
Abstract
Activation of phosphatidylinositol-3-kinase (PI3K) and downstream signalling by AKT/mammalian target of rapamycin (mTOR) modulates cellular processes such as increased cell growth, cell proliferation and increased cell migration as well as deregulated apoptosis and oncogenesis. The PI3K/AKT/mTOR pathway (particularly Class I PI3K isoforms) is frequently activated in a variety of solid tumours and haematological malignancies, making PI3K an attractive therapeutic target in oncology. Inhibitors of PI3K also have the potential to restore sensitivity to other modalities of treatments when administered as part of combination regimens. Although many PI3K inhibitors have reached different stages of clinical development, only two (idelalisib and copanlisib) have been currently approved for use in the treatment of B cell lymphoma and leukaemias. While these two agents are effective clinically, their use is associated with a number of serious class-related as well as drug-specific adverse effects. Some of these are immune-mediated and include cutaneous reactions, severe diarrhoea with or without colitis, hepatotoxicity and pneumonitis. They also induce various metabolic abnormalities such as hyperglycaemia and hypertriglyceridaemia. Not surprisingly, therefore, many new PI3K inhibitors with a varying degree of target selectivity have been synthesised in expectations of improved safety and efficacy, and are currently under clinical investigations for use in a variety of solid tumours as well as haematological malignancies. However, evidence from early clinical trials, reviewed herein, suggests that these newer agents are also associated not only with class-related but also other serious and unexpected adverse effects. Their risk/benefit evaluations have resulted in a number of them being discontinued from further development. Cumulative experience with the use of PI3K inhibitors under development suggests that, compared with their use as monotherapy, combining them with other anticancer therapies may be a more effective strategy in improving current standard-of-care and clinical outcomes in cancers beyond haematological cancers. For example, combination of alpelisib with fulvestrant has recently demonstrated unexpectedly superior efficacy compared to fulvestrant alone. Furthermore, the immunomodulatory activity of PI3Kδ and PI3Kγ inhibitors also provides unexpected opportunities for their use in cancer immunotherapy, as is currently being tested in several clinical trials.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Phosphatidylinositol-3-kinase (PI3K) inhibitors are a welcome addition to therapies for treating various lymphomas and leukaemias, but the only two currently approved PI3K inhibitors carry risks of serious and potentially fatal adverse events. |
New PI3K inhibitors under development are anticipated to offer exciting prospects for treating other malignancies, especially when used in combination with other anticancer modalities. |
Available evidence suggests, however, that these new agents are also associated with class-related as well as other serious safety issues, impacting adversely on their risk/benefit evaluation. |
1 Introduction
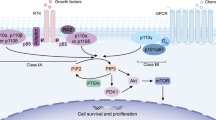
The phosphatidylinositol (PI)-3-kinase (PI3K)-based pathway is an intracellular signalling pathway that responds to a number of hormones and growth factors. Its downstream mediators include AKT and mammalian target of rapamycin (mTOR), both of which are serine–threonine protein kinases. These three components, PI3K, AKT and mTOR, are connected so intricately that they are often regarded as constituting a single unique pathway, the PI3K/AKT/mTOR pathway [1]. Activation of this pathway modulates cellular processes such as increased cell growth, cell proliferation and survival, and increased cell migration as well as deregulated apoptosis and oncogenesis [1, 2]. The reader is referred to a review by Fruman et al. [3] for a comprehensive account of this pathway and its significance for human disease. In this review, we focus only on the PI3K node of this pathway.
Structurally, PI3K enzymes share a common core motif, consisting of a C2 domain, a helical domain and a catalytic (kinase) domain. In mammals, these enzymes are divided into three classes (I, II and III) based on their coding genes, distinct structures and substrate preference [4,5,6]. Class I PI3Ks are further divided into two subclasses: subclass IA (includes isoforms PI3Kα, PI3Kβ and PI3Kδ) and subclass IB (includes only isoform PI3Kγ) [7,8,9]. Genes (and their protein catalytic effectors) for the three class IA PI3Ks are designated PIK3CA (p110α), PIK3CB (p110β) and PIK3CD (p110δ) and that for the only class 1B PI3K is designated as PIK3CG (p110γ). Class IA PI3Ks are constitutive heterodimers of their corresponding 110 kDa catalytic subunits (p110) with one of the five regulatory adaptor proteins (p85α, p55α, p50α, p85β or p55γ, collectively called ‘p85s’) that recruits the p110 to intracellular locations of tyrosine kinase activation. Class 1B PI3K complexes with the p101 or p84 adapter protein [10].
The three isoforms of subclass IA are usually (but not always) activated by activation of receptors with protein tyrosine kinase activity, whereas the isoform in subclass IB is usually activated by activation of G protein-coupled receptors (GPCRs) [5, 11, 12]. Activation of tyrosine kinase receptors by their ligand growth factors results in autophosphorylation on tyrosine residues, which results in recruitment of PI3Ks to the cellular membrane [1, 2, 4]. PI3Ks are a family of lipid kinases that phosphorylate inositol lipids at the 3ʹ-position of the inositol ring, generating PI-3-phosphate (PI(3)P1), PI-3,4-bisphosphate (PI(3,4)P2) and PI-3,4,5-trisphosphate (PI(3,4,5)P3). PI(3,4,5)P3 acts as a docking site for AKT [2, 10, 13] for further downstream signalling. Physiologically, the PI3K activity is counter-balanced by phosphatase and tensin homolog (referred to as PTEN). The main lipid substrate of PTEN is PI(3,4,5)P3, converting it to PI(4,5)P2, thereby terminating/deactivating PI3K/AKT-mediated signalling [1, 14]. Thus, PTEN acts as a negative regulator of PI3K/AKT signalling.
The four class I isoforms differ in terms of their tissue distribution and function and play a central role in the signalling pathway. PI3Kα is ubiquitously expressed and essential for angiogenesis, insulin signalling and maintaining glucose homeostasis. PI3Kβ is also ubiquitously expressed but plays a non-redundant role in phagocytosis and reactive oxygen species (ROS) production in macrophages and neutrophils and is involved in the development of thrombotic diseases by activating platelets. PI3Kδ and PI3Kγ are expressed mainly in the leukocytes [11] and are involved in inflammatory and autoimmune diseases [10, 11, 15]. PI3Kδ signalling is critical for activation, proliferation and survival of B cells. It has an important role in their homing to, and retention in, lymphoid tissues. The critical role of p110δ in B cells has led to the development of highly p110δ-specific inhibitors for treatment of B cell malignancies. In addition, PI3Kδ has important roles in T cells and regulates adaptive immune system, whereas PI3Kγ is more important in the innate immune response. Accumulating evidence suggests that inhibition of PI3Kδ leads to activation of immune response through its selective inhibitory effect on regulatory T cells over helper T cells [16, 17]. As discussed in Sect. 3.1, this activation of immune response has important clinical implications with regard to the toxicity associated with PI3Kδ-selective inhibitors.
Activation of the PI3K/AKT/mTOR pathway is often associated with a variety of solid tumours and haematological malignancies [2, 18,19,20,21]. Common mechanisms of PI3K/AKT/mTOR pathway activation include (1) activating mutations and/or amplification of PIK3CA gene, which encodes for the catalytic subunit PI3Kα; (2) mutations or loss of PTEN; (3) mutation and/or amplification of genes encoding tyrosine receptor kinases such as epidermal growth factor receptor (EGFR) and human epidermal growth factor receptor 2 (HER2); and (4) mutation and/or amplification of AKT [22]. PI3Kα plays important roles in oncogenesis and has been detected with persistent mutations and amplification in most human cancers, including breast, ovarian and colorectal cancers [23,24,25,26]. PTEN is also frequently mutated in several advanced human cancers [24, 25] and loss of PTEN activity leads to permanent activation of the PI3K/AKT pathway [27]. Dysregulation of the class 1A PI3K signalling pathway is also associated with the development of resistance to a variety of anticancer therapies, including chemotherapy, radiotherapy, hormone therapy and targeted agents [25, 28,29,30].
