mediator of tissue remodeling and fibrosis and its inhibition can reverse the process of fibrosis. Fibrogenesis Tissue Repair. 2012;5(Suppl 1):S24.
Raghu G, Scholand MB, De Andrade J, Lancaster L, Mageto YN, Goldin JG, et al. Safety and efficacy of anti-CTGF monoclonal antibody FG-3019 for treatment of idiopathic pulmonary fibrosis (IPF): results of phase 2 clinical trial two years after initiation [abstract]. Am J Respir Crit Care Med. 2014;189:A1426.
Pinnell SR, Martin GR. The cross-linking of collagen and elastin: enzymatic conversion of lysine in peptide linkage to alpha-aminoadipic-delta-semialdehyde (allysine) by an extract from bone. Proc Natl Acad Sci USA. 1968;61(2):708–16.
Siegel RC, Pinnell SR, Martin GR. Cross-linking of collagen and elastin: properties of lysyl oxidase. Biochemistry. 1970;9(23):4486–92.
Barry-Hamilton V, Spangler R, Marshall D, McCauley S, Rodriguez HM, Oyasu M, et al. Allosteric inhibition of lysyl oxidase-like-2 impedes the development of a pathologic microenvironment. Nat Med. 2010;16(9):1009–17.
Chien JW, Richards TJ, Gibson KF, Zhang Y, Lindell KO, Shao L, et al. Serum lysyl oxidase-like 2 levels and idiopathic pulmonary fibrosis disease progression. Eur Respir J. 2014;43(5):1430–8.
Border WA, Noble NA. Transforming growth factor beta in tissue fibrosis. N Engl J Med. 1994;331(19):1286–92.
Pardali K, Moustakas A. Actions of TGF-beta as tumor suppressor and pro-metastatic factor in human cancer. Biochim Biophys Acta. 2007;1775(1):21–62.
Munger JS, Harpel JG, Gleizes PE, Mazzieri R, Nunes I, Rifkin DB. Latent transforming growth factor-beta: structural features and mechanisms of activation. Kidney Int. 1997;51(5):1376–82.
Munger JS, Huang X, Kawakatsu H, Griffiths MJ, Dalton SL, Wu J, et al. The integrin alpha v beta 6 binds and activates latent TGF beta 1: a mechanism for regulating pulmonary inflammation and fibrosis. Cell. 1999;96(3):319–28.
Puthawala K, Hadjiangelis N, Jacoby SC, Bayongan E, Zhao Z, Yang Z, et al. Inhibition of integrin alpha(v)beta6, an activator of latent transforming growth factor-beta, prevents radiation-induced lung fibrosis. Am J Respir Crit Care Med. 2008;177(1):82–90.
Horan GS, Wood S, Ona V, Li DJ, Lukashev ME, Weinreb PH, et al. Partial inhibition of integrin alpha(v)beta6 prevents pulmonary fibrosis without exacerbating inflammation. Am J Respir Crit Care Med. 2008;177(1):56–65.
Shea BS, Tager AM. Role of the lysophospholipid mediators lysophosphatidic acid and sphingosine 1-phosphate in lung fibrosis. Proc Am Thorac Soc. 2012;9(3):102–10.
Pradere JP, Klein J, Gres S, Guigne C, Neau E, Valet P, et al. LPA1 receptor activation promotes renal interstitial fibrosis. J Am Soc Nephrol. 2007;18(12):3110–8.
Tager AM, LaCamera P, Shea BS, Campanella GS, Selman M, Zhao Z, et al. The lysophosphatidic acid receptor LPA1 links pulmonary fibrosis to lung injury by mediating fibroblast recruitment and vascular leak. Nat Med. 2008;14(1):45–54.
Castelino FV, Seiders J, Bain G, Brooks SF, King CD, Swaney JS, et al. Amelioration of dermal fibrosis by genetic deletion or pharmacologic antagonism of lysophosphatidic acid receptor 1 in a mouse model of scleroderma. Arthritis Rheum. 2011;63(5):1405–15.
Sakai N, Chun J, Duffield JS, Wada T, Luster AD, Tager AM. LPA1-induced cytoskeleton reorganization drives fibrosis through CTGF-dependent fibroblast proliferation. FASEB J. 2013;27(5):1830–46.
Hirokawa K. Autoimmunity and aging. Concepts Immunopathol. 1985;1:251–88.
Prelog M. Aging of the immune system: a risk factor for autoimmunity? Autoimmun Rev. 2006;5(2):136–9.
Donahoe M, Valentine VG, Chien N, Gibson KF, Raval JS, Saul M, et al. Autoantibody-targeted treatments for acute exacerbations of idiopathic pulmonary fibrosis. PLoS One. 2015;10(6):e0127771.
Daoussis D, Liossis SN, Tsamandas AC, Kalogeropoulou C, Paliogianni F, Sirinian C, et al. Effect of long-term treatment with rituximab on pulmonary function and skin fibrosis in patients with diffuse systemic sclerosis. Clin Exp Rheumatol. 2012;30(2 Suppl 71):S17–22.
Leon RJ, Gonsalvo A, Salas R, Hidalgo NC. Rituximab-induced acute pulmonary fibrosis. Mayo Clin Proc. 2004;79(7):949, 53.
Chaumais MC, Garnier A, Chalard F, Peuchmaur M, Dauger S, Jacqz-Agrain E, et al. Fatal pulmonary fibrosis after rituximab administration. Pediatr Nephrol. 2009;24(9):1753–5.
Rathi M, Ramachandran R, Gundlapalli S, Agarwal R, Das A, Kumar V, et al. Rituximab induced pulmonary fibrosis in a patient with lupus nephritis. Lupus. 2012;21(10):1131–4.
Ryter SW, Choi AM. Therapeutic applications of carbon monoxide in lung disease. Curr Opin Pharmacol. 2006;6(3):257–62.
Kohmoto J, Nakao A, Kaizu T, Tsung A, Ikeda A, Tomiyama K, et al. Low-dose carbon monoxide inhalation prevents ischemia/reperfusion injury of transplanted rat lung grafts. Surgery. 2006;140(2):179–85.
Hoetzel A, Dolinay T, Vallbracht S, Zhang Y, Kim HP, Ifedigbo E, et al. Carbon monoxide protects against ventilator-induced lung injury via PPAR-gamma and inhibition of Egr-1. Am J Respir Crit Care Med. 2008;177(11):1223–32.
Hoetzel A, Schmidt R, Vallbracht S, Goebel U, Dolinay T, Kim HP, et al. Carbon monoxide prevents ventilator-induced lung injury via caveolin-1. Crit Care Med. 2009;37(5):1708–15.
Chiang N, Shinohara M, Dalli J, Mirakaj V, Kibi M, Choi AM, et al. Inhaled carbon monoxide accelerates resolution of inflammation via unique proresolving mediator-heme oxygenase-1 circuits. J Immunol. 2013;190(12):6378–88.
Zhou Z, Song R, Fattman CL, Greenhill S, Alber S, Oury TD, et al. Carbon monoxide suppresses bleomycin-induced lung fibrosis. Am J Pathol. 2005;166(1):27–37.
Han MK, Zhou Y, Murray S, Tayob N, Noth I, Lama VN, et al. Lung microbiome and disease progression in idiopathic pulmonary fibrosis: an analysis of the COMET study. Lancet Respir Med. 2014;2(7):548–56.
Molyneaux PL, Cox MJ, Willis-Owen SA, Mallia P, Russell KE, Russell AM, et al. The role of bacteria in the pathogenesis and progression of idiopathic pulmonary fibrosis. Am J Respir Crit Care Med. 2014;190(8):906–13.
Wootton SC, Kim DS, Kondoh Y, Chen E, Lee JS, Song JW, et al. Viral infection in acute exacerbation of idiopathic pulmonary fibrosis. Am J Respir Crit Care Med. 2011;183(12):1698–702.
Wuyts WA, Willems S, Vos R, Vanaudenaerde BM, De Vleeschauwer SI, Rinaldi M, et al. Azithromycin reduces pulmonary fibrosis in a bleomycin mouse model. Exp Lung Res. 2010;36(10):602–14.
Shulgina L, Cahn AP, Chilvers ER, Parfrey H, Clark AB, Wilson EC, et al. Treating idiopathic pulmonary fibrosis with the addition of co-trimoxazole: a randomised controlled trial. Thorax. 2013;68(2):155–62.
Leung T, Manser E, Tan L, Lim L. A novel serine/threonine kinase binding the Ras-related RhoA GTPase which translocates the kinase to peripheral membranes. J Biol Chem. 1995;270(49):29051–4.
Riento K, Ridley AJ. ROCKs: multifunctional kinases in cell behaviour. Nat Rev Mol Cell Biol. 2003;4(6):446–56.
Street CA, Bryan BA. Rho kinase proteins—pleiotropic modulators of cell survival and apoptosis. Anticancer Res. 2011;31(11):3645–57.
Zhou Y, Huang X, Hecker L, Kurundkar D, Kurundkar A, Liu H, et al. Inhibition of mechanosensitive signaling in myofibroblasts ameliorates experimental pulmonary fibrosis. J Clin Invest. 2013;123(3):1096–108.
Wang P, Tang F, Li R, Zhang H, Chen S, Liu P, et al. Contribution of different Nox homologues to cardiac remodeling in two-kidney two-clip renovascular hypertensive rats: effect of valsartan. Pharmacol Res. 2007;55(5):408–17.
Masamune A, Kikuta K, Watanabe T, Satoh K, Hirota M, Shimosegawa T. Hypoxia stimulates pancreatic stellate cells to induce fibrosis and angiogenesis in pancreatic cancer. Am J Physiol Gastrointest Liver Physiol. 2008;295(4):G709–17.
Hecker L, Vittal R, Jones T, Jagirdar R, Luckhardt TR, Horowitz JC, et al. NADPH oxidase-4 mediates myofibroblast activation and fibrogenic responses to lung injury. Nat Med. 2009;15(9):1077–81.
Amara N, Goven D, Prost F, Muloway R, Crestani B, Boczkowski J. NOX4/NADPH oxidase expression is increased in pulmonary fibroblasts from patients with idiopathic pulmonary fibrosis and mediates TGFbeta1-induced fibroblast differentiation into myofibroblasts. Thorax. 2010;65(8):733–8.
Kuroda J, Ago T, Matsushima S, Zhai P, Schneider MD, Sadoshima J. NADPH oxidase 4 (Nox4) is a major source of oxidative stress in the failing heart. Proc Natl Acad Sci USA. 2010;107(35):15565–70.
Sancho P, Mainez J, Crosas-Molist E, Roncero C, Fernandez-Rodriguez CM, Pinedo F, et al. NADPH oxidase NOX4 mediates stellate cell activation and hepatocyte cell death during liver fibrosis development. PLoS One. 2012;7(9):e45285.
Thannickal VJ, Fanburg BL. Activation of an H2O2-generating NADH oxidase in human lung fibroblasts by transforming growth factor beta 1. J Biol Chem. 1995;270(51):30334–8.
Cheng G, Cao Z, Xu X, van Meir EG, Lambeth JD. Homologs of gp91phox: cloning and tissue expression of Nox3, Nox4, and Nox5. Gene. 2001;269(1–2):131–40.
Thannickal VJ. Aging, antagonistic pleiotropy and fibrotic disease. Int J Biochem Cell Biol. 2010;42(9):1398–400.
Thannickal VJ. Mechanisms of pulmonary fibrosis: role of activated myofibroblasts and NADPH oxidase. Fibrogenesis Tissue Repair. 2012;5(Suppl 1):S23.
Sanders YY, Liu H, Liu G, Thannickal VJ. Epigenetic mechanisms regulate NADPH oxidase-4 expression in cellular senescence. Free Radic Biol Med. 2015;79:197–205.
Hecker L, Logsdon NJ, Kurundkar D, Kurundkar A, Bernard K, Hock T, et al. Reversal of persistent fibrosis in aging by targeting Nox4–Nrf2 redox imbalance. Sci Transl Med. 2014;6(231):231ra47.
Salminen A, Kaarniranta K. AMP-activated protein kinase (AMPK) controls the aging process via an integrated signaling network. Ageing Res Rev. 2012;11(2):230–41.
Rangarajan S, Liu Y, Park DW, Zmijewska A, Abraham E, Thannickal VJ, et al. AMP-activated protein kinase activation diminishes the severity of experimental lung fibrosis [abstract]. Am J Respir Crit Care Med. 2015;191:A3474.
Mihaylova MM, Shaw RJ. The AMPK signalling pathway coordinates cell growth, autophagy and metabolism. Nat Cell Biol. 2011;13(9):1016–23.
Burkewitz K, Zhang Y, Mair WB. AMPK at the nexus of energetics and aging. Cell Metab. 2014;20(1):10–25.
Daniels CE, Wilkes MC, Edens M, Kottom TJ, Murphy SJ, Limper AH, et al. Imatinib mesylate inhibits the profibrogenic activity of TGF-beta and prevents bleomycin-mediated lung fibrosis. J Clin Invest. 2004;114(9):1308–16.
Daniels CE, Lasky JA, Limper AH, Mieras K, Gabor E, Schroeder DR, et al. Imatinib treatment for idiopathic pulmonary fibrosis: randomized placebo-controlled trial results. Am J Respir Crit Care Med. 2010;181(6):604–10.
Vittal R, Horowitz JC, Moore BB, Zhang H, Martinez FJ, Toews GB, et al. Modulation of prosurvival signaling in fibroblasts by a protein kinase inhibitor protects against fibrotic tissue injury. Am J Pathol. 2005;166(2):367–75.
Vittal R, Zhang H, Han MK, Moore BB, Horowitz JC, Thannickal VJ. Effects of the protein kinase inhibitor, imatinib mesylate, on epithelial/mesenchymal phenotypes: implications for treatment of fibrotic diseases. J Pharmacol Exp Ther. 2007;321(1):35–44.
Garneau-Tsodikova S, Thannickal VJ. Protein kinase inhibitors in the treatment of pulmonary fibrosis. Curr Med Chem. 2008;15(25):2632–40.
Grimminger F, Gunther A, Vancheri C. The role of tyrosine kinases in the pathogenesis of idiopathic pulmonary fibrosis. Eur Respir J. 2015;45(5):1426–33.
Hu M, Che P, Han X, Cai GQ, Liu G, Antony V, et al. Therapeutic targeting of Src kinase in myofibroblast differentiation and pulmonary fibrosis. J Pharmacol Exp Ther. 2014;351(1):87–95.
Bartel DP. MicroRNAs: genomics, biogenesis, mechanism, and function. Cell. 2004;116(2):281–97.
Guo H, Ingolia NT, Weissman JS, Bartel DP. Mammalian microRNAs predominantly act to decrease target mRNA levels. Nature. 2010;466(7308):835–40.
Cui H, Xie N, Thannickal VJ, Liu G. The code of non-coding RNAs in lung fibrosis. Cell Mol Life Sci. 2015;72(18):3507–19.
Cushing L, Kuang PP, Qian J, Shao F, Wu J, Little F, et al. miR-29 is a major regulator of genes associated with pulmonary fibrosis. Am J Respir Cell Mol Biol. 2011;45(2):287–94.
Yang T, Liang Y, Lin Q, Liu J, Luo F, Li X, et al. miR-29 mediates TGFbeta1-induced extracellular matrix synthesis through activation of PI3K–AKT pathway in human lung fibroblasts. J Cell Biochem. 2013;114(6):1336–42.
Liu G, Friggeri A, Yang Y, Milosevic J, Ding Q, Thannickal VJ, et al. miR-21 mediates fibrogenic activation of pulmonary fibroblasts and lung fibrosis. J Exp Med. 2010;207(8):1589–97.
van Rooij E, Kauppinen S. Development of microRNA therapeutics is coming of age. EMBO Mol Med. 2014;6(7):851–64.
Morry J, Ngamcherdtrakul W, Gu S, Goodyear SM, Castro DJ, Reda MM, et al. Dermal delivery of HSP47 siRNA with NOX4-modulating mesoporous silica-based nanoparticles for treating fibrosis. Biomaterials. 2015;66:41–52.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Funding
No specific sources of funding were used to support the writing of this manuscript.
Conflicts of Interest
Tracy Luckhardt participated in an Advisory Board for Intermune Inc. (the manufacturer of pirfenidone) in June 2014 and received an honorarium and travel expenses. Sunad Rangarajan, Morgan Locy and Victor Thannickal have no conflicts of interest to report.
Rights and permissions
About this article

Cite this article
Rangarajan, S., Locy, M.L., Luckhardt, T.R. et al. Targeted Therapy for Idiopathic Pulmonary Fibrosis: Where To Now?. Drugs 76, 291–300 (2016). https://doi.org/10.1007/s40265-015-0523-6
Published:
Issue Date:
DOI: https://doi.org/10.1007/s40265-015-0523-6