
Abstract
Hyperphosphataemia can be induced by three main conditions: a massive acute phosphate load, a primary increase in renal phosphate reabsorption, and an impaired renal phosphate excretion due to acute or chronic renal insufficiency. Renal excretion is so efficient in normal subjects that balance can be maintained with only a minimal rise in serum phosphorus concentration even for a large phosphorus load. Therefore, acute hyperphosphataemia usually resolves within few hours if renal function is intact. The most frequent cause of chronic hyperphosphataemia is chronic renal failure. Hyperphosphataemia in chronic kidney disease (CKD) is associated with increased cardiovascular morbidity and mortality. Lowering the phosphate load and maintaining serum phosphorus levels within the normal range are considered important therapeutic goals to improve clinical outcomes in CKD patients. Treatment consists of diminishing intestinal phosphate absorption by a low phosphate diet and phosphate binders. In CKD patients on dialysis an efficient dialysis removal of phosphate should be ensured. Dietary restriction of phosphorus while maintaining adequate protein intake is not sufficient to control serum phosphate levels in most CKD patients; therefore, the prescription of a phosphate binder is required. Aluminium-containing agents are efficient but no longer widely used because of their toxicity. Calcium-based salts are inexpensive, effective and most widely used, but there is now concern about their association with hypercalcaemia, parathyroid gland suppression, adynamic bone disease, and vascular and extraosseous calcification. The average daily dose of calcium acetate or carbonate prescribed in the randomised controlled trials to control hyperphosphataemia in dialysis patients ranges between 1.2 and 2.3 g of elemental calcium. Such doses are greater than the recommended dietary calcium intake and can lead to a positive calcium balance. Although large amounts of calcium salts should probably be avoided, modest doses (<1 g of elemental calcium) may represent a reasonable initial approach to reduced serum phosphorus levels. A non-calcium-based binder can then be added when large doses of binder are required. At present, there are three types of non-calcium-based phosphate binders available: sevelamer, lanthanum carbonate and magnesium salts. Each of these compounds is as effective as calcium salts in lowering serum phosphorus levels depending on an adequate prescribed dose and adherence of the patient to treatment. Sevelamer is the only non-calcium-containing phosphate binder that does not have potential for systemic accumulation and presents pleiotropic effects that may impact on cardiovascular disease. In contrast, lanthanum carbonate and magnesium salts are absorbed in the gut and their route of excretion is biliary for lanthanum and urinary for magnesium. There are insufficient data to establish the comparative superiority of non-calcium binding agents over calcium salts for such important patient-level outcomes as all-cause mortality and cardiovascular end points. Moreover, full adoption of sevelamer and lanthanum by government drug reimbursement agencies in place of calcium salts would lead to a large increase in health-care expenditure. Therefore, the choice of phosphate binder should be individualised, considering the clinical context, the costs, and the individual tolerability the concomitant effects on other parameters of mineral metabolism, such as serum calcium and parathyroid hormone, besides those on serum phosphorus.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
1 Introduction
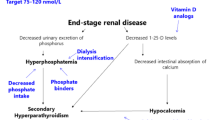
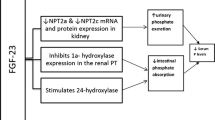
The serum phosphorus concentration depends on dietary phosphorus intake, intestinal absorption, bone turnover, distribution of phosphorus into intracellular compartments and renal phosphorus excretion. Dietary phosphorus is adsorbed throughout the small intestine by a passive paracellular transport and by a vitamin D-dependent active transport through the sodium-dependent phosphate transporter 2b (NPT2b) [1, 2]. The passive absorption is nonsaturable, so that the greater the dietary intake, the much higher is the net absorption. The vitamin D-dependent absorption is of secondary importance and accounts for about 20 % of total absorption under conditions of normal phosphorus intake [3]. Overall, 60–80 % of dietary phosphorus is adsorbed in the gut [4]. Given the relative lack of regulation of intestinal phosphate absorption, the kidney ultimately plays a key role in the regulation of phosphate balance. Renal excretion is so efficient in normal subjects that balance can be maintained with only a minimal rise in serum phosphorus concentration even if the dietary intake is increased up to 130 mmol/day (4,000 mg/day). The increase of renal phosphate excretion in response to the increased dietary intake depends on the inhibition of sodium-phosphate co-transporters 2a and 2c (NPT2a and NPT2c) in the luminal membrane of renal tubules that allow reabsorption of filtered phosphate [5, 6]. The inhibition of sodium-phosphate co-transporters is mainly due to the increased secretion of fibroblast growth factor 23 (FGF23), which is stimulated by the high dietary phosphorus intake [6–10]. FGF23 is secreted by osteocytes and osteoblasts in response to oral phosphate loading or increased serum 1,25-dihydroxyvitamin D levels [11]. High FGF23 levels in response to high phosphate intake induce greater urinary fractional excretion of phosphate and reduce the efficiency of phosphate absorption in the gut by lowering 1,25-dihydroxyvitamin D levels [8–10], thus preventing wide serum phosphorus fluctuations. The increase in renal phosphate excretion in response to elevated dietary intake can also be due to the stimulation of parathyroid hormone (PTH) secretion by the fall in serum ionised calcium induced by transient hyperphosphataemia. PTH reduces expression of NPT2a and NPT2c in the proximal renal tubules, thereby diminishing phosphate reabsorption and increasing urinary phosphate excretion [5, 6]. The kidney is able to excrete large amounts of phosphorus, allowing only small increases in serum phosphorus concentrations when the intake is distributed over the course of the day. If, however, an acute phosphate load is given over several hours, transient hyperphosphataemia will ensue. The diagnostic approach to hyperphosphataemia involves the identification of the reason that phosphate entry into the extracellular fluid exceeds the rate at which it can be excreted.
2 Causes of Hyperphosphatemia
There are three general circumstances that can induce hyperphosphataemia: massive acute phosphate load, a primary increase in renal phosphate reabsorption and renal failure.
2.1 Massive Acute Phosphate Load
Delivery of a large load of exogenous or endogenous phosphate over a short time can overwhelm renal capacity to excrete phosphate. Since phosphate is the major intracellular anion, any cause of marked tissue breakdown can lead to release of intracellular phosphate into the extracellular fluid. Ingestion of phosphate-containing laxatives such as the use of Fleet’s phospho-soda, for example, in preparation for colonoscopy is a not uncommon cause of severe exogenous hyperphosphataemia, which can lead to toxic effects, such as acute phosphate nephropathy or death [12–16]. Patients with chronic renal failure and those dehydrated are more susceptible to these complications. Endogenous hyperphosphataemia can result from severe tissue necrosis due to tumour lysis syndrome, rhabdomyolysis, and massive haemolysis or transfusion of stored blood [17, 18]. Other causes of release of intracellular phosphate into the extracellular fluid are lactic acidosis and diabetic ketoacidosis [19]. Hyperphosphataemia can induce potentially symptomatic hypocalcaemia because of calcium-phosphate precipitation in the tissues. Thus, clinical manifestation of acute hyperphosphataemia includes tetany and seizures due to hypocalcaemia, soft tissue calcification due calcium-phosphate precipitation and acute phosphate nephropathy. Common metabolic complications of tumour lysis syndrome are hyperkalaemia, hyperuricaemia (which may lead to acute renal failure) and azotaemia due to cytotoxic therapy-induced destruction of tumours characterised by rapid cell turnover.
2.2 Increased Tubular Reabsorption of Phosphate
Hyperphosphataemia can result from reduced renal phosphate excretion due to a primary increase in proximal phosphate reabsorption in patients with normal renal function. Hyperphosphataemia with inappropriately low fractional excretion of phosphate results mainly from deficiency of PTH or FGF23 or resistance to their actions. Hyperphosphataemia results from increased tubular reabsorption in the following conditions:
2.2.1 Hypoparathyroidism
Either deficient PTH secretion or renal resistance to PTH (pseudohypoparathyroidism) results in increased phosphate reabsorption and leads to hyperphosphataemia. Hyperphosphataemia in hypoparathyroidism is associated with hypocalcaemia caused by reduced bone resorption and intestinal calcium absorption (due to the decrease of serum calcitriol levels secondary to low PTH levels). Treatment with calcium and active vitamin D (calcitriol) corrects hypocalcaemia and lowers the serum phosphate concentration. How this occurs is not fully clarified, but the reduction of tubular phosphate reabsorption observed with calcium and vitamin D therapy is likely to be induced by the increase in FGF23 levels secondary to the elevation of serum calcium and calcitriol concentrations [20].
2.2.2 Vitamin D Toxicity
Vitamin D increases intestinal absorption of both phosphate and calcium. Vitamin D toxicity is generally characterised by the increase of both serum phosphorus and calcium, and low serum PTH levels. Hyperphosphataemia results from both increased intestinal phosphate absorption and a decrease in renal function induced by hypercalcaemia.
2.2.3 Familial Tumoral Calcinosis
Familial tumoral calcinosis is a rare disorder characterised by hyperphosphataemia due to an increase in proximal tubular phosphate transport, often in association with increased serum calcitriol levels [21]. The underlying defect first described was a mutation in the GALNT3 gene, which encodes a glycosyltransferase [22]. Mutations in FGF23 and Klotho genes have also been implicated [23–25]. Mutations in either the GALNT3 or FGF23 gene can lead to deficiency of circulating FGF23, which is a promoter of renal phosphate excretion. Klotho is a cofactor necessary for binding of FGF23 to its receptor [26, 27]. In the kidney, Klotho is required to allow the phosphaturic action of FGF23. Lack of FGF23 activity results in hyperphosphataemia and elevated 1,25 dihydroxyvitamin D levels. Serum calcium and PTH concentrations are typically within the normal range in patients with familial tumoral calcinosis. However, increased intestinal calcium absorption due to increased calcitriol levels in concert with hyperphosphataemia causes massive calcium and phosphate deposition in the skin and subcutaneous tissues, for which the disease is named [21, 28].
2.2.4 Acromegaly
Mild hyperphosphataemia occurs in some patients with acromegaly and is due to direct stimulation of phosphate reabsorption by growth hormone and insulin-like growth factor 1 [29].
2.3 Renal Failure
Phosphate is freely filtered by the glomerulus, is mainly reabsorbed in the proximal tubule (about 80–95 % of the filtered load) and 5–20 % of the filtered phosphate is normally excreted in the urine. An acute or chronic reduction in glomerular filtration rate (GFR) will initially diminish phosphate filtration and excretion. Nevertheless, serum phosphorus is maintained within the normal range even in advanced renal failure by the decrease of proximal phosphate reabsorption under the influence of increased secretion of PTH and FGF23 [30, 31]. However, when the GFR falls below 20–25 mL/min, despite the maximal suppression of phosphate reabsorption, urinary excretion is not usually able to counterbalance dietary phosphate intake. At this time, hyperphosphataemia must occur to increase the filtered load and re-establish phosphate balance.
2.4 Pseudohyperphosphataemia
Pseudohyperphosphataemia is a laboratory artefact caused by colorimetric interference with analytical methods that may occur in patients with hyperglobulinaemia (due to multiple myeloma, Waldenstrom’s macroglobulinaemia or monoclonal gammopathy), hyperlipidaemia, haemolysis and hyperbilirubinaemia [32]. Other possible causes of spurious hyperphosphataemia are therapy with liposomal amphotericin B and blood sample contamination with heparin or tissue plasminogen activator [33–35]. In these situations, serum phosphorus can be determined accurately by use of a different analyser [33].
3 Treatment of Hyperphosphataemia
The therapeutic approach differs in acute and chronic hyperphosphataemia. Acute severe hyperphosphataemia, in particular when associated with symptomatic hypocalcaemia (tetany, seizures), can be life-threatening. An acute phosphate load can be rapidly cleared if renal function is normal, and hyperphosphataemia usually tends to resolve within 6–12 h. Hyperphosphataemia can be treated by increasing renal phosphate excretion with extracellular volume expansion by saline infusion. A side effect of this treatment can be a further reduction of the serum calcium concentration by dilution. Haemodialysis may be indicated in hyperphosphataemic patients with symptomatic hypocalcaemia, particularly if renal function is impaired.
Treatment of chronic hyperphosphataemia consists of dietary restriction of phosphate intake and reduction of intestinal phosphate absorption using phosphate binder agents. In hyperphosphataemic patients with normal renal function, as in those affected by tumoral calcinosis, urinary phosphate excretion can be increased by the chronic administration of acetazolamide [36].
3.1 Treatment of Hyperphosphataemia of Chronic Kidney Disease (CKD)
The overall phosphate balance is positive in patients with CKD when the GFR falls below 20–25 mL/min and therapeutic strategies are aimed at correcting this. The therapeutic strategy includes reducing phosphorus intake by dietary modifications, reducing intestinal absorption using phosphate-binders and, in patients with CKD on dialysis, enhancing phosphate removal with more efficient dialysis treatments.
3.1.1 Rationale for Controlling Hyperphosphataemia and Target Serum Levels
Hyperphosphataemia is an important and inevitable clinical manifestation of declining kidney function. The rationale for controlling serum phosphorus is based on epidemiological evidence suggesting that hyperphosphataemia is an important risk factor not only for secondary hyperparathyroidism but also for progression of renal failure, development of cardiovascular disease and mortality [37–41]. Serum phosphorus has been documented as an independent predictor of mortality in both CKD stage 3–5 (patients with an estimated GFR below 60 mL/min/1.73 m2) and 5D (patients receiving dialysis) [39–41]. Hyperphosphataemia is associated with an increased risk of vascular, valvular and other soft tissue calcification in patients with CKD [38]. High serum phosphorus can directly and indirectly increase PTH secretion and decrease calcitriol synthesis, leading to the development of secondary hyperparathyroidism, bone abnormalities and extraskeletal calcification [37, 42]. Although no direct interventional evidence is available to show lowering serum phosphorus can improve morbidity or mortality in CKD patients, controlling serum phosphorus is widely accepted as a biochemical endpoint with probable clinical benefit, and the reduction of high serum phosphorus levels is expected to improve not only bone disease but also potentially reduce cardiovascular morbidity and mortality in CKD patients.
The more recent guidelines (better known as “Kidney Disease Improving Global Outcome guidelines” or “KDIGO guidelines”) [43] suggest maintaining serum phosphorus in the normal range in patients with CKD stages 3–5 and lowering elevated phosphorus levels toward the normal range in patients with CKD stage 5D. In a recent prospective observational cohort study, the reduction of serum phosphorus was associated with improved outcomes, even in normophosphataemic patients [44]. Higher serum phosphorus levels, although within the normal range, were associated with a stepwise increase in mortality in a large observational study on CKD stage 3–4 patients (average GFR 32 mL/min/1.73 m2) [45]. Therefore, some authors have emphasised the importance of an early and effective control of phosphate load in the early stages of CKD before hyperphosphataemia develops to prevent the increase in PTH and FGF23 and help maintain near-normal phosphorus levels for longer as CKD progresses [46]. This early treatment has been suggested as a means of potentially reducing the risk of development of renal secondary hyperparathyroidism and all of the adverse clinical consequences of poorly controlled mineral and bone disorder in CKD [47].
The use of phosphate-restricted diets in combination with oral phosphate binders has become a well-established approach in the management of patients with CKD stages 3–5 (including CKD stage 5D), and this strategy has been recommended by the recent guidelines, with appropriate education and counselling to ensure adequate protein intake and avoid extra-phosphate load from food additives [43, 48, 49]. In CKD patients on dialysis an efficient dialysis removal of phosphate should be ensured and, when the usual dialysis prescription may be insufficient to substantially reduce phosphorus levels, the increase in frequency or duration of dialysis should be considered [43].
The present review is mainly aimed to discuss the therapeutic approach to hyperphosphataemia of chronic renal failure using oral phosphate binders.
4 Oral Phosphate Binders: Comparative Efficacy, Tolerability and Safety
A wide range of phosphate binders is currently available for the treatment of hyperphosphataemia in CKD patients (Table 1). These agents are generally divided into two main classes: calcium-based binders (calcium carbonate and acetate) and calcium-free binders (aluminium hydroxide, lanthanum carbonate, magnesium carbonate, sevelamer hydrochloride and sevelamer carbonate). Current phosphate binders are all effective in lowering phosphorus, so the main considerations for selection should be their other characteristics, including absorbability (ideally non-absorbed), adequate gastrointestinal tolerability, and cost or cost-effectiveness. These characteristics depend on the chemical properties of the compound.
Aluminium hydroxide is a potent phosphate binder, but concern about skeletal, haematological and neurological toxicity led to a preferential use of calcium salts (carbonate and acetate) in the 1990s. The KDIGO guidelines recommend avoiding the long-term use of aluminium-containing phosphate binders in patients with CKD stages 3–5D [43]. The recommendations of the KDIGO guidelines for the treatment of hyperphosphataemia in CKD patients are summarised in Table 2.
4.1 Calcium Acetate and Calcium Carbonate
Calcium acetate and calcium carbonate are often considered current standard therapy, since they very effectively lower serum phosphorus levels [43, 50–52], are well tolerated, inexpensive and readily available: they are consequently often used as a benchmark in comparative clinical studies. The use of calcium salts is generally associated with higher serum calcium, more frequent episodes of hypercalcaemia and lower serum PTH levels when compared with the use of placebo or calcium-free phosphate binders [53]. A recent meta-analysis [53] that evaluated the randomised controlled trials of phosphate binders in CKD patients showed no significant difference in end-of-treatment serum phosphorus levels with calcium acetate in comparison to calcium carbonate. Similarly, the incidence of hypercalcaemia and the end-of-treatment PTH levels were not different in patients treated with calcium acetate compared to calcium carbonate, despite the lower calcium content of calcium acetate (250 mg/g of elemental calcium compared to 400 mg/g of calcium carbonate). Moreover, the incidence of adverse gastrointestinal events was not different using calcium carbonate or calcium acetate [53]. Thus, these two calcium-containing binders can be considered comparable for efficacy in control of hyperphosphataemia, effects on mineral metabolism parameters and tolerability. The average daily dose of calcium acetate or carbonate prescribed in the randomised controlled trials to control hyperphosphataemia in dialysis patients ranged between 1.2 and 2.3 g of elemental calcium [54–57]. Such doses are greater than the recommended dietary calcium intake [58] and can lead to a positive calcium balance [59–61], which can increase the risk of hypercalcaemia linked to low-turnover bone disease and vascular calcification [62–65]. Thus, it is advisable to limit calcium intake from phosphate binders to a maximum daily amount of 1 g of elemental calcium. In particular, the KDIGO guidelines recommend restricting the dose of calcium-based phosphate binders in the presence of persistent or recurrent hypercalcaemia, arterial calcification, adynamic bone disease and/or persistently low serum PTH levels (Table 2) [43]. In clinical practice, when large doses of phosphate binders are required to control serum phosphorus, the combination of low doses of calcium acetate or carbonate with a calcium-free phosphate binder seems to be an effective and preferable approach compared to monotherapy with calcium salts to control hyperphosphataemia, while avoiding calcium overload [66, 67].
4.2 Sevelamer Hydrochloride and Carbonate
Sevelamer, a polymeric amine, is the only non-absorbable oral phosphate binder currently available to control hyperphosphataemia [43, 68, 69]. It has the advantage of avoiding the risk related to accumulation in CKD patients, which may be particularly important in anuric dialysis patients. The first formulation of sevelamer to be approved was sevelamer hydrochloride, while sevelamer carbonate became available only recently to lower serum phosphorus while avoiding the risk of metabolic acidosis associated with sevelamer hydrochloride [69]. Several studies have documented that sevelamer hydrochloride effectively lowers serum phosphorus [68, 70] and achieves a phosphate control comparable to calcium acetate and calcium carbonate in haemodialysis patients [54–56, 70–72]. A recent systematic review concluded that sevelamer significantly decreases the risk of hypercalcaemia compared with calcium salts [53]. In most randomised studies, treatment with sevelamer was associated with lower serum calcium and higher serum PTH compared with calcium salts [54–56, 70–72].
Sevelamer hydrochloride was documented to be as effective as calcium acetate/magnesium carbonate combination in controlling serum phosphorus to a target level of 1.1–1.8 mmol/L (3.5–5.5 mg/dL) in a 24-week study [73]. The average serum phosphorus level achieved after 24 weeks was 1.7 mmol/L (5.27 mg/dL) in the calcium/magnesium group (average dosage: calcium acetate 3 g/day plus magnesium carbonate 1.6 g/day) in comparison with 1.8 mmol/L (5.5 mg/dL) in the sevelamer group (average sevelamer dosage 6.4 g/day). No significant differences in ionised serum calcium were seen between the groups. PTH decreased during the course of the study in both groups [from a mean baseline level of 48 pmol/L (450 pg/mL) to 36 pmol/L (337 pg/mL) in the calcium/magnesium group and from 47 pmol/L (439 pg/mL) to 41 pmol/L (384 pg/mL) in the sevelamer group], but the decrease was more marked in the calcium/magnesium group [73].
Sevelamer carbonate has shown comparable efficacy and safety to sevelamer hydrochloride in dialysis patients [69, 74] and is indicated to lower serum phosphorus also in hyperphosphataemic CKD stage 3–5 patients not on dialysis [75]. Dose titration of sevelamer can help patients with either CKD stages 3–5 or CKD stage 5D achieve a high rate of phosphate control (Table 3). Delmez et al. [76] documented that about 70 % of haemodialysis patients treated with sevelamer hydrochloride or sevelamer carbonate maintained serum phosphorus levels within 1.1–1.8 mmol/L (3.5–5.5 mg/dL) over 8 weeks of treatment. Fishbane et al. [74] similarly reported that 64 % of haemodialysis patients taking sevelamer hydrochloride thrice daily (mean daily dose 9.1 g) and 54 % taking sevelamer carbonate powder once daily (mean daily dose 9.2 g) were able to maintain phosphorus levels of 1.1–1.8 mmol/L (3.5–5.5 mg/dL) after 24 weeks. Ketteler et al. [75] showed that 75 % of CKD stage 4 and 70 % of CKD stage 5 patients had phosphorus within 0.9–1.5 mmol/L (2.7–4.6 mg/dL) after 8 weeks of sevelamer carbonate (mean ending prescribed dose 7.8 g/day).
Navaneethan et al. [53] reported in their meta-analysis that included all the randomised controlled trials published until April 2009 a significantly higher incidence of gastrointestinal adverse events with sevelamer in comparison to calcium salts. Gastrointestinal side effects reported with sevelamer included vomiting, nausea, diarrhoea, dyspepsia, abdominal bloating and constipation.
4.3 Lanthanum Carbonate
Lanthanum carbonate is a non-calcium-based phosphate binder supplied as a chewable tablet of three dosage strengths (500, 750 and 1,000 mg of elemental lanthanum) that has been shown to be effective in reducing phosphorus in short-term clinical trials compared to placebo or previous phosphate-binder therapy in stage 3–4 CKD patients and in patients on dialysis [77–80]. Sprague et al. [80] showed that 44.6 % of CKD stage 3–4 patients achieved phosphorus <1.5 mmol/L (<4.6 mg/dL) after 8 weeks (mean dose dosage of lanthanum carbonate 2,645 mg/day) compared to 26.5 % in the placebo group (not statistically different). Serum phosphorus levels below 1.8 mmol/L (<5.5 mg/dL) were achieved in 48–65 % of dialysis patients during short-term clinical trials using median doses of about 3 g/day [70–72] (Table 3). In a longer term study in dialysis patients, 46 % of patients in the lanthanum carbonate group (maximum daily dose of 3,000 mg elemental lanthanum) achieved control of serum phosphorus levels [<1.9 mmol/L (<5.9 mg/dL)] compared with 49 % in the standard therapy group (p = NS) after 2 years of treatment [81]. However, there was a higher frequency of hypercalcaemia (20.2 vs. 0.4 %) and suppressed PTH levels in the calcium carbonate group compared with that in those receiving lanthanum carbonate therapy [81]. In another study, the long-term use of lanthanum carbonate (up to 3 years) was shown to maintain stable phosphorus levels similar to those with calcium salts or pre-existing phosphate binders [82]. Thus, lanthanum carbonate can be considered as effective as calcium carbonate in controlling serum phosphorus in either haemodialysis or peritoneal dialysis patients [81–83]. However, dropouts due to adverse events were higher in the lanthanum arm (14 %) than in the arm using other binders (5 %) [81]. Findings from a meta-analysis showed that, compared with calcium-based agents, lanthanum significantly decreased end-of-treatment serum calcium and calcium-phosphorus product levels, but with similar end-of-treatment phosphorus levels [53]. While most data on lanthanum carbonate and sevelamer reported that both have comparable efficacy to calcium-based binders, their efficacy in lowering serum phosphorus was directly compared in only two randomised studies [84, 85]. The primary analysis of the study of Sprague et al. [84] showed a significant and comparable reduction of serum phosphorus of about 0.5 mmol/L (1.5 mg/dL) after 4 weeks of treatment with either fixed doses of lanthanum (2,250–3,000 mg/day) or sevelamer hydrochloride (4,800–6,400 mg/day). Kasai et al. [85] showed that lanthanum (945 mg/day on average) or sevelamer hydrochloride (2,971 mg/day on average) were similarly effective in controlling serum calcium and phosphate levels in 42 Japanese dialysis patients. The number of pills prescribed to control serum phosphorus was lower in the lanthanum group.
The reduction of tablet burden with the use of lanthanum carbonate tablets with higher dosage strengths was documented as an effective measure to achieved a better control of serum phosphorus in nonresponders [79]. Moreover, patients and physicians reported significantly higher levels of satisfaction with lanthanum compared with previous phosphate binders due mainly to the reduced tablet burden (using higher dosage strengths) required to obtain serum phosphorus control [79]. Therefore, the reduction of pill burden could be a way to improve adherence to therapy in CKD patients.
Lanthanum carbonate treatment may be associated with some gastrointestinal side effects [77, 82, 83]. The relative risk of gastrointestinal adverse events in the randomised trials was not different with lanthanum carbonate in comparison to placebo or calcium carbonate [53]. Experimental data have shown that lanthanum accumulates in tissues, such as liver, brain and bone [86, 87]. However, long-term studies have found no evidence of liver, bone or central nervous system toxicity after 1–3 years of lanthanum treatment [81]. No clinically relevant safety concern was documented in a small select number of dialysis patients treated for up to 6 years [82]. Lanthanum use has been shown to substantially increase plasma lanthanum levels in dialysis patients compared with calcium use over 1 year, with plasma levels decreasing significantly over the subsequent 2-year period following discontinuation [87]. In the same group of patients, the bone content of lanthanum increased significantly over 1 year of treatment and remained similarly elevated for 2 years following discontinuation. This finding suggests that the removal of lanthanum from bone occurs very slowly after discontinuation of therapy. Even though lanthanum bone accumulation was not associated with suppression of bone turnover [86], the long-term impact of lanthanum accumulation on bone health is still unknown, since bone biopsy data on a large number of patients following long-term treatment with lanthanum carbonate are lacking. Indeed, past experience with aluminium hydroxide suggests caution in the use of any binder because it is likely to accumulate in the presence of impaired renal function.
4.4 Magnesium Salts
Magnesium-based phosphate binders have been used since the mid-1980s and were initially introduced to replace aluminium-containing binders. Magnesium hydroxide is effective in lowering serum phosphorus [88, 89] but the administration of large doses (2–3 g/day) has been complicated by side effects, such as diarrhoea and hyperkalaemia [98], which have limited the widespread use as a phosphate binder. Magnesium carbonate is better tolerated than magnesium hydroxide, and it was studied in the 1990s to reduce the intake of calcium as a phosphate binder. In a crossover-designed study in haemodialysis patients, magnesium carbonate was added at a starting dose of 750 mg/day (214 mg elemental magnesium) and calcium carbonate was reduced by 50 % [90]. The magnesium dose was up-titrated to achieve a phosphorus level below 1.9 mmol/L (6 mg/dL), and the dialysate magnesium was decreased from 0.75 mmol/L (1.5 mEq/L) to 0.25 mmol/L (0.5 mEq/L). Overall, phosphorus was well controlled without an increase in serum magnesium concentrations [90]. However, magnesium-based phosphate binders have not been widely adopted since calcium therapy was perceived to be more than adequate and effective.
Recently, magnesium has received renewed interest as an alternative to calcium for its potential beneficial effects on bone health and vascular processes in CKD patients [91]. Several magnesium-containing regimens have been investigated in a limited number of small clinical studies. Tzanakis et al. [92], in a 6-month randomised study, documented that magnesium carbonate (mean daily dose of 1,552 mg, range 750–2,250) was able to reduced serum phosphorus as effectively as calcium carbonate (2,839 mg/day, range 1,260–3,780), while the development of hypermagnesaemia was prevented by decreasing the dialysate magnesium concentration to 0.3 mmol/L (0.6 mEq/L).
Spiegel et al. [93] reported an effective phosphate control with combinations of magnesium carbonate and calcium carbonate. In this randomised trial magnesium carbonate associated with calcium carbonate was as effective as calcium acetate in lowering serum phosphorus in haemodialysis patients on a standard 0.375 mmol/L (0.75 mEq/L) magnesium bath, while the net calcium ingestion was significantly less (on average 908 vs. 1,743 mg/day) in the magnesium-treated group [93]. The percentage of patients achieving the serum phosphorus target was higher in the magnesium group (70.6 vs. 62.5 %). The serum magnesium did increase in the magnesium-treated group, although it was well tolerated.
A formulation of magnesium carbonate and calcium acetate (film-coated tablets containing calcium acetate 435 mg and magnesium carbonate 235 mg) was recently authorised for use as a phosphate binder. In a randomised 24-week trial in 204 patients dialysed against a 0.5 mmol/L magnesium dialysate, magnesium carbonate/calcium acetate was compared to sevelamer hydrochloride. Phosphorus control was overall no inferior in the magnesium group [1.70 ± 0.5 mmol/L (5.3 ± 1.5 mg/dL) vs. 1.77 ± 0.6 mmol/L (5.5 ± 1.9 mg/dL)] [73]. Although the serum magnesium [1.3 ± 0.25 vs. 1.0 ± 0.18 mmol/L (3.15 ± 0.6 vs. 2.5 ± 0.4 mg/dL), p < 0.001] and serum potassium (5.8 ± 0.8 vs. 5.4 ± 0.8 mmol/L, p < 0.001) at the end of the study were higher in the magnesium carbonate/calcium acetate group, the medication was well tolerated without serious short-term adverse events, and the number of patients with adverse events was comparable in both treatment groups. Average daily study medication intake was slightly but significantly higher in the sevelamer group at week 25 (8.1 vs. 7.3 pills).
Another agent in development is iron–magnesium hydroxycarbonate, which can also provide good phosphate control and is generally well tolerated [94]. In a double-blind, three-arm, parallel-group study comparing two doses of iron–magnesium hydrocarbonate (fermagate) (1 g three times daily or 2 g three times daily with placebo) a significant and dose-dependent reduction in the serum phosphorus level was observed [from a mean baseline phosphorus level of 2.15 mmol/L (6.7 mg/dL) to a mean phosphorus level of 1.7 and 1.5 mmol/L (5.2 and 4.6 mg/dL), respectively] [94]. The 1-g dose was well tolerated, while the 2-g dose resulted in a statistically higher number of adverse events (mainly gastrointestinal side effects) resulting in drug discontinuation [94].
Although several studies have shown that magnesium carbonate is generally well tolerated, further large studies are needed to establish the efficacy and safety of magnesium in CKD and, probably, to re-evaluate its appropriate concentration in haemodialysis and continuous ambulatory peritoneal dialysis fluids [91, 92, 95]. Hypermagnesaemia is not typically a major issue as it can be readily mitigated through dialysis. There is, however, a lack of clinical data on longer term effects of chronic magnesium elevation. It has been documented that magnesium accumulates in bone in dialysis patients, and this may contribute to mineralisation defects and osteomalacia [96, 97]. Moreover, the suppressive effect of magnesium on PTH could promote the development of low-turnover bone disease [95].
5 Patient-Level Outcomes with Phosphate Binders
Even though numerous epidemiological data have shown a positive association, although not a causal link, between higher serum phosphorus levels and the relative risk of mortality, independent of CKD stage [38–41, 43], the benefits of lowering serum phosphorus values for patient-level clinical outcomes (for example, hospitalisation, bone fracture, cardiovascular events and mortality) have not yet been studied. However, several randomised studies have compared the effects of different phosphate binders on mortality or surrogate markers of cardiovascular disease, such as the progression of vascular calcification. In these comparative clinical studies calcium carbonate and/or calcium acetate are generally used as reference binder therapy.
5.1 Sevelamer Hydrochloride and Carbonate
Six randomised clinical trials have been conducted to compare the progression of coronary calcification in CKD patients treated with sevelamer hydrochloride or calcium salts. Five studies, Treat to Goal (TTG) [54], Renagel in New Dialysis (RIND) [55], Calcium Acetate-Renagel Evaluation-2 (CARE-2) [56], Bone Relationship with Inflammation and Coronary Calcification (BRiC) [57] and a recent Japanese study [98], enrolled dialysis patients, whereas the study of Russo et al. [99] included CKD patients not on dialysis. In the CARE-2 study atorvastatin was added to both arms as required to reach a comparable low-density lipoprotein (LDL) cholesterol target [<1.8 mmol/L (<70 mg/dL)] [56]. Four of these studies showed a reduction of calcification progression with sevelamer [54, 55, 98, 99], and two of them did not [56, 57]. Russo et al. [99] showed differences in vascular calcification progression in 90 CKD 3–5 patients randomised to receive dietary phosphate restriction alone or dietary phosphate restriction in association with calcium carbonate or sevelamer as a phosphate binder. The highest progression of calcification was reported with dietary restriction alone. The progression was moderate with calcium carbonate (2 g/day) and lower with sevelamer (1,600 mg/day). Sevelamer hydrochloride and calcium carbonate were equally effective in controlling serum phosphorus. At the end of 2-year follow-up, there were no differences in the mean levels of serum phosphorus [1.55 vs. 1.5 mmol/L (4.8 vs. 4.7 mg/dL)], calcium [2.25 and 2.28 mmol/L (9.0 and 9.1 mg/dL)] and PTH [15 vs. 20 pmol/L (143 vs. 187 pg/mL)] between patients treated with sevelamer or calcium carbonate [99]. The BRiC study was hampered by several significant confounders, including small sample size (49 patients randomised to calcium acetate and 52 patients to sevelamer), differences in baseline calcium artery scores between the two study arms, the use of high dialysate calcium concentrations (1.75 mmol/L) in most patients, resulting in a positive calcium balance, and multiple interventions during the course of the study based on bone biopsy results [57]. All the studies performed in dialysis patients were affected by a high frequency of dropouts (from 27 to 68 %) [54–57]. Moreover, the enrolment of patients with a wide range of calcium score might have affected the results, since the rate of progression of vascular calcification may depend on the amount of vascular calcification present at the beginning of the therapeutic intervention. Indeed, in a study of coronary artery calcification in patients new to haemodialysis, vascular calcification in incident patients with a zero coronary artery calcium score at baseline did not progress at all over an 18-month treatment period with sevelamer or a calcium-containing binder [55]. Taken together, the data on vascular calcification overall are discrepant and considered of low quality by the working group that drew up the KDIGO guidelines [43].
Few studies have compared the effects of sevelamer and calcium salts on bone health. Salusky et al. [100] did not find differences between calcium and sevelamer-hydrochloride in bone turnover or mineralisation, and the same number of children developed adynamic disease. Ferreira et al. [101], in a 54-week randomised study that enrolled 119 dialysis patients (68 patients underwent follow-up bone biopsies after 1 year), reported that bone turnover increased in the sevelamer group compared with that in calcium-treated patients (p < 0.02). The turnover worsened by becoming higher in 12 % of sevelamer and 3 % of calcium groups; on the other hand, it worsened by becoming lower (development of adynamic disease) in 17 % of calcium patients and 9 % of sevelamer patients. Turnover improved in 26 % of calcium and 15 % of sevelamer patients. Change in bone volume was almost the same in both groups. The BRiC study showed no differences in bone remodelling between sevelamer or calcium groups [57], although a selection and survival bias as well as the use of lower dialysate calcium in patients with low bone turnover may have contributed to this lack of difference between the treatment groups [57]. In summary, when patients are considered as a group, only minor overall changes in bone histology were documented in response to sevelamer compared with calcium-containing phosphate binders.
Several studies emphasised the pleiotropic effects of sevelamer. Sevelamer consistently improves the lipid profile by reducing the LDL-cholesterol levels compared with calcium salts [54, 55, 57, 73]. It can also improve calcification and inflammatory mediators including C-reactive protein [102, 103], interleukin-6 [104] and fetuin-A, a negative acute phase protein involved in bone remodelling in the foetus, having an inhibitory function on calcium phosphate deposition [104, 105]. A recent study has also shown greater reductions in FGF23 with sevelamer compared to calcium salts in patients with CKD stages 3–5D [106]. These pleiotropic actions of sevelamer might induce beneficial effects in CKD patients in addition to those arising from the reduction in serum phosphorus [107].
It was assumed that the potential benefits of sevelamer on the bone and vascular axis, and its pleiotropic effects, could translate into significant clinical benefits. This hypothesis was initially supported by the results of the RIND study, which showed a lower mortality rate in patients new to dialysis treated with sevelamer compared with that observed with calcium [55]. However, the Dialysis Clinical Outcomes Revisited (DCOR) study found no differences in mortality in the overall study population of prevalent dialysis patients (2,103 patients randomised) treated with sevelamer or calcium salts for 20 months on average [108]. The lower mortality rates with sevelamer in an elderly subgroup over 65 years and in those with at least 2 years’ follow-up suggest potential differences in clinical outcomes between non-calcium and calcium-based phosphate binders [108]. However, further studies on a large number of patients and with longer follow-up need to confirm these impressions. A recent meta-analysis of cardiovascular mortality and coronary artery calcification did not find differences between CKD patients treated with calcium-based or non-calcium-based phosphate binders [109]. Similarly, another systematic review showed that there was no significant decrease in all-cause mortality or hospitalisation in patients treated with sevelamer compared with those treated with calcium-based agents [46]. On the contrary, in a recent randomised, multicentre, non-blinded pilot study that enrolled 212 CKD stage 3–4 patients, Di Iorio et al. [110] documented a significantly lower mortality rate in patients treated with sevelamer hydrochloride (mean dose 2,184 mg/day) compared to that observed in patients treated with calcium carbonate (mean dose 2,950 mg/day) over a 3-year follow-up. Patients treated with sevelamer showed lower on-treatment average serum phosphorus [1.4 vs. 1.5 mmol/L (4.37 vs. 4.72 mg/dL)], LDL cholesterol [2.2 vs. 2.8 mmol/L (86 vs. 109 mg/dL)] and PTH levels [22 vs. 34 pmol/L (209 vs. 317 pg/mL)], and these effects may have accounted for at least part of the difference in the all-cause mortality and composite end point (mortality and dialysis start) [110]. Although this pilot study has a number of shortcomings, including being underpowered with an inadequate sample size to estimate the difference in mortality, it might represent a valid aid for designing a properly powered randomised controlled trial to clearly demonstrate the potential beneficial effects of sevelamer.
5.2 Lanthanum Carbonate
No prospective randomised study has been expressly designed to assess the effects of lanthanum carbonate on hard clinical outcomes. In the 2-year extension period of a phase 3 randomised study that compared lanthanum carbonate to standard phosphate binder therapy in 1,354 dialysis patients, overall mortality was not different in the lanthanum group and standard therapy group [111]. Recently, a randomised controlled pilot study has been conducted to compare the effect of lanthanum carbonate and calcium carbonate on the progression of vascular calcification in 45 haemodialysis patients [112]. Lanthanum carbonate was associated with a reduced progression of aortic calcification compared with calcium carbonate over 18 months, but only 17 lanthanum carbonate patients and 13 calcium carbonate patients completed the study.
Three studies compared the effects of lanthanum carbonate with those of calcium carbonate on bone histomorphometry. In the study of D’Haese et al. [113], an improvement in turnover was seen in 36–45 % of patients receiving lanthanum and in 20–23 % of those receiving calcium. The turnover worsened in 30 % with calcium treatment (20 % developed adynamic disease) and in 12 % with lanthanum treatment (6 % developed adynamic disease). Mineralisation changes were similar in both treatment groups. Overall, the results favoured lanthanum carbonate treatment. The study of Malluche et al. [114] evaluated 2 years of treatment. Paired bone biopsy samples for histomorphometric analysis were available at baseline and at 1 year in 32 lanthanum carbonate-treated dialysis patients and in 33 patients receiving standard care, and at baseline and 2 years in 32 lanthanum carbonate-treated patients and in 24 patients receiving standard phosphate binder therapy. At 1 year, turnover worsened in 45 % of the calcium group and in 42 % of the lanthanum group, and improved in 3 % of the calcium group and in 12 % of the lanthanum group. At 2 years, the turnover had worsened in 72 % of the calcium group (29 % decreasing toward adynamic lesions) and in 40 % of the lanthanum group (23 % decreasing toward adynamic lesions), with improvement being similar in both groups. Therefore, at 2 years, the results showed a better turnover with lanthanum carbonate treatment. Spasovski et al. [87] studied 20 new dialysis patients randomly treated with lanthanum carbonate or calcium salts. None of the lanthanum-treated patients developed low bone turnover after 1 year of treatment, in contrast to three patients developing adynamic bone disease in the calcium group [87]. In summary, the studies on bone histology overall documented a lower risk of developing low-bone turnover with lanthanum carbonate compared with calcium-containing phosphate binders.
5.3 Magnesium Salts
It has recently been hypothesised that magnesium-based phosphate binders could reduce hyperphosphataemia and improve vascular calcification, based on observational studies that documented an inverse correlation between serum magnesium levels, progression of vascular calcification and mortality in dialysis patients [91, 95]. While these observational studies suggest a theoretical benefit for moderately high magnesium levels, such as those induced by treatment with magnesium-based phosphate binders, so far no large prospective studies have assessed the effects of magnesium salts on vascular calcification and on cardiovascular morbidity and mortality in CKD patients.
6 Discussion and Conclusions
Hyperphosphataemia is an inevitable consequence of advanced chronic renal failure and is present in the majority of CKD patients on dialysis. Hyperphosphataemia is an important risk factor for the development and progression of secondary hyperparathyroidism and is associated with increased cardiovascular morbidity and mortality among CKD patients [37–41]. Lowering the phosphate load and maintaining serum phosphorus levels within the normal range are considered important therapeutic goals to improve clinical outcomes in CKD patients [43, 46, 47].
The first therapeutic strategy to control serum phosphorus is dietary restriction of phosphate intake, including phosphate-containing additives [43, 48, 49]. The limitation of low-phosphate diets in dialysis patients is the risk of lowering protein intake below the recommended intake of 1.2 g/kg/day and inducing protein malnutrition [115, 116]. The 2012 KDIGO guidelines suggest lowering protein intake to 0.8 g/kg/day in CKD stage 4–5 patients [117]. The mean daily dietary protein intake in a large number of CKD stage 3 and 4 patients was documented to be, respectively, 1.22 and 1.13 g/kg of ideal body weight [118], a value above the Institute of Medicine requirements for healthy adults and the KDIGO recommendation for CKD patients not on dialysis [117, 119]. Therefore, the reduction of protein intake within the recommended requirements in association with the restriction of those protein sources with the highest phosphorus content can be an effective measure to control serum phosphorus in patients with advanced CKD not on dialysis. Indeed, a randomised trial in 392 CKD stage 4–5 patients (mean estimated GFR 18 mL/min) documented that a low-protein diet (average protein intake of 0.72 g/kg/day) resulted in mean serum phosphorus levels 0.1 mmol/L (0.32 mg/dL) lower than a moderate protein diet (0.92 g/kg/day) and less frequent need of phosphate binders [120]. However, the proportion of patients adherent to protein prescription was lower in the low-protein group [120]. A very-low-protein diet supplemented with keto and amino acids was shown to be effective in reducing serum phosphorus and was associated with the use of fewer concurrent phosphate binders in 53 CKD patients with an eGFR <30 mL/min/1.73 m2 [121]. However, a very-low-protein diet can hardly be prescribed to the majority of patients. In patients with less advanced CKD (stage 3), the reduction of dietary phosphorus intake significantly decreases urinary phosphate excretion but has a modest effect on serum phosphorus [122, 123].
In patients on dialysis, an efficient removal of phosphorus should be ensured, and the increase of duration or frequency of dialysis treatment should be considered in presence of persistent hyperphosphataemia [43]. However, dietary restriction of phosphorus and current dialysis modalities are not sufficiently effective to maintain serum phosphorus levels within the recommended range so that the majority of dialysis patients require treatment with oral phosphate binders [43, 44, 124, 125].
Current phosphate binders are all effective in lowering phosphorus, so the considerations to be made to select a binder should be based on other characteristics, such as absorbability, adequate gastrointestinal tolerability, and cost or cost-effectiveness. Calcium-based phosphate binders have good clinical efficacy, are inexpensive and are the most widely used binders in clinical practice [124]. However, in patients with reduced or absent kidney function, there is the risk of positive calcium balance [59–61], hypercalcaemia [53], parathyroid gland suppression with development of low bone turnover [93, 105], and vascular and extraosseous calcification [55, 62–64]. Therefore, it is advisable to reduce calcium intake from phosphate binders, particularly in the presence of low PTH levels, a tendency to develop hypercalcaemia, vascular calcification, use of high calcium concentrations in the dialysate (>1.25 mmol/L) and concomitant use of active vitamin D compounds (calcitriol, paricalcitol) that increase the intestinal calcium absorption [43, 59–61]. Although large doses of calcium salts should probably be avoided, modest doses (<1 g of elemental calcium) may be reasonable. Thus, the prescription of low doses of calcium acetate or carbonate may represent a reasonable initial approach to control serum phosphorus in both non-dialysis and dialysis CKD patients. Average daily calcium carbonate doses of 2–3 g (800–1,200 mg of elemental calcium) are generally effective to control serum phosphorus in CKD stage 3–5 patients [99, 110]. However, larger dosages of calcium-containing binders (1.2–2.5 g/day of elemental calcium) are generally required to control hyperphosphataemia in dialysis patients using monotherapy [44–57, 63, 66, 124, 125]. In patients requiring large doses of binders, the addition of a non-calcium-based phosphate binder to low doses of calcium salts may be useful to control hyperphosphataemia and minimise the risk of calcium overload [66, 67, 125]. In clinical practice the combination of two binders is common [124, 125] and probably allows a higher tolerability and compliance compared to monotherapy [66].
Currently, three kinds of non-calcium-based phosphate binders are commercially available: sevelamer, lanthanum carbonate and magnesium salts. Each of these compounds is effective in lowering serum phosphorus levels depending on an adequate prescribed dose and adherence of the patient to treatment. Sevelamer and lanthanum carbonate can achieve appropriate serum phosphorus targets to varying degrees in CKD patients. Recent studies of small numbers of cases have shown that also magnesium salts effectively reduce phosphorus levels (Table 3).
Another important aspect to be considered when choosing a phosphate binder is long-term safety. Sevelamer is the only non-calcium-containing phosphate binder that does not have potential to cause systemic accumulation and presents pleiotropic effects that may impact positively on cardiovascular disease. In contrast, lanthanum carbonate and magnesium salts are absorbed in the gut and their route of excretion is biliary for lanthanum and urinary for magnesium. Lanthanum is known to accumulate mainly in bone and liver, but, even if toxicity has not been documented so far, the risk of long-term effects of chronic accumulation have not yet been entirely established. There are also limited data on the long-term effects of accumulation of magnesium in bone.
Overall, there are insufficient data to establish the superiority of non-calcium binding agents over calcium-containing phosphate binders for such important patient-level outcomes as all-cause mortality and cardiovascular morbidity. Moreover, full adoption of sevelamer and lanthanum by government drug reimbursement agencies in place of calcium salts would lead to a large increase in health care expenditure [126]. Even assuming that sevelamer can be associated with a clinical benefit, Manns et al. [126] documented that the cost per quality-adjusted life years (QALY) gained for treating all dialysis patients with sevelamer, compared with calcium-based phosphate binders, was too high and unattractive. Recently, Bernard et al. [127] evaluated the cost-effectiveness of sevelamer compared to calcium-based binders as the first-line treatment of hyperphosphataemia in dialysis patients in the UK. Data on hospitalisations, resource utilisation and survival were derived from the DCOR study [108]. The use of sevelamer resulted in a gain of 0.7 life years and 0.44 QALY. Total per-patient costs were higher for sevelamer, resulting in an incremental cost of £22,157 per QALY gained and £13,427 per life years gained. The incremental cost effectiveness ratio (ICER) was £50,356 per QALY gained, a value non-attractive from an economic point of view to propose sevelamer as a first-line therapy. Assigning to lanthanum a health gain similar to that of sevelamer, it can be roughly estimated that the ICER for lanthanum is similarly high.
Thus, the choice of phosphate binder should be individualised, considering the clinical context, the individual tolerability (the use of phosphate binders is associated with side effects, especially gastrointestinal, and generally dose-dependent) and the concomitant effects on other parameters of mineral metabolism besides those on serum phosphorus (such as serum calcium and PTH). Calcium-based binders should be considered as the preferred first-line option because of their cost-effectiveness.
The compliance of patients to treatment is another aspect to consider in the choice of a phosphate binder. A not negligible proportion of dialysis patients have uncontrolled serum phosphorus because of poor adherence to therapy [128–130]. Phosphate binders accounted for about one-half of the daily pill burden [129, 130], and no adherence to phosphate binders was reported to be greater than that of other drugs, such as antihypertensive drugs [128]. The percentage of patients considered adherent to binder therapy ranges between 72 and 98 % in different randomised trials [71, 73, 74, 77, 131]. The high pill burden from phosphate binders may affect patients’ adherence to therapy and their ability to maintain optimal serum phosphorus levels [128]. Educational strategies can also help to improve the control of hyperphosphataemia in CKD patients and should be one of the methods of improving achievement of serum phosphate targets [132, 133].
The primary advantage for more recently developed phosphate binders (lanthanum carbonate and sevelamer) is a decrease in hypercalcaemia in CKD patients. Existing trials using patient-focused end points are inadequate to inform clinical recommendations for any phosphate binder, and further research using a randomised controlled trial design is required before advocating that newer agents are superior to existing lower cost interventions. Trials of direct comparisons between calcium salts, sevelamer and lanthanum carbonate in CKD patients with primary outcomes of all-cause and cardiovascular mortality, fractures, hospitalisation and parathyroidectomy are desirable.
References
Marks J, Debnam ES, Unwin RJ. Phosphate homeostasis and the renal–gastrointestinal axis. Am J Physiol Ren Physiol. 2010;299:F285–96.
Sabbagh Y, Giral H, Caldas Y, et al. Intestinal phosphate transport. Adv Chronic Kidney Dis. 2011;18:85–90.
Uribarri J. Phosphorus homeostasis in normal health and in chronic kidney disease patients with special emphasis on dietary phosphorus intake. Semin Dial. 2007;20:295–301.
Gutierrez OM, Wolf M. Dietary phosphorus restriction in advanced chronic kidney disease: merits, challenges, and emerging strategies. Semin Dial. 2010;23:401–6.
Murer H, Lötscher M, Kaissling B, et al. Renal brush border membrane Na/Pi-cotransport: molecular aspects in PTH-dependent and dietary regulation. Kidney Int. 1996;49:1769–73.
Gattineni J, Bates C, Twombley K, et al. FGF23 decreases renal NaPi-2a and NaPi-2c expression and induces hypophosphatemia in vivo predominantly via FGF receptor 1. Am J Physiol Renal Physiol. 2009;297:F282–91.
Bergwitz C, Juppner H. Regulation of phosphate homeostasis by PTH, vitamin D, and FGF23. Annu Rev Med. 2010;61:91–100.
Ferrari SL, Bonjour JP, Rizzoli R. Fibroblast growth factor-23 relationship to dietary phosphate and renal phosphate handling in healthy young men. J Clin Endocrinol Metab. 2005;90:1519–24.
Burnett SM, Gunawardene SC, Bringhurst FR, et al. Regulation of C-terminal and intact FGF-23 by dietary phosphate in men and women. J Bone Miner Res. 2006;21:1187–96.
Antoniucci DM, Yamashita T, Portale AA. Dietary phosphorus regulates serum fibroblast growth factor-23 concentrations in healthy men. J Clin Endocrinol Metab. 2006;91:3144–9.
Riminucci M, Collins MT, Fedarko NS, et al. FGF-23 in fibrous dysplasia of bone and its relationship to renal phosphate wasting. J Clin Investig. 2003;112:683–92.
Beloosesky Y, Grinblat J, Weiss A, et al. Electrolyte disorders following oral sodium phosphate administration for bowel cleansing in elderly patients. Arch Intern Med. 2003;163:803–8.
Curran MP, Plosker GL. Oral sodium phosphate solution: a review of its use as a colorectal cleanser. Drugs. 2004;64:1697–714.
Casais MN, Rosa-Diez G, Pérez S, et al. Hyperphosphatemia after sodium phosphate laxatives in low risk patients: prospective study. World J Gastroenterol. 2009;15:5960–5.
Markowitz GS, Stokes MB, Radhakrishnan J, et al. Acute phosphate nephropathy following oral sodium phosphate bowel purgative: an underrecognized cause of chronic renal failure. J Am Soc Nephrol. 2005;16:3389–96.
Ori Y, Rozen-Zvi B, Chagnac A, et al. Fatalities and severe metabolic disorders associated with the use of sodium phosphate enemas: a single center’s experience. Arch Intern Med. 2012;172:263–5.
Arrambide K, Toto RD. Tumor lysis syndrome. Semin Nephrol. 1993;13:273–80.
Llach F, Felsenfeld AJ, Haussler MR. The pathophysiology of altered calcium metabolism in rhabdomyolysis-induced acute renal failure. Interactions of parathyroid hormone, 25-hydroxycholecalciferol, and 1,25-dihydroxycholecalciferol. N Engl J Med. 1981;305:117–23.
Kebler R, McDonald FD, Cadnapaphornchai P. Dynamic changes in serum phosphorus levels in diabetic ketoacidosis. Am J Med. 1985;79:571–6.
Collins MT, Lindsay JR, Jain A, et al. Fibroblast growth factor-23 is regulated by 1alpha,25-dihydroxyvitamin D. J Bone Miner Res. 2005;20:1944–50.
Quigley R, Baum M. Effects of growth hormone and insulin-like growth factor I on rabbit proximal convoluted tubule transport. J Clin Investig. 1991;88:368–74.
Mitnick PD, Goldfarb S, Slatopolsky E, et al. Calcium and phosphate metabolism in tumoral calcinosis. Ann Intern Med. 1980;92:482–7.
Topaz O, Shurman DL, Bergman R, et al. Mutations in GALNT3, encoding a protein involved in O-linked glycosylation, cause familial tumoral calcinosis. Nat Genet. 2004;36:579–81.
Chefetz I, Heller R, Galli-Tsinopoulou A, et al. A novel homozygous missense mutation in FGF23 causes Familial Tumoral Calcinosis associated with disseminated visceral calcification. Hum Genet. 2005;118:261–6.
Araya K, Fukumoto S, Backenroth R, et al. A novel mutation in fibroblast growth factor 23 gene as a cause of tumoral calcinosis. J Clin Endocrinol Metab. 2005;90:5523–37.
Ichikawa S, Imel EA, Kreiter ML, et al. A homozygous missense mutation in human KLOTHO causes severe tumoral calcinosis. J Clin Investig. 2007;117:2684–91.
Kurosu H, Ogawa Y, Miyoshi M, et al. Regulation of fibroblast growth factor-23 signaling by klotho. J Biol Chem. 2006;281:6120–3.
Urakawa I, Yamazaki Y, Shimada T, et al. Klotho converts canonical FGF receptor into a specific receptor for FGF23. Nature. 2006;444:770–4.
Slavin RE, Wen J, Kumar D, et al. Familial tumoral calcinosis. A clinical, histopathologic, and ultrastructural study with an analysis of its calcifying process and pathogenesis. Am J Surg Pathol. 1993;17:788–802.
Wolf M. Update on fibroblast growth factor 23 in chronic kidney disease. Kidney Int. 2012;89:737–47.
Isakova T, Wahl P, Vargas GS, et al. Fibroblast growth factor 23 is elevated before parathyroid hormone and phosphate in chronic kidney disease. Kidney Int. 2011;79:1370–8.
Larner AJ. Pseudohyperphosphatemia. Clin Biochem. 1995;28:391–3.
Lane JW, Rehak NN, Hortin GL, et al. Pseudohyperphosphatemia associated with high-dose liposomal amphotericin B therapy. Clin Chim Acta. 2008;387:145–9.
Schiller B, Virk B, Blair M, et al. Spurious hyperphosphatemia in patients on hemodialysis with catheters. Am J Kidney Dis. 2008;52:617–20.
Ball CL, Tobler K, Ross BC, et al. Spurious hyperphosphatemia due to sample contamination with heparinized saline from an indwelling catheter. Clin Chem Lab Med. 2004;42:107–8.
Yamaguchi T, Sugimoto T, Imai Y, et al. Successful treatment of hyperphosphatemic tumoral calcinosis with long-term acetazolamide. Bone. 1995;16:247S–50S.
Slatopolsky E, Brown A, Dusso A. Role of phosphorus in the pathogenesis of secondary hyperparathyroidism. Am J Kidney Dis. 2001;37(Suppl 2):S54–7.
Mathew S, Tustison KS, Sugatani T, Chaudhary LR, Rifas L, Hruska KA. The mechanism of phosphorus as a cardiovascular risk factor in CKD. J Am Soc Nephrol. 2008;19:1092–105.
Kestenbaum B, Sampson JN, Rudser KD, Patterson DJ, Seliger SL, Young B, Sherrard DJ, Andress DL. Serum phosphate levels and mortality risk among people with chronic kidney disease. J Am Soc Nephrol. 2005;16:520–8.
Voormolen N, Noordzij M, Grootendorst DC, et al. High plasma phosphate as a risk factor for decline in renal function and mortality in pre-dialysis patients. Nephrol Dial Transplant. 2007;22:2909–16.
Block GA, Klassen PS, Lazarus JM, Ofsthun N, Lowrie EG, Chertow GM. Mineral metabolism, mortality, and morbidity in maintenance hemodialysis. J Am Soc Nephrol. 2004;15:2208–18.
Hruska KA, Teitelbaum SL. Mechanisms of disease: renal osteodystrophy. N Engl J Med. 1995;333:166–75.
Disease Kidney, Improving Global Outcomes (KDIGO) CKD-MBD Work Group. KDIGO clinical practice guideline for the diagnosis, evaluation, prevention, and treatment of chronic kidney disease-mineral and bone disorder (CKD-MBD). Kidney Int. 2009;76(Suppl 113):S1–130.
Isakova T, Gutierrez OM, Chang Y, Shah A, Tamez H, Smith K, Thadhani R, Wolf M. Phosphorus binders and survival on hemodialysis. J Am Soc Nephrol. 2009;20:388–96.
Eddington H, Hoefield R, Sinha S, et al. Serum phosphate and mortality in patients with chronic kidney disease. Clin J Am Soc Nephrol. 2010;5:2251–7.
Martin KJ, Gonzalez EA. Prevention and control of phosphate retention/hyperphosphatemia in CKD-MBD: what is normal, when to start, and how to treat? Clin J Am Soc Nephrol. 2011;6:440–6.
Molony DA, Stephens BW. Derangements in phosphate metabolism in chronic kidney diseases/end-stage renal disease: therapeutic considerations. Adv Chronic Kidney Dis. 2011;18:120–31.
Benini O, D’Alessandro C, Gianfaldoni D, Cupisti A. Extra-phosphate load from food additives in commonly eaten foods: a real and insidious danger for renal patients. J Ren Nutr. 2011;21:303–8.
Cupisti A, Benini O, Ferretti V, et al. Novel differential measurement of natural and added phosphorus in cooked ham with or without preservatives. J Ren Nutr. 2012;22:533–40.
Slatopolsky E, Weerts C, Norwood K, et al. Long-term effects of calcium carbonate and 2.5 mEq/liter calcium dialysate on mineral metabolism. Kidney Int. 1989;36:897–903.
Emmett M, Sirmon MD, Kirkpatrick WG, et al. Calcium acetate control of serum phosphorus in hemodialysis patients. Am J Kidney Dis. 1991;17:544–50.
Rudnicki M, Hyldstrup L, Petersen LJ, et al. Effect of oral calcium on noninvasive indices of bone formation and bone mass in hemodialysis patients: a randomized double-blind placebo-controlled study. Miner Electrolyte Metab. 1994;20:130–4.
Navaneethan SD, Palmer SC, Craig JC, et al. Benefits and harms of phosphate binders in CKD: a systematic review of randomized controlled trials. Am J Kidney Dis. 2009;54:619–37.
Chertow GM, Burke SK, Raggi P, Treat to Goal Working Group. Sevelamer attenuates the progression of coronary and aortic calcification in hemodialysis patients. Kidney Int. 2002;62:245–52.
Block GA, Spiegel DM, Ehrlich J, et al. Effects of sevelamer and calcium on coronary artery calcification in patients new to hemodialysis. Kidney Int. 2005;68:1815–24.
Qunibi W, Moustafa M, Muenz LR, CARE-2 Investigators, et al. A 1-year randomized trial of calcium acetate versus sevelamer on progression of coronary artery calcification in hemodialysis patients with comparable lipid control: the Calcium Acetate Renagel Evaluation-2 (CARE-2) study. Am J Kidney Dis. 2008;51:952–65.
Barreto DV, Barreto FC, De Carvalho AB, et al. Phosphate binder impact on bone remodeling and coronary calcification: results from the BRiC study. Nephron Clin Pract. 2008;110:273–83.
Ross AC, Manson JE, Abrams SA, et al. The 2011 report on dietary reference intakes for calcium and vitamin D from the Institute of Medicine: what clinicians need to know. J Clin Endocrinol Metab. 2011;96:53–8.
Bushinsky DA. Contribution of intestine, bone, kidney, and dialysis to extracellular fluid calcium content. Clin J Am Soc Nephrol. 2010;5(Suppl 1):S12–22.
Gotch FA, Levin N, Kotanko P. Calcium balance in dialysis is best managed by adjusting dialysate calcium guided by kinetic modeling of the interrelationship between calcium intake, dose of vitamin D analogues and the dialysate calcium concentration. Blood Purif. 2010;29:163–76.
Bosticardo G, Malberti F, Basile C, et al. Optimizing the dialysate calcium concentration in bicarbonate haemodialysis. Nephrol Dial Transplant. 2012;27:2489–96.
Goodman WG, Goldin J, Kuizon BD, et al. Coronary-artery calcification in young adults with end-stage renal disease who are undergoing dialysis. N Engl J Med. 2000;342:1478–83.
Goodman WG, London G, Amann K, et al. Vascular calcification in chronic kidney disease. Am J Kidney Dis. 2004;43:572–9.
Giachelli CM. Vascular calcification mechanisms. J Am Soc Nephrol. 2004;15:2959–64.
Yang H, Curinga G, Giachelli CM. Elevated extracellular calcium levels induce smooth muscle cell matrix mineralization in vitro. Kidney Int. 2004;66:246768.
Koiwa F, Onoda N, Kato H, et al. Prospective randomized multicenter trial of sevelamer hydrochloride and calcium carbonate for the treatment of hyperphosphatemia in hemodialysis patients in Japan. Ther Apher Dial. 2005;9:340–6.
Ouellet G, Cardinal H, Mailhot M, et al. Does concomitant administration of sevelamer and calcium carbonate modify the control of phosphatemia? Ther Apher Dial. 2010;14:172–7.
Slatopolsky EA, Burke SK, Dillon MA. Renagel, a nonabsorbed calcium- and aluminum-free phosphate binder, lowers serum phosphorus and parathyroid hormone. The RenaGel Study Group. Kidney Int. 1999;55:299–307.
Fan S, Ross C, Mitra S, et al. A randomized, crossover design study of sevelamer carbonate powder and sevelamer hydrochloride tablets in chronic kidney disease patients on haemodialysis. Nephrol Dial Transplant. 2009;24:3794–9.
Chertow GM, Dillon M, Burke SK, et al. A randomized trial of sevelamer hydrochloride (RenaGel) with and without supplemental calcium. Strategies for the control of hyperphosphatemia and hyperparathyroidism in hemodialysis patients. Clin Nephrol. 1999;51:18–26.
Braun J, Asmus HG, Holzer H, Brunkhorst R, Krause R, Schulz W, Neumayer HH, Raggi P, Bommer J. Long-term comparison of a calcium-free phosphate binder and calcium carbonate-phosphorus metabolism and cardiovascular calcification. Clin Nephrol. 2004;62:104–15.
Block GA, Raggi P, Bellasi A, et al. Mortality effect of coronary calcification and phosphate binder choice in incident hemodialysis patients. Kidney Int. 2007;71:438–41.
De Francisco AL, Leidig M, Covic AC, et al. Evaluation of calcium acetate/magnesium carbonate as a phosphate binder compared with sevelamer hydrochloride in haemodialysis patients: a controlled randomized study (CALMAG study) assessing efficacy and tolerability. Nephrol Dial Transplant. 2010;25:3707–17.
Fishbane S, Delmez J, Suki WN, et al. A randomized, parallel, open-label study to compare once-daily sevelamer carbonate powder dosing with thrice-daily sevelamer hydrochloride tablet dosing in CKD patients on hemodialysis. Am J Kidney Dis. 2010;55:307–15.
Ketteler M, Rix M, Fan S, et al. Efficacy and tolerability of sevelamer carbonate in hyperphosphatemic patients who have chronic kidney disease and are not on dialysis. Clin J Am Soc Nephrol. 2008;3:1125–30.
Delmez J, Block G, Robertson J, et al. A randomized, double-blind, crossover design study of sevelamer hydrochloride and sevelamer carbonate in patients on hemodialysis. Clin Nephrol. 2007;68:386–91.
Hutchison AJ, Maes B, Vanwalleghem J, et al. Efficacy, tolerability, and safety of lanthanum carbonate in hyperphosphatemia: a 6-month, randomized, comparative trial versus calcium carbonate. Nephron Clin Pract. 2005;100:c8–19.
Hutchison AJ, Laville M. Switching to lanthanum carbonate monotherapy provides effective phosphate control with a low tablet burden. Nephrol Dial Transplant. 2008;23:3677–84.
Mehrotra R, Martin KJ, Fishbane S, et al. Higher strength lanthanum carbonate provides serum phosphorus control with a low tablet burden and is preferred by patients and physicians: a multicenter study. Clin J Am Soc Nephrol. 2008;3:1437–45.
Sprague SM, Abboud H, Qiu P, et al. Lanthanum carbonate reduces phosphorus burden in patients with CKD stages 3 and 4: a randomized trial. Clin J Am Soc Nephrol. 2009;4:178–85.
Finn WF. Lanthanum carbonate versus standard therapy for the treatment of hyperphosphatemia: safety and efficacy in chronic maintenance hemodialysis patients. Clin Nephrol. 2006;65:191–202.
Hutchison AJ, Maes B, Vanwalleghem J, et al. Long-term efficacy and tolerability of lanthanum carbonate: results from a 3-year study. Nephron Clin Pract. 2006;102:c61–71.
Lee YK, Choi HY, Shin SK, et al. Effect of lanthanum carbonate on phosphate control in continuos ambulatory peritoneal dialysis patients in Korea: a randomized prospective study. Clin Nephrol. 2013;79:136–42.
Sprague SM, Ross EA, Nath SD, et al. Lanthanum carbonate vs. sevelamer hydrochloride for the reduction of serum phosphorus in hemodialysis patients: a crossover study. Clin Nephrol. 2009;72:252–8.
Kasai S, Sato K, Murata Y, et al. Randomized crossover study of the efficacy and safety of sevelamer hydrochloride and lanthanum carbonate in Japanese patients undergoing hemodialysis. Ther Apher Dial. 2012;16(4):341–9.
Slatopolsky E, Liapis H, Finch J. Progressive accumulation of lanthanum in the liver of normal and uremic rats. Kidney Int. 2005;68:2809–13.
Spasovski GB, Sikole A, Gelev S, et al. Evolution of bone and plasma concentration of lanthanum in dialysis patients before, during 1 year of treatment with lanthanum carbonate and after 2 years of follow-up. Nephrol Dial Transplant. 2006;21:2217–24.
Guillot AP, Hood VL, Runge CF, et al. The use of magnesium-containing phosphate binders in patients with end-stage renal disease on maintenance hemodialysis. Nephron. 1982;30:114–7.
Spiegel DM. Magnesium in chronic kidney disease: unanswered questions. Blood Purif. 2011;31:172–6.
Moriniere P, Vinatier I, Westeel PF, et al. Magnesium hydroxide as a complementary aluminium-free phosphate binder to moderate doses of oral calcium in uraemic patients on chronic haemodialysis: lack of deleterious effect on bone mineralisation. Nephrol Dial Transplant. 1988;3:651–6.
Delmez JA, Kelber J, Norword KY, et al. Magnesium carbonate as a phosphorus binder: a prospective, controlled, crossover study. Kidney Int. 1996;49:163–7.
Tzanakis IP, Papadaki AN, Wei M, et al. Magnesium carbonate for phosphate control in patients on hemodialysis. A randomized controlled trial. Int Urol Nephrol. 2008;40:193–201.
Spiegel DM, Farmer B, Smits G, et al. Magnesium carbonate is an effective phosphate binder for chronic hemodialysis patients: a pilot study. J Ren Nutr. 2007;17:416–22.
McIntyre CW, Pai P, Warwick G, et al. Iron–magnesium hydroxycarbonate (Fermagate): a novel noncalcium-containing phosphate binder for the treatment of hyperphosphatemia in chronic hemodialysis patients. Clin J Am Soc Nephrol. 2009;4:401–9.
Navarro-Gonzalez JF, Mora-Fernandez C, et al. Clinical implications of disordered magnesium homeostasis in chronic renal failure and dialysis. Semin Dial. 2009;22:37–44.
Alfrey AC, Miller NL. Bone magnesium pools in uremia. J Clin Investig. 1973;52:3019–27.
Gonella M, Ballanti P, Della Rocca C, et al. Improved bone morphology by normalizing serum magnesium in chronically hemodialyzed patients. Miner Electrolyte Metab. 1988;14:240–5.
Kakuta T, Tanaka R, Hyodo T. Effect of sevelamer and calcium-based phosphate binders on coronary artery calcification and accumulation of circulating advanced glycation end products in hemodialysis patients. Am J Kidney Dis. 2011;57:422–31.
Russo D, Miranda I, Ruocco C, et al. The progression of coronary artery calcification in predialysis patients on calcium carbonate or sevelamer. Kidney Int. 2007;72:1255–61.
Salusky IB, Goodman WG, Sahney S, et al. Sevelamer controls parathyroid hormone-induced bone disease as efficiently as calcium carbonate without increasing serum calcium levels during therapy with active vitamin D sterols. J Am Soc Nephrol. 2005;16:2501–8.
Ferreira A, Frazao JM, Monier-Faugere MC, et al. Effects of sevelamer hydrochloride and calcium carbonate on renal osteodystrophy in hemodialysis patients. J Am Soc Nephrol. 2008;19:405–12.
Shantouf R, Budoff MJ, Ahmadi N, et al. Effects of sevelamer and calcium-based phosphate binders on lipid and inflammatory markers in hemodialysis patients. Am J Nephrol. 2008;28:275–9.
Navarro-González JF, Mora-Fernández C, Muros de Fuentes M, et al. Effect of phosphate binders on serum inflammatory profile, soluble CD14, and endotoxin levels in hemodialysis patients. Clin J Am Soc Nephrol. 2011;6:2272–9.
Caglar K, Yilmaz MI, Saglam M, et al. Short-term treatment with sevelamer increases serum fetuin-A concentration and improves endothelial dysfunction in chronic kidney disease stage 4 patients. Clin J Am Soc Nephrol. 2008;3:61–8.
Brandenburg VM, Schlieper G, Heussen N, et al. Serological cardiovascular and mortality risk predictors in dialysis patients receiving sevelamer: a prospective study. Nephrol Dial Transplant. 2010;25:2672–9.
Oliveira RB, Cancela AL, Graciolli FG, et al. Early control of PTH and FGF-23 in normophosphatemic CKD patients: a new target in CKD-MBD therapy? Clin J Am Soc Nephrol. 2010;5:286–91.
Striker GE. Beyond phosphate binding: the effect of binder therapy on novel biomarkers may have clinical implications for the management of chronic kidney disease patients. Kidney Int. 2009;76(Suppl 114):S1–2.
Suki WN, Zabaneh R, Cangiano JL, et al. Effects of sevelamer and calcium-based phosphate binders on mortality in hemodialysis patients. Kidney Int. 2007;72:1130–7.
Jamal SA, Fitchett D, Lok CE, et al. The effects of calcium-based versus non-calcium-based phosphate binders on mortality among patients with chronic kidney disease: a meta-analysis. Nephrol Dial Transplant. 2009;24:3168–74.
Di Iorio B, Bellasi A, Russo D, et al. Mortality in kidney disease patients treated with phosphate binders: a randomized study. Clin J Am Soc Nephrol. 2012;7:487–93.
Wilson R, Zhang P, Smyth M, et al. Assessment of survival in a 2-year comparative study of lanthanum carbonate versus standard therapy. Curr Med Res Opin. 2009;25:3021–8.
Toussaint ND, Lau KK, Polkinghorne KR, Kerr PG, et al. Attenuation of aortic calcification with lanthanum carbonate versus calciumbased phosphate binders in haemodialysis: a pilot randomized controlled trial. Nephrology (Carlton). 2011;16:290–8.
D’Haese PC, Spasovski GB, Sikole A, et al. A multicenter study on the effects of lanthanum carbonate (Fosrenol) and calcium carbonate on renal bone disease in dialysis patients. Kidney Int. 2003;85:S73–8.
Malluche HH, Siami GA, Swanepoel C, et al. Improvements in renal osteodystrophy in patients treated with lanthanum carbonate for two years. Clin Nephrol. 2008;70:284–95.
Kopple JD. The National Kidney Foundation K/DOQI clinical practice guidelines for dietary protein intake for chronic dialysis patients. Am J Kidney Dis. 2001;37:S66–70.
Shinaberger CS, Greenland S, Kopple JD, et al. Is controlling phosphorus by decreasing dietary protein intake beneficial or harmful in persons with chronic kidney disease? Am J Clin Nutr. 2008;88:1511–8.
KDIGO 2012 clinical practice guideline for the evaluation and management of chronic kidney disease: management of progression and complications of CKD. Kidney Int. 2013;3;S73–90.
Moore LW, Byham-Gray LD, Scott Parrot J, et al. The mean dietary protein intake at different stages of chronic kidney disease is higher than current guidelines. Kidney Int. 2013. doi:10.1038/ki.2012.420. [Epub ahead of print].
Trumbo P, Schlicker S, Yates AA, et al. Dietary reference intakes for energy, carbohydrate, fiber, fat, fatty acids, cholesterol, protein and amino acids. J Am Diet Assoc. 2002;102:1621–30.
Cianciaruso B, Pota A, Pisani A, et al. Metabolic effects of two low protein diets in chronic kidney disease stage 4–5: a randomized controlled trial. Am J Kidney Dis. 2008;23:636–44.
Mircescu G, Garneata L, Stancu SH, et al. Effects of a supplemented hypoproteic diet in chronic kidney disease. J Ren Nutr. 2007;17:179–88.
Murtaugh MA, Filipowicz R, Baird BC, et al. Dietary phosphorus intake and mortality in moderate chronic kidney disease: NHANES III. Nephrol Dial Transplant. 2012;27:990–6.
Newsome B, Ix JH, Tighiouart H, et al. Effect of protein restriction on serum and urine phosphate in the modification of diet in renal disease (MDRD) study. Am J Kidney Dis. 2013. doi:10.1053/j.ajkd.2013.01.007.
Lopes AA, Tong L, Thumma J, et al. Phosphate binder use and mortality among hemodialysis patients in the dialysis outcomes and practice patterns study (DOPPS): evaluation of possible confounding by nutritional status. Am J Kidney Dis. 2012;60:90–101.
Soroka SD, Beard KM, Mendelssohn DC, et al. Mineral metabolism management in canadian peritoneal dialysis patients. Clin Nephrol. 2012;75:410–5.
Manns B, Klarenbach S, Lee H, et al. Economic evaluation of sevelamer in patients with end-stage renal disease. Nephrol Dial Transplant. 2007;22:2867–78.
Bernard L, Mendelssohn D, Dunn E, et al. A modeled economic evaluation of sevelamer for treatment of hyperphosphatemia associated with chronic kidney disease among patients on dialysis in the United Kingdom. J Med Econ. 2013;16:1–9.
Karamanidou C, Clatworthy J, Weinman J, et al. A systematic review of the prevalence and determinants of nonadherence to phosphate binding medication in patients with end-stage renal disease. BMC Nephrol. 2008;9:2.
Arenas MD, Malek T, Gil MT, et al. Challenge of phosphorus control in hemodialysis patients: a problem of adherence? J Nephrol. 2010;23:525–34.
Chiu YW, Teitelbaum I, Misra M, et al. Pill burden, adherence, hyperphosphatemia, and quality of life in maintenance dialysis patients. Clin J Am Soc Nephrol. 2009;4:1089–96.
Chertow GM, Raggi P, McCarthy JT, et al. The effect of sevelamer and calcium acetate on proxies of atherosclerotic and arteriosclerotic vascular disease in hemodialysis patients. Am J Nephrol. 2003;23:307–14.
Caldeira D, Amaral T, David C, et al. Educational strategies to reduce serum phosphorus in hyperphosphatemic patients with chronic kidney disease: systematic review with meta-analysis. J Ren Nutr. 2011;21:285–94.
Gardulf A, Pålsson M, Nicolay U. Education for dialysis patients lowers long-term phosphate levels and maintains health-related quality of life. Clin Nephrol. 2011;75:319–27.
Acknowledgments
No funding was provided for preparation of the manuscript. The author declares that he has received lecture fees from AMGEN.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Malberti, F. Hyperphosphataemia: Treatment Options. Drugs 73, 673–688 (2013). https://doi.org/10.1007/s40265-013-0054-y
Published:
Issue Date:
DOI: https://doi.org/10.1007/s40265-013-0054-y