Therefore, PI3Ks are an attractive therapeutic target in oncology and their inhibitors also have the potential to restore sensitivity to other modalities of treatments when administered as part of combination regimens [30, 31]. The first generation of PI3K inhibitors (such as Wortmannin and LY294002) bind to all PI3K isoforms in class I and are referred to as ‘pan–PI3K inhibitors’. Second-generation inhibitors are more potent and have greater isoform-selective activity. Appreciation of the potential advantages of combined inhibition of two oncogenic targets has also led to the synthesis of compounds with dual activity (at PI3K and mTOR), referred to as ‘dual PI3K/mTOR inhibitors’ (third-generation inhibitors). The molecular and pharmacological rationale supporting the synthesis of these dual inhibitors is a high degree of sequence homology shared by the catalytic sites of PI3K and mTOR, and therefore, the anticipation that this combined activity may lead to much stronger inhibition of the whole PI3K/AKT/mTOR pathway. A number of pan–PI3K inhibitors targeting all four isoforms of class I PI3K as well as isoform-selective inhibitors and dual PI3K/mTOR inhibitors are currently under clinical development [14, 20, 32,33,34,35].
This review summarises some of the serious adverse effects associated with PI3K inhibitors that have been approved as well as those under development. The only PI3K inhibitors currently approved are idelalisib, which selectively inhibits PI3Kδ and copanlisib, a highly selective and potent inhibitor of PI3Kδ and PI3Kα. We emphasise that we have deliberately chosen to discuss only the PI3K inhibitors and not the inhibitors of other nodes in the PI3K/AKT/mTOR pathway. Our review is based on drug evaluation reports and prescribing information provided by the US Food and Drug Administration (FDA) [36] and European Medicines Agency (EMA) [37], complemented by review of published literature where indicated. It is worth emphasising that the frequency and severity of an adverse effect varies across different trials, especially when there are different patient populations or indications under investigation and different treatment regimens investigated in relatively small study populations [38]. Therefore, only broadly applicable estimates of frequency of various adverse effects are provided in what follows. Locally approved prescribing information as well as guidelines and consensus statements from expert groups and societies should be consulted to evaluate and manage these adverse effects.
2 Pharmacology and Indications of Currently Approved Phosphatidylinositol-3-Kinase (PI3K) Inhibitors
As of 31 July 2018, only two PI3K inhibitors, idelalisib (coded CAL-101 or GS-1101) and copanlisib (coded BAY 80-6946), have been approved.
Idelalisib is available as tablets for oral use and was approved in July 2014 by the FDA and in September 2014 by the EMA. It is indicated for use in a well-defined setting and subsets of patients with relapsed chronic lymphocytic leukaemia (CLL), relapsed follicular B cell non-Hodgkin lymphoma and relapsed small lymphocytic lymphoma. The indications approved by the EMA are more specific in terms of patient selection.
Copanlisib is available as intravenous formulation for infusion and was approved in September 2017 by the FDA. It has not yet been approved by the EMA. It is indicated for use in the treatment of adult patients with relapsed follicular lymphoma who have received at least two prior systemic therapies.
2.1 Primary Pharmacodynamics
Both idelalisib and copanlisib are isoform selective but their selectivity varies substantially. Their pharmacological mechanisms of action are summarised below:
-
Idelalisib is a selective inhibitor of PI3Kδ kinase, which is expressed in normal and malignant B cells. Its concentration of drug producing 50% inhibition (IC50) values for PI3Kα, PI3Kβ, PI3Kγ and PI3Kδ isoforms are 820, 56.5, 89 and 2.5 nmol/L, respectively. It also inhibits several cell signalling pathways, including B cell receptor (BCR) signalling and the C-X-C chemokine receptor (CXCR) 4 and CXCR5 signalling, which are involved in trafficking and homing of B cells to the lymph nodes and bone marrow.
-
Copanlisib is an inhibitor of PI3Kδ as well as PI3Kα isoforms, both of which are expressed in normal and malignant B cells. Its IC50 values for PI3Kα, PI3Kβ, PI3Kγ and PI3Kδ isoforms are 0.5, 3.7, 6.4 and 0.7 nmol/L, respectively. It also inhibits several key cell signalling pathways, including BCR signalling, CXCR12-mediated chemotaxis of malignant B cells and nuclear factor (NF)-κB signalling in lymphoma cell lines.
2.2 Pharmacokinetics and Drug Interactions
2.2.1 Idelalisib
Following oral administration of a single dose of idelalisib in the fasted state, the median time to maximum concentration (tmax) is observed at 1.5 h. Administration of idelalisib with a high-fat meal increases its exposure (area under the concentration–time curve [AUC]) 1.4-fold. Idelalisib is metabolised to its major metabolite GS-563117 via aldehyde oxidase and cytochrome P450 (CYP) 3A, the metabolite being inactive. Idelalisib also undergoes minor metabolism by UDP glucuronyltransferase (UGT) 1A4. Its terminal elimination half-life is 8.2 h.
Idelalisib exposure is unaffected by age, sex, race or body weight. Its pharmacokinetics in a paediatric population have not been studied. Clinical studies show that idelalisib exposure is unaffected by severe renal impairment but its AUC is increased up to 1.7-fold in subjects with mild hepatic impairment. Patients with baseline hepatic impairment should be monitored for signs of idelalisib toxicity and its dose modified as appropriate.
Rifampicin, a strong inducer of CYP3A4 and P-glycoprotein (P-gp), decreased the geometric mean idelalisib AUC by 75% and the geometric mean maximum concentration (Cmax) by 58%. Ketoconazole (a strong CYP3A4 and P-gp inhibitor) increased the geometric mean AUC of idelalisib by 1.8-fold without any change in the geometric mean Cmax. It is therefore recommended that coadministration of idelalisib be avoided with strong CYP3A4 and P-gp inducers and that patients taking concomitant CYP3A inhibitors should be monitored for signs of idelalisib toxicity and its dose modified as appropriate.
In vitro studies have shown that idelalisib inhibits CYP2C8, CYP2C19, CYP3A and UGT1A1 and induces CYP2B6, whereas GS-563117 inhibits CYP2C8, CYP2C9, CYP2C19, CYP3A and UGT1A1. Clinically, idelalisib is a strong inhibitor of CYP3A4; it increased the geometric mean midazolam Cmax by 2.4-fold and the geometric mean AUC by 5.4-fold. Therefore, coadministration of idelalisib with CYP3A-sensitive substrates should be avoided. Indeed, there is a post-marketing report of severe diazepam-induced altered mental status and respiratory failure resulting in hospitalisation following coadministration of diazepam (a CYP3A4 substrate) with idelalisib; this resolved on stopping both drugs and the patient tolerated a combination of lorazepam (not a CYP3A4 substrate) and idelalisib [39]. Clinical studies have not revealed any significant effect on pharmacokinetics of rosuvastatin (an organic-anion-transporting polypeptide [OATP] 1B1 and OATP1B3 substrate) or digoxin (a P-gp substrate).
2.2.2 Copanlisib
More than 90% of copanlisib metabolism is mediated by CYP3A and < 10% by CYP1A1. Its terminal elimination half-life is 39.1 (range 14.6–82.4) h. The M-1 metabolite of copanlisib accounts for 5% of total radioactivity AUC and its pharmacological activity against PI3Kα and PI3Kδ is comparable with that of the parent compound. Pharmacokinetics of copanlisib are not affected to any clinically significant extent by age, sex, race, smoking status, body weight, mild hepatic impairment or mild to moderate renal impairment. The pharmacokinetics of copanlisib in patients with moderate to severe hepatic impairment, severe renal impairment or end-stage renal disease with or without dialysis have not been studied.
Since copanlisib is principally metabolised by CYP3A4, it is recommended that the use of copanlisib be avoided in combination with strong CYP3A4 inducers or inhibitors. Concomitant use of copanlisib with such agents has been shown to decrease or increase, respectively, copanlisib AUC and Cmax values. If concurrent use of a CYP3A4 inhibitor cannot be avoided, either the dose of copanlisib must be reduced or the patient carefully monitored for toxicity.
Copanlisib does not inhibit any of the major CYP isoforms, UGT, dihydropyrimidine dehydrogenase (DPD), P-gp or various transporters at therapeutic concentrations. Neither does it induce CYP1A2, CYP2B6 and CYP3A.
3 Serious and Life-Threatening Adverse Effects
Although PI3K inhibitors are associated with a whole range of frequent adverse effects such as nausea, vomiting, fatigue, cough, fever, headache and anorexia, these are not usually serious or severe enough to discontinue therapy. Clinically, they are easily managed and are not discussed in this review. We discuss here only the serious adverse effects that present clinical challenges associated with these agents.
Serious adverse reactions were reported in 65 (59%) patients treated with idelalisib plