
Abstract
Tedizolid phosphate is a novel oxazolidinone prodrug (converted to the active form tedizolid by phosphatases in vivo) that has been developed and recently approved (June 2014) by the United States FDA for the treatment of acute bacterial skin and skin structure infections (ABSSSIs) caused by susceptible Gram-positive pathogens, including methicillin-resistant Staphylococcus aureus (MRSA). Tedizolid is an oxazolidinone, but differs from other oxazolidinones by possessing a modified side chain at the C-5 position of the oxazolidinone nucleus which confers activity against certain linezolid-resistant pathogens and has an optimized C- and D-ring system that improves potency through additional binding site interactions. The mechanism of action of tedizolid is similar to other oxazolidinones and occurs through inhibition of bacterial protein synthesis by binding to 23S ribosomal RNA (rRNA) of the 50S subunit of the ribosome. As with other oxazolidinones, the spontaneous frequency of resistance development to tedizolid is low. Tedizolid is four- to eightfold more potent in vivo than linezolid against all species of staphylococci, enterococci, and streptococci, including drug-resistant phenotypes such as MRSA and vancomycin-resistant enterococci (VRE) and linezolid-resistant phenotypes. Importantly, tedizolid demonstrates activity against linezolid-resistant bacterial strains harboring the horizontally transmissible cfr gene, in the absence of certain ribosomal mutations conferring reduced oxazolidinone susceptibility. With its half-life of approximately 12 h, tedizolid is dosed once daily. It demonstrates linear pharmacokinetics, has a high oral bioavailability of approximately 90 %, and is primarily excreted by the liver as an inactive, non-circulating sulphate conjugate. Tedizolid does not require dosage adjustment in patients with any degree of renal dysfunction or hepatic dysfunction. Studies in animals have demonstrated that the pharmacodynamic parameter most closely associated with the efficacy of tedizolid is fAUC0–24h/MIC. In non-neutropenic animals, a dose-response enhancement was observed with tedizolid and lower exposures were required compared to neutropenic cohorts. Two Phase III clinical trials have demonstrated non-inferiority of a once-daily tedizolid 200 mg dose for 6–10 days versus twice-daily 600 mg linezolid for the treatment of ABSSSIs. Both trials used the primary endpoint of early clinical response at 48–72 h; however, one trial compared oral formulations while the other initiated therapy with the parenteral formulation and allowed oral sequential therapy following initial clinical response. Throughout its development, tedizolid has demonstrated that it is well tolerated and animal studies have shown a lower propensity for neuropathies with long-term use than its predecessor linezolid. Data from the two completed Phase III clinical trials demonstrated that the studied tedizolid regimen (200 mg once daily for 6 days) had significantly less impact on hematologic parameters as well as significantly less gastrointestinal treatment-emergent adverse effects (TEAEs) than its comparator linezolid. As with linezolid, tedizolid is a weak, reversible MAO inhibitor; however, a murine head twitch model validated to assess serotonergic activity reported no increase in the number of head twitches with tedizolid even at doses that exceeded the C max in humans by up to 25-fold. Tyramine and pseudoephedrine challenge studies in humans have also reported no meaningful MAO-related interactions with tedizolid. With its enhanced in vitro activity against a broad-spectrum of Gram-positive aerobic bacteria, convenient once-daily dosing, a short 6-day course of therapy, availability of both oral and intravenous routes of administration, and an adverse effect profile that appears to be more favorable than linezolid, tedizolid is an attractive agent for use in both the hospital and community settings. Tedizolid is currently undergoing additional Phase III clinical trials for the treatment of hospital-acquired bacterial pneumonia (HABP) and ventilated nosocomial pneumonia (VNP).
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Tedizolid’s potent in vitro activity against a broad-spectrum of Gram-positive organisms, high bioavailability with oral administration, long half-life allowing once-daily dosing, lack of dosage adjustment in patients with renal or hepatic dysfunction, short (6-day) course of therapy, and availability of both oral and intravenous routes of administration, make tedizolid an attractive antimicrobial for use in both hospital and community settings. |
While tedizolid does possess many qualities of the ideal antimicrobial, until further clinical trials in other types of infections (HABP/VNP) are completed, its role in therapy may be limited to ABSSSIs presently treated with linezolid. |
Pharmacoeconomic considerations will also help to further elucidate tedizolid’s place in the clinician’s arsenal of antimicrobials against Gram-positive infections. |
1 Introduction
Over 50 years have elapsed since the first isolates of methicillin-resistant Staphylococcus aureus (MRSA) were described by Patricia Jevons only 2 years after the initial clinical use of methicillin [1–3]. Despite an increase in the prevalence of MRSA and other multidrug-resistant (MDR) Gram-positive bacterial pathogens, the development of new antimicrobials, particularly for oral use, to combat these challenging infections has dwindled and the number of effective treatments for them has decreased [4]. Vancomycin has long been the “gold standard” for treatment of serious MRSA infections with more than 50 years of clinical use in this setting; however, there is ongoing debate as to whether or not it has become obsolete [5]. With the emergence of less susceptible strains to vancomycin (hVISA; heteroresistant vancomycin intermediate resistant Staphylococcus aureus), increased treatment failures in MRSA infections with vancomycin MIC ≥2 mg/L, and increased nephrotoxicity with high-dose vancomycin therapy, its role as first-line therapy is being challenged and researchers are searching for novel antimicrobials to combat these serious infections [5].
Linezolid, the first oxazolidinone marketed, provided an effective alternative to vancomycin with several advantages. With the availability of both oral and intravenous routes of administration, a broad Gram-positive spectrum including staphylococci (including MRSA), streptococci and enterococci (including vancomycin-resistant Enterococcus faecium-VRE), and slow emergence of resistance, linezolid has established itself as a reliable option for the treatment of infections caused by Gram-positive pathogens [6]. Linezolid, however, is not without its own limitations. Linezolid is administered twice daily, which can make compliance in an outpatient setting more difficult. It can cause reversible hematologic effects, such as thrombocytopenia and bone marrow suppression, as well as neuropathies, with prolonged duration of use (>10 days). It has also been associated with serotonin toxicity due to its inhibitory effects on the monoamine oxidase enzyme (MAO) when used concomitantly with other drugs that increase circulating serotonin, including the commonly prescribed selective serotonin reuptake inhibitors (SSRIs). While resistance to linezolid has been slow to develop (and usually involving 23S rRNA mutations), recently it was discovered that a multidrug resistance gene, cfr, confers resistance to linezolid. This is a disturbing finding as this gene is horizontally transferable, meaning there is potential for its widespread dissemination, as well as its widespread presence in the environment and the fact that it can be transferred not just from species to species but genus to genus (e.g., staphylococci to enterococci) [2, 3].
Tedizolid (previously known as TR-700 or torezolid) is a second-generation oxazolidinone. Tedizolid has been demonstrated to possess enhanced in vitro potency compared to linezolid, and data from comparative clinical trials using short-course therapy (i.e., 6 days) suggests that it has a more favorable hematologic tolerability profile. It has also been reported that tedizolid possesses good oral bioavailability and only requires once-daily administration. Perhaps most importantly, tedizolid appears to maintain its activity against staphylococci containing the cfr gene (in the absence of certain ribosomal mutations conferring reduced oxazolidinone susceptibility), which will prove to be of greater benefit if this resistance mechanism becomes widely disseminated [2, 7, 8].
Tedizolid (developed as the phosphate prodrug) has been recently approved by the United States FDA (June 2014), for the treatment of acute bacterial skin and skin structure infections (ABSSSIs) caused by susceptible Gram-positive pathogens, including MRSA. In addition, tedizolid is currently under review in Europe (European Medicines Agency) and Canada (Health Canada). Tedizolid is also being studied in Phase III clinical trials for use in patients with hospital-acquired bacterial pneumonia (HABP) and ventilated nosocomial pneumonia (VNP). This article reviews available published data on tedizolid, including relevant chemistry, mechanisms of action, mechanisms of resistance, microbiology, pharmacokinetics, pharmacodynamics, and efficacy and safety data from both animal and clinical trials. A comprehensive literature search was conducted using PUBMED for all material containing the names “tedizolid (DA-7157)”, “tedizolid phosphate (DA-7218)”, and any of “TR-700”, “TR-701”, or “torezolid”. These results were supplemented with tedizolid bibliographies obtained from Cubist Pharmaceuticals.
2 Chemistry
Tedizolid phosphate (formerly TR-701) is an inactive prodrug that is rapidly and extensively converted to the active moiety, tedizolid [8–11]. As mentioned earlier, tedizolid is a novel oxazolidinone, a class of synthetic antimicrobials that derive their name from the common 3-(3-fluorophenyl)-oxazolidinone ring core present in both linezolid and tedizolid [12].
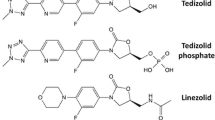
Tedizolid is structurally similar to linezolid (Fig. 1). The structure activity relationships (SAR) described below are summarized in Fig. 2. The 5-R configuration on the A-ring is necessary for antibacterial activity as is the N-aryl B-ring. Fluorination of the B-ring improves antibacterial activity and is present in linezolid and tedizolid, as well as other similar compounds in clinical development [13]. It was originally thought that the acetamide arm found in linezolid at the C-5 position of the oxazolidinone ring was required for optimal antibacterial activity; however, it has now been reported that this is not the case and many compounds with other C-5 substituents have demonstrated increased potency in vitro [12–14]. Tedizolid possesses a hydroxymethyl group at the C-5 position, which differs from its predecessor linezolid, which contains the aforementioned acetamide group. Locke et al. [12, 15] reported that tedizolid, and other oxazolidinones with the hydroxymethyl or 1,2,3-triazole C-5 substituent, maintained their potency against strains possessing the cfr gene (perhaps due to less steric hindrance), while the acetamide substituent at C-5 resulted in a two- to eightfold reduction in activity. The phosphate group of the prodrug tedizolid phosphate confers advantages over the parent molecule. The addition of the phosphate helps to improve both the water solubility and bioavailability of the drug. In addition to the C-5 modification, tedizolid also differs from linezolid in that tedizolid contains a biaryl ring system, in contrast to the morpholine ring found in linezolid. The optimization of the C- and D-ring system, pyridine and tetrazole rings respectively, in tedizolid is responsible for its enhanced potency relative to linezolid because tedizolid is able to form additional binding interactions with the upper region of the peptidyltransferase center (PTC) of the 50S ribosomal subunit [14].

