
Abstract
Asthma remains a major health problem with significant morbidity, mortality and economic costs. In asthma, airway remodelling, which refers to all the microscopic structural changes seen in the airway tissue, has been recognised for many decades and remains one of the defining characteristics of the disease; however, it is still poorly understood. The detrimental pathophysiological consequences of some features of remodelling, like increased airway smooth muscle mass and subepithelial fibrosis, are well documented. However, whether targeting these by therapy would be beneficial is unknown. Although the prevailing thinking is that remodelling is an abnormal response to persistent airway inflammation, recent evidence, especially from studies of remodelling in asthmatic children, suggests that the two processes occur in parallel. The effects of asthma therapy on airway remodelling have not been studied extensively due to the challenges of obtaining airway tissue in the context of clinical trials. Corticosteroids remain the cornerstone of asthma therapy, and their effects on remodelling have been better studied than other drugs. Bronchial thermoplasty is the only asthma therapy to primarily target remodelling, although how it results in the apparent clinical benefits seen is not exactly clear. In this article we discuss the mechanisms of airway remodelling in asthma and review the effects of conventional and novel asthma therapies on the process.
Similar content being viewed by others
Avoid common mistakes on your manuscript.
Although airway remodelling is one of the defining characteristics of asthma, several aspects remain poorly understood, including its causes, natural history and, most importantly, the exact role it plays in the pathophysiology of asthma. |
Studying the effects of various asthma therapies on airway remodelling has been limited by the lack of non-invasive markers and uncertainty about the potential for reversibility in response to treatment and whether that would have any clinical or physiological benefit. |
1 Introduction
Asthma is a common chronic inflammatory airway disease affecting an estimated 300 million worldwide, which is expected to increase by one-third by 2025 [1]. It is associated with significant morbidity and mortality, especially in patients with the severe form of the disease, who account for 5–10 % of all patients in both the adult and the paediatric populations. Asthma is a heterogeneous condition characterised by various symptoms of cough, wheeze, chest tightness and breathlessness, which are closely associated with features of abnormal airway physiology, namely reversible airflow obstruction and airway hyper-responsiveness (AHR). These symptoms can undergo episodes of sudden deterioration, beyond day-to-day variation; these are termed exacerbations and are responsible for a significant proportion of the burden attributed to the disease [2]. Additionally, at the microscopic tissue level, asthma is also characterised by airway inflammation (hence the recent definition of asthma as primarily an inflammatory disease) and airway remodelling, a collective term describing all the microscopic structural changes seen in the airway tissue.
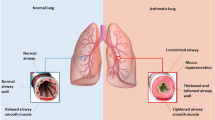
The pathogenesis of asthma is complex and far from being fully understood, with many aspects remaining controversial and debatable. Salter wrote in 1860 “Asthma is a disease about whose pathology more various and discrepant ideas prevail than about any other disease that could be named” [3]. Surprising, more than 150 years later, this statement remains largely true. This is mainly due to the extremely wide heterogeneity of various aspects of the disease, resulting in the lack of a unifying pathophysiological mechanism and, consequently, asthma is increasingly being regarded as a syndrome rather than a single disease entity. There is universal agreement that the two fundamental pathological domains of asthma pathogenesis are airway inflammation, with the involvement of a wide range of inflammatory cells, and airway remodelling changes, which include increased airway smooth muscle (ASM) mass, mucus gland hypertrophy, new vessel formation in the submucosa, subepithelial fibrosis due to excessive deposition of matrix proteins in the lamina reticularis, and epithelial changes including epithelial fragility and increased mucus secretion by goblet cells (Fig. 1). However, the relative contribution of these two domains or the exact nature of their relationship has not been well defined. Chronic inflammation in various pathological conditions other than asthma leads to structural changes in the tissue; as such, inflammation has long been thought of as the primary cause of airway remodelling in asthma, and hence should be the target of any therapy that aims at modifying remodelling. Consistent with this view, different aspects of remodelling have been clearly shown to be caused or worsened by inflammation. For example, subepithelial fibrosis, one of the most studied features of remodelling in asthma, has been closely associated with eosinophils-derived cytokines, namely interleukin (IL)-5 and transforming growth factor (TGF)-β [4, 5]. However, mounting evidence, especially from studies on the paediatric asthmatic population, has shown that the remodelling, at least in part, is not simply a consequence of inflammation. While airway inflammation and remodelling were shown in children aged 1–3 years with preschool wheeze, this was not seen in symptomatic infants with reversible airflow obstruction [6–8]. Supporting this, Saglani et al. [9] showed, in a neonatal murine model of allergic asthma, that both epithelial and extracellular matrix (ECM) remodelling occur simultaneously. This was also suggested by Kariyawasam et al. [10], who, in a group of asthmatics, showed that both inflammation and remodelling occurred 24 h after allergic challenge and only features of the latter persisted at 7 days. All this supports the argument that, in the natural history of asthma, airway inflammation and remodelling might occur in parallel and that remodelling can be a consequence of airway stimuli acting directly on the airway structural cells. Indeed, bronchoconstriction resulting from methacholine, a cholinergic drug normally used to measure AHR, was shown to induce remodelling in asthmatics without causing inflammation [11]. Furthermore, viruses have also been implicated in remodelling. Rhinovirus, which can cause asthma exacerbations, induces ECM remodelling in vitro and in animal models of asthma [12].

Endobronchial biopsy samples from subjects with asthma showing features of airway remodelling. Sections a and b (stained with alpha-smooth muscle actin antibodies) show epithelial metaplasia with loss of cilia, increased airway smooth muscle mass, myofibroblast hyperplasia (a), increased reticular basement membrane (RBM) thickness (a) and neoangiogenesis (b). Section c (stained with collagen III antibodies) shows excessive deposition of collagen III in the lamina propria and in the reticular basement membrane. Section d (stained with MUC5AC antibodies) shows goblet cell hyperplasia with increased stored mucin. ASM airway smooth muscle, RBM reticular basement membrane
To date, no asthma drug therapy has been used to primarily target airway remodelling. Anti-inflammatory drugs remain the most important drug category used in asthma, and in spite of recent advances in new highly targeted biological therapy in late phase development, there is still a desperate need for new effective asthma therapies. In this review article we summarize the current understanding of the role of airway remodelling in the pathogenesis of asthma and discuss the effects of both established and novel asthma therapies on remodelling (summarized in Table 1). Databases used for the literature search included the PubMed/MEDLINE database, the Cochrane library database and clinical trials databases ‘Current Controlled Trials’ and ClinicalTrials.gov. Keywords used included ‘asthma’, ‘airway remodelling’, ‘airway remodeling’ and ‘treatment’. Only articles in English with abstracts were reviewed.
2 How to Measure Airway Remodelling
This remains one of the major difficulties in studying airway remodelling in asthma. Asthma phenotypes (a phenotype is defined as observable characteristics of a disease that are determined from the interaction between genetics and the environment) are the end result of a long chain of biological events. In this paradigm, individuals with the susceptible genotypes, under the influence of factors that affect gene expression, have molecular and cellular changes that result in histological changes (i.e. remodelling), leading to airway physiological changes and other patients’ phenotypic characteristics. Consequently, in this model, remodelling is directly connected to physiology and thus one can use a lung function test as a surrogate measure for remodelling; however, this connection between airway remodelling and function has been difficult to prove. Although some features of airway remodelling have been linked with AHR and airflow obstruction, direct causality remains unproven [13, 14].
Biological samples used for studying remodelling, beside ex vivo tissue cultures and animal models of asthma, include lung tissue obtained from lung resections or post mortems, or much smaller airway biopsies obtained through flexible bronchoscopy. Specimens obtained from post mortems and lung resections have contributed significantly to our understanding of the disease, especially fatal asthma; however, they have obvious limitations. Surgical resections carry significant morbidity and thus, ethically, they cannot be conducted solely for research purposes, while post mortem samples are scarce and of limited value. Two types of biopsies can be obtained through flexible bronchoscopy for the purpose of studying remodelling: transbronchial and endobronchial biopsies. Transbronchial biopsies are obtained from distal small airways and include alveolar tissue, but carry a significant risk of pneumothorax or excessive bleeding and therefore are not widely performed.
The most common histological samples obtained for studying remodelling are endobronchial biopsies (EBBs), which are also obtained using flexible bronchoscopy but from large proximal airways under direct bronchoscopic vision [15]. They are comparatively safe, even in severe asthmatics, and can be used to assess the dynamics of remodelling and inflammation in the context of asthma drug trials. Nonetheless, EBBs also have significant limitations to be considered when used in measuring airway remodelling. These include (1) the inability of EBBs to represent changes in the whole airway, especially small airways, which have a crucial role in asthma pathogenesis, as EBBs are only obtained from the surface of carina of large airways; (2) large intra-subject variability, which is also affected by the sampling process; and finally (3) the possibility of reference space bias [16]. The lack of standardized guidelines for the methodology of assessing airway remodelling in biopsies creates another source of bias. Some measures of remodelling like reticular basement membrane (RBM) thickness have been well established, as most studies use the method described and validated by Sullivan et al. [17, 18], while other measures have been less well defined, e.g. RBM-ASM distance. Stereological methods have been suggested for cell counts; however, they are complicated and time consuming, and their superiority is less proven when using EBBs. Bronchoalveolar lavage (BAL) is very useful in assessing inflammatory changes in asthma; and although remodelling mediators in asthma have been studied in BAL, its value in measuring remodelling is limited [19, 20]. The same principle also limits the value of using induced sputum and exhaled breath condensate, beyond measuring mediators, to assess remodelling [21, 22].
Non-invasive imaging techniques for measuring airway remodelling in obstructive lung diseases have recently gained significant interest. The modalities of imaging involved include computed tomography (CT) and optical coherence tomography (OCT) [23]. In clinical settings, CT is not infrequently performed in patients with asthma, especially in patients with the severe form of the disease. This is usually done to either diagnose associated conditions like allergic bronchopulmonary aspergillosis or bronchiectasis, or to rule out other pathologies that can present with asthma-like symptoms, e.g. interstitial lung diseases. Beyond this, qualitative CT was proven to be a sensitive tool in investigating the structural changes in both the large and the small airway seen in asthma. Several studies have demonstrated increased bronchial wall thickness and reduced lumen area in asthmatics compared with healthy subjects [24]. In asthma, right upper lobe apical segmental bronchus (RB1) thickening correlates with AHR and spirometry [25, 26]. Gupta et al. [24] showed, in severe asthmatics, RB1 percentage wall area (WA %) was associated with airflow limitation and neutrophilic inflammation. Changes in CT-derived large airway measurements in response to treatments have been shown with corticosteroids and anti-IL5, indicating that CT is a valid dynamic biomarker of remodelling [27, 28]. CT has also been used to assess small airway in asthma indirectly using lung density measurements. Studies have showed significant air trapping in asthma, which correlated with severity, AHR and airflow obstruction [29, 30].
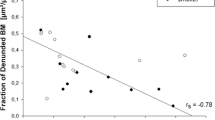
To our knowledge, in asthma, only three studies have attempted to link changes seen on CT to those observed in histological samples of the airways in adults. Kasahara et al. [31] reported in 49 asthmatics strong correlation between RBM thickness and CT-measured bronchial wall thickness. Moreover, Aysola et al. [26] showed that the increased airway wall thickness seen on CT of severe asthmatics (n = 32) positively correlated with epithelial thickness. In another smaller study, where the airway biopsies of 11 asthmatics were retrospectively analysed along with their CT parameters, airway CT-derived indices correlated with measured ASM and sub-epithelial areas, airway inflammation and subepithelial fibrosis [32]. In children with severe asthma, a significant correlation between bronchial wall thickness measured by CT and RBM thickness has been reported by de Blic et al. (n = 37, r = 0.34); however, this could not be further confirmed in a subsequent study by Saglani et al. (n = 27, r s = 0.066) [33, 34].
In OCT, near-infrared light is used to generate cross-sectional images. It uses shorter wavelength and greater frequency than ultrasound, thus giving sections with much higher resolution (10–20 μm) although with lower tissue penetration (2–3 mm) [35]. This theoretically would allow OCT to detect microscopic tissue changes, for example in ASM or RBM. OCT has been well established in cardiovascular research; however, its role in airway diseases is still emerging. Coxson et al. [36], in a group of smokers (n = 44), demonstrated that OCT was superior to CT in detecting changes in the airway wall that better correlated with changes in spirometry. In asthma, a recent report showed that OCT performed in patients undergoing bronchial thermoplasty (BT) was able to detect ultrastructural changes in airway remodelling, including reduction in ASM mass [37]. Although these results are very exciting, they are only preliminary, and more studies on the use OCT in airway diseases are needed.
3 Components and Mechanisms of Airway Remodelling in Asthma
3.1 Airway Smooth Muscle (ASM) Remodelling
ASM cells are normally present in most airways where they form bundles arranged geodesically around the airway lumen. As a result of this geodesic arrangement, ASM contraction leads to both narrowing and shortening of the airways. This is important in asthma where, as a consequence of remodelling, airways are stiffer and resistant to shortening, hence most of the force generated from ASM contraction translates into excessive airway narrowing [38]. The physiological function of ASM is not clear. While some argue that ASM has a role in the maintenance of bronchial tone, clearance of respiratory secretions and regulation of ventilation, others consider ASM a vestigial component of the airway, with no function [39, 40]. Increased ASM mass was the first remodelling feature to be observed in asthma. Huber and Koesser [41] in 1922 reported increased ASM mass in patients who died from asthma when compared with controls who died from other conditions. This has since been confirmed in patients with asthma of all severities and in small and large airways. Until the significance of inflammation had been suggested, asthma was primarily considered an ASM dysfunction.
Increased ASM mass in asthma is due to hyperplasia and possibly hypertrophy, although the evidence for the latter is incongruent [17, 42]. ASM hypertrophy was shown in some studies but not in others [17, 42, 43]. The origins of ASM hyperplasia in asthma are the subject of great debate. Increased proliferative capacity of asthmatic ASM cells in vitro has been shown in some, but not all, studies investigating the issue and was never demonstrated in vivo in asthma [44–46]. Other cells have been suggested as possible origins of ASM hyperplasia; these include tissue-resident mesenchymal cells, epithelial cells (which can undergo mesenchymal transition), and fibrocytes, which are monocyte-like blood cells that originate in the bone marrow and are capable of entering the tissue and transforming into contractile mesenchymal cells. However, the evidence for each is far from conclusive [47].
The significance of ASM contraction in asthma is obvious, as it is the effector of reversible airway obstruction and of AHR. Most bronchodilator therapy in asthma acts by relaxing ASM, resulting in improvements in symptoms and lung function. Furthermore, evidence is consistent that ASM in asthma is inherently hypercontractile [48]. Ex vivo gel-contraction assays have shown that ASM cells from asthmatics contract more than ASM from healthy donors [49, 50]. Although the mechanisms driving this hypercontractility are not very clear, several have been suggested. Sutcliff et al. [49] have shown increased oxidative stress burden in asthmatic ASM, with excessive production of reactive oxygen species as a result of nicotinamide adenine dinucleotide phosphate oxidase type 4 (NOX4) over-expression. This was associated with airflow obstruction, AHR and ASM hypercontractility as measured by gel-contraction assay. The latter was abolished, in vitro, by adding an inhibitor of NOX4. Calcium signalling has also been implicated in ASM hypercontractility. Several studies have demonstrated altered calcium homeostasis in asthma, with increased cytosolic calcium levels, which, given the central role of calcium in ASM contraction, would potentially result in ASM hypercontractility [51]. Furthermore, as suggested by animal studies, ASM hypercontractility could also be mediated through abnormalities in the RhoA/Rho kinase signalling pathway, leading to increased calcium sensitivity [52]. Finally, increased expression of the contractile protein α-actin, resulting in the transformation of ASM cells to a hypercontractile phenotype, has been shown, in in vivo and in vitro coculture, to be closely linked to mast cell infiltration of ASM, a known feature of asthma [53].
Increased ASM mass is a structural predictor of disordered airway physiology in asthma [17, 54]. Additionally, ASM is an important source of inflammatory mediators and matrix proteins. Adding asthmatic serum to ASM cells in culture was shown to result in excessive ECM protein production [55]. Several reports suggest that ASM cells in asthma are pro-inflammatory, with the expression of various immunomodulatory mediators, including IL-1, IL-6, IL-8 and prostaglandin E2 [56, 57]. From all the above evidence, it is clear that ASM is pivotal in the pathogenesis of asthma, with roles going far beyond being the regulator of bronchomotor tone.
3.2 Mesenchymal Fibrosis
ECM changes have been extensively investigated in asthma. The airway ECM is mainly composed of various types of macromolecules, including proteoglycans, collagens, elastic fibres and other non-proteoglycan glycoproteins like fibronectin, laminin and tenascin. They are secreted mainly by mesenchymal cells, and they provide structural and biological support to the airway tissue. The ECM dynamic environment is normally controlled by matrix metalloproteinases (MMPs), which are proteolytic enzymes that degrade ECM proteins, and their inhibitors, termed tissue inhibitors of metalloproteinases (TIMPs).
ECM protein content is abnormal in asthma, with studies showing excessive laminin, fibronectin, tenascin, proteoglycans and the collagens I, III and V, and reduced content of collagen IV and elastic fibres [58–60]. Although excessive ECM protein deposition occurs diffusely in the airway tissue, it is most intense in the reticular lamina, which is normally a thin layer in the lamina propria just below the true epithelial basement membrane. This subepithelial fibrosis is normally referred to, as in this review, as RBM thickness, although this is obviously a misnomer. Increased RBM thickness has been reported in both adults and children with asthma and throughout the severity spectrum. It has been linked to airway thickness (seen on CT), AHR and obstructive lung function [31, 61, 62]. Increased RBM thickness has been strongly associated with T-helper type 2 (Th2) inflammatory cells and mediators. This was completely absent in murine models of allergic asthma, which are depleted from IL-5 or eosinophils [63]. The action of inflammatory cells on ECM protein deposition is mediated mainly by TGF-β, which is a profibrotic cytokine that induces mesenchymal cells ECM protein production [64]. Abnormalities of both MMPs and TIMPs have been reported in asthma, although their exact role in ECM remodelling in asthma is not fully understood [65].
3.3 Vascular Remodelling
Bronchial vascular remodelling features in asthma include new vessel formation, increased blood flow and vascular leakage. Under a favourable pro-angiogenic environment, as in asthma, the bronchial vasculature is able to proliferate by sprouting from pre-existing vessels. This submucosal neoangiogenesis is a recognized feature of remodelling in adults and children with asthma and increases with the disease severity [66]. It has been shown to be negatively associated with post-bronchodilator forced expiratory volume in 1 s (FEV1) and, additionally, a link with AHR has been suggested by some, but not all, studies [67, 68]. Vascular endothelial growth factor (VEGF) is the most significant mediator of neoangiogenesis in asthma. VEGF is secreted by a range of cells, including Th2 cells, eosinophils, macrophages and ASM cells. Increased levels of VEGF in asthma have been associated with severity and airflow limitation [68]. Anti-VEGF has been successfully used to ameliorate vascular remodelling, but only in a murine model of asthma [69].
Increased airway vascular flow in asthma is caused by both increased vessel numbers and vasodilatation induced by various inflammatory mediators. However, the functional and physiological consequences of this are not fully known. Increased permeability is also a feature of vascular changes in asthma and is driven by various mediators, including histamine, prostaglandins, leukotrines and VEGF. This leads to airway oedema and contributes to airway inflammation and obstruction [70].
3.4 The Epithelium in Asthma
The airway epithelium plays a crucial complex role in disease and in health, as it is the only part of the airway structure that has direct contact with the external environment, and, through it, allergens and other stimuli influence the airways. Epithelial cells have a significant modulatory influence on airway inflammation and on underlying mesenchymal changes. These influences are far beyond its function as a simple mechanical barrier [71]. Epithelial cells have a central role in linking innate and adaptive immunological responses in the normal human lung. This is mainly through activation of pattern recognition receptors (PRRs), expressed by epithelial cells, by pathogen-associated molecular patterns (PAMPs) from microorganisms or by damage-associated molecular patterns (DAMPs), usually resulting from cellular damage. Upon activation, epithelial cells secrete a range of chemokines and cytokines, including CCL2, CCL20, TSLP, IL-25, IL-33 and granulocyte macrophage colony-stimulating factor (GM-CSF), resulting in the attraction and activation of dendritic cells and monocytes, which in turn act as antigen-presenting cells to immature T cells, which then differentiate into Th2 lineage. Interestingly, early in childhood, the threshold of PRR activation might be influenced by exposure to such stimuli as cigarette smoke or respiratory syncytial virus infection, which then theoretically leads to upregulation of the epithelial response to allergen and possibly the development of allergic airway diseases, although this remains to be proven [72, 73]. Airway epithelial cells can also influence lung inflammation through their apoptotic function. Juncadella et al. [74] recently used an asthma murine model to demonstrate that epithelial cells have an apoptotic function that is dependent on the GTPase Rac1 and this has an anti-inflammatory effect medicated by IL10.
In asthma, the respiratory epithelium is abnormal; however, the pathological and clinical relevance of this is not yet fully resolved. Several studies have shown that the asthmatic epithelium is fragile, with abnormal tight and adherence junctions. This fragility is partly inherent, as it is present in differentiated epithelial cultures, which are devoid of inflammatory cells and mediators, and partly caused by elements of airway inflammation [75–77]. This defective barrier function leads to worsening of airway inflammation by enhancing the exposure of dendritic cells to allergens in the airway lumen [78]. Moreover, the asthmatic epithelium reacts abnormally to viral infections by having impaired apoptosis, a normal protective biological reaction in infected epithelial cells. This allows viruses to further replicate, leading to worsening of infection [79]. Defective wound repair is also evident in the asthmatic epithelium [80]. This is associated with increased epidermal growth factor receptor (EGFR) expression, which in turn is linked with excess TGF-β production, which is known to exacerbate inflammation and ECM remodelling [81].
Epithelial mesenchymal transition (EMT) is another biological mechanism in asthma, through which the epithelium can directly influence the cellular composition of the underlying lamina propria. EMT describes a biological process in which epithelial cells lose adhesion and polarity and migrate into the lamina propria and acquire mesenchymal cell properties such as increased ECM protein production and prolonged survival. EMT is known to play a significant role in fibrotic conditions, including idiopathic pulmonary fibrosis; however, in asthma, the evidence for the significance of EMT is limited [82]. TGF-β, which is increased in asthma, has been shown to increase EMT in normal human epithelial cultures [83, 84]. Epithelial cells from asthmatics have been shown, in culture, to undergo EMT in response to TGF-β markedly more than cells of non-asthmatics [85]. Moreover, in a murine asthma model, EMT was shown to occur in response to prolonged allergen exposure [86]. Mediated by TGF-β, eosinophils have been shown to induce EMT when co-cultured with human epithelial cells and also when placed in the trachea of a murine model [87]. Despite all this evidence, EMT has yet to be demonstrated in asthma in vivo.
3.5 Mucus-Related Changes
The respiratory airways produce mucus that is composed of heterogeneous constituents normally homogenised together in a gel-like form by the effect of containing mucins, which are glycosylated proteins that play a significant role in the pathogenesis of asthma. The biochemical characteristics of mucins include a very high molecular weight and heavy glycosylation, with carbohydrates forming 50–90 % of their total mass. Airway mucins are generally divided into two types: (1) membrane-bound mucins that are attached to the cell membranes with a large extracellular domain, and (2) secreted mucins, which are gel forming [88]. Secreted mucins have cysteine-rich terminal domains that, through disulfide bonds, form dimers and then long-chain polymers, which give the airway mucus its viscous and elastic (gel-like) properties. In the airways, secreted gel-forming mucins are produced by luminal secretary cells, named goblet cells, and by submucosal glands, which are mainly present in large cartilaginous airways. Although more than 20 mucin (MUC) genes have been identified in humans, only two mucins are significantly expressed in the airway mucus: MUC5AC, mainly produced by goblet cells and MUC5B, which is produced by mucus glands [89].
In asthma, mucus-related abnormalities include mucus hypersecretion, increased goblet cell numbers, enlarged mucus glands and abnormal ciliary function. Post mortem studies have shown that mucus plugging is frequent in patients dying of status asthmaticus [90]. Increases in both mucus production and mucin gene expression, namely MUC5AC and MUC5B, are known in asthma and are associated with AHR, high airway resistance and deterioration of symptoms and lung function [91, 92]. Th2 inflammation has been shown to contribute to mucus hyper-production [93, 94]. Mucus hyperviscosity due to increased cells, plasma proteins, mucins and nucleic acids, together with ciliary dysfunction (both of which have been shown in asthma) result in defective mucociliary clearance [95]. This has many detrimental effects in asthma, including contributing to airflow obstruction and delayed clearance of allergens and pathogens from the airway, thus allowing increased epithelial exposure to those stimuli. Epithelial metaplasia resulting in goblet cell hyperplasia is known to occur in asthma and is associated with mucus hypersecretion. Goblet cells lack cilia and thus their hyperplasia reduces the epithelial area covered by cilia and consequently leads to mucus clearance delay. Mucus gland hypertrophy, although not as well studied as the other features of remodelling due to the paucity of mucus glands in endobronchial biopsies, is present in asthma and contributes to increased airway wall thickness and the airflow obstruction.
3.6 Role of Inflammatory Cells
Mast cells and immunoglobulin E (IgE) have a central role in the classical Th2 allergic asthma paradigm [96]. Briefly, in allergic asthma, antigens are presented by antigen-presenting cells, mostly dendritic cells, to naive T cells, which differentiate into Th2 phenotype and start to produce IL-4, IL-5 and IL-13. IL-4 and IL-13 stimulate B-lymphocytes to produce IgE, which, upon binding to its specific high-affinity receptor (FcεRI) on mast cells, and after sensitization, leads to their activation, migration and degranulation, releasing various inflammatory mediators and cytokines [97]. These include mediators of the early allergic response like histamine, leukotrine C4, prostaglandin D2 and platelet-activating factors, which lead to bronchoconstriction, increased mucus production, mucosal oedema and AHR. Of note, prostaglandin D2 also contributes to the pathogenesis of allergic asthma through its action on its receptor DP2/chemoattractant receptor homologous molecule expressed on Th2 cells (CRTh2) on Th2 lymphocytes and eosinophils facilitating their chemotaxis and activation [98]. Mast cells also produce cytokines and chemokines like IL-4, IL-13 and IL-5, which mediate the late allergic response, mainly through their action on other inflammatory cells. Directly relevant to airway remodelling, mast cells, after activation, also synthesize both TGF-β and basic fibroblast growth factor (FGF), which are strongly pro-fibrogenic, and the serine proteases tryptase, chymase, and carboxy-peptidase, which act on fibroblasts, leading to their proliferation and increased collagen I production. Additionally, mast cell infiltration of ASM bundles, a feature seen in asthma, has been liked with BHR, increased α-actin expression and ASM hypercontractility [53]. These infiltrating mast cells are predominantly of a chymase phenotype, which is known to strongly promote Th2 inflammation. Although some early in vitro studies suggested a role for mast cells in increased ASM mass, this has not been supported by more recent clinical studies [45, 99].
IgE also contributes to asthma pathophysiology by acting directly on basophils and dendritic cells, which also express FcεRI. Moreover, it stimulates B lymphocytes, monocytes, macrophages, and dendritic cells though the low-affinity IgE receptor (FcεRII or CD23) [100]. Although IgE may affect remodelling indirectly through the aforementioned allergic cascade, there is recent evidence to support direct effect on structural effector cells. Roth et al. [101], using a human ASM culture, showed increased proliferation and collagen deposition upon stimulation by activated IgE. Mast cells also contribute directly to epithelial remodelling. FcεRI-mediated amphiregulin production, a ligand for EGF, by mast cells is up-regulated in asthma, leads to increased mucin gene expression in epithelial cells with subsequent increased mucus production, and correlates with goblet cell hyperplasia [102]. Moreover, ex vivo, mast cell-derived amphiregulin has been shown to induce the proliferation of primary human lung fibroblast and ASM cells [103, 104].
Eosinophils have also been linked to airway remodelling. In animal models of asthma and in asthmatics, the association of subepithelial fibrosis and submucosal eosinophils has been well documented [105]. Cytotoxic proteins, which are stored and secreted by eosinophils upon activation, have been shown to directly cause epithelial damage [106]. The nature of the relationship between other inflammatory cells and airway remodelling in asthma is far less known. Th2 lymphocytes have been linked to ASM proliferation and remodelling; however, all the evidence for this is from in vitro studies [107].
4 Effect of Asthma Therapy on Airway Remodelling
4.1 Glucocorticoids
With the exception of patients with mild disease, where only as-needed inhaled short-acting β2-agonists suffice to control symptoms, glucocorticoid (GC) therapy remains the cornerstone of asthma management, both in stable disease state and in exacerbations. The first controlled trial showing the effectiveness of GCs in improving asthma symptoms dates back to the 1950s. Inhaled corticosteroids (ICS) were first introduced in the early 1970s remains the most significant preventive therapy for asthma as recognised by all current guidelines [108]. Arguably, since the introduction of ICS, no new asthma therapy has had a similar transfiguring effect on chronic asthma; hence, ICS are the benchmark against which all new therapies are compared in both efficacy and adverse event profiles.
GCs have been clearly shown to ameliorate asthma symptoms, improve lung function and reduce exacerbation. The main mode of action of GCs in asthma is through their anti-inflammatory effect. This is mainly through their direct effect in different white blood cells including T cells, mast cells and eosinophils. GCs suppress these leukocytes by inhibiting chemotaxis and adhesion, thus reducing recruitment, reducing production of inflammatory cytokines, inhibiting phagocytosis and inducing apoptosis. Airway structural cells, as will be discussed, are also influenced by GCs and that might also contribute to the therapeutic effect. In general, GCs exert their action through influencing transcription factors, which are proteins that bind to specific promoter regions of the DNA called glucocorticoid response elements (GREs), and thus control protein expression [109]. Initially GCs cross the cell membrane and bind with a specific intracellular GC receptor. This leads to translocation of the receptor to the cell nucleus, where it undergoes dimerization and then binds to corticosteroid-responsive genes, altering their transcription and subsequently influencing protein synthesis. This process is called transactivation. Transactivation has a major role in metabolic homeostasis (e.g. regulation of glucose and lipid metabolism) [109]. Furthermore, activated GC receptors can also alter gene expression indirectly, in an inhibitory process termed transrepression, by combining, as a monomer, with other transcription factors such as activator protein (AP)-1, nuclear factor (NF)-κB and smad3 and thus preventing them from binding with their corresponding genes. Examples of genes encoding for pro-inflammatory proteins and inhibited through transrepression include tumour necrosis factor (TNF)-α, IL-1β, GM-CSF and nitric oxide oxidase. Relevant to airway remodelling, GCs, through the action on smad3, inhibit the TGF-β-mediated deposition of matrix proteins such as collagen, fibronectin, elastin and proteoglycans and inhibition of various metalloproteinases, thus reducing TGF-β pro-fibrotic effect.
In recent years, it has become more apparent that GCs can also exert effects through non-transcriptional non-genomic mechanisms, although the significance of this in asthma therapy remains to be explored [110]. Non-genomic effects are rapid in onset, taking only seconds to start, and last only for a short period, thus their role might be more beneficial in asthma exacerbations. One important example is the inhibitory action on phospholipase A2, resulting in the reduction of arachidonic acid, the precursor for prostaglandin D2 and cysteinyl leukotrienes, which are important inflammatory and bronchoconstriction mediators in asthma. Another non-genomic action is through combining with membrane-bound GC receptors, leading to inhibition of T-cell activation [111]. GCs can also alter ion movement through the cell membrane, which might inhibit mast cell degranulation as shown in an animal model [112]. There is currently a wide consensus that the beneficiary anti-inflammatory effect of GCs results mainly from transrepression and non-transcriptional mechanisms, while side effects are mainly mediated by transactivation [109, 113–116].
The effect of GCs on airway remodelling is complex and still not well understood. This is despite the fact that most of the studies investigating the effect of asthma therapy on remodelling involved GCs (Tables 1, 2). As the main anti-inflammatory drugs used in asthma, and believing the simplistic paradigm of “remodelling is the result of an aberrant response to inflammation”, GCs were clearly the ideal candidate to study remodelling in asthma and explore its relation to inflammation. The influence of GCs on airway remodelling is not only attributed to the effect on inflammatory cells, but, as evidence supports, is also due to mechanisms involving direct action of GCs on non-inflammatory effector structural cells and various cytokines.
4.1.1 Glucocorticoid (GC) Effect on Extracellular Matrix Remodelling
Due to the ease of assessing ECM remodelling by measuring RBM thickness in EBBs, this has been extensively studied in the context of GC therapy in asthma. GCs can potentially affect ECM protein deposition indirectly by their action on inflammatory cells, such as eosinophils, and inflammatory mediators that are known to play an effector role in the process. They can also affect ECM remodelling directly by acting on the regulators of ECM protein degradation MMPs and TIMPs, although the evidence for this is scarce [117, 118]. However, studies have produced conflicting results, with some showing reduction of RBM thickness with GC therapy, whilst others have not. Ward et al. [119], in a double-blind randomized controlled trial (RCT), demonstrated mean reduction in RBM thickness of 1.9 μm following 12 months of treatment with high-dose inhaled fluticasone propionate. This improvement was not seen with 3 months of treatment. Using a multiple regression model, the authors concluded that this reduction in RBM thickness at 12 months contributed significantly to the improvement of AHR. Sont et al. [120] compared treatment targeted at reducing AHR versus guideline-based treatment alone in 75 mild-moderate asthmatics. The AHR-guided treatment group received a significantly higher ICS dose and this was associated with significant improvements in exacerbations, lung function and RBM thickness. Other studies showed RBM thickness reduction with high-dose ICS after a shorter duration of treatment [58, 121–124]. Nonetheless, multiple studies have failed to show any significant RBM thickness effect from GC treatment (Table 2) [125–128].
4.1.2 GC Effect on the Asthmatic Epithelium
Whether GCs improve or even worsen epithelial damage and other epithelial abnormalities in asthma remains a matter of conflict, with contradicting evidence. Although some studies have shown amelioration of some of the epithelial changes, not a single study has shown complete restoration of the epithelium to its normal state in response to any asthma therapy. Dorscheid et al. [129] used a healthy murine model to show that treatment with dexamethasone results in increased epithelial apoptosis and shedding. Furthermore, the authors also demonstrated that epithelial damage caused by allergen exposure in sensitized mice did not improve with dexamethasone treatment. This epithelial apoptotic effect of GCs was also shown in several studies using epithelial cultures; however, this is yet to be shown to have a significant effect in vivo in asthma [130, 131]. The effect of GCs on epithelial repair has also been studied on animal models and using epithelial cultures. Using guinea pig tracheal epithelium, budesonide was shown not to affect epithelial repair [132]. Moreover, in a wound repair cultured airway epithelial model, dexamethasone was shown to improve the repair potential significantly compared with salbutamol-treated or control cultures [133]. On the other hand, in another in vitro study, Dorscheid et al. [134] showed that GCs impaired epithelial cell migration and wound repair after mechanical injury. Recently, this was further supported by a study showing that corticosteroid impairment of epithelial repair is medicated through GC-induced leukine zipper (GILZ). GILZ, which was shown to be induced by dexamethasone, is known to have an anti-inflammatory effect; however, in this study, it was shown to impair epithelial repair through the inhibition of the mitogen-activated protein kinase extracellular signal-regulated kinase (MAPK-ERK) signalling pathway. Amphiregulin expression by mast cells, which results in goblet cell hyperplasia, is not affected by GC therapy [102].
Although in vivo studies of effects of asthma therapies on the airway epithelium are rare, most studies assessing GCs have shown a beneficial effect. In a group of six severe asthmatics, 10 years of treatment with ICS resulted in improvement in epithelial damage [125]. Similar results were seen with 3 months of treatment with inhaled budesonide [135].
4.1.3 GC Effect on ASM
GCs have been shown to reduce human ASM proliferation directly in vitro [136]. Several cellular mechanisms have been suggested to drive this effect of GCs, including the stimulation of p21 gene expression, an important regulator of cell cycle progression, and the inhibitory action on both cyclin D1 expression and retinoblastoma protein phosphorylation [137, 138]. The inhibitory effect of GCs on ASM proliferation was shown to be defective in asthma and that is mediated by an abnormal interaction between C/EBPα and the GC receptor [139]. In culture, fluticasone propionate, through its effect on NF-κB signalling, has been found to prevent myofibroblast differentiation, a possible precursor of ASM cells [140]. In a murine asthma model, Leung et al. [141] showed that intratracheal administration of ciclesonide or fluticasone, both powerful GCs, resulted in the inhibition of features of inflammation and remodelling, including ASM hyperplasia. However, GCs altering ASM remodelling in asthma in vivo is yet to be shown.
4.1.4 GC Effect on Vascular Remodelling
Unsurprisingly, GCs have been shown to improve all aspects of vascular remodelling in asthma, including increased blood flow, neoangiogenesis and vascular leakage [142]. Orsida et al. [143] showed that asthmatics on high-dose ICS had reduced the number of vessels/mm2 of lamina propria. Hoshino et al. [144] further supported this in a placebo-controlled study of the effect of 6 months’ treatment with inhaled beclomethasone dipropionate (BDP) 800 μg/daily or placebo in a group of 28 asthmatics. Beside improvements in FEV1 and AHR, BDP treatment also resulted in a significant reduction in vessel number and percent vascularity in the lamina propria compared with placebo144. These vascular improvements with ICS treatment are dose dependant and have been shown in other studies using high doses of ICS (Table 2) [69, 121, 145]. This reduction in vascularity is thought to be mediated by the effect of GCs on VEGF expression in the airway tissue [69].
4.2 β2-Adrenergic Receptor Agonists
Studies examining the effect of β2-adrenergic receptor agonists on airway remodelling are lacking. Long-acting β2-adrenergic receptor agonists (LABAs) persist in the airway tissue for long periods (about 12 h) due to their lipophilic nature, and they induce sustained bronchodilatation. LABAs are always used in combination with ICS in asthma and are never used as monotherapy due to their proven association with severe exacerbations and asthma death [146]. Repeated bronchoconstriction per se has been proven to result in epithelial and ECM remodelling, thus LABAs, by acting as bronchodilators, could potentially prevent that [11]. Todorova et al. [147] showed in vitro that the combination of formoterol, a LABA, and budesonide resulted in the total inhibition of serum-induced proteoglycan production by human lung fibroblasts. Formoterol on its own had no effect at all on the process, while budesonide alone inhibited proteoglycan production by only 44 %. This synergetic effect of LABAs and GCs on ECM remodelling was shown to be mediated by their combined effect on metalloproteolytic balance [148].
4.3 Leukotriene Modifiers
Leukotrienes are inflammatory mediators that play an important role in the pathogenesis of asthma. They are lipid eicosanoids derived from arachidonic acid through the enzymatic action of 5-lipooxygenase. Although leukotrienes are mostly produced by myeloid cells, other cells are capable of up-taking the intermediate leukotriene LTA4 and transforming it into other biologically active leukotrienes, e.g. LTC4 in a process called transcellular biosynthesis [149]. The effects of the cysteinyl leukotrienes (CysLT) LTC4, LTD4, and LTE4 in asthma are mediated mainly through the G-protein coupled receptors CysLT1, which when activated lead to eosinophilic inflammation, bronchoconstriction, mucus production and airway oedema [150]. Furthermore, the non-CysLT LTB4, through different receptors, has also been implicated in asthma, with roles in leukocyte chemotaxis and AHR [151, 152].
Although leukotrienes are important effectors in the inflammatory component of asthma, their role in airway remodelling is less clear. In vitro LTC4 has been shown to increase rat lung fibroblast collagen synthesis [153]. This profibrotic effect has also been suggested by the finding of increased CysLT levels in idiopathic pulmonary fibrosis patients [154]. Moreover, from both animal and ex vivo studies, there is evidence of CysLT promoting ASM proliferation by augmenting the mitogenic effect of EGF and insulin-like growth factor [155, 156].
Leukotriene modifiers are now well established in the management of asthma. Three such drugs are currently approved by the US FDA, montelukast and zafirlukast, both CysLT1 antagonists, and zileuton, a 5-lipooxygenase inhibitor. In asthma, they have been shown to improve eosinophilic inflammation, lung function, AHR, asthma control and exacerbations. However, as recognised by most current guidelines, ICS are clearly superior in efficacy; hence, the current role of leukotriene modifiers is mainly as an add-on therapy to ICS [157–159]. Studies assessing the effect of leukotriene modifiers on airway remodelling are rare (Table 1 ). Montelukast improved epithelial desquamation in animal models [160]. Henderson et al. [161], using a murine asthma model, showed that montelukast, in addition to its anti-eosinophilic effect, reduced features of remodelling like mucus plugging, ASM hyperplasia and subepithelial fibrosis. Montelukast was even shown to reverse allergen-induced airway remodelling in another murine model [162]. More recently montelukast has been shown to reduce ECM protein deposition in the small distal airways in sensitized guinea pigs [163]. However, the direct effect of leukotriene antagonists on airway remodelling in asthma has yet to be studied in asthmatics.
4.4 Methylxanthines
Methylxanthines exert both a bronchodilator and anti-inflammatory effects on the airways and are used in asthma, mainly as an add-on maintenance therapy option and occasionally in the acute treatment of severe asthma exacerbations where they are given intravenously under close monitoring, usually in the intensive care unit. The exact mechanism of action of methylxanthines is not fully understood, although the non-specific inhibition of phosphodiesterase enzyme is strongly suspected of driving most of the clinical therapeutic effects. Inhibiting phosphodiesterase type VI isoenzyme has been shown to relax human ASM and also to have a direct anti-inflammatory effect [164, 165]. In asthma, methylxanthines were shown to reduce inflammatory cells and improve both lung function and AHR [166–169]. Moreover, methylxanthines also increase corticosteroid responsiveness through their stimulatory action on histone deacetylase-2 [170]. However, the use of methylxanthines in asthma has always been limited by their significant adverse event profile and narrow therapeutic index.
The effect of methylxanthines on airway remodelling is unknown. Phosphodiesterase antagonism is unlikely to have any direct effect on airway remodelling. Although methylxanthines have been shown to reduce eosinophils, which are important effector cells in airway remodelling, no study, in our knowledge, has studied the effect of methylxanthines on airway remodelling in asthma [166].
4.5 Cholinergic Antagonists
Short-acting anticholinergics have an established role in the treatment of asthma. Acetylcholine is the principle autonomic parasympathetic neurotransmitter released by postganglionic neurones in the airways. Through its action on the muscarinic acetylcholine (M3) receptors located on ASM cells, mucus glands and vascular endothelium, acetylcholine leads to ASM contraction, increased mucus secretion and airway oedema, all of which directly contribute to airflow obstruction [171]. Excess acetylcholine results not only from an increased parasympathetic tone, a known feature of chronic asthma, but is also secreted by inflammatory and epithelial cells. Beyond the acute effects of acetylcholine described above, muscarinic stimulation also plays a role in airway remodelling in asthma. In vitro prolonged activation of muscarinic receptors has been shown to increase contractile protein expression and proliferation of primary cultured fibroblasts [172–174]. M3 activation has also been shown to potentiate the proliferative effect of pro-mitogenic factors on cultured ASM [173].
Most of the studies on the effects of anticholinergics on airway remodelling in asthma have used tiotropium, a long-acting antimuscarinic with an established role in chronic obstructive pulmonary disease (COPD) and which has recently been shown to be beneficial in asthma. In two large placebo-controlled RCTs involving severe asthmatics, tiotropium in addition to high-dose ICS and a LABA, resulted in significant improvement in FEV1 and prolongation of time to severe asthma exacerbation [175, 176]. Furthermore, in patients with milder asthma, tiotropium was also shown to improve lung function and symptoms and was comparable to LABAs [177].
The effect of tiotropium on airway remodelling has been mainly studied in animal models of asthma. Gosens et al. [178], using a guinea pig model of allergic asthma, demonstrated that administration of inhaled tiotropium during ovalbumin sensitization significantly prevented the development of features of ASM remodelling seen in the non-treated animals, including increased ASM mass, increased contractile protein expression and hypercontractility. Using the same guinea pig asthma model, Bos et al. [179], in addition to showing similar effects on ASM remodelling, showed that tiotropium also prevented mucus gland hypertrophy and reduced the numbers of both goblet cell, as measured by (MUC5AC)-positive cells, and eosinophils. Ohta et al. [180], in a murine chronic asthma model following treatment with tiotropium, showed reductions in all the following: airway inflammation, Th2 cytokines and TGF-β levels, ASM mass, airway fibrosis, goblet cell metaplasia and AHR. Though only in vitro, tiotropium has resulted in reduction of collagen synthesis by primary human lung fibroblasts [181]. It has also been suggested that it reduces respiratory syncytial virus replication in epithelial cells, with possible attenuation of the inflammation and remodelling that results from the viral infection [182].
Tiotropium has been shown to reduce lung function decline in COPD, a disease where structural changes in the airways play a significant role in the pathogenesis. This clearly shows a potential role for anticholinergics beyond simple bronchodilatation. However, in asthma, the effect of anticholinergics on airway remodelling, whether directly or indirectly through their anti-inflammatory action, is not fully explored and clearly needs further study.
4.6 Anti-Immunoglobulin E (IgE) Therapy
As discussed earlier, IgE has a central role in the pathogenesis of allergic asthma. Corticosteroids, although they have a broad anti-inflammatory action, do not reduce IgE production by B lymphocytes and can even increase circulatory IgE levels [183]. Omalizumab, the only licensed biological treatment for asthma, has been shown in several RCTs to reduce asthma exacerbations, improve asthma-related quality of life and decrease corticosteroid requirements [184, 185]. Omalizumab is a recombinant monoclonal IgG1 anti-IgE antibody that works by binding to the Fc region on the IgE, which normally binds to the IgE receptor FcεRI on mast cells. This means that it only binds and deactivates circulating IgE and does not affect mast cell-bound IgE. In several studies, omalizumab treatment led to significant improvement of airway inflammation, with reduction of a range on inflammatory cells in the submucosa, including eosinophils, basophils, T lymphocytes and B lymphocytes. Furthermore, it was also shown to improve both early and late allergic responses [186, 187]. Some of the anti-inflammatory effect of omalizumab is independent of mast cells as it down-regulates FcεRI expression not only on mast cells, but also on basophils and dendritic cells [188, 189]. Although the effect of omalizumab on inflammation is significant, the evidence for its influence on the other aspects of asthma, including remodelling, has been either disappointing or not well defined. Studies examining the effect of omalizumab treatment on AHR have shown mixed results, while the improvements in lung function have been disappointing [186, 190]. Omalizumab leads to the reduction of a number of remodelling mediators like endothelin-1, TGF-β and TNF-α; however, its exact effect on remodelling is not fully known. Roth et al. [101] showed that IgE-induced ASM proliferation and collagen production, in ASM culture, were completely inhibited by the drug. This provided the possible biological mechanisms explaining the results of an earlier study, which looked at the effect of 16 weeks’ omalizumab treatment on right apical segmental bronchus thickness as measured by CT versus placebo. In addition to improvement in sputum eosinophils, omalizumab treatment (n = 16) significantly decreased percent wall area and wall thickness and increased lumen area compared with placebo (n = 14) [191]. An effect of omalizumab on mesenchymal changes was also demonstrated by Riccio et al. [192], who reported that, in a small group of patients with severe persistent allergic asthma, 12 months of omalizumab treatment led to a significant mean reduction of RBM thickness.
Treatments directed against the IgE low-affinity CD23 receptor have not gone beyond early phase studies; hence, their clinical efficacy or effect on airway remodelling remains to be investigated [193].
4.7 Interleukin (IL)-5 Antagonists
Given the central role of IL-5 in eosinophil biology, targeting this cytokine has been regarded as logical option in various eosinophilic diseases, including asthma. IL-5 is the most significant cytokine in the modulation of eosinophils; hence, it is also known as eosinophil colony-stimulating factor, as it is important for all aspects of eosinophil survival and function. IL-5 is produced by Th2 lymphocytes, basophils, eosinophils and natural killer cells. It is essential for eosinophil proliferation, differentiation, maturation, chemotaxis, migration and survival in the tissues. Early clinical trials with the anti-IL-5 monoclonal antibody mepolizumab have been disappointing, largely due to failure to select patients with the appropriate asthma phenotype, i.e. eosinophilic asthmatics [194–196]. Later studies examining the effect of mepolizumab in patients with severe eosinophilic asthma have shown significant clinical benefits [27, 197]. The largest study, the multicenter DREAM trial, has shed more light on the role of eosinophils in asthma. Treatment for 1 year with mepolizumab reduced severe asthma exacerbations by more than 50 % [198]. A more recent meta-analysis confirmed that mepolizumab, in severe eosinophilic asthma, significantly ameliorates asthma-related quality of life and reduces exacerbations [199]. Mepolizumab has profound effect on blood, submucosal and sputum eosinophil counts [200].
Anti-IL-5 affects remodelling in asthma through its anti-eosinophilic action [201]. Using a murine asthma model, Tanaka et al. [202] showed that administration of anti-IL-5 prior to allergen exposure led to the prevention of TGF-mediated peribronchial and subepithelial fibrosis. Only a few clinical studies have examined the effect of anti-IL-5 on remodelling in asthma. In an RCT involving atopic asthmatics, Flood-Page et al. [4] showed that, compared with placebo, three infusions of mepolizumab resulted in the reduction of tenascin, lumican and procollagen III deposited in the subepithelial area, with improvement of RBM thickness. There was also reduction in the profibrotic mediator TGF-β in the BAL and reduction of its corresponding messenger RNA (mRNA) from eosinophils. Haldar et al. [27, 203], after 12 months of treatment with mepolizumab, showed, besides improvements in exacerbations and eosinophil counts in blood and sputum, reductions in total airway area and airway wall area on CT compared with placebo which, interestingly, returned to the pre-treatment baseline 1 year post mepolizumab cessation. Reslizumab is another anti-IL-5 monoclonal IgG antibody currently being evaluated in phase III asthma trials. Other methods of antagonising IL-5 include anti-IL-5 receptor antibodies (anti-IL-5Rα and IL-5-βc) and small interfering RNA techniques; however, the effect of these treatments on airway remodelling is still unknown.
4.8 Anti-IL-13, Anti-IL-17, Anti-Tumor Necrosis Factor (TNF)-Alpha and Tyrosine Kinase Inhibitors
IL-13 is a pleiotropic Th2 cytokine that plays a significant role in the development of airway inflammation and remodelling in asthma. It is produced by activated Th2 cells, dendritic cells and mast cells, and induces goblet cell hyperplasia, IgE production by B-lymphocytes and ECM protein deposition through fibroblast activation [204]. In several animal models, anti-IL-13 has clearly been shown to improve Th2 inflammation, airway remodelling and AHR [205–207]. In a large double-blind RCT (n = 219) in patients with poorly controlled asthma, lebrikizumab (a humanized antibody that directly deactivates IL-13) has been shown to improve lung function [208]. This effect was more pronounced in patients who had high serum levels of periostin, an ECM protein produced by fibroblasts and epithelial cells under the influence of IL-13. Periostin affects epithelial cell adhesion, thus contributing to epithelial fragility, and also leads to increased activation of lung fibroblasts, leading to mesenchymal fibrosis. Tralokinumab, another anti-IL-13 antibody, has been shown in a phase II RCT to improve FEV1 when administered to patients with moderate-to-severe uncontrolled asthma [209]. The signalling of both IL-4 (another Th2 cytokine with important remodelling and inflammatory effects in asthma) and IL-13 could be inhibited downstream by targeting the alpha subunit of the IL-4 receptor (IL-4α), which is a common overlapping part of both IL-4 and IL-13 receptors [210, 211]. Recently, Wenzel et al. [212] showed that treating patients with persistent moderate-to-severe eosinophilic asthma with dupilumab, a human monoclonal antibody to IL-4α, in an ICS and LABA withdrawal study, resulted in improved lung function and the reduction of both Th2-associated markers and asthma exacerbations.
IL-17 is a cytokine mainly produced by Th17 cells, specialised lymphocytes that are distinct from Th1 and Th2 cells, and contributes to adaptive immunity, especially against extracellular bacteria. Furthermore, IL-17 is also a pro-inflammatory cytokine involved in the pathogenesis of different autoimmune and chronic inflammatory conditions, including asthma. IL-17 is involved in the recruitment and activation of both neutrophils and eosinophils. In asthma, IL-17 expression is increased and is associated with disease severity and AHR. Moreover, IL-17 might also contribute to remodelling in asthma through its direct action on structural cells as suggested by some in vitro studies showing an effect on epithelial proliferation and mucin production [213, 214]. Brodalumab (a human anti-IL-17 receptor monoclonal antibody) is the only IL-17 antagonist to have been tested in a clinical trial in asthma. Busse et al. [215], in patients with moderate-severe asthma, reported no improvement in lung function or asthma control with 12 weeks’ treatment with brodalumab; however, pre-specified subgroup analysis showed improvement in asthma control in patients with high FEV1 reversibility.
TNF-α is a pro-inflammatory cytokine secreted by a range of cells, including mast cells, eosinophils and macrophages. Airway high TNF-α signalling was demonstrated in patients with refractory asthma compared with both patients with milder asthma and non-asthmatic healthy controls [216, 217]. TNF-α has been demonstrated to drive vascular remodelling in a murine model of airway inflammation [218]. Anti-TNF-α has an established role in the treatment of chronic inflammatory conditions like rheumatoid arthritis, Crohn’s disease and psoriasis. An initial small clinical trial of the anti-TNF-α agent etanercept, a soluble TNF-α receptor, in patients with refractory asthma showed promising results, with improvement in FEV1, AHR and asthma-related quality-of-life scores; however, a subsequent larger RCT using golimumab, a monoclonal antibody against TNF-α, showed no improvement in FEV1 or exacerbation rate and the trial was discontinued early due to an unfavourable risk/benefit profile [217, 219]. Unfortunately, airway remodelling was never measured as an endpoint in any clinical study involving anti-TNF-α therapy in asthma.
Tyrosine kinase is an enzyme that plays an important role in signal transduction and regulates various cellular activities. Due to their effect on apoptosis, angiogenesis and cellular division, tyrosine kinase inhibitors are used as a biological treatment of various cancers. Signalling of some airway remodelling mediators is through receptor tyrosine kinases, which could be blocked by such therapy with direct effect on airway remodelling. EGFR, which is increased in expression in asthma and correlates with severity, has been linked with epithelial and mesenchymal remodelling changes and could be targeted by tyrosine kinase inhibitors [220–222]. In murine asthma models, treatment with erlotinib, an EGFR tyrosine kinase inhibitor, resulted in reduction in ASM mass, RBM thickness, collagen deposition and AHR [220, 223]. Stem cell factor is important in mast cell biology and could also be blocked by the tyrosine kinase imatinib. Using another murine model, Rhee et al. [224] showed that imatinib reduces ASM thickness. A double-blind RCT on the use of imatinib in severe refractory asthma is currently on-going [225]. Masitinib is another tyrosine kinase inhibitor that could potentially be used in the treatment of asthma. In addition to inhibiting stem cell factor receptor (c-kit), it also targets platelet-derived growth factor receptor. In a phase IIa RCT involving 44 patients with severe corticosteroid-dependent asthma, 16 weeks of treatment with masitinib resulted in improvement in asthma control [226]. A phase III multicentre RCT using masitinib in patients with severe persistent asthma who are on maintenance oral GCs is currently recruiting [227].
4.9 Statins, Metformin and Vitamin D
Statins are inhibitors of the 3-hydroxy-3-methylglutaryl-coenzyme-A (HMG-CoA) reductase enzyme and are used for therapeutically inhibiting cholesterol biosynthesis and reducing cholesterol levels. It has been suggested that statins have a beneficial effect in asthma. However, a systematic review concluded that although statins had an anti-inflammatory effect in asthma, they did not significantly affect lung function or symptoms [228]. On the other hand, a recent retrospective review of obese patients with severe asthma showed that receiving statins was associated with better symptom control, suggesting a role for statins in this specific asthma phenotype [229]. The effects of statins on inflammation and remodelling have been studied mainly using simvastatin in animal models of allergic asthma. Simvastatin has been shown to inhibit epithelial IL-13-inducable pro-inflammatory cytokine production [230]. Ahmad et al. [231] showed that simvastatin in a murine model resulted in reduction in inflammatory cell infiltration, mucus hypersecretion, epithelial injury, collagen deposition and AHR. This was shown to be mediated by the influence of simvastatin on nitric oxide metabolism. Zeki et al. [232], in a similar model, showed reduction in goblet cell hyperplasia and lung arginase, an important mediator of vascular remodelling in asthma. In vitro, simvastatin was shown to inhibit ASM proliferation, which was mediated through RhoA inactivation; and to reduce TGF-β1-induced fibronectin production by human airway fibroblasts [233, 234].
Metformin is a biguanide used in the treatment of type 2 diabetes mellitus. It possesses anti-inflammatory and antioxidant properties and has been shown to improve tissue injury and remodelling in models of various diseases [235, 236]. In a murine asthma model, through the activation of AMP-activated protein kinase (AMPK), metformin resulted in the attenuation of eosinophilic inflammation, peribronchial fibrosis and ASM hypertrophy. Moreover, the authors also demonstrated, in vitro, decreased TGF-β1-induced fibronectin expression in cultured fibroblasts, which was also mediated through AMPK activation [237].
Vitamin D and its deficiency has been implicated in airway remodelling in asthma. Gupta et al. [238] demonstrated that vitamin D levels are significantly lower in children with severe therapy-resistant asthma and this was associated with increased ASM mass, poor lung function and worse asthma control. In vitro, vitamin D has been shown to inhibit platelet-derived growth factor-mediated human ASM cell proliferation through the inhibition of phosphorylation of both checkpoint kinase-1 and the retinoblastoma protein [239]. This negative effect of vitamin D on ASM proliferation could also result from the inhibition of NF-κB signalling, which has also been demonstrated in human ASM cells [240].
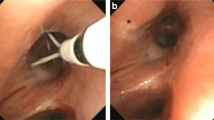
4.10 Bronchial Thermoplasty
BT is the only FDA-approved asthma therapy that directly targets airway remodelling. It is a novel non-drug technique for patients with severe persistent asthma who do not achieve control despite maximal medical therapy, where temperature-controlled radio frequency (RF) energy is targeted at proximal airways to reduce ASM mass, resulting in clinical benefit. In cardiology, RF has long been used to treat conduction defects and arrhythmias by targeting specific areas of the cardiac muscle and inducing thermal cell death [241]. The effect of RF on ASM has initially been seen in patients with lung cancer treated with the technique. The first proof of concept study was published by Danek et al. [242] in 2004, where applying BT to dogs resulted in reduction of the ASM layer, which was largely replaced by a mature collagen layer. This was accompanied by and correlated with significant reduction in AHR. The first study of BT in humans was published the year after, where the treatment was applied to nine patients who were undergoing lung resection for proven or suspected lung cancer. At 3 weeks before the planned resection, BT was applied only to the segments and lobes to be removed. This was a feasibility and safety study, and it showed that BT was safe and resulted in 50 % reduction in ASM, with the effects of the treatment limited to the airway and peribronchial regions [243]. The first clinical study in patients with asthma was reported by Cox et al. [244], who showed that, in 16 patients with mild-to-moderate asthma, BT resulted in improvement in AHR that was sustained for up to 2 years. They also showed improvement in symptom-free days and peak flow measurements monitored in the first 12 weeks post treatment. However, no change in spirometry was seen. Three RCTs assessing BT in asthmatics followed: the AIR, RISA and AIR2 trials. The AIR trial was an unblinded RCT (n = 116) assessing BT in patients with moderate-severe persistent asthma. After 12 months, BT resulted in improvements in peak flow measurements, mild exacerbations, symptom-free days, Asthma Quality of Life Questionnaire (AQLQ) and Asthma Control Questionnaire (ACQ) scores, and rescue medication use; however, no effect on AHR or spirometry was seen [245]. In a smaller unblinded RCT (RISA trail), Pavord et al. [246], in a group with severe asthma, compared BT (n = 15) and usual care (n = 17). They showed similar results, with improvements in symptoms and use of rescue medications. Additionally, they also showed improvement in FEV1. This was at the expense of increased short-term asthma-related morbidity246.
Finally, the AIR2 trial was the largest study on the use of BT in patients with severe asthma. This was a multicentre, randomized, double-blind, sham-controlled clinical trial involving 288 patients [247]. BT significantly improved AQLQ score, the primary outcome, compared with sham treatment. Moreover, in the period between 6 and 52 weeks post BT, the BT group had reduction in severe exacerbations, emergency department (ED) visits and days missed from work/school. Most of the patients in the BT group were followed up in the second year following treatment, revealing the same rate of exacerbations, adverse events, and hospitalization as in the first year, although this was not compared with the sham group [248]. Reduced rates of both severe exacerbations and ED visits for respiratory symptoms were further demonstrated in 85.3 % of the BT group patients, who were followed up annually for 5 years [249]. All BT studies have shown short-term adverse events in the form of lower respiratory tract infections and pneumonia, lobar collapse and increased hospitalization (6 % more in the BT group than in the sham group in AIR2), but these are limited to the first 6 weeks post treatment [249].
The exact effect of BT on airway structure and how that leads to clinical benefit is unknown. All the evidence for the effect of BT on remodelling is from healthy dogs and cancer patients undergoing resection where it was shown to reduce ASM mass, but whether this is the mechanism in asthma is uncertain. The inconsistent effect on AHR and the absence of spirometry improvement is curious. It has been suggested that BT might be ablating a form of ASM pacemaker in the proximal airways with effect propagating distally, although this is no more than a hypothesis [250]. Examples of alternative mechanisms of action of BT include influencing mucus production or altering behaviour of other mesenchymal cells are possible but need to be assessed by future studies. Interestingly, a recently reported case series showed reduction in ASM, although the number of participants was too small (three patients) to make any informative conclusions [251]. Several studies are currently evaluating the effect of BT on airway tissue but their results are yet to be published [252, 253].
5 Conclusion
New asthma therapies are desperately needed, especially for patients with severe disease. In recent years, airway remodelling in asthma has regained significant interest so as to help develop therapies that target the process; however, remodelling is complex and not fully understood. Beside GCs, the effect of various asthma treatments on remodelling has not been well studied. BT, the only therapy used to treat remodelling, has unfortunately only resulted in modest clinical benefits. The relative contribution of the heterogeneous elements of airway remodelling to the pathogenesis and pathophysiology of asthma is unknown; this remains a major gap in our knowledge that hinders development of anti-remodelling therapies. As a result of the lack of longitudinal studies examining remodelling in asthma, both the relevance and the fluidity of different aspects of remodelling are still obscure. Which aspects of remodelling should be targeted and at what stage in the natural history of the process remains unanswered.
Heterogeneity in the clinical and inflammatory characteristics of asthma has been well recognised and has been influencing asthma drug development [254]. Many recent asthma drug trials have been conducted in patients with specific phenotypes, e.g. anti-IL-13 in patients with Th2 biological markers, thus developing a personalized medicine approach to asthma treatment [208]. However, airway remodelling has never been taken into account in the models used to identify asthma phenotypes. Whether doing this would help discern new asthma phenotypes or possibly new endotypes, such as an airway remodelling-predominant endophenotype (an endotype is defined as a subtype of a condition, with distinct molecular and pathobiological mechanisms), and whether this would subsequently help inform future asthma drug developments remains to be answered. Furthermore, whether future interventions should be directed at preventing airway remodelling, perhaps in childhood, or should aim for the amelioration of established remodelling needs to be fully understood.
References
World Health Organization. Global surveillance, prevention and control of chronic respiratory diseases: a comprehensive approach. http://www.who.int/respiratory/publications/global_surveillance/en/index.html. 2007. Accessed 10 Feb 2014.
Jackson DJ, Sykes A, Mallia P, Johnston SL. Asthma exacerbations: origin, effect, and prevention. J Allergy Clin Immunol. 2011;128:1165–74.
Salter HH. On asthma, its pathology and treatment. London: J Churchill; 1860.
Flood-Page P, Menzies-Gow A, Phipps S, Ying S, Wangoo A, Ludwig MS, Barnes N, Robinson D, Kay AB. Anti-IL-5 treatment reduces deposition of ECM proteins in the bronchial subepithelial basement membrane of mild atopic asthmatics. J Clin Invest. 2003;112:1029–36.
Humbles AA, Lloyd CM, McMillan SJ, Friend DS, Xanthou G, McKenna EE, Ghiran S, Gerard NP, Yu C, Orkin SH, Gerard C. A critical role for eosinophils in allergic airways remodeling. Science. 2004;305:1776–9.
Lowe LA, Simpson A, Woodcock A, Morris J, Murray CS, Custovic A, NAC Manchester Asthma and Allergy Study Group. Wheeze phenotypes and lung function in preschool children. Am J Respir Crit Care Med. 2005;171:231–7.
Saglani S, Malmstrom K, Pelkonen AS, Malmberg LP, Lindahl H, Kajosaari M, Turpeinen M, Rogers AV, Payne DN, Bush A, Haahtela T, Makela MJ, Jeffery PK. Airway remodeling and inflammation in symptomatic infants with reversible airflow obstruction. Am J Respir Crit Care Med. 2005;171:722–7.
Saglani S, Payne DN, Zhu J, Wang Z, Nicholson AG, Bush A, Jeffery PK. Early detection of airway wall remodeling and eosinophilic inflammation in preschool wheezers. Am J Respir Crit Care Med. 2007;176:858–64.
Saglani S, Mathie SA, Gregory LG, Bell MJ, Bush A, Lloyd CM. Pathophysiological features of asthma develop in parallel in house dust mite-exposed neonatal mice. Am J Respir Cell Mol Biol. 2009;41:281–9.
Kariyawasam HH, Aizen M, Barkans J, Robinson DS, Kay AB. Remodeling and airway hyperresponsiveness but not cellular inflammation persist after allergen challenge in asthma. Am J Respir Crit Care Med. 2007;175:896–904.
Grainge CL, Lau LC, Ward JA, Dulay V, Lahiff G, Wilson S, Holgate S, Davies DE, Howarth PH. Effect of bronchoconstriction on airway remodeling in asthma. N Engl J Med. 2011;364:2006–15.
Kuo C, Lim S, King NJ, Bartlett NW, Walton RP, Zhu J, Glanville N, Aniscenko J, Johnston SL, Burgess JK, Black JL, Oliver BG. Rhinovirus infection induces expression of airway remodelling factors in vitro and in vivo. Respirology. 2011;16:367–77.
McParland BE, Macklem PT, Pare PD. Airway wall remodeling: friend or foe? J Appl Physiol. 2003;95:426–34.
Royce SG, Tan L, Koek AA, Tang ML. Effect of extracellular matrix composition on airway epithelial cell and fibroblast structure: implications for airway remodeling in asthma. Ann Allergy Asthma Immunol. 2009;102:238–46.
Bergeron C, Tulic MK, Hamid Q. Tools used to measure airway remodelling in research. Eur Respir J. 2007;29:596–604.
Woodruff PG, Innes AL. Quantitative morphology using bronchial biopsies. Eur Respir Rev. 2006;15:157–61.
Benayoun L, Druilhe A, Dombret MC, Aubier M, Pretolani M. Airway structural alterations selectively associated with severe asthma. Am J Respir Crit Care Med. 2003;167:1360–8.
Sullivan P, Stephens D, Ansari T, Costello J, Jeffery P. Variation in the measurements of basement membrane thickness and inflammatory cell number in bronchial biopsies. Eur Respir J. 1998;12:811–5.
Veraldi KL, Gibson BT, Yasuoka H, Myerburg MM, Kelly EA, Balzar S, Jarjour NN, Pilewski JM, Wenzel SE, Feghali-Bostwick CA. Role of insulin-like growth factor binding protein-3 in allergic airway remodeling. Am J Respir Crit Care Med. 2009;180:611–7.
Redington AE, Madden J, Frew AJ, Djukanovic R, Roche WR, Holgate ST, Howarth PH. Transforming growth factor-beta 1 in asthma. Measurement in bronchoalveolar lavage fluid. Am J Respir Crit Care Med. 1997;156:642–7.
Zietkowski Z, Skiepko R, Tomasiak MM, Bodzenta-Lukaszyk A. Endothelin-1 in exhaled breath condensate of stable and unstable asthma patients. Respir Med. 2008;102:470–4.
Kanazawa H, Yoshikawa T. Up-regulation of thrombin activity induced by vascular endothelial growth factor in asthmatic airways. Chest. 2007;132:1169–74.
Coxson HO, Lam S. Quantitative assessment of the airway wall using computed tomography and optical coherence tomography. Proc Am Thorac Soc. 2009;6:439–43.
Gupta S, Siddiqui S, Haldar P, Entwisle JJ, Mawby D, Wardlaw AJ, Bradding P, Pavord ID, Green RH, Brightling CE. Quantitative analysis of high-resolution computed tomography scans in severe asthma subphenotypes. Thorax. 2010;65:775–81.
Siddiqui S, Gupta S, Cruse G, Haldar P, Entwisle J, Mcdonald S, Whithers PJ, Hainsworth SV, Coxson HO, Brightling C. Airway wall geometry in asthma and nonasthmatic eosinophilic bronchitis. Allergy. 2009;64:951–8.
Aysola RS, Hoffman EA, Gierada D, Wenzel S, Cook-Granroth J, Tarsi J, Zheng J, Schechtman KB, Ramkumar TP, Cochran R, Xueping E, Christie C, Newell J, Fain S, Altes TA, Castro M. Airway remodeling measured by multidetector CT is increased in severe asthma and correlates with pathology. Chest. 2008;134:1183–91.
Haldar P, Brightling CE, Hargadon B, Gupta S, Monteiro W, Sousa A, Marshall RP, Bradding P, Green RH, Wardlaw AJ, Pavord ID. Mepolizumab and exacerbations of refractory eosinophilic asthma. N Engl J Med. 2009;360:973–84.
Niimi A, Matsumoto H, Amitani R, Nakano Y, Sakai H, Takemura M, Ueda T, Chin K, Itoh H, Ingenito EP, Mishima M. Effect of short-term treatment with inhaled corticosteroid on airway wall thickening in asthma. Am J Med. 2004;116:725–31.
Ueda T, Niimi A, Matsumoto H, Takemura M, Hirai T, Yamaguchi M, Matsuoka H, Jinnai M, Muro S, Chin K, Mishima M. Role of small airways in asthma: investigation using high-resolution computed tomography. J Allergy Clin Immunol. 2006;118:1019–25.
Busacker A, Newell JD Jr, Keefe T, Hoffman EA, Granroth JC, Castro M, Fain S, Wenzel S. A multivariate analysis of risk factors for the air-trapping asthmatic phenotype as measured by quantitative CT analysis. Chest. 2009;135:48–56.
Kasahara K, Shiba K, Ozawa T, Okuda K, Adachi M. Correlation between the bronchial subepithelial layer and whole airway wall thickness in patients with asthma. Thorax. 2002;57:242–6.
Montaudon M, Lederlin M, Reich S, Begueret H, Tunon-de-Lara JM, Marthan R, Berger P, Laurent F. Bronchial measurements in patients with asthma: comparison of quantitative thin-section CT findings with those in healthy subjects and correlation with pathologic findings. Radiology. 2009;253:844–53.
Saglani S, Papaioannou G, Khoo L, Ujita M, Jeffery PK, Owens C, Hansell DM, Payne DN, Bush A. Can HRCT be used as a marker of airway remodelling in children with difficult asthma? Respir Res. 2006;7:46.
de Blic J, Tillie-Leblond I, Emond S, Mahut B, Dang Duy TL, Scheinmann P. High-resolution computed tomography scan and airway remodeling in children with severe asthma. J Allergy Clin Immunol. 2005;116:750–4.
Gutierrez-Chico JL, Alegria-Barrero E, Teijeiro-Mestre R, Chan PH, Tsujioka H, de Silva R, Viceconte N, Lindsay A, Patterson T, Foin N, Akasaka T, di Mario C. Optical coherence tomography: from research to practice. Eur Heart J Cardiovasc Imaging. 2012;13:370–84.
Coxson HO, Quiney B, Sin DD, Xing L, McWilliams AM, Mayo JR, Lam S. Airway wall thickness assessed using computed tomography and optical coherence tomography. Am J Respir Crit Care Med. 2008;177:1201–6.
Lam S, Lee AMD, Lane P, Ohtani K, MacAulay C, Varfolomeva N, Hui L, Coxson HO, Fitzgerald J. Optical coherence tomography imaging of asthmatic airways before and after thermoplasty. Am J Respir Crit Care Med. 2013;187:A6014.
Bates JH, Martin JG. A theoretical study of the effect of airway smooth muscle orientation on bronchoconstriction. J Appl Physiol. 1985;1990(69):995–1001.
Mitzner W. Airway smooth muscle: the appendix of the lung. Am J Respir Crit Care Med. 2004;169:787–90.
Amrani Y, Panettieri RA. Airway smooth muscle: contraction and beyond. Int J Biochem Cell Biol. 2003;35:272–6.
Huber H, Koesser K. The pathology of bronchial asthma. Arch Intern Med. 1922;30:689–760.
Woodruff PG, Dolganov GM, Ferrando RE, Donnelly S, Hays SR, Solberg OD, Carter R, Wong HH, Cadbury PS, Fahy JV. Hyperplasia of smooth muscle in mild to moderate asthma without changes in cell size or gene expression. Am J Respir Crit Care Med. 2004;169:1001–6.
Begueret H, Berger P, Vernejoux JM, Dubuisson L, Marthan R, Tunon-de-Lara JM. Inflammation of bronchial smooth muscle in allergic asthma. Thorax. 2007;62:8–15.
Johnson PR, Roth M, Tamm M, Hughes M, Ge Q, King G, Burgess JK, Black JL. Airway smooth muscle cell proliferation is increased in asthma. Am J Respir Crit Care Med. 2001;164:474–7.
Kaur D, Hollins F, Saunders R, Woodman L, Sutcliffe A, Cruse G, Bradding P, Brightling C. Airway smooth muscle proliferation and survival is not modulated by mast cells. Clin Exp Allergy. 2010;40:279–88.
Ward JE, Harris T, Bamford T, Mast A, Pain MC, Robertson C, Smallwood D, Tran T, Wilson J, Stewart AG. Proliferation is not increased in airway myofibroblasts isolated from asthmatics. Eur Respir J. 2008;32:362–71.
Berair R, Saunders R, Brightling CE. Origins of increased airway smooth muscle mass in asthma. BMC Med. 2013;11:145-7015-11-145.
Berair R, Hollins F, Brightling C. Airway smooth muscle hypercontractility in asthma. J Allergy (Cairo). 2013;2013:185971.
Sutcliffe A, Hollins F, Gomez E, Saunders R, Doe C, Cooke M, Challiss RA, Brightling CE. Increased nicotinamide adenine dinucleotide phosphate oxidase 4 expression mediates intrinsic airway smooth muscle hypercontractility in asthma. Am J Respir Crit Care Med. 2012;185:267–74.
Matsumoto H, Moir LM, Oliver BG, Burgess JK, Roth M, Black JL, McParland BE. Comparison of gel contraction mediated by airway smooth muscle cells from patients with and without asthma. Thorax. 2007;62:848–54.
Mahn K, Ojo OO, Chadwick G, Aaronson PI, Ward JPT, Lee TH. Ca2+ homeostasis and structural and functional remodelling of airway smooth muscle in asthma. Thorax. 2010;65:547–52.
Chiba Y, Takada Y, Miyamoto S, MitsuiSaito M, Karaki H, Misawa M. Augmented acetylcholine-induced, rho-mediated Ca2+ sensitization of bronchial smooth muscle contraction in antigen-induced airway hyperresponsive rats. Br J Pharmacol. 1999;127:597–600.
Woodman L, Siddiqui S, Cruse G, Sutcliffe A, Saunders R, Kaur D, Bradding P, Brightling C. Mast cells promote airway smooth muscle cell differentiation via autocrine up-regulation of TGF-beta 1. J Immunol. 2008;181:5001–7.
Gunst SJ, Panettieri RA Jr. Point: alterations in airway smooth muscle phenotype do/do not cause airway hyperresponsiveness in asthma. J Appl Physiol. 1985;2012(113):837–9.
Johnson PR, Black JL, Carlin S, Ge Q, Underwood PA. The production of extracellular matrix proteins by human passively sensitized airway smooth-muscle cells in culture: the effect of beclomethasone. Am J Respir Crit Care Med. 2000;162:2145–51.
Damera G, Tliba O, Panettieri RA Jr. Airway smooth muscle as an immunomodulatory cell. Pulm Pharmacol Ther. 2009;22:353–9.
Damera G, Panettieri RA Jr. Does airway smooth muscle express an inflammatory phenotype in asthma? Br J Pharmacol. 2011;163:68–80.
Laitinen A, Altraja A, Kampe M, Linden M, Virtanen I, Laitinen LA. Tenascin is increased in airway basement membrane of asthmatics and decreased by an inhaled steroid. Am J Respir Crit Care Med. 1997;156:951–8.
Roberts CR, Burke AK. Remodelling of the extracellular matrix in asthma: proteoglycan synthesis and degradation. Can Respir J. 1998;5:48–50.
Slats AM, Janssen K, van Schadewijk A, van der Plas DT, Schot R, van den Aardweg JG, de Jongste JC, Hiemstra PS, Mauad T, Rabe KF, Sterk PJ. Expression of smooth muscle and extracellular matrix proteins in relation to airway function in asthma. J Allergy Clin Immunol. 2008;121:1196–202.
Milanese M, Crimi E, Scordamaglia A, Riccio A, Pellegrino R, Canonica GW, Brusasco V. On the functional consequences of bronchial basement membrane thickening. J Appl Physiol. 1985;2001(91):1035–40.
Shiba K, Kasahara K, Nakajima H, Adachi M. Structural changes of the airway wall impair respiratory function, even in mild asthma. Chest. 2002;122:1622–6.
Cho JY, Miller M, Baek KJ, Han JW, Nayar J, Lee SY, McElwain K, McElwain S, Friedman S, Broide DH. Inhibition of airway remodeling in IL-5-deficient mice. J Clin Invest. 2004;113:551–60.
McMillan SJ, Xanthou G, Lloyd CM. Manipulation of allergen-induced airway remodeling by treatment with anti-TGF-beta antibody: effect on the smad signaling pathway. J Immunol. 2005;174:5774–80.
Royce SG, Cheng V, Samuel CS, Tang ML. The regulation of fibrosis in airway remodeling in asthma. Mol Cell Endocrinol. 2012;351:167–75.
Salvato G. Quantitative and morphological analysis of the vascular bed in bronchial biopsy specimens from asthmatic and non-asthmatic subjects. Thorax. 2001;56:902–6.
Hashimoto M, Tanaka H, Abe S. Quantitative analysis of bronchial wall vascularity in the medium and small airways of patients with asthma and COPD. Chest. 2005;127:965–72.
Siddiqui S, Sutcliffe A, Shikotra A, Woodman L, Doe C, McKenna S, Wardlaw A, Bradding P, Pavord I, Brightling C. Vascular remodeling is a feature of asthma and nonasthmatic eosinophilic bronchitis. J Allergy Clin Immunol. 2007;120:813–9.
Feltis BN, Wignarajah D, Reid DW, Ward C, Harding R, Walters EH. Effects of inhaled fluticasone on angiogenesis and vascular endothelial growth factor in asthma. Thorax. 2007;62:314–9.
Goldie RG, Pedersen KE. Mechanisms of increased airway microvascular permeability: role in airway inflammation and obstruction. Clin Exp Pharmacol Physiol. 1995;22:387–96.
Tam A, Wadsworth S, Dorscheid D, Man SF, Sin DD. The airway epithelium: more than just a structural barrier. Ther Adv Respir Dis. 2011;5:255–73.
Gern JE. Rhinovirus and the initiation of asthma. Curr Opin Allergy Clin Immunol. 2009;9:73–8.
Ahanchian H, Jones CM, Chen YS, Sly PD. Respiratory viral infections in children with asthma: do they matter and can we prevent them? BMC Pediatr. 2012;12:147-2431-12-147.
Juncadella IJ, Kadl A, Sharma AK, Shim YM, Hochreiter-Hufford A, Borish L, Ravichandran KS. Apoptotic cell clearance by bronchial epithelial cells critically influences airway inflammation. Nature. 2013;493:547–51.
Ahdieh M, Vandenbos T, Youakim A. Lung epithelial barrier function and wound healing are decreased by IL-4 and IL-13 and enhanced by IFN-gamma. Am J Physiol Cell Physiol. 2001;281:C2029–38.
de Boer WI, Sharma HS, Baelemans SM, Hoogsteden HC, Lambrecht BN, Braunstahl GJ. Altered expression of epithelial junctional proteins in atopic asthma: possible role in inflammation. Can J Physiol Pharmacol. 2008;86:105–12.
Xiao C, Puddicombe SM, Field S, Haywood J, Broughton-Head V, Puxeddu I, Haitchi HM, Vernon-Wilson E, Sammut D, Bedke N, Cremin C, Sones J, Djukanovic R, Howarth PH, Collins JE, Holgate ST, Monk P, Davies DE. Defective epithelial barrier function in asthma. J Allergy Clin Immunol. 2011;128:549-56.e1-12.
Jiang A, Bloom O, Ono S, Cui W, Unternaehrer J, Jiang S, Whitney JA, Connolly J, Banchereau J, Mellman I. Disruption of E-cadherin-mediated adhesion induces a functionally distinct pathway of dendritic cell maturation. Immunity. 2007;27:610–24.
Bucchieri F, Puddicombe SM, Lordan JL, Richter A, Buchanan D, Wilson SJ, Ward J, Zummo G, Howarth PH, Djukanovic R, Holgate ST, Davies DE. Asthmatic bronchial epithelium is more susceptible to oxidant-induced apoptosis. Am J Respir Cell Mol Biol. 2002;27:179–85.
Kicic A, Sutanto EN, Stevens PT, Knight DA, Stick SM. Intrinsic biochemical and functional differences in bronchial epithelial cells of children with asthma. Am J Respir Crit Care Med. 2006;174:1110–8.
Puddicombe SM, Polosa R, Richter A, Krishna MT, Howarth PH, Holgate ST, Davies DE. Involvement of the epidermal growth factor receptor in epithelial repair in asthma. FASEB J. 2000;14:1362–74.
Hackett TL. Epithelial-mesenchymal transition in the pathophysiology of airway remodelling in asthma. Curr Opin Allergy Clin Immunol. 2012;12:53–9.
Heijink IH, Postma DS, Noordhoek JA, Broekema M, Kapus A. House dust mite-promoted epithelial-to-mesenchymal transition in human bronchial epithelium. Am J Respir Cell Mol Biol. 2010;42:69–79.
Torrego A, Hew M, Oates T, Sukkar M, Fan Chung K. Expression and activation of TGF-beta isoforms in acute allergen-induced remodelling in asthma. Thorax. 2007;62:307–13.
Hackett TL, Warner SM, Stefanowicz D, Shaheen F, Pechkovsky DV, Murray LA, Argentieri R, Kicic A, Stick SM, Bai TR, Knight DA. Induction of epithelial-mesenchymal transition in primary airway epithelial cells from patients with asthma by transforming growth factor-beta 1. Am J Respir Crit Care Med. 2009;180:122–33.
Johnson JR, Roos A, Berg T, Nord M, Fuxe J. Chronic respiratory aeroallergen exposure in mice induces epithelial-mesenchymal transition in the large airways. PLoS One. 2011;6:e16175.
Yasukawa A, Hosoki K, Toda M, Miyake Y, Matsushima Y, Matsumoto T, Boveda-Ruiz D, Gil-Bernabe P, Nagao M, Sugimoto M, Hiraguchi Y, Tokuda R, Naito M, Takagi T, D’Alessandro-Gabazza CN, Suga S, Kobayashi T, Fujisawa T, Taguchi O, Gabazza EC. Eosinophils promote epithelial to mesenchymal transition of bronchial epithelial cells. PLoS One. 2013;8:e64281.
Rose MC, Voynow JA. Respiratory tract mucin genes and mucin glycoproteins in health and disease. Physiol Rev. 2006;86:245–78.
Thornton DJ, Rousseau K, McGuckin MA. Structure and function of the polymeric mucins in airways mucus. Annu Rev Physiol. 2008;70:459–86.
Dunnill MS. The pathology of asthma, with special reference to changes in the bronchial mucosa. J Clin Pathol. 1960;13:27–33.
Ulrik CS. Outcome of asthma: longitudinal changes in lung function. Eur Respir J. 1999;13:904–18.
Jinnai M, Niimi A, Ueda T, Matsuoka H, Takemura M, Yamaguchi M, Otsuka K, Oguma T, Takeda T, Ito I, Matsumoto H, Mishima M. Induced sputum concentrations of mucin in patients with asthma and chronic cough. Chest. 2010;137:1122–9.
Bry K, Whitsett JA, Lappalainen U. IL-1beta disrupts postnatal lung morphogenesis in the mouse. Am J Respir Cell Mol Biol. 2007;36:32–42.
Shim JJ, Dabbagh K, Ueki IF, Dao-Pick T, Burgel PR, Takeyama K, Tam DC, Nadel JA. IL-13 induces mucin production by stimulating epidermal growth factor receptors and by activating neutrophils. Am J Physiol Lung Cell Mol Physiol. 2001;280:L134–40.
Thomas B, Rutman A, Hirst RA, Haldar P, Wardlaw AJ, Bankart J, Brightling CE, O’Callaghan C. Ciliary dysfunction and ultrastructural abnormalities are features of severe asthma. J Allergy Clin Immunol. 2010;126(722–729):e2.
Bradding P, Walls AF, Holgate ST. The role of the mast cell in the pathophysiology of asthma. J Allergy Clin Immunol. 2006;117:1277–84.
Bradding P, Holgate ST. Immunopathology and human mast cell cytokines. Crit Rev Oncol Hematol. 1999;31:119–33.
Hirai H, Tanaka K, Yoshie O, Ogawa K, Kenmotsu K, Takamori Y, Ichimasa M, Sugamura K, Nakamura M, Takano S, Nagata K. Prostaglandin D2 selectively induces chemotaxis in T helper type 2 cells, eosinophils, and basophils via seven-transmembrane receptor CRTH2. J Exp Med. 2001;193:255–61.
Berger P, Perng DW, Thabrew H, Compton SJ, Cairns JA, McEuen AR, Marthan R, Tunon De Lara JM, Walls AF. Tryptase and agonists of PAR-2 induce the proliferation of human airway smooth muscle cells. J Appl Physiol. 1985;2001(91):1372–9.
Gould HJ, Sutton BJ. IgE in allergy and asthma today. Nat Rev Immunol. 2008;8:205–17.
Roth M, Zhong J, Zumkeller C, S’ng CT, Goulet S, Tamm M. The role of IgE-receptors in IgE-dependent airway smooth muscle cell remodelling. PLoS One. 2013;8:e56015.
Okumura S, Sagara H, Fukuda T, Saito H, Okayama Y. Fcepsilon RI-mediated amphiregulin production by human mast cells increases mucin gene expression in epithelial cells. J Allergy Clin Immunol. 2005;115:272–9.
Wang SW, Oh CK, Cho SH, Hu G, Martin R, Demissie-Sanders S, Li K, Moyle M, Yao Z. Amphiregulin expression in human mast cells and its effect on the primary human lung fibroblasts. J Allergy Clin Immunol. 2005;115:287–94.
Shim JY, Park SW, Kim DS, Shim JW, Jung HL, Park MS. The effect of interleukin-4 and amphiregulin on the proliferation of human airway smooth muscle cells and cytokine release. J Korean Med Sci. 2008;23:857–63.
Wenzel SE, Schwartz LB, Langmack EL, Halliday JL, Trudeau JB, Gibbs RL, Chu HW. Evidence that severe asthma can be divided pathologically into two inflammatory subtypes with distinct physiologic and clinical characteristics. Am J Respir Crit Care Med. 1999;160:1001–8.
Motojima S, Frigas E, Loegering DA, Gleich GJ. Toxicity of eosinophil cationic proteins for guinea pig tracheal epithelium in vitro. Am Rev Respir Dis. 1989;139:801–5.
Ramos-Barbon D, Fraga-Iriso R, Brienza NS, Montero-Martinez C, Verea-Hernando H, Olivenstein R, Lemiere C, Ernst P, Hamid QA, Martin JG. T Cells localize with proliferating smooth muscle alpha-actin + cell compartments in asthma. Am J Respir Crit Care Med. 2010;182:317–24.
GINA Report. Global strategy for asthma management and prevention. http://www.ginasthma.org . 2012. Accessed 3 Feb 2014.
Heitzer MD, Wolf IM, Sanchez ER, Witchel SF, DeFranco DB. Glucocorticoid receptor physiology. Rev Endocr Metab Disord. 2007;8:321–30.
Alangari AA. Genomic and non-genomic actions of glucocorticoids in asthma. Ann Thorac Med. 2010;5:133–9.
Lowenberg M, Verhaar AP, van den Brink GR, Hommes DW. Glucocorticoid signaling: a nongenomic mechanism for T-cell immunosuppression. Trends Mol Med. 2007;13:158–63.
Zhou J, Liu DF, Liu C, Kang ZM, Shen XH, Chen YZ, Xu T, Jiang CL. Glucocorticoids inhibit degranulation of mast cells in allergic asthma via nongenomic mechanism. Allergy. 2008;63:1177–85.
Kagoshima M, Wilcke T, Ito K, Tsaprouni L, Barnes PJ, Punchard N, Adcock IM. Glucocorticoid-mediated transrepression is regulated by histone acetylation and DNA methylation. Eur J Pharmacol. 2001;429:327–34.
Reichardt HM, Kaestner KH, Tuckermann J, Kretz O, Wessely O, Bock R, Gass P, Schmid W, Herrlich P, Angel P, Schutz G. DNA binding of the glucocorticoid receptor is not essential for survival. Cell. 1998;93:531–41.
Reichardt HM, Tuckermann JP, Gottlicher M, Vujic M, Weih F, Angel P, Herrlich P, Schutz G. Repression of inflammatory responses in the absence of DNA binding by the glucocorticoid receptor. EMBO J. 2001;20:7168–73.
Freishtat RJ, Nagaraju K, Jusko W, Hoffman EP. Glucocorticoid efficacy in asthma: is improved tissue remodeling upstream of anti-inflammation. J Investig Med. 2010;58:19–22.
Mattos W, Lim S, Russell R, Jatakanon A, Chung KF, Barnes PJ. Matrix metalloproteinase-9 expression in asthma: effect of asthma severity, allergen challenge, and inhaled corticosteroids. Chest. 2002;122:1543–52.
Hoshino M, Takahashi M, Takai Y, Sim J. Inhaled corticosteroids decrease subepithelial collagen deposition by modulation of the balance between matrix metalloproteinase-9 and tissue inhibitor of metalloproteinase-1 expression in asthma. J Allergy Clin Immunol. 1999;104:356–63.
Ward C, Pais M, Bish R, Reid D, Feltis B, Johns D, Walters EH. Airway inflammation, basement membrane thickening and bronchial hyperresponsiveness in asthma. Thorax. 2002;57:309–16.
Sont JK, Willems LN, Bel EH, van Krieken JH, Vandenbroucke JP, Sterk PJ. Clinical control and histopathologic outcome of asthma when using airway hyperresponsiveness as an additional guide to long-term treatment. The AMPUL Study Group. Am J Respir Crit Care Med. 1999;159:1043–51.
Chetta A, Zanini A, Foresi A, Del Donno M, Castagnaro A, D’Ippolito R, Baraldo S, Testi R, Saetta M, Olivieri D. Vascular component of airway remodeling in asthma is reduced by high dose of fluticasone. Am J Respir Crit Care Med. 2003;167:751–7.
Trigg CJ, Manolitsas ND, Wang J, Calderon MA, McAulay A, Jordan SE, Herdman MJ, Jhalli N, Duddle JM, Hamilton SA. Placebo-controlled immunopathologic study of four months of inhaled corticosteroids in asthma. Am J Respir Crit Care Med. 1994;150:17–22.
Olivieri D, Chetta A, Del Donno M, Bertorelli G, Casalini A, Pesci A, Testi R, Foresi A. Effect of short-term treatment with low-dose inhaled fluticasone propionate on airway inflammation and remodeling in mild asthma: a placebo-controlled study. Am J Respir Crit Care Med. 1997;155:1864–71.
Hoshino M, Nakamura Y, Sim JJ, Yamashiro Y, Uchida K, Hosaka K, Isogai S. Inhaled corticosteroid reduced lamina reticularis of the basement membrane by modulation of insulin-like growth factor (IGF)-I expression in bronchial asthma. Clin Exp Allergy. 1998;28:568–77.
Lundgren R, Soderberg M, Horstedt P, Stenling R. Morphological studies of bronchial mucosal biopsies from asthmatics before and after ten years of treatment with inhaled steroids. Eur Respir J. 1988;1:883–9.
Jeffery PK, Godfrey RW, Adelroth E, Nelson F, Rogers A, Johansson SA. Effects of treatment on airway inflammation and thickening of basement membrane reticular collagen in asthma. A quantitative light and electron microscopic study. Am Rev Respir Dis. 1992;145:890–9.
Boulet LP, Turcotte H, Laviolette M, Naud F, Bernier MC, Martel S, Chakir J. Airway hyperresponsiveness, inflammation, and subepithelial collagen deposition in recently diagnosed versus long-standing mild asthma. Influence of inhaled corticosteroids. Am J Respir Crit Care Med. 2000;162:1308–13.
Bergeron C, Hauber HP, Gotfried M, Newman K, Dhanda R, Servi RJ, Ludwig MS, Hamid Q. Evidence of remodeling in peripheral airways of patients with mild to moderate asthma: effect of hydrofluoroalkane-flunisolide. J Allergy Clin Immunol. 2005;116:983–9.
Dorscheid DR, Low E, Conforti A, Shifrin S, Sperling AI, White SR. Corticosteroid-induced apoptosis in mouse airway epithelium: effect in normal airways and after allergen-induced airway inflammation. J Allergy Clin Immunol. 2003;111:360–6.
White SR, Dorscheid DR. Corticosteroid-induced apoptosis of airway epithelium: a potential mechanism for chronic airway epithelial damage in asthma. Chest. 2002;122:278S–84S.
Dorscheid DR, Wojcik KR, Sun S, Marroquin B, White SR. Apoptosis of airway epithelial cells induced by corticosteroids. Am J Respir Crit Care Med. 2001;164:1939–47.
Erjefalt JS, Erjefalt I, Sundler F, Persson CG. Effects of topical budesonide on epithelial restitution in vivo in guinea pig trachea. Thorax. 1995;50:785–92.
Wadsworth SJ, Nijmeh HS, Hall IP. Glucocorticoids increase repair potential in a novel in vitro human airway epithelial wounding model. J Clin Immunol. 2006;26:376–87.
Liu J, Zhang M, Niu C, Luo Z, Dai J, Wang L, Liu E, Fu Z. Dexamethasone inhibits repair of human airway epithelial cells mediated by glucocorticoid-induced leucine zipper (GILZ). PLoS One. 2013;8:e60705.
Laitinen LA, Laitinen A, Haahtela T. A comparative study of the effects of an inhaled corticosteroid, budesonide, and a beta 2-agonist, terbutaline, on airway inflammation in newly diagnosed asthma: a randomized, double-blind, parallel-group controlled trial. J Allergy Clin Immunol. 1992;90:32–42.
Stewart AG, Fernandes D, Tomlinson PR. The effect of glucocorticoids on proliferation of human cultured airway smooth muscle. Br J Pharmacol. 1995;116:3219–26.
Cha HH, Cram EJ, Wang EC, Huang AJ, Kasler HG, Firestone GL. Glucocorticoids stimulate p21 gene expression by targeting multiple transcriptional elements within a steroid responsive region of the p21waf1/cip1 promoter in rat hepatoma cells. J Biol Chem. 1998;273:1998–2007.
Fernandes D, Guida E, Koutsoubos V, Harris T, Vadiveloo P, Wilson JW, Stewart AG. Glucocorticoids inhibit proliferation, cyclin D1 expression, and retinoblastoma protein phosphorylation, but not activity of the extracellular-regulated kinases in human cultured airway smooth muscle. Am J Respir Cell Mol Biol. 1999;21:77–88.
Roth M, Johnson PR, Borger P, Bihl MP, Rudiger JJ, King GG, Ge Q, Hostettler K, Burgess JK, Black JL, Tamm M. Dysfunctional interaction of C/EBPalpha and the glucocorticoid receptor in asthmatic bronchial smooth-muscle cells. N Engl J Med. 2004;351:560–74.
Cazes E, Giron-Michel J, Baouz S, Doucet C, Cagnoni F, Oddera S, Korner M, Dasic G, Testi R, Azzarone B, Canonica GW. Novel anti-inflammatory effects of the inhaled corticosteroid fluticasone propionate during lung myofibroblastic differentiation. J Immunol. 2001;167:5329–37.
Leung SY, Eynott P, Nath P, Chung KF. Effects of ciclesonide and fluticasone propionate on allergen-induced airway inflammation and remodeling features. J Allergy Clin Immunol. 2005;115:989–96.
Chetta A, Olivieri D. Role of inhaled steroids in vascular airway remodelling in asthma and COPD. Int J Endocrinol. 2012;2012:397693.
Orsida BE, Li X, Hickey B, Thien F, Wilson JW, Walters EH. Vascularity in asthmatic airways: relation to inhaled steroid dose. Thorax. 1999;54:289–95.
Hoshino M, Takahashi M, Takai Y, Sim J, Aoike N. Inhaled corticosteroids decrease vascularity of the bronchial mucosa in patients with asthma. Clin Exp Allergy. 2001;31:722–30.
Orsida BE, Ward C, Li X, Bish R, Wilson JW, Thien F, Walters EH. Effect of a long-acting beta2-agonist over three months on airway wall vascular remodeling in asthma. Am J Respir Crit Care Med. 2001;164:117–21.
Nelson HS, Weiss ST, Bleecker ER, Yancey SW, Dorinsky PM, SMART Study Group. The Salmeterol Multicenter Asthma Research Trial: a comparison of usual pharmacotherapy for asthma or usual pharmacotherapy plus salmeterol. Chest. 2006;129:15–26.
Todorova L, Gurcan E, Miller-Larsson A, Westergren-Thorsson G. Lung fibroblast proteoglycan production induced by serum is inhibited by budesonide and formoterol. Am J Respir Cell Mol Biol. 2006;34:92–100.
Todorova L, Gurcan E, Westergren-Thorsson G, Miller-Larsson A. Budesonide/formoterol effects on metalloproteolytic balance in TGFbeta-activated human lung fibroblasts. Respir Med. 2009;103:1755–63.
Peters-Golden M, Henderson WR Jr. Leukotrienes. N Engl J Med. 2007;357:1841–54.
Hallstrand TS, Henderson WR Jr. An update on the role of leukotrienes in asthma. Curr Opin Allergy Clin Immunol. 2010;10:60–6.
Asanuma F, Kuwabara K, Arimura A, Furue Y, Fleisch JH, Hori Y. Effects of leukotriene B4 receptor antagonist, LY293111Na, on antigen-induced bronchial hyperresponsiveness and leukocyte infiltration in sensitized guinea pigs. Inflamm Res. 2001;50:136–41.
Tager AM, Bromley SK, Medoff BD, Islam SA, Bercury SD, Friedrich EB, Carafone AD, Gerszten RE, Luster AD. Leukotriene B4 receptor BLT1 mediates early effector T cell recruitment. Nat Immunol. 2003;4:982–90.
Phan SH, McGarry BM, Loeffler KM, Kunkel SL. Binding of leukotriene C4 to rat lung fibroblasts and stimulation of collagen synthesis in vitro. Biochemistry. 1988;27:2846–53.
Ozaki T, Hayashi H, Tani K, Ogushi F, Yasuoka S, Ogura T. Neutrophil chemotactic factors in the respiratory tract of patients with chronic airway diseases or idiopathic pulmonary fibrosis. Am Rev Respir Dis. 1992;145:85–91.
Panettieri RA, Tan EM, Ciocca V, Luttmann MA, Leonard TB, Hay DW. Effects of LTD4 on human airway smooth muscle cell proliferation, matrix expression, and contraction In vitro: differential sensitivity to cysteinyl leukotriene receptor antagonists. Am J Respir Cell Mol Biol. 1998;19:453–61.
Cohen P, Noveral JP, Bhala A, Nunn SE, Herrick DJ, Grunstein MM. Leukotriene D4 facilitates airway smooth muscle cell proliferation via modulation of the IGF axis. Am J Physiol. 1995;269:L151–7.
Chauhan BF, Ducharme FM. Anti-leukotriene agents compared to inhaled corticosteroids in the management of recurrent and/or chronic asthma in adults and children. Cochrane Database Syst Rev. 2012;5:CD002314.
Bousquet J, Demoly P, Humbert M. Montelukast in guidelines and beyond. Adv Ther. 2009;26:575–87.
Joos S, Miksch A, Szecsenyi J, Wieseler B, Grouven U, Kaiser T, Schneider A. Montelukast as add-on therapy to inhaled corticosteroids in the treatment of mild to moderate asthma: a systematic review. Thorax. 2008;63:453–62.
Muz MH, Deveci F, Bulut Y, Ilhan N, Yekeler H, Turgut T. The effects of low dose leukotriene receptor antagonist therapy on airway remodeling and cysteinyl leukotriene expression in a mouse asthma model. Exp Mol Med. 2006;38:109–18.
Henderson WR Jr, Tang LO, Chu SJ, Tsao SM, Chiang GK, Jones F, Jonas M, Pae C, Wang H, Chi EY. A role for cysteinyl leukotrienes in airway remodeling in a mouse asthma model. Am J Respir Crit Care Med. 2002;165:108–16.
Henderson WR Jr, Chiang GK, Tien YT, Chi EY. Reversal of allergen-induced airway remodeling by CysLT1 receptor blockade. Am J Respir Crit Care Med. 2006;173:718–28.
Gobbato NB, de Souza FC, Fumagalli SB, Lopes FD, Prado CM, Martins MA, Tiberio Ide F, Leick EA. Antileukotriene reverts the early effects of inflammatory response of distal parenchyma in experimental chronic allergic inflammation. Biomed Res Int. 2013;2013:523761.
Rabe KF, Magnussen H, Dent G. Theophylline and selective PDE inhibitors as bronchodilators and smooth muscle relaxants. Eur Respir J. 1995;8:637–42.
Lagente V, Pruniaux MP, Junien JL, Moodley I. Modulation of cytokine-induced eosinophil infiltration by phosphodiesterase inhibitors. Am J Respir Crit Care Med. 1995;151:1720–4.
Sullivan P, Bekir S, Jaffar Z, Page C, Jeffery P, Costello J. Anti-inflammatory effects of low-dose oral theophylline in atopic asthma. Lancet. 1994;343:1006–8.
Horiguchi T, Tachikawa S, Kasahara J, Doi M, Shiga M, Miyazaki J, Sasaki Y, Hirose M, Imazu M. Suppression of airway inflammation by theophylline in adult bronchial asthma. Respiration. 1999;66:124–7.
Hendeles L, Harman E, Huang D, O’Brien R, Blake K, Delafuente J. Theophylline attenuation of airway responses to allergen: comparison with cromolyn metered-dose inhaler. J Allergy Clin Immunol. 1995;95:505–14.
Magnussen H, Reuss G, Jorres R. Theophylline has a dose-related effect on the airway response to inhaled histamine and methacholine in asthmatics. Am Rev Respir Dis. 1987;136:1163–7.
Barnes PJ. Histone deacetylase-2 and airway disease. Ther Adv Respir Dis. 2009;3:235–43.
Gosens R, Zaagsma J, Meurs H, Halayko AJ. Muscarinic receptor signaling in the pathophysiology of asthma and COPD. Respir Res. 2006;7:73.
Matthiesen S, Bahulayan A, Kempkens S, Haag S, Fuhrmann M, Stichnote C, Juergens UR, Racke K. Muscarinic receptors mediate stimulation of human lung fibroblast proliferation. Am J Respir Cell Mol Biol. 2006;35:621–7.
Gosens R, Nelemans SA, Grootte Bromhaar MM, McKay S, Zaagsma J, Meurs H. Muscarinic M3-receptors mediate cholinergic synergism of mitogenesis in airway smooth muscle. Am J Respir Cell Mol Biol. 2003;28:257–62.
Oenema TA, Smit M, Smedinga L, Racke K, Halayko AJ, Meurs H, Gosens R. Muscarinic receptor stimulation augments TGF-beta1-induced contractile protein expression by airway smooth muscle cells. Am J Physiol Lung Cell Mol Physiol. 2012;303:L589–97.
Kerstjens HA, Engel M, Dahl R, Paggiaro P, Beck E, Vandewalker M, Sigmund R, Seibold W, Moroni-Zentgraf P, Bateman ED. Tiotropium in asthma poorly controlled with standard combination therapy. N Engl J Med. 2012;367:1198–207.
Kerstjens HA, Disse B, Schroder-Babo W, Bantje TA, Gahlemann M, Sigmund R, Engel M, van Noord JA. Tiotropium improves lung function in patients with severe uncontrolled asthma: a randomized controlled trial. J Allergy Clin Immunol. 2011;128:308–14.
Peters SP, Kunselman SJ, Icitovic N, Moore WC, Pascual R, Ameredes BT, Boushey HA, Calhoun WJ, Castro M, Cherniack RM, Craig T, Denlinger L, Engle LL, DiMango EA, Fahy JV, Israel E, Jarjour N, Kazani SD, Kraft M, Lazarus SC, Lemanske RF Jr, Lugogo N, Martin RJ, Meyers DA, Ramsdell J, Sorkness CA, Sutherland ER, Szefler SJ, Wasserman SI, Walter MJ, Wechsler ME, Chinchilli VM, Bleecker ER, National Heart, Lung, and Blood Institute Asthma Clinical Research Network. Tiotropium bromide step-up therapy for adults with uncontrolled asthma. N Engl J Med. 2010;363:1715–26.
Gosens R, Bos IS, Zaagsma J, Meurs H. Protective effects of tiotropium bromide in the progression of airway smooth muscle remodeling. Am J Respir Crit Care Med. 2005;171:1096–102.
Bos IS, Gosens R, Zuidhof AB, Schaafsma D, Halayko AJ, Meurs H, Zaagsma J. Inhibition of allergen-induced airway remodelling by tiotropium and budesonide: a comparison. Eur Respir J. 2007;30:653–61.
Ohta S, Oda N, Yokoe T, Tanaka A, Yamamoto Y, Watanabe Y, Minoguchi K, Ohnishi T, Hirose T, Nagase H, Ohta K, Adachi M. Effect of tiotropium bromide on airway inflammation and remodelling in a mouse model of asthma. Clin Exp Allergy. 2010;40:1266–75.
Haag S, Matthiesen S, Juergens UR, Racke K. Muscarinic receptors mediate stimulation of collagen synthesis in human lung fibroblasts. Eur Respir J. 2008;32:555–62.
Iesato K, Tatsumi K, Saito K, Ogasawara T, Sakao S, Tada Y, Kasahara Y, Kurosu K, Tanabe N, Takiguchi Y, Kuriyama T, Shirasawa H. Tiotropium bromide attenuates respiratory syncytial virus replication in epithelial cells. Respiration. 2008;76:434–41.
Zieg G, Lack G, Harbeck RJ, Gelfand EW, Leung DY. In vivo effects of glucocorticoids on IgE production. J Allergy Clin Immunol. 1994;94:222–30.
Strunk RC, Bloomberg GR. Omalizumab for asthma. N Engl J Med. 2006;354:2689–95.
Holgate ST, Chuchalin AG, Hebert J, Lotvall J, Persson GB, Chung KF, Bousquet J, Kerstjens HA, Fox H, Thirlwell J, Cioppa GD, Omalizumab 011 International Study Group. Efficacy and safety of a recombinant anti-immunoglobulin E antibody (omalizumab) in severe allergic asthma. Clin Exp Allergy. 2004;34:632–8.
Djukanovic R, Wilson SJ, Kraft M, Jarjour NN, Steel M, Chung KF, Bao W, Fowler-Taylor A, Matthews J, Busse WW, Holgate ST, Fahy JV. Effects of treatment with anti-immunoglobulin E antibody omalizumab on airway inflammation in allergic asthma. Am J Respir Crit Care Med. 2004;170:583–93.
Fahy JV, Fleming HE, Wong HH, Liu JT, Su JQ, Reimann J, Fick RB Jr, Boushey HA. The effect of an anti-IgE monoclonal antibody on the early- and late-phase responses to allergen inhalation in asthmatic subjects. Am J Respir Crit Care Med. 1997;155:1828–34.
MacGlashan DW Jr, Bochner BS, Adelman DC, Jardieu PM, Togias A, McKenzie-White J, Sterbinsky SA, Hamilton RG, Lichtenstein LM. Down-regulation of Fc(epsilon)RI expression on human basophils during in vivo treatment of atopic patients with anti-IgE antibody. J Immunol. 1997;158:1438–45.
Prussin C, Griffith DT, Boesel KM, Lin H, Foster B, Casale TB. Omalizumab treatment downregulates dendritic cell FcepsilonRI expression. J Allergy Clin Immunol. 2003;112:1147–54.
Boulet LP, Chapman KR, Cote J, Kalra S, Bhagat R, Swystun VA, Laviolette M, Cleland LD, Deschesnes F, Su JQ, DeVault A, Fick RB Jr, Cockcroft DW. Inhibitory effects of an anti-IgE antibody E25 on allergen-induced early asthmatic response. Am J Respir Crit Care Med. 1997;155:1835–40.
Hoshino M, Ohtawa J. Effects of adding omalizumab, an anti-immunoglobulin E antibody, on airway wall thickening in asthma. Respiration. 2012;83:520–8.
Riccio AM, Dal Negro RW, Micheletto C, De Ferrari L, Folli C, Chiappori A, Canonica GW. Omalizumab modulates bronchial reticular basement membrane thickness and eosinophil infiltration in severe persistent allergic asthma patients. Int J Immunopathol Pharmacol. 2012;25:475–84.
Rosenwasser LJ, Meng J. Anti-CD23. Clin Rev Allergy Immunol. 2005;29:61–72.
Berair R, Pavord ID. Rationale and clinical results of inhibiting interleukin-5 for the treatment of severe asthma. Curr Allergy Asthma Rep. 2013;13(5):469-76.
Flood-Page P, Swenson C, Faiferman I, Matthews J, Williams M, Brannick L, Robinson D, Wenzel S, Busse W, Hansel TT, Barnes NC, International Mepolizumab Study Group. A study to evaluate safety and efficacy of mepolizumab in patients with moderate persistent asthma. Am J Respir Crit Care Med. 2007;176:1062–71.
Flood-Page PT, Menzies-Gow AN, Kay AB, Robinson DS. Eosinophil’s role remains uncertain as anti-interleukin-5 only partially depletes numbers in asthmatic airway. Am J Respir Crit Care Med. 2003;167:199–204.
Nair P, Pizzichini MM, Kjarsgaard M, Inman MD, Efthimiadis A, Pizzichini E, Hargreave FE, O’Byrne PM. Mepolizumab for prednisone-dependent asthma with sputum eosinophilia. N Engl J Med. 2009;360:985–93.
Pavord ID, Korn S, Howarth P, Bleecker ER, Buhl R, Keene ON, Ortega H, Chanez P. Mepolizumab for severe eosinophilic asthma (DREAM): a multicentre, double-blind, placebo-controlled trial. Lancet. 2012;380:651–9.
Liu Y, Zhang S, Li DW, Jiang SJ. Efficacy of anti-interleukin-5 therapy with mepolizumab in patients with asthma: a meta-analysis of randomized placebo-controlled trials. PLoS One. 2013;8:e59872.
Leckie MJ, ten Brinke A, Khan J, Diamant Z, O’Connor BJ, Walls CM, Mathur AK, Cowley HC, Chung KF, Djukanovic R, Hansel TT, Holgate ST, Sterk PJ, Barnes PJ. Effects of an interleukin-5 blocking monoclonal antibody on eosinophils, airway hyper-responsiveness, and the late asthmatic response. Lancet. 2000;356:2144–8.
Foster PS, Hogan SP, Ramsay AJ, Matthaei KI, Young IG. Interleukin 5 deficiency abolishes eosinophilia, airways hyperreactivity, and lung damage in a mouse asthma model. J Exp Med. 1996;183:195–201.
Tanaka H, Komai M, Nagao K, Ishizaki M, Kajiwara D, Takatsu K, Delespesse G, Nagai H. Role of interleukin-5 and eosinophils in allergen-induced airway remodeling in mice. Am J Respir Cell Mol Biol. 2004;31:62–8.
Haldar P, Brightling CE, Singapuri A, Hargadon B, Gupta S, Monteiro W, Bradding P, Green RH, Wardlaw AJ, Ortega H, Pavord ID. Outcomes after cessation of mepolizumab therapy in severe eosinophilic asthma: a 12-month follow-up analysis. J Allergy Clin Immunol. 2014;133:921–3.
Wills-Karp M, Luyimbazi J, Xu X, Schofield B, Neben TY, Karp CL, Donaldson DD. Interleukin-13: central mediator of allergic asthma. Science. 1998;282:2258–61.
Yang G, Volk A, Petley T, Emmell E, Giles-Komar J, Shang X, Li J, Das AM, Shealy D, Griswold DE, Li L. Anti-IL-13 monoclonal antibody inhibits airway hyperresponsiveness, inflammation and airway remodeling. Cytokine. 2004;28:224–32.
Tomlinson KL, Davies GC, Sutton DJ, Palframan RT. Neutralisation of interleukin-13 in mice prevents airway pathology caused by chronic exposure to house dust mite. PLoS One. 2010; 5. doi:10.1371/journal.pone.0013136.
Blease K, Jakubzick C, Westwick J, Lukacs N, Kunkel SL, Hogaboam CM. Therapeutic effect of IL-13 immunoneutralization during chronic experimental fungal asthma. J Immunol. 2001;166:5219–24.
Corren J, Lemanske RF, Hanania NA, Korenblat PE, Parsey MV, Arron JR, Harris JM, Scheerens H, Wu LC, Su Z, Mosesova S, Eisner MD, Bohen SP, Matthews JG. Lebrikizumab treatment in adults with asthma. N Engl J Med. 2011;365:1088–98.
Piper E, Brightling C, Niven R, Oh C, Faggioni R, Poon K, She D, Kell C, May RD, Geba GP, Molfino NA. A phase II placebo-controlled study of tralokinumab in moderate-to-severe asthma. Eur Respir J. 2013;41:330–8.
Bergeron C, Page N, Barbeau B, Chakir J. Interleukin-4 promotes airway remodeling in asthma: regulation of procollagen I (alpha1) gene by interleukin-4. Chest. 2003;123:424S.
Gavett SH, O’Hearn DJ, Karp CL, Patel EA, Schofield BH, Finkelman FD, Wills-Karp M. Interleukin-4 receptor blockade prevents airway responses induced by antigen challenge in mice. Am J Physiol. 1997;272:L253–61.
Wenzel S, Ford L, Pearlman D, Spector S, Sher L, Skobieranda F, Wang L, Kirkesseli S, Rocklin R, Bock B, Hamilton J, Ming JE, Radin A, Stahl N, Yancopoulos GD, Graham N, Pirozzi G. Dupilumab in persistent asthma with elevated eosinophil levels. N Engl J Med. 2013;368:2455–66.
Chen Y, Thai P, Zhao YH, Ho YS, DeSouza MM, Wu R. Stimulation of airway mucin gene expression by interleukin (IL)-17 through IL-6 paracrine/autocrine loop. J Biol Chem. 2003;278:17036–43.
Inoue D, Numasaki M, Watanabe M, Kubo H, Sasaki T, Yasuda H, Yamaya M, Sasaki H. IL-17A promotes the growth of airway epithelial cells through ERK-dependent signaling pathway. Biochem Biophys Res Commun. 2006;347:852–8.
Busse WW, Holgate S, Kerwin E, Chon Y, Feng J, Lin J, Lin SL. Randomized, double-blind, placebo-controlled study of brodalumab, a human anti-IL-17 receptor monoclonal antibody, in moderate to severe asthma. Am J Respir Crit Care Med. 2013;188:1294–302.
Howarth PH, Babu KS, Arshad HS, Lau L, Buckley M, McConnell W, Beckett P, Al Ali M, Chauhan A, Wilson SJ, Reynolds A, Davies DE, Holgate ST. Tumour necrosis factor (TNFalpha) as a novel therapeutic target in symptomatic corticosteroid dependent asthma. Thorax. 2005;60:1012–8.
Berry MA, Hargadon B, Shelley M, Parker D, Shaw DE, Green RH, Bradding P, Brightling CE, Wardlaw AJ, Pavord ID. Evidence of a role of tumor necrosis factor alpha in refractory asthma. N Engl J Med. 2006;354:697–708.
Baluk P, Yao LC, Feng J, Romano T, Jung SS, Schreiter JL, Yan L, Shealy DJ, McDonald DM. TNF-alpha drives remodeling of blood vessels and lymphatics in sustained airway inflammation in mice. J Clin Invest. 2009;119:2954–64.
Wenzel SE, Barnes PJ, Bleecker ER, Bousquet J, Busse W, Dahlen SE, Holgate ST, Meyers DA, Rabe KF, Antczak A, Baker J, Horvath I, Mark Z, Bernstein D, Kerwin E, Schlenker-Herceg R, Lo KH, Watt R, Barnathan ES, Chanez P, T03 Asthma Investigators. A randomized, double-blind, placebo-controlled study of tumor necrosis factor-alpha blockade in severe persistent asthma. Am J Respir Crit Care Med. 2009;179:549–58.
Le Cras TD, Acciani TH, Mushaben EM, Kramer EL, Pastura PA, Hardie WD, Korfhagen TR, Sivaprasad U, Ericksen M, Gibson AM, Holtzman MJ, Whitsett JA, Hershey GK. Epithelial EGF receptor signaling mediates airway hyperreactivity and remodeling in a mouse model of chronic asthma. Am J Physiol Lung Cell Mol Physiol. 2011;300:L414–21.
Heijink IH, van Oosterhout A, Kapus A. Epidermal growth factor receptor signalling contributes to house dust mite-induced epithelial barrier dysfunction. Eur Respir J. 2010;36:1016–26.
Hamilton LM, Puddicombe SM, Dearman RJ, Kimber I, Sandstrom T, Wallin A, Howarth PH, Holgate ST, Wilson SJ, Davies DE. Altered protein tyrosine phosphorylation in asthmatic bronchial epithelium. Eur Respir J. 2005;25:978–85.
Kung YC, Lin CC, Liaw SF, Lin MW, Chang FT. Effects of erlotinib on pulmonary function and airway remodeling after sensitization and repeated allergen challenge in Brown-Norway rats. Respir Physiol Neurobiol. 2011;175:349–56.
Rhee CK, Kim JW, Park CK, Kim JS, Kang JY, Kim SJ, Kim SC, Kwon SS, Kim YK, Park SH, Lee SY. Effect of imatinib on airway smooth muscle thickening in a murine model of chronic asthma. Int Arch Allergy Immunol. 2011;155:243–51.
Harvard Clinical Research Institute; National Heart, Lung, and Blood Institute (NHLBI). Effects of cKit inhibition by imatinib in patients with severe refractory asthma (KIA). http://clinicaltrials.gov/ct2/show/study/NCT01097694. 2013. Accessed 07 March 2014.
Humbert M, de Blay F, Garcia G, Prud’homme A, Leroyer C, Magnan A, Tunon-de-Lara JM, Pison C, Aubier M, Charpin D, Vachier I, Purohit A, Gineste P, Bader T, Moussy A, Hermine O, Chanez P. Masitinib, a c-kit/PDGF receptor tyrosine kinase inhibitor, improves disease control in severe corticosteroid-dependent asthmatics. Allergy. 2009;64:1194–201.
AB Science. A phase 3 study to compare efficacy and safety of masitinib to placebo in treatment of patients with severe persistent asthma treated with oral. http://www.clinicaltrials.gov/ct2/show/NCT01449162. 2012. Accessed April 2014.
Yuan C, Zhou L, Cheng J, Zhang J, Teng Y, Huang M, Adcock IM, Barnes PJ, Yao X. Statins as potential therapeutic drug for asthma? Respir Res. 2012;13:108-9921-13-108.
Zeki AA, Oldham J, Wilson M, Fortenko O, Goyal V, Last M, Last A, Patel A, Last JA, Kenyon NJ. Statin use and asthma control in patients with severe asthma. BMJ Open 2013; 3. doi:10.1136/bmjopen-2013-003314.
Zeki AA, Thai P, Kenyon NJ, Wu R. Differential effects of simvastatin on IL-13-induced cytokine gene expression in primary mouse tracheal epithelial cells. Respir Res. 2012;13:38.
Ahmad T, Mabalirajan U, Sharma A, Aich J, Makhija L, Ghosh B, Agrawal A. Simvastatin improves epithelial dysfunction and airway hyperresponsiveness: from asymmetric dimethyl-arginine to asthma. Am J Respir Cell Mol Biol. 2011;44:531–9.
Zeki AA, Bratt JM, Rabowsky M, Last JA, Kenyon NJ. Simvastatin inhibits goblet cell hyperplasia and lung arginase in a mouse model of allergic asthma: a novel treatment for airway remodeling? Transl Res. 2010;156:335–49.
Takeda N, Kondo M, Ito S, Ito Y, Shimokata K, Kume H. Role of RhoA inactivation in reduced cell proliferation of human airway smooth muscle by simvastatin. Am J Respir Cell Mol Biol. 2006;35:722–9.
Schaafsma D, McNeill KD, Mutawe MM, Ghavami S, Unruh H, Jacques E, Laviolette M, Chakir J, Halayko AJ. Simvastatin inhibits TGFbeta1-induced fibronectin in human airway fibroblasts. Respir Res. 2011;12:113-9921-12-113.
Bergheim I, Luyendyk JP, Steele C, Russell GK, Guo L, Roth RA, Arteel GE. Metformin prevents endotoxin-induced liver injury after partial hepatectomy. J Pharmacol Exp Ther. 2006;316:1053–61.
Zhao X, Zmijewski JW, Lorne E, Liu G, Park YJ, Tsuruta Y, Abraham E. Activation of AMPK attenuates neutrophil proinflammatory activity and decreases the severity of acute lung injury. Am J Physiol Lung Cell Mol Physiol. 2008;295:L497–504.
Park CS, Bang BR, Kwon HS, Moon KA, Kim TB, Lee KY, Moon HB, Cho YS. Metformin reduces airway inflammation and remodeling via activation of AMP-activated protein kinase. Biochem Pharmacol. 2012;84:1660–70.
Gupta A, Sjoukes A, Richards D, Banya W, Hawrylowicz C, Bush A, Saglani S. Relationship between serum vitamin D, disease severity, and airway remodeling in children with asthma. Am J Respir Crit Care Med. 2011;184:1342–9.
Damera G, Fogle HW, Lim P, Goncharova EA, Zhao H, Banerjee A, Tliba O, Krymskaya VP, Panettieri RA Jr. Vitamin D inhibits growth of human airway smooth muscle cells through growth factor-induced phosphorylation of retinoblastoma protein and checkpoint kinase 1. Br J Pharmacol. 2009;158:1429–41.
Song Y, Hong J, Liu D, Lin Q, Lai G. 1,25-Dihydroxyvitamin D3 inhibits nuclear factor kappa B activation by stabilizing inhibitor IkappaBalpha via mRNA stability and reduced phosphorylation in passively sensitized human airway smooth muscle cells. Scand J Immunol. 2013;77:109–16.
Manolis AS, Vassilikos V, Maounis TN, Chiladakis J, Cokkinos DV. Radiofrequency ablation in pediatric and adult patients: comparative results. J Interv Card Electrophysiol. 2001;5:443–53.
Danek CJ, Lombard CM, Dungworth DL, Cox PG, Miller JD, Biggs MJ, Keast TM, Loomas BE, Wizeman WJ, Hogg JC, Leff AR. Reduction in airway hyperresponsiveness to methacholine by the application of RF energy in dogs. J Appl Physiol. 1985;2004(97):1946–53.
Miller JD, Cox G, Vincic L, Lombard CM, Loomas BE, Danek CJ. A prospective feasibility study of bronchial thermoplasty in the human airway. Chest. 2005;127:1999–2006.
Cox G, Miller JD, McWilliams A, Fitzgerald JM, Lam S. Bronchial thermoplasty for asthma. Am J Respir Crit Care Med. 2006;173:965–9.
Cox G, Thomson NC, Rubin AS, Niven RM, Corris PA, Siersted HC, Olivenstein R, Pavord ID, McCormack D, Chaudhuri R, Miller JD, Laviolette M, AIR Trial Study Group. Asthma control during the year after bronchial thermoplasty. N Engl J Med. 2007;356:1327–37.
Pavord ID, Cox G, Thomson NC, Rubin AS, Corris PA, Niven RM, Chung KF, Laviolette M, RISA Trial Study Group. Safety and efficacy of bronchial thermoplasty in symptomatic, severe asthma. Am J Respir Crit Care Med. 2007;176:1185–91.
Castro M, Rubin AS, Laviolette M, Fiterman J, De Andrade Lima M, Shah PL, Fiss E, Olivenstein R, Thomson NC, Niven RM, Pavord ID, Simoff M, Duhamel DR, McEvoy C, Barbers R, Ten Hacken NH, Wechsler ME, Holmes M, Phillips MJ, Erzurum S, Lunn W, Israel E, Jarjour N, Kraft M, Shargill NS, Quiring J, Berry SM, Cox G, AIR2 Trial Study Group. Effectiveness and safety of bronchial thermoplasty in the treatment of severe asthma: a multicenter, randomized, double-blind, sham-controlled clinical trial. Am J Respir Crit Care Med. 2010;181:116–24.
Castro M, Rubin A, Laviolette M, Hanania NA, Armstrong B, Cox G, AIR2 Trial Study Group. Persistence of effectiveness of bronchial thermoplasty in patients with severe asthma. Ann Allergy Asthma Immunol. 2011;107:65–70.
Wechsler ME, Laviolette M, Rubin AS, Fiterman J, Lapa e Silva JR, Shah PL, Fiss E, Olivenstein R, Thomson NC, Niven RM, Pavord ID, Simoff M, Hales JB, McEvoy C, Slebos DJ, Holmes M, Phillips MJ, Erzurum SC, Hanania NA, Sumino K, Kraft M, Cox G, Sterman DH, Hogarth K, Kline JN, Mansur AH, Louie BE, Leeds WM, Barbers RG, Austin JH, Shargill NS, Quiring J, Armstrong B, Castro M, Asthma Intervention Research 2 Trial Study Group. Bronchial thermoplasty: Long-term safety and effectiveness in patients with severe persistent asthma. J Allergy Clin Immunol. 2013;132:1295–302.
Jesudason EC. Airway smooth muscle: an architect of the lung? Thorax. 2009;64:541–5.
Gordon IO, Husain AN, Charbeneau J, Krishnan JA, Hogarth DK. Endobronchial biopsy: a guide for asthma therapy selection in the era of bronchial thermoplasty. J Asthma. 2013;50:634–41.
Arcispedale Santa Maria Nuova-IRCCS. Bronchial thermoplasty: effect on neuronal and chemosensitive component of the bronchial mucosa. 2013. http://www.clinicaltrials.gov/ct2/show/NCT01839591. Accessed March 2014.
Fundació Institut de Recerca de l’Hospital de la Santa Creu i Sant Pau. Study of physiopathological mechanisms and results of treatment with bronchial thermoplasty in severe asthma. 2013. http://www.clinicaltrials.gov/ct2/show/NCT01974921. Accessed March 2014.
Wenzel SE. Asthma phenotypes: the evolution from clinical to molecular approaches. Nat Med. 2012;18:716–25.
Acknowledgments
CEB is funded by AirPROM (FP7 270194). This article is supported by the National Institute for Health Research (NIHR) Leicester Respiratory Biomedical Research Unit. The views expressed are those of the authors and not necessarily those of the National Health Service (NHS), the NIHR or the Department of Health. CEB has received grant funding and consultancy from Novartis, GSK, Genentech/Roche, Chiesi and Medimmune/AstraZeneca. RB declares no conflict of interest.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article

Cite this article
Berair, R., Brightling, C.E. Asthma Therapy and Its Effect on Airway Remodelling. Drugs 74, 1345–1369 (2014). https://doi.org/10.1007/s40265-014-0250-4
Published:
Issue Date:
DOI: https://doi.org/10.1007/s40265-014-0250-4