Chemical structures of tedizolid and linezolid

Structure-activity relationships for tedizolid phosphate
3 Mechanism of Action
Like linezolid, tedizolid exhibits its antibacterial effects through the inhibition of protein synthesis, specifically through binding to the 23S ribosomal RNA (rRNA) of the 50S subunit of the ribosome. This binding prevents the formation of the 70S ribosomal initiation complex and therefore prevents translation of bacterial proteins [8, 9]. More recent studies utilizing in vivo cross-linking of radiolabelled oxazolidinone photoaffinity probes have demonstrated that these drugs specifically bind at the A site of the PTC, further defining the specific mode of action through blocking the incoming aminoacyl-tRNA complex from binding to the A site [16]. The unique mechanism of action employed by these antimicrobials makes cross-resistance to other antibiotics, such as beta-lactams and glycopeptides, unlikely [9, 16, 17].
The formation of additional target site interactions at the PTC has been associated with increased antimicrobial potency. A model of tedizolid binding suggests that the C- and D-rings of tedizolid add two hydrogen bonds to the sugar backbone residues A2451 and U2584. As observed in Fig. 1, linezolid lacks the D-ring found in tedizolid and it is hypothesized that these additional site interactions contribute to the increase in antimicrobial potency that is observed with tedizolid [16, 18]. Data from an SAR study by Locke et al. [15] support this hypothesis by demonstrating that between three oxazolidinones containing a C-5 acetamide substituent, one of which was linezolid, the most potent of the three was a compound with an identical D-ring to that found in tedizolid and the least potent was linezolid. The middle potency compound also contained a D-ring, albeit different than the one present in tedizolid, highlighting the contribution of the D-ring to the potency advantage seen in tedizolid over linezolid. Additionally, the smaller hydroxymethyl C-5 substituent on the A ring (Fig. 2) in tedizolid is hypothesized to explain its retained potency over cfr positive bacterial strains, due to less steric hindrance with the methylated A2503 bacterial rRNA residue than observed with the acetamide arm of linezolid at this position [16, 19].
4 Mechanism of Resistance
The spontaneous frequency of resistance to oxazolidinone antimicrobials is relatively low when compared to other antimicrobial agents. This is in large part due to the fact that most bacteria possess multiple copies of rRNA, necessitating mutations in multiple copies of the 23S rRNA central loop of domain V gene target to increase MICs [3, 16, 20]. While rare in the clinical setting, resistance to linezolid was first observed in 1999 when two strains of linezolid-resistant E. faecium were identified in a compassionate use program [20, 21]. More troubling are reports of S. aureus and S. epidermidis containing the horizontally transferrable plasmid-borne ribosomal methyltransferase gene, cfr [19, 21]. Acquisition of the cfr gene results in resistance to linezolid, as well as other antimicrobials such as phenicols, lincosamides, pleuromutilins, streptogramin A, and 16-member-ring macrolide antibiotics [19–22]. The potential for widespread dissemination of this resistance element due to its potential for horizontal transmission (with low fitness cost) is of concern [20]. Mutations in the genes encoding the ribosomal proteins L3 and L4 associated with the PTC have also been associated with linezolid resistance, while tedizolid has been shown to demonstrate activity against these mutations (although the MICs are increased) even when they are combined with the cfr gene [23, 24].
A study by Locke et al. [25] reported that following 30 serial passages in the presence of tedizolid, the MIC for MSSA ATCC 29213 (methicillin-susceptible S. aureus) remained constant at 0.5 mg/L while it increased eightfold from 0.25 to 2.0 mg/L for MRSA ATCC 33591. Serial passage in the presence of linezolid for the two strains resulted in 64- and 32-fold increases in MIC values, respectively, highlighting that the potential of S. aureus to develop resistance upon prolonged exposure to tedizolid might be lower than for linezolid. Tedizolid also required multiple mutations in the 23S rRNA in order for the initial stepwise MIC increases for MRSA ATCC 33591 to occur, in contrast to linezolid where single mutations were identified in both strains resulting in two- to fourfold changes in MIC with five to eight serial passages. The same study also reported that spontaneous mutations conferring reduced susceptibility to tedizolid are less frequent that those conferring reduced susceptibility to linezolid [25]. The spontaneous frequencies of mutation of MSSA ATCC 29213 and MRSA ATCC 33591 resulting in reduced susceptibility to tedizolid at twice the MIC were 1.1 × 10−10 and 1.9 × 10−10, respectively, approximately 16-fold lower than the corresponding linezolid frequencies for both strains [25].
Another study by Shaw et al. [18] reported the activity of tedizolid against linezolid-resistant strains demonstrating that tedizolid maintained MIC values between 0.5 and 1 mg/L against MRSA strains that possessed the cfr gene. This is an important finding due to the possibility that tedizolid will achieve sufficient tissue concentrations to treat these linezolid-resistant infections. Researchers trying to explain tedizolid’s activity versus linezolid-resistant MRSA stated that optimization of the C- and D-rings allowed interaction with more highly conserved regions of the PTC binding site [12, 18]. In addition, the less bulky hydroxymethyl substituent on the C-5 group of the A-ring of tedizolid was less sterically hindered than the acetamide group of linezolid by the methylation of the A2503 residue encoded for by the cfr gene. However, more studies are needed to determine the contributions of C and D- ring optimization as well as A-ring substitutions of tedizolid to its 16-fold potency advantage over linezolid for these strains. Studies have confirmed the in vitro activity of tedizolid against linezolid-resistant MRSA possessing the cfr gene, in the absence of certain chromosomal linezolid-resistant mutations. Ribosomal protein or S23 rRNA mutations co-occurring with the cfr mechanism, however, frequently resulted in MIC values above the currently accepted susceptibility breakpoint [19, 23].
5 Microbiology
The in vitro activities of tedizolid and its comparators against key Gram-positive and Gram-negative organisms are presented in Tables 1, 2, 3. The MIC values presented therein are derived from available in vivo studies conducted on tedizolid the data of which were pooled (for all isolates) and reviewed [26–46]. Comparator data were pooled from these same studies and are included in the tables when such data were available. The range presented is made up of the lowest and highest MIC values reported in the literature cited. All in vitro studies cited in this review utilized methods described by the Clinical Laboratory Standards Institute (CLSI).
Table 1 shows the activities of tedizolid and the comparators, linezolid and vancomycin, against common aerobic Gram-positive organisms, including certain drug-resistant phenotypes. Tedizolid is four- to eightfold (based on MIC50 and MIC90 values) more potent than linezolid against all species of staphylococci, enterococci, and streptococci examined, including drug-resistant phenotypes such as MRSA [both community-acquired (CA-MRSA) and healthcare-acquired (HA-MRSA)] and VRE. Tedizolid continues to be four- to eightfold (based on MIC50 and MIC90 values) more active than linezolid versus MRSA phenotypes as well as hVISA, VISA, and VRSA (vancomycin-resistant S. aureus) [36, 40].
While resistance to linezolid remains rare (<1%) in multicenter, international surveillance programs, cfr gene mediated linezolid resistance in MRSA has been reported as well as resistance due to mutations in the 23S rRNA binding site [19, 25, 37, 47–49]. Jones et al. [49] examined the activity of tedizolid against linezolid-resistant Gram-positive cocci with well-characterized resistance mechanisms. Against 39 staphylococcal isolates with the G2576T 23S rRNA mutation, the most commonly observed mechanism of linezolid-resistance encountered clinically [25], tedizolid was fourfold more potent than linezolid with an MIC90 of 8 mg/L compared to 32 mg/L for linezolid. For enterococci containing the same mutation, tedizolid was eightfold more potent than linezolid with an MIC90 of 2 mg/L compared to 16 mg/L in linezolid. It should be cautioned that both agents, linezolid and tedizolid, using current breakpoints, would be considered resistant versus these phenotypes. The study also reported MICs for four clinical isolates of cfr-carrying staphylococci and found tedizolid to be 32-fold more potent than linezolid in this small sample. In a study by Betriu et al. [28], the activities of tedizolid and linezolid against staphylococcal blood isolates collected in Spain, including five isolates from the aforementioned outbreak, were tested. The researchers reported that tedizolid was 32-fold more potent than linezolid against these strains with all seven strains tested inhibited at 0.5 mg/L (considered susceptible to tedizolid). Another study by Rodriguez-Avial et al. [50] also examined the activity of tedizolid against linezolid-resistant staphylococcal strains, for which the genetic basis of resistance was not determined. The study found that tedizolid inhibited the five linezolid-resistant S. aureus strains tested at a concentration of 0.5 mg/L (considered susceptible to tedizolid). Against the 164 strains of linezolid-resistant coagulase-negative staphylococci examined, tedizolid demonstrated an MIC50 value of 4 mg/L and an MIC90 value of 8 mg/L, making it at least 16-fold more potent than linezolid against these organisms.
Table 2 displays the activities of tedizolid and linezolid against Gram-negative aerobes. Presently, available data are limited to key respiratory pathogens Haemophilus influenzae and Moraxella cattarhalis. Tedizolid is twofold more potent than linezolid versus these pathogens. More comparative data with linezolid are required with tedizolid against other Gram-negative aerobes, such as Enterobacteriaceae.
Table 3 displays the activity of tedizolid and linezolid versus anaerobes. Tedizolid is eight- to 16-fold more active than linezolid versus Gram-positive anaerobic bacteria. Tedizolid is ~twofold more active than linezolid versus anaerobic Gram-negative bacilli (Table 3).
The in vivo activity of tedizolid against Mycobacterium tuberculosis and Nocardia brasiliensis has also been investigated. A study by Vera-Cabrera et al. [51] reported an MIC50 of 0.25 mg/L and an MIC90 of 0.5 mg/L for tedizolid against M. tuberculosis,while linezolid’s MIC values were 1 and 2 mg/L respectively. These MIC values for tedizolid were unchanged when MDR strains resistant to both isoniazid and rifampin were examined. The same study reported MIC values of 1 mg/L for both the MIC50 and MIC90 of tedizolid against N. brasiliensis. A study by Molina-Torres et al. [52] reported similar decreases in mycobacterium growing in a THP-1 monocytic cell line at 3 days between tedizolid (1.3 log10) and two antibiotics with proven activity against tuberculosis, rifampin (1.4 log10), and moxifloxacin (1 log10).
Current MIC susceptibility breakpoints are available for tedizolid (US product monograph 2014), including staphylococci, MIC breakpoints of ≤0.5 mg/L for susceptible, ≤1 mg/L for intermediate, and ≥2 mg/L for resistant were suggested. For streptococci and enterococci, a susceptible-only breakpoint of ≤0.5 mg/L is proposed.
6 Pharmacokinetics
The results of Phase 1 pharmacokinetic studies involving both oral and intravenous administration of tedizolid phosphate to healthy volunteers are summarized in Table 4 [10, 53–56]. These data include the study of two prodrug formulations including tedizolid phosphate disodium and the marketed tedizolid phosphate-free acid. Based on the demonstration of comparable pharmacokinetics, further discussion refers to both formulations as tedizolid phosphate [53].
Flanagan et al. [57] determined the absolute bioavailability of tedizolid in eight healthy adult subjects who received 200 mg of tedizolid phosphate intravenously and orally in a two-way crossover design. The mean area under the concentration-time curve from zero to infinity (AUC0–inf) after oral administration was 26.67 ± 6.03 mg·h/L, while the AUC0–inf after intravenous administration was 29.02 ± 6.14 mg·h/L with an absolute bioavailability of 91.4 ± 6.8 %. Tanaka et al. [58] reported an oral bioavailability of 82.6 % (90 % confidence interval 77.9–87.6%) in eight healthy Japanese male volunteers. Finally a study of the effects of food on the pharmacokinetics of oral tedizolid phosphate found that food delayed absorption; however, the AUC was unaltered, supporting administration without regard to meals [53].
Flanagan et al. [59] conducted a population pharmacokinetic study of oral tedizolid phosphate in 188 patients with complicated skin and skin structure infections. The data described a two-compartment model with linear pharmacokinetics for doses of 200, 300, and 400 mg once daily for 5–7 days. These data are consistent with earlier studies by Flanagan et al. [53] and Bien et al. [56] who also reported linear pharmacokinetics for tedizolid phosphate with single doses ranging from 200 to 1200 mg orally and from 100 to 400 mg intravenously.
The maximum plasma concentration, area under the curve over 24 h, half-life, volume of distribution, and clearance of tedizolid phosphate reported in several Phase 1 pharmacokinetic studies involving both oral and intravenous administration are summarized in Table 4 [10, 53–56]. Oral doses (200 mg) of tedizolid produced mean maximum plasma concentrations of 1.8–2.4 mg/L at 2–3 h, whereas intravenous administration over 60 min resulted in maximum concentrations of 2.62–3.45 mg/L. The mean half-life of tedizolid following oral administration was 9.2–11.8 h, similar to the 10.8–12.4 h observed with the intravenous formulation. The mean volume of distribution was 86.9–108 L (V d/F, where F is oral bioavailability) with oral administration, compared with 67.1–80.1 L (V d) with intravenous administration. The total clearance with oral (CL/F) and intravenous (CL) tedizolid was 5.73–8.4 and 5.41–5.87 L/h, respectively.
Sahre et al. [60] measured the protein binding of tedizolid using two microdialysis probes implanted into the thighs of 12 healthy adults. The primary purpose of this study was to assess the penetration of tedizolid into tissues relevant to skin/soft tissue infections. Single 600-mg doses of tedizolid phosphate produced a plasma-free AUC0–24h of 7.3 ± 1.9 mg·h/L total AUC0–24h of 57.1 ± 14.7 mg·h/L and calculated protein binding of 87.3 ± 1.3 %. The ratios of free AUC in tissue to plasma were 1.1 ± 0.2 and 1.2 ± 0.2 for adipose and muscle tissue, respectively. Housman et al. [55], using an ultrafiltration method for determination, reported a similar protein binding of 89.4 ± 1.6 % for tedizolid. The primary purpose of this study was to assess the penetration of tedizolid into tissues relevant in lung infection by investigating the penetration of tedizolid into the epithelial lining fluid (ELF) and alveolar macrophages (AM) of lungs in 20 healthy volunteers. The penetration using the AUC0–24h in ELF and AM, compared with the free AUC0–24h in plasma, were approximately 40 and 20 mg·h/L, respectively [55].
Tedizolid is primarily excreted by the liver as an inactive, non-circulating sulphate conjugate with only 18 % excreted unchanged in the urine [61, 62]. Flanagan et al. [63] evaluated the pharmacokinetics of tedizolid phosphate in subjects with advanced renal impairment with an estimated glomerular filtration rate (eGFR) less than 30 mL/min/1.73 m2 but not undergoing hemodialysis. Eight study subjects (mean eGFR of 18.3 mL/min/1.73 m2) and eight controls (eGFR >80 mL/min/1.73 m2) received a 200-mg intravenous dose of tedizolid phosphate. The mean half-life of tedizolid was 12.8 ± 2.28 h in those with renal impairment compared with 12.3 ± 2.04 h in the controls. Other pharmacokinetic parameters were similarly unchanged with AUC0–inf values of 29.99 ± 8.97 and 32.43 ± 9.53 mg·h/L in the renal impairment and control groups, respectively. The investigators concluded that tedizolid does not require dosage adjustment in patients with any degree of renal dysfunction. This study also reported that tedizolid pharmacokinetics were not influenced by hemodialysis and that dialysis did not significantly remove the drug [63].
The pharmacokinetics of a single 200-mg oral dose of tedizolid phosphate in subjects with hepatic impairment was also examined by Flanagan et al. [63]. Study groups included moderate (Child-Pugh 7–9) or severe (Child-Pugh 10–15) hepatic impairment and matched controls for age, weight, and sex. Tedizolid pharmacokinetics were reported to be minimally altered in hepatic impairment (AUC0–24h of 34.82 ± 20.87 mg·h/L in those with the severe hepatic impairment compared with 24.38 ± 8.03 mg·h/L in controls). The investigators concluded that tedizolid does not require dosage adjustment in patients with any degree of hepatic dysfunction. Further study in this patient population is required to determine the tolerability with multiple dosing and whether dosage adjustments are required especially in those with both severe renal and hepatic disease. Preliminary studies in other populations, including obese, elderly, and adolescent subjects, have suggested that tedizolid phosphate dose adjustments are not required (as only minor differences in pharmacokinetics are observed); however, the small number of participants in these trials warrants further study [64–70].
7 Pharmacodynamics
The pharmacodynamics of tedizolid were studied in murine thigh infection and pneumonia models. Through the integration of pharmacokinetics, in vitro potency (i.e., MIC) and microbiological and/or clinical response, pharmacodynamic studies are important to identify exposure targets most closely linked with positive therapeutic outcomes [71–73]. In both infection models, the pharmacodynamic index most notably associated with tedizolid activity was free AUC0–24h divided by the MIC (fAUC0–24h/MIC) [71–73].
Louie et al. [73] conducted an initial dose fraction study against MRSA in a neutropenic murine thigh infection model. Neutropenic mice were inoculated with MRSA ATCC 33591 and subsequently treated with tedizolid at doses of 0–100 mg/kg/day administered as single doses at time zero, two equally divided doses every 12 h, or four equally divided doses every 6 h. Bacterial colony counts in the posterior thigh muscle were done before treatment and after 24 h. Using measured plasma concentrations and an estimated protein binding of 80 %, the fAUC0–24h/MIC ratio had the highest correlation (r 2 = 0.984) with treatment effect compared with the maximum free plasma concentration divided by the MIC (fCmax/MIC) (r 2 = 0.757) and percent of time that free plasma concentrations exceeded the MIC (f%T > MIC) (r 2 = 0.624). A fAUC0–24h/MIC of approximately 50 was associated with bacteriostasis whereas a value of 106 was associated with 1 log10 CFU/g of bacterial kill.
The pharmacodynamics of tedizolid were further examined in a neutropenic murine pneumonia model using four isolates of MSSA and seven isolates of MRSA [72]. Mice were treated with either tedizolid or linezolid 2 h after pulmonary challenge. Based on an estimated protein binding of 85 %, the mean fAUC0–24h/MIC associated with bacteriostasis was 19 for linezolid and 20 for tedizolid. A 1 log10 CFU reduction in bacterial burden in lung tissue was associated with fAUC0–24h/MIC values of 46 and 35 for linezolid and tedizolid, respectively. The significant values of 20 and 35 for bacteriostasis and 1 log10 CFU/g bacterial kill, respectively, were somewhat lower than those identified by the previous study by Louie et al. [73], even after adjusting their results for the differences in estimated protein binding (i.e., 38 and 80, respectively, using a protein binding of 85 %).
The activity and pharmacodynamics of tedizolid against MSSA and MRSA (tedizolid MICs 0.25–0.5 mg/L) have also been studied in immunocompetent animal infection models. Keel et al. [74] examined the pharmacodynamics of human-simulated exposures of tedizolid 200 mg in an immunocompetent murine thigh infection model. These exposures resulted in bacterial reductions of 1.04–1.80 log10 CFU/mL after 24 h. Similar reductions in bacterial density were observed in the comparator group with human-simulated exposures to linezolid. The finding of dose-response enhancement in the presence of granulocytes in a murine thigh infection model was also evaluated by Drusano et al. [75], who reported a 16-fold increase in antibacterial effects after 24 h (which increased at the 48- and 72-h timepoints) in non-neutropenic versus neutropenic animals. Thus it is clear that the presence of granulocytes significantly (25-fold on average) increases the activity of tedizolid [71]. Since neutropenic patients were excluded from clinical efficacy studies (as is the ethical standard for these types of initial clinical trials), tedizolid carries a warning that safety and efficacy in patients with neutropenia (neutrophil counts <1,000 cells/mm3) have not been adequately evaluated [76].
Flanagan et al. [59] recently performed a population pharmacokinetic, exposure-response, and target attainment study. Based on Phase 3 data evaluating 200-mg once-daily tedizolid for acute bacterial skin and skin structure infections (ABSSSI), these investigators reported no relationships in various efficacy outcomes and estimated tedizolid exposure. Target attainment simulations were performed for the 200 mg tedizolid dose used in clinical trials. Target attainment simulations performed for the 200 mg tedizolid dose indicated a 98.31 % probability of attaining the target measure (fAUC/MIC = 3) against Staphylococcus aureus with MIC ≤ 0.5 mg/L. The simulated probability of target attainment is due the 70.70 % for an MIC value of 1 mg/L and approaches 0 for MIC values of 2 mg/L or more. When they performed safety data modeling using once-daily doses up to 400 mg of tedizolid, they reported a small increase in the probability of an adverse event with increasing model-estimated tedizolid exposure (this effect was not observed with the 200-mg dose). There were no trends in neutrophil or platelet counts with increasing tedizolid exposure. The researchers conclude based on these data that 200 mg tedizolid once daily was the optimum dose for treatment of ABSSSI. Based on these data it is clear that fAUC/MIC is the tedizolid pharmacodynamic parameter associated with bacterial eradication and clinical efficacy.
8 Animal Studies
The efficacy of tedizolid in various types of infections, as well as the safety of long-term administration, has been evaluated using animal models.
Choi et al. [77], using a murine infection model, have described the activity of tedizolid against both penicillin-susceptible and penicillin-resistant Streptococcus pneumoniae (PRSP) [77]. In the study, the authors examined the efficacy of both tedizolid and linezolid in two different infection models, a PRSP lethal systemic infection and a penicillin-susceptible S. pneumoniae (PSSP) pneumonia infection. In the lethal systemic infection model a single dose of tedizolid or linezolid, either orally or intravenously, was administered to mice 1 h post-infection with a sufficient amount of bacteria to kill 100 % of the control mice. Following oral administration, the ED50 of tedizolid, using survival data 7 days after infection, was approximately twofold lower than linezolid for each of the four strains tested; however, the 95 % confidence intervals for each strain did overlap. After intravenous administration, tedizolid’s ED50 values ranged from three- to sixfold lower than linezolid, and these results were statistically significant for each strain tested. In the PSSP pneumonia model, a clinical isolate found to be highly virulent in mice when administered into the lungs was used to compare the in vivo efficacy of oral tedizolid and linezolid. Starting 4 h post-infection, the infected mice received either oral tedizolid (QD) or linezolid (BID) over a range of doses for 48 h, and survival was recorded daily for 15 days and compared to untreated infected control mice. A 100 % survival rate was achieved with tedizolid at a dose of 10 mg/kg/day, while linezolid required a 20 mg/kg dose twice daily to achieve this same result. The calculated ED50 values at day 15 for tedizolid and linezolid, respectively, were 2.80 and 8.09 mg/kg/day, with no overlap in the 95 % confidence intervals for the two antibiotics. The authors suggested that the enhanced potency of tedizolid in the mouse pneumonia model might reflect better penetration into the pulmonary epithelial lining fluid (ELF), as well as increases in in vitro activity, as was found in a separate study in healthy human volunteers [55]. Another study utilizing a mouse pneumonia model examined the efficacy of tedizolid, linezolid, and vancomycin against infection with MRSA and found tedizolid and linezolid to be statistically superior to vancomycin in reducing bacterial density at doses that simulated human ELF exposures [78].
As briefly alluded to, Keel et al.[74] examined the efficacies of 3-day human simulated exposures of tedizolid (200 mg once daily) and linezolid (600 mg twice daily) against S. aureus in an immunocompetent murine thigh infection model. The study utilized five strains of S. aureus (four MRSA and one MSSA) and efficacy was determined by evaluating the reduction in mean bacterial density. The mean bacterial density of the control mice prior to initiation of dosing was 6.89 log10 colony forming units (CFU) and increased to 7.34, 6.94, and 7.08 log10 CFU after 24, 48, and 72 h, respectively. The human simulated tedizolid regimen caused 1.04–1.8 and 2.68–3.72 log10 CFU reductions at 24 and 72 h, respectively, against all of the isolates tested in the model. Similarly, the linezolid human simulated regimen caused 2.02 and 3.76 log10 CFU reduction at 24 and 72 h, respectively.
Long-term therapy with linezolid has been associated with both peripheral and optic neuropathy [6]. Hosako et al. [79] examined the neurotoxic potential of tedizolid following long-term administration to rats to determine the likelihood of this toxicity in humans. In this study, several cohorts of male and female rats received either tedizolid (n = 366), over a range of doses, or vehicle control (n = 120) once daily for 1, 3, 6, or 9 months. The doses used in the study were chosen to yield exposures up to tenfold greater than achieved in human patients with recommended dosing. The authors reported that no tedizolid-related effects were observed in assessments of survival, food consumption, functional observational battery testing (which included 45 different observations), ophthalmic evaluations, brain weights and measurements, and both macro- and microscopic neuropathological findings at 1, 3, 6, and 9 months. Additionally, average steady-state tedizolid exposures were approximately sixfold higher than those observed in humans. The results of this rigorous study showed no evidence of neuropathy in rats after long-term administration, even at high tedizolid exposures. While the applicability of these data to clinical practice remain to be seen, these results may suggest a lower risk for neuropathy with tedizolid than with linezolid, but more data is needed.
9 Clinical Trials
Currently, one Phase II and two Phase III clinical trials have evaluated the efficacy and safety (safety will be discussed in Sect. 11) of tedizolid for the treatment of acute bacterial skin and skin-structure infections (ABSSSIs). The results of these clinical trials are summarized in Table 5.
The efficacy and safety of tedizolid in patients with complicated skin and skin-structure infections (cSSSIs) was evaluated in a Phase II randomized, double-blind, dose-ranging study (Table 5) [59]. In the study, three different doses (200, 300, or 400 mg) of oral, once-daily tedizolid phosphate were administered to patients aged 18–75 years for a duration of 5–7 days. In addition to the aforementioned age requirement, the study included male or female in- and outpatients diagnosed with a cSSSI caused by a suspected or confirmed Gram-positive pathogen. Infections under study were abscesses (with at least 2 cm of surrounding induration or requiring incision and drainage), surgical or post-traumatic wounds, and deep, extensive cellulitis. In order to be included in the study patients also were required to possess at least two signs and symptoms of infection and at least one additional sign of systemic infection, unless the patients had a lesion greater than 5 cm in diameter. Key exclusion criteria for the study were hepatic disease [aspartate aminotransferase (AST) or alanine aminotransferase (ALT) >3 × upper limit of normal (ULN)], decreased renal function (CrCl <52 mL/min), and neutropenia. Patients with diagnoses of diabetic foot infections, gangrene, osteomyelitis, or endocarditis, as well as any life-threatening condition or bacteremia, were also excluded from participating. In total 188 patients received at least one dose of the study drug, with 63, 63, and 62 patients in the 200-, 300-, and 400-mg groups, respectively, with all three dosage groups being well balanced across all demographic data. As reported in Table 5, the primary outcome of the study was to determine the clinical response rate of each dose group at the test-of-cure (TOC) visit (7–14 days post-treatment) in the clinically evaluableFootnote 1 (CE) and clinical modified-intent-to-treat (cMITT)Footnote 2 populations. Clinical response rates for the 200-, 300-, and 400-mg groups in the CE population were 98.2, 94.4, and 94.4 %, respectively, at the TOC visit. In the cMITT population, response rates at the same visit were 88.9, 88.9, and 85.5 % for the 200-, 300-, and 400-mg groups, respectively. The secondary objectives of the study, including the results, are also reported in Table 5. Safety will be discussed further in Sect. 10, but no patient discontinued therapy in the study due to an adverse effect.
The ESTABLISH-1 clinical trial was a Phase III randomized, double-blind, multicenter, multinational, noninferiority trial that compared the efficacy and safety of 6-day, 200 mg, once-daily oral tedizolid phosphate to 10-day, 600 mg, twice-daily oral linezolid for the treatment of adults with ABSSSIs [80, 81]. The study was the first trial performed according to the draft guidance document describing the evaluation of new treatments for ABSSSIs released by the US FDA in 2010 [82, 83]. The study included both male and female patients aged 18 years or older with cellulitis/erysipelas, major cutaneous abscess, or wound infection surrounded by erythema with a minimum total lesion surface area of 75 cm2. Inclusion also required a documented or suspected Gram-positive pathogen as well as at least 1 local and 1 regional or 1 systemic sign of infection. Key criteria excluding patients were uncomplicated ABSSSIs, suspected or documented Gram-negative pathogen, use of systemic or topical antibiotics with Gram-positive activity within 96 h before the first dose of study drug, and previous treatment failure of the same infection site. In total, 667 patients from 54 study centers were enrolled and randomized on a 1:1 basis to treatment with either tedizolid (n = 332) or linezolid (n = 335) in the regimens previously mentioned, with both groups being similar with respect to demographic data. As reported in Table 5, the primary outcome assessed by the study was the early clinical response (no increase in lesion surface area from baseline and oral temperature of ≤37.6 °C, confirmed by a second temperature measurement within 24 h) in the intention-to-treat (ITT)Footnote 3 population. Early clinical response rates were 79.5 % (95 % CI 74.8–83.7) for tedizolid and 79.4 % (95 % CI 74.7–83.6) for linezolid, yielding an absolute treatment difference of 0.1 % (95 % CI −6.1 to 6.2). The lower limit of the 95 % CI was above the −10 % cut-off that was the predefined requirement for non-inferiority. A total of 8.1 and 10.4 %, respectively, for tedizolid and linezolid were true non-responders with either the spread of skin/skin structure lesions and/or temperatures >37.6 °C at the 48- to 72-h assessment. The secondary outcomes and results are reported in Table 5. Of note, all of the secondary analyses also demonstrated non-inferiority of tedizolid versus linezolid in each of the outcomes measured. Two patients in each treatment group discontinued due to adverse effects, which will be discussed further in the next section.
A second Phase III multicenter, multinational, clinical trial, ESTABLISH-2, was completed to elaborate on the potential role for intravenous tedizolid in a strategy for the management of ABSSSIs [82, 84]. ESTABLISH-2 was a randomized, double-blind, non-inferiority trial comparing intravenous once-daily tedizolid phosphate (200 mg for 6 days) to intravenous twice-daily linezolid (600 mg twice daily for 10 days), with an optional switch to oral therapy after at least one day of IV study drug. Inclusion and exclusion criteria were similar to ESTABLISH-1. In total 666 patients at 58 centers in nine countries were enrolled and randomly assigned on a 1:1 basis to either treatment with tedizolid (n = 332) or linezolid (n = 334) in the regimens previously outlined with both treatment groups being similar with respect to their baseline demographic data. The primary outcome was clinical response, which was a 20 % or more reduction in lesion area from baseline. In the tedizolid group 85.2 % of patients and in the linezolid group 83.6 % of patients achieved early clinical response at the 48- to 72-h assessment, for an absolute difference of 2.6 % (95 % CI −3.0 to 8.2). As was the case in the previous Phase III trial, a −10 % cut-off for the lower limit of the 95 % CI of the difference of the primary endpoint was the predefined requirement for non-inferiority, which was met. Along with these results, the secondary objectives and results are reported in Table 5. Clinical success rates were similar between both groups at all subsequent points in time, and both groups had similar microbiologic outcomes. Because the study allowed for the optional switch to oral sequential therapy, it is important to note that the mean time to oral switch was similar between tedizolid (1.7 days) and linezolid (1.8 days). A total of five patients, one in the tedizolid group and four in the linezolid group, discontinued the study drug due to adverse effects.
The similar design and outcome measures of the previously mentioned trials lend themselves to integration and pooling of the data. Based on the pooled data from both Phase III studies, 6 days of once-daily tedizolid 200 mg treatment was demonstrated to be non-inferior to 10 days of twice-daily linezolid 600 mg at all time points evaluated [85, 86]. In addition, Cubist Pharmaceuticals has recently (June 2014) initiated a Phase III clinical trial (clincaltrials.gov NCT02019420) comparing once-daily tedizolid phosphate for 7 days versus twice-daily linezolid for 10 days in patients with hospital-acquired bacterial pneumonia (HABP) and ventilated nosocomial pneumonia (VNP).
10 Adverse Effects
The safety, tolerability, and adverse effect profile of tedizolid has been evaluated throughout its development, including early studies in healthy volunteers as well as in several Phase II and III clinical trials [53, 57, 59, 63, 80, 82, 87–91].
The safety and tolerability of tedizolid was examined in a placebo-controlled, multiple-ascending dose study involving 40 healthy male and female volunteers who received 21 days of oral tedizolid administered in doses of 200, 300, or 400 mg daily or 600 mg linezolid administered twice daily [53]. Thirty-six of the 40 subjects enrolled completed the study. One of eight subjects in the tedizolid 200 mg group, none of eight subjects in the tedizolid 300 mg group, two of eight in the tedizolid 400 mg group, and one of eight in the linezolid 600 mg group discontinued therapy due to meeting predefined thresholds in laboratory parameters. One subject receiving oral tedizolid 200 mg demonstrated an ALT value more than twice the ULN after 11 days. A reticulocyte count less than 75 % the lower limit of normal (LLN) was observed after 18 days of 400 mg tedizolid once daily in one subject and after 18 days of linezolid 600 mg twice daily in another. The final withdrawal was observed after 19 days of 400 mg once-daily tedizolid due to a white cell count less than 75 % the LLN. It is important to note that these predefined thresholds were conservative and that all of the subject withdrawals occurred well after the 6-day duration of tedizolid therapy. Additionally, all laboratory parameters returned to normal after discontinuation of study drug in those withdrawn. Overall, tedizolid was well tolerated with the most commonly reported adverse effects being gastrointestinal (nausea, stomach discomfort) of mild to moderate severity [87].
Due to the potential for patients to develop peripheral and optic neuropathies while receiving linezolid therapy, a Phase I study by Fang et al. [88] examined the effects of 10-day oral tedizolid 200 mg therapy on neurologic and ophthalmologic assessments in 72 healthy volunteers [88]. No subjects discontinued the study due to adverse effects and the study reported no changes suggestive of peripheral or optic neuropathy in anyone receiving study drug. The most common treatment-emergent adverse effects (TEAEs) reported were headache, musculoskeletal pain, nausea, and diarrhea consistent with other studies previously completed.
A Phase II study by Prokocimer et al. [80], described in an earlier section, evaluated the safety of 5- to 7-day oral tedizolid therapy in patients with cSSSIs. As previously mentioned, no patient discontinued the study drug due to an adverse effect. TEAEs were reported in 69.1 % of patients receiving at least one dose of study drug, with the majority graded mild (72.3 %) or moderate (24.6 %) in severity. Five patients (2.7 %) experienced a serious adverse event, although none were considered potentially drug related. Hematologic parameters were unaltered in the study, consistent with earlier studies in healthy adults demonstrating no changes after 5 to 7 days of oral once-daily dosing [87]. Results from electrocardiogram (ECG) data revealed no patients with a Bazett-corrected QT interval >500 ms, consistent with results from a separate Phase I study by Fang et al. [89], which reported no meaningful changes in ECG data after single doses of oral tedizolid 200 or 1,200 mg.
As previously described, two Phase III studies involving both oral and IV once-daily tedizolid 200 mg have been completed [80, 82]. Due to the similar design of these two trials, an integrated safety analysis comparing tedizolid to linezolid was completed by Fang et al. [90]. In total, 1,333 patients were randomized into the two studies with 664 receiving tedizolid and 669 linezolid. Overall TEAE rates were similar between the groups, with 42.7 and 43.2 % of patients experiencing a TEAE for tedizolid and linezolid, respectively. Rates of TEAEs leading to discontinuation of study drug and serious TEAEs were also similar between the two drugs. The incidence of gastrointestinal TEAEs, however, was significantly (p = 0.0018) lower (16.0 vs. 23.0 %) for tedizolid versus linezolid. Importantly, the incidence of low platelet counts was significantly (p = 0.0175) lower for tedizolid, with 2.1 vs. 4.5 % of patients’ platelet counts dropping below 75 % the LLN for tedizolid and linezolid, respectively. Recently Lodise et al. [91] characterized the platelet profile of patients receiving tedizolid compared to linezolid over the course of treatment using pooled data from the ESTABLISH clinical trials. The occurrences of clinically defined and statistical analysis plan-specified reduced platelet counts were both assessed at study days 7–9, days 11–13, and the post-therapy evaluation (PTE) visit. At days 7–9, incidences of reduced platelet counts were low and largely similar between groups. The only notable difference was a lower incidence of thrombocytopenia (platelet counts <150,000 cells/mm3) among patients who received tedizolid (3.2 %) compared with linezolid (5.6 %). At days 11–13, patients who received tedizolid had lower incidences of platelet counts <150,000 (−5.9 %), <112,500 (−2.4 %), and <100,000 (−1.9 %) cells/mm3 than patients in the linezolid group. Similar differences were noted at the PTE visit. The researchers found that the findings from ESTABLISH-1 and ESTABLISH-2 suggested that 6 days of tedizolid 200 mg daily conferred a low potential for reduced platelet counts among patients with ABSSSI. These findings support the notion that tedizolid may have less impact on hematologic parameters than its predecessor linezolid.
11 Drug Interactions
As previously discussed, tedizolid is primarily excreted by the liver as an inactive, non-circulating sulphate, and thus the potential for drug interactions with the cytochrome P450 system is minimal due to its low affinity for these enzymatic pathways [76]. There have been case reports of patients experiencing serotonergic side effects and/or serotonin toxicity, a potentially fatal condition, when linezolid, a weak reversible monoamine oxidase (MAO) inhibitor, has been taken concomitantly with a serotonin agonist such as selective serotonin reuptake inhibitors (SSRIs) and, as such, its label contraindicates use with many serotonergic agents [6, 92]. To determine the extent of MAO inhibition by tedizolid, and thus its potential for drug–drug and drug-food interactions, Flanagan et al. [93, 94] conducted several studies in both animals and healthy volunteers. Tedizolid demonstrated weak, reversible inhibition of MAO in vitro with mean IC50 values of 8.7 and 5.7 uM for MAO-A and MAO-B, respectively. For comparison, both tedizolid and linezolid, whose corresponding IC50 values were 46.0 and 2.1 μM, are three orders of magnitude less potent than the MAO inhibitor clorgyline [93]. In a 5-hydroxytryptophan murine head twitch model validated to assess serotonergic activity, tedizolid did not increase head twitch response at any dose examined, even at concentrations that exceeded the 200 mg/day tedizolid C max in humans by up to 25-fold. In contrast, linezolid, at a dose that resulted in concentration similar to the C max in humans receiving 600 mg twice daily, did significantly (p < 0.05) elevate the number of head twitches in the mice [93, 94].
The potential tedizolid interaction with oral tyramine or pseudoephedrine was evaluated in two randomized, double-blind, placebo-controlled, steady-state crossover studies involving healthy human volunteers [94, 95]. In the tyramine sensitivity study the dose of tyramine required to increase systolic blood pressure by ≥30 mmHg (TYR30) was used to evaluate potential MAO interactions. Only one out of seven subjects receiving tedizolid exceeded the clinically meaningful threshold of 2 for the tyramine sensitivity factor (TSF), calculated as TYR30 with placebo divided by TYR30 with drug, compared to eight out of ten in those receiving linezolid. Additionally, the lowest TYR30 dose of any tedizolid subject was 275 mg, while a typical tyramine-rich meal is expected to contain no more than 40 mg of tyramine [93]. The pseudoephedrine challenge study found no significant difference in mean maximum increases in blood pressure and heart rate between tedizolid and placebo. The potential confounding pharmacokinetic interaction with tedizolid and pseudoephedrine reported that no meaningful interactions occur supporting the previous results [95]. While the results of the animal and human studies suggest low probability for MAO related adverse events with tedizolid therapy, the use of tedizolid outside of controlled trials will provide clarification with regard to clinically meaningful interactions [8].
12 Place in Therapy
With its recent FDA approval for the treatment ABSSSIs, tedizolid offers clinicians another alternative in the fight against infections caused by MDR Gram-positive pathogens. In addition, recent work suggests that linezolid can be used as a surrogate for tedizolid susceptibility testing [96]. Tedizolid’s potent in vitro activity against a broad-spectrum of Gram-positive organisms, high bioavailability with oral administration, long half-life allowing once-daily dosing, lack of dosage adjustment in patients with renal or hepatic dysfunction, short (6-day) course of therapy, and availability of both oral and IV routes of administration, make tedizolid an attractive antimicrobial for use in both hospital and community settings. Tedizolid also displays low potential to interact with serotonergic agents, MAOIs, adrenergic agents, and foods with high tyramine content. In addition, its apparent reduced myelosuppressive activity will provide clinicians with a potentially safer oxazolidinone. Additionally, the ability of tedizolid to retain some activity against certain linezolid-resistant pathogens, such as strains harboring the cfr gene, may prove to be invaluable if this form of linezolid resistance continues to spread. While tedizolid does possess many qualities of the ideal antimicrobial, until further clinical trials in other types of infections (HABP/VNP) are completed, its role in therapy may be limited to ABSSSIs presently treated with linezolid. Pharmacoeconomic considerations will also help to further elucidate tedizolid’s place in the clinician’s arsenal of antimicrobials against Gram-positive infections.
Notes
CE population: Patients receiving at least 5 days of study therapy, having a TOC assessment, and no other confounding events or factors. Twenty-eight patients were excluded from the CE group due to indeterminate clinical status (N = 16), visit window violation (N = 4), concomitant medication (N = 4), confounding medical event (N = 1), and Gram-negative pathogen (N = 1). Two patients had multiple reasons.
cMITT population: All randomized patients with a diagnosis of cSSSI (including ten patients who discontinued the study drug: 200 mg N = 4, 300 mg N = 3, 400 mg N = 3).
ITT population: All patients randomized to treatment.
References
Moellering RC Jr. MRSA: the first half century. J Antimicrob Chemother. 2012;67(1):4–11.
Moellering RC Jr. Tedizolid: a novel oxazolidinone for Gram-positive infections. Clin Infect Dis. 2014;58(Suppl 1):S1–3.
Stryjewski ME, Corey GR. Methicillin-resistant Staphylococcus aureus: an evolving pathogen. Clin Infect Dis. 2014;58(Suppl 1):S10–9.
Rybak JM, Barber KE, Rybak MJ. Current and prospective treatments for multidrug-resistant gram-positive infections. Expert Opin Pharmacother. 2013;14(14):1919–32.
Rodvold KA, McConeghy KW. Methicillin-resistant Staphylococcus aureus therapy: past, present, and future. Clin Infect Dis. 2014;58(Suppl 1):S20–7.
Herrmann DJ, Peppard WJ, Ledeboer NA, et al. Linezolid for the treatment of drug-resistant infections. Expert Rev Anti Infect Ther. 2008;6(6):825–48.
Burke SL, Rose WE. New pharmacological treatments for methicillin-resistant Staphylococcus aureus infections. Expert Opin Pharmacother. 2014;15(4):483–91.
Kisgen JJ, Mansour H, Unger NR, et al. Tedizolid: a new oxazolidinone antimicrobial. Am J Health Syst Pharm. 2014;71(8):621–33.
Kanafani ZA, Corey GR. Tedizolid (TR-701): a new oxazolidinone with enhanced potency. Expert Opin Investig Drugs. 2012;21(4):515–22.
Shen K, Yoshikawa K, Tanaka T, et al. Pharmacokinetic profile of tedizolid phosphate, an antibiotic prodrug, in healthy Japanese adults [abstract no. PS925 plus poster]. In: 43rd Society of Critical Care Medicine Critical Care Congress: San Francisco; 2014.
Tanaka T, Hayashi Y, Okumura K, et al. Pharmacokinetics of 7-day multiple-dose tedizolid phosphate in healthy Japanese subjects in a phase 1 placebo-controlled study [abstract no. P1723 plus poster]. In: 24th European Congress of Clinical Microbiology and Infectious Diseases: Barcelona; 2014.
Locke JB, Finn J, Hilgers M, et al. Structure-activity relationships of diverse oxazolidinones for linezolid-resistant Staphylococcus aureus strains possessing the cfr methyltransferase gene or ribosomal mutations. Antimicrob Agents Chemother. 2010;54(12):5337–43.
Barbachyn MR, Ford CW. Oxazolidinone structure-activity relationships leading to linezolid. Angew Chem Int Ed Engl. 2003;42(18):2010–23.
Michalska K, Karpiuk I, Krol M, et al. Recent development of potent analogues of oxazolidinone antibacterial agents. Bioorg Med Chem. 2013;21(3):577–91.
Locke J, Finn J, Hilgers M, et al. Structure-activity relationships of diverse oxazolidinones for linezolid-resistant S. aureus strains possessing the cfr methyl transferase gene or ribsomal mutations [abstract no. C1-1432 plus poster]. In: 50th Annual Interscience Conference on Antimicrobial Agents and Chemotherapy: Boston; 2010.
Shaw KJ, Barbachyn MR. The oxazolidinones: past, present, and future. Ann N Y Acad Sci. 2011;1241:48–70.
Leach KL, Swaney SM, Colca JR, et al. The site of action of oxazolidinone antibiotics in living bacteria and in human mitochondria. Mol Cell. 2007;26(3):393–402.
Shaw KJ, Poppe S, Schaadt R, et al. In vitro activity of TR-700, the antibacterial moiety of the prodrug TR-701, against linezolid-resistant strains. Antimicrob Agents Chemother. 2008;52(12):4442–7.
Locke J, Zuill D, Scharn CR, et al. Identification and characterization of linezolid-resistant USA300 Staphylococcus aureus isolates collected from a New York city medical center possessing the cfr multidrug resistant gene. Antimicrob Agents Chemother 2014;58(11):6949–52.
Locke JB, Zurenko GE, Shaw KJ, et al. Tedizolid for the management of human infections: in vitro characteristics. Clin Infect Dis. 2014;58(Suppl 1):S35–42.
Mendes RE, Deshpande LM, Jones RN. Linezolid update: stable in vitro activity following more than a decade of clinical use and summary of associated resistance mechanisms. Drug Resist Update. 2014;17(1–2):1–12.
Baos E, Candel FJ, Merino P, et al. Characterization and monitoring of linezolid-resistant clinical isolates of Staphylococcus epidermidis in an intensive care unit 4 years after an outbreak of infection by cfr-mediated linezolid-resistant Staphylococcus aureus. Diag Microbiol Infect Dis. 2013;76(3):325–9.
Cercenado E, Marin M, Gama B, et al. In vitro activity of tedizolid and radezolid against linezolid-resistant Gram-positive clinical isolates with genetically characterized resistance mechanisms [abstract no. C2-142 plus poster]. In: 52nd Annual Interscience Conference on Antimicrobial Agents and Chemotherapy: San Francisco; 2012.
Locke JB, Morales G, Hilgers M, et al. Elevated linezolid resistance in clinical cfr-positive Staphylococcus aureus isolates is associated with co-occurring mutations in ribosomal protein L3. Antimicrob Agents Chemother. 2010;54(12):5352–5.
Locke JB, Hilgers M, Shaw KJ. Novel ribosomal mutations in Staphylococcus aureus strains identified through selection with the oxazolidinones linezolid and torezolid (TR-700). Antimicrob Agents Chemother. 2009;53(12):5265–74.
Yum JH, Choi SH, Yong D, et al. Comparative in vitro activities of torezolid (DA-7157) against clinical isolates of aerobic and anaerobic bacteria in South Korea. Antimicrob Agents Chemother. 2010;54(12):5381–6.
Prokocimer P, Bien P, Deanda C, et al. In vitro activity and microbiological efficacy of tedizolid (TR-700) against Gram-positive clinical isolates from a phase 2 study of oral tedizolid phosphate (TR-701) in patients with complicated skin and skin structure infections. Antimicrob Agents Chemother. 2012;56(9):4608–13.
Betriu C, Morales G, Rodriguez-Avial I, et al. Comparative activities of TR-700 (torezolid) against staphylococcal blood isolates collected in Spain. Antimicrob Agents Chemother. 2010;54(5):2212–5.
Brown SD, Traczewski MM. Comparative in vitro antimicrobial activities of torezolid (TR-700), the active moiety of a new oxazolidinone, torezolid phosphate (TR-701), determination of tentative disk diffusion interpretive criteria, and quality control ranges. Antimicrob Agents Chemother. 2010;54(5):2063–9.
Schaadt R, Sweeney D, Shinabarger D, et al. In vitro activity of TR-700, the active ingredient of the antibacterial prodrug TR-701, a novel oxazolidinone antibacterial agent. Antimicrob Agents Chemother. 2009;53(8):3236–9.
Zhanel GG, Calic D, Schweizer F, et al. New lipoglycopeptides: a comparative review of dalbavancin, oritavancin and telavancin. Drugs. 2010;70(7):859–86.
Deane J, Simenauer A, Shaw K, et al. In vitro activity profile of tedizolid (TZD) and correlation with linezolid (LZD) activity against recent staphylococcal isolates [abstract no. E-1479 plus poster]. In: 52nd Annual Interscience Conference on Anitmicrobial Agents and Chemotherapy: San Francisco; 2012.
Deane J, Opiela C, Shah D, et al. Activity of tedizolid (TZD) and linezolid (LZD) against key bacterial pathogens associated with respiratory, skin/wound infections, and bacteremia [abstract no. C2-090 plus poster]. In: 53rd Annual Interscience Conference on Antimicrobial Agents and Chemotherapy: Denver; 2013.
Bien P, Locke J, Zuill D, et al. Longitudinal comparison of the in vitro activities of tedizolid and linezolid against Staphylococcus aureus clinical isolates from Europe (2009–2013) [abstract no. P1674 plus poster]. In: 24th European Congress of Clinical Microbiology and Infectious Diseases: Barcelona; 2014.
Deane J, Simenauer A, Shaw K, et al. Comparison of tedizolid (TZD) in vitro activity with that of other key Gram-postive agents against recent enterococcal and streptococcal isolates [abstract no. E-778 plus poster]. In: 52nd Annual Interscience Conference on Antimicrobial Agents and Chemotherapy: San Francisco; 2012.
Golden A, Baxter M, Nichol K, et al. In vitro activity of tedizolid against Canadian clinical Gram-postive pathogens, including hVISA and the CDC NARSA strains [abstract no. E-143 plus poster]. In: 53rd Annual Interscience Conference on Antimicrobial Agents and Chemotherapy: Denver; 2013.
Chen H, Yang Q, Zhang R, et al. In vitro antimicrobial activity of the novel oxazolidinone tedizolid and comparators agents against Staphylococcus aureus and linezolid-resistant Gram-positive cocci pathogens: a multicenter study in China. Intern J Antimicrob Agents. 2014;44(3):276–7.
Urbina O, Ferrandez O, Espona M, et al. Potential role of tedizolid phosphate in the treatment of acute bacterial skin infections. Drug Des Devel Ther. 2013;7:243–65.
Hong S, Choi S, Lim W, et al. In vitro activity of tedizolid against gram-positive cocci isolated from skin and soft tissue infections in Korea: a multicenter study [abstract no. F-1604]. In: 54th Annual Interscience Conference on Antimicrobial Agents and Chemotherapy (ICAAC): Washington, DC; 2014.
Barber KE, McRoberts JP, Raut A, et al. Evaluation of tedizolid against multi-drug resistant Staphylococcus aureus and enterococci [abstract no. F-1606]. In: 54th Annual Interscience Conference on Antimicrobial Agents and Chemotherapy (ICAAC): Washington, DC; 2014.
Kozlov R, Sukhorukova M, Edelstein M, et al. Tedizolid comparative in vitro activity against Staphylococcus aureus strains isolated in Russia. [abstract no. F-1605]. In: 54th Annual Interscience Conference on Antimicrobial Agents and Chemotherapy (ICAAC): Washington, DC; 2014.
Lee Y, Hong S, Choi S, et al. In vitro activity of tedizolid against gram-positive bacteria associated with nosocomial pneumonia in Korea: a multicenter study [abstract no. F-1619]. In: 54th Annual Interscience Conference on Antimicrobial Agents and Chemotherapy (ICAAC): Washington, DC; 2014.
Bien PA, Bensaci M, Prokocimer P. Results of the surveillance of tedizolid activity and resistance (STAR) program: in vitro susceptibility of gram-positive pathogens collected in 2009 to 2012 from the United States and Europe [abstract no. C-829]. In: 54th Interscience Conference on Antimicrobial Agents and Chemotherapy (ICAAC): Washington, DC: 2014.
Golden AR, Adam HJ, Baxter M, et al. Activity of tedizolid against gram-positive cocci from Canadian hospitals: CANWARD 2013 and 2014 [abstract no. F-1603]. In: 54th Interscience Conference on Antimicrobial Agents and Chemotherapy (ICAAC): Washington, DC; 2014.
Hackel M, Lynch T, Alder J, et al. In vitro activity of tedizolid and key comparators against Staphylococcus aureus isolated from Latin America, Eastern Europe, and the Pacific Rim: 2013 [abstract no. C-828]. In: 54th Interscience Conference on Antimicrobial Agents and Chemotherapy (ICAAC: Washington, DC; 2014.
Barber KE, McRoberts JP, Raut A, et al. Evaluation of tedizolid against multi-drug resistant Staphylococcus aureus isolated from Latin America, Eastern Europe, and the Pacific Rim: 2013 [abstract no. C-828]. In: 54th Interscience Conference on Antimicrobial Agents and Chemotherapy (ICAAC): Washington, DC; 2014.
Sanchez Garcia M, De la Torre MA, Morales G, et al. Clinical outbreak of linezolid-resistant Staphylococcus aureus in an intensive care unit. JAMA. 2010;303(22):2260–4.
Livermore DM, Mushtaq S, Warner M, et al. Activity of oxazolidinone TR-700 against linezolid-susceptible and -resistant staphylococci and enterococci. J Antimicrob Chemother. 2009;63(4):713–5.
Jones RN, Moet GJ, Sader HS, et al. TR-700 in vitro activity against and resistance mutation frequencies among Gram-positive pathogens. J Antimicrob Chemother. 2009;63(4):716–20.
Rodriguez-Avial I, Culebras E, Betriu C, et al. In vitro activity of tedizolid (TR-700) against linezolid-resistant staphylococci. J Antimicrob Chemother. 2012;67(1):167–9.
Vera-Cabrera L, Gonzalez E, Rendon A, et al. In vitro activities of DA-7157 and DA-7218 against Mycobacterium tuberculosis and Nocardia brasiliensis. Antimicrob Agents Chemother. 2006;50(9):3170–2.
Molina-Torres CA, Barba-Marines A, Valles-Guerra O, et al. Intracellular activity of tedizolid phosphate and ACH-702 versus Mycobacterium tuberculosis infected macrophages. Ann Clin Microbiol Antimicrob 2014;13. (13-0711-13-13).
Flanagan SD, Bien PA, Munoz KA, et al. Pharmacokinetics of tedizolid following oral administration: single and multiple dose, effect of food, and comparison of two solid forms of the prodrug. Pharmacotherapy. 2014;34(3):240–50.
Dreskin H, Munoz K, Fang E, et al. Safety and pharmacokinetics of single oral administration of tedizolid phosphate in healthy elderly subjects and adult control subjects [abstract no. A-1293 plus poster]. In: 52nd Annual Interscience Conference on Antimicrobial Agents and Chemotherapy: San Francisco; 2012.
Housman ST, Pope JS, Russomanno J, et al. Pulmonary disposition of tedizolid following administration of once-daily oral 200-milligram tedizolid phosphate in healthy adult volunteers. Antimicrob Agents Chemother. 2012;56(5):2627–34.
Bien P, Prokocimer P, Munoz K, et al. Absolute bioavailability of TR-701 FA and pharmacokinetics after single and multiple dose intravenous administration in healthy adult subjects [abstract no. A1-013 plus poster]. In: 50th Annual Interscience Conference on Antimicrobial Agents and Chemotherapy: Boston; 2010.
Flanagan S, Fang E, Munoz KA, et al. Single and multiple-dose pharmacokinetics and absolute bioavailability of tedizolid. Pharmacother. 2014;34(9):891–900.
Tanaka T, Hayashi Y, Okumura K, et al. Oral bioavailability of tedizolid in healthy Japanese subjects in a phase 1 study [abstract no. P1718 plus poster]. In: 24th European Congress of Clinical Microbiology and Infectious Diseases: Barcelona; 2014.
Flanagan S, Passarell J, Lu Q, et al. Tedizolid population pharmacokinetics exposure response and target attainment. Antimicrob Agents Chemother 2014 [Aug 18 Epub].
Sahre M, Sabarinath S, Grant M, et al. Skin and soft tissue concentrations of tedizolid (formerly torezolid), a novel oxazolidinone, following a single oral dose in healthy volunteers. Int J Antimicrob Agents. 2012;40(1):51–4.
Dreskin H, Boyea T, Barker J, et al. The absorption, metabolism, and excretion of orally administered [14C]-TR-701 FA in healthy subjects [abstract no. A2-033 plus poster]. 51st Annual Interscience Conference on Antimicrobial Agents and Chemotherapy; 17–20 Sep 2011; Chicago.
Ong V, Flanagan S, Fang E, et al. Absorption, distribution, metabolism, and excretion of the novel antibacterial prodrug tedizolid phosphate. Drug Metab Dispos. 2014;42(8):1275–84.
Flanagan S, Minassian SL, Morris D, et al. Pharmacokinetics of tedizolid in subjects with renal of hepatic impairment. Antimicrob Agents Chemother 2014;58(11):6471–6.
Flanagan S, Fang E, Dreskin H, et al. Pharmacokinetics of tedizolid in adolescent and elderly subjects and subjects with renal or hepatic impairment [abstract no. 707 plus poster]. In: IDWeek 2013: a joint meeting of IDSA, SHEA, HIVMA, and PIDS: San Francisco; 2013.
Flanagan S, Boyea T, Dreskin H, et al. A phase 1 study of orally administered tedizolid phosphate in subjects with moderate or severe hepatic impairment [abstract no. A-1295 plus poster]. In: 52nd Annual Interscience Conference on Antimicrobial Agents and Chemotherapy: San Francisco; 2012.
Flanagan S, Minassian S, Passarell J, et al. Tedizolid plasma pharmacokinetics are comparable in obese and nonobese patients and healthy subjects [abstract no. P1703 plus poster]. In: 24th European Congress of Clinical Microbiology and Infectious Diseases: Barcelona; 2014.
Dreskin H, Munoz K, Bradley J, et al. Safety and pharmacokinetics after single oral and IV administration of tedizolid phosphate in adolescent patients [abstract no. A-1292 plus poster]. In: 52nd Annual Interscience Conference on Antimicrobial Agents and Chemotherapy: San Francisco; 2012.
Bradley J, Arrieta A, Capparelli E, et al. A phase 1, open-label, multi-center, single-dose, pharmacokinetic, safety and tolerance study of oral tedizolid phosphate in 12 to 17 year old patients [abstract no. P-1420 plus poster]. In: 22nd European Congress of Clinical Microbiology and Infectious Diseases: London; 2012.
Capparelli E, Flanagan S, Bradley J, et al. Population pharmacokinetics (PK) of oral and intravenously administered tedizolid phosphate in adolescent patients [abstract no. P1460 plus poster]. In: IDWeek 2012: a joint meeting of IDSA, SHEA, HIVMA, and PIDS: San Diego; 2012.
Flanagan S, Passarell J, Fiedler-Kelly J, et al. Tedizolid dose adjustments based on patient characteristics are not warranted [abstract no. A-691]. In: 54th Interscience Conference on Antimicrobial Agents and Chemotherapy (ICAAC): Washington, DC; 2014.
Lodise TP, Drusano GL. Use of pharmacokinetic/pharmacodynamic systems analyses to inform dose selection of tedizolid phosphate. Clin Infect Dis. 2014;58(Suppl 1):S28–34.
Lepak AJ, Marchillo K, Pichereau S, et al. Comparative pharmacodynamics of the new oxazolidinone tedizolid phosphate and linezolid in a neutropenic murine Staphylococcus aureus pneumonia model. Antimicrob Agents Chemother. 2012;56(11):5916–22.
Louie A, Liu W, Kulawy R, et al. In vivo pharmacodynamics of torezolid phosphate (TR-701), a new oxazolidinone antibiotic, against methicillin-susceptible and methicillin-resistant Staphylococcus aureus strains in a mouse thigh infection model. Antimicrob Agents Chemother. 2011;55(7):3453–60.
Keel RA, Tessier PR, Crandon JL, et al. Comparative efficacies of human simulated exposures of tedizolid and linezolid against Staphylococcus aureus in the murine thigh infection model. Antimicrob Agents Chemother. 2012;56(8):4403–7.
Drusano GL, Liu W, Kulawy R, et al. Impact of granulocytes on the antimicrobial effect of tedizolid in a mouse thigh infection model. Antimicrob Agents Chemother. 2011;55(11):5300–5.
Cubist Pharmaceuticals. Sivextro prescribing information. 2014; http://sivextro.com/pdf/PrescribingInformation.pdf. Accessed 28 July 2014.
Choi S, Im W, Bartizal K. Activity of tedizolid phosphate (TR-701) in murine models of infection with penicillin-resistant and penicillin-sensitive Streptococcus pneumoniae. Antimicrob Agents Chemother. 2012;56(9):4713–7.
Tessier PR, Keel RA, Hagihara M, et al. Comparative in vivo efficacies of epithelial lining fluid exposures of tedizolid, linezolid, and vancomycin for methicillin-resistant Staphylococcus aureus in a mouse pneumonia model. Antimicrob Agents Chemother. 2012;56(5):2342–6.
Hosako H, Radovsky A, Draganov D, et al. Lack of neuropathy after long-term tedizolid phosphate administration in rats [abstract no. A-017b plus poster]. In: 53rd Annual Interscience Conference on Antimicrobial Agents and Chemotherapy: Denver; 2013.
Prokocimer P, De Anda C, Fang E, et al. Tedizolid phosphate vs linezolid for treatment of acute bacterial skin and skin structure infections: the ESTABLISH-1 randomized trial. JAMA. 2013;309(6):559–69.
Deanda C, Fang E, Green S, et al. Comparison of investigator assessed and programmatic clinical outcomes of tedizolid phosphate vs. linezolid in a phase 3 study in patients with ABSSSI [abstract no. L1-1665 plus poster]. In: 52nd Annual Interscience Conference on Antimicrobial Agents and Chemotherapy: San Francisco; 2012.
Moran GJ, Fang E, Corey GR, et al. Tedizolid for 6 days versus linezolid for 10 days for acute bacterial skin and skin-structure infections (ESTABLISH-2): a randomised, double-blind, phase 3, non-inferiority trial. Lancet Infect Dis. 2014;14(8):696–705.
Itani KM, Shorr AF. FDA guidance for ABSSSI trials: implications for conducting and interpreting clinical trials. Clin Infect Dis. 2014;58(Suppl 1):S4–9.
Fang E, De Anda C, Das A, et al. Efficacy and safety results from the ESTABLISH-2 ABSSSI study comparing IV and oral tedizolid phosphate and linezolid [abstract no. LB 2964 plus poster]. In: 23rd European Congress of Clinical Microbiology and Infectious Diseases: Berlin; 2013.
Barie P, Fang E, Minassian S, et al. Tedizolid versus linezolid in patients with wound infection or major abscess: pooled analysis of two phase 3 double-blind studies [abstract no. P-28 plus poster]. In: 34th Annual Meeting of the Surgical Infection Society: Baltimore; 2012.
De Anda C, Fang E, Das A, et al. Integrated results from two phase 3 studies comparing tedizolid phosphate 6 days vs. linezolid 10 days in patients with ABSSSI [abstract no. L-203 plus poster]. In: 53rd Annual Interscience Conference on Antimicrobial Agents and Chemotherapy: Denver; 2013.
Das D, Tulkens PM, Mehra P, et al. Tedizolid phosphate for the management of acute bacterial skin and skin structure infections: safety summary. Clin Infect Dis. 2014;58(Suppl 1):S51–7.
Fang E, Munoz K,Prokocimer P. Neurologic and ophthalmologic safety results with 10-day dosing of tedizolid phosphate [abstract no. 916 plus poster]. In: 43rd Society of Critical Care Medicine Critical Care Congress: San Francisco; 2014.
Fang E, Litwin J, Lewis W, et al. Effects of oral tedizolid phosphate on QTcF and other electrocardiogram (ECG) parameters [abstract no. P1648 plus poster]. In: 23rd European Congress of Clinical Microbiology and Infectious Diseases: Berlin; 2013.
Fang E, De Anda C,Prokocimer P. Safety and tolerability of tedizolid phosphate, a novel oxazolidinone, versus linezolid in two phase 3 studies in skin/skin structure infections [abstract no. PS295 plus poster]. In: 18th Asian Pacific Society of Respirology Meeting: Yokohama; 2013.
Lodise TP, Fang E, Minassian SL, et al. Platelet profile in patients with acute bacterial skin and skin structure infections receiving tedizolid or linezolid: Findings from the phase 3 ESTABLISH clinical trials. Antimicrob Agents Chemother 2014;58(12):7198–204.
Pfizer. Zyvoxam: Product Monograph. 2013; http://www.pfizer.ca/en/our_products/products/monograph/143. Accessed 23 June 2014.
Flanagan S, Bartizal K, Minassian SL, et al. In vitro, in vivo, and clinical studies of tedizolid to assess the potential for peripheral or central monoamine oxidase interactions. Antimicrob Agents Chemother. 2013;57(7):3060–6.
Flanagan S, Minassian S, Fang E, et al. Lack of MAO inhibition by tedizolid phosphate in clinical and nonclinical studies [abstract no. A-1295a plus poster]. In: 52nd Annual Interscience Conference on Antimicrobial Agents and Chemotherapy: San Francisco; 2012.
Flanagan S, Minassian S, Munoz K, et al. Lack of pharmacokinetic drug interactions of tedizolid phosphate with pseudoephedrine in healthy subjects [abstract no. P 921 plus poster]. In: 23rd European Congress of Clinical Microbiology and Infectious Disease: Berlin; 2013.
Zurenko G, Bien P, Bensaci M, et al. Use of linezolid susceptibility test results as a surrogate for the susceptibility of gram-positive pathogens to tedizolid a new oxazolidinone. Ann Clin Microbiol Antimicrob. 2014;13(1):46.
Acknowledgments
The authors are grateful to Cubist Pharmaceuticals, Inc. for their assistance with literature retrieval.
Conflict of interest
Dr. Zhanel has received research grants from Cubist Pharmaceuticals, Inc. Drs Love, Adam, Golden, Zelenitsky, Schweizer, Gorityala, Lagace-Weins, Rubinstein, Gin, Walkty, Gilmour, Hoban, Lynch, and Karlowsky have no conflicts of interest to declare.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article

Cite this article
Zhanel, G.G., Love, R., Adam, H. et al. Tedizolid: A Novel Oxazolidinone with Potent Activity Against Multidrug-Resistant Gram-Positive Pathogens. Drugs 75, 253–270 (2015). https://doi.org/10.1007/s40265-015-0352-7
Published:
Issue Date:
DOI: https://doi.org/10.1007/s40265-015-0352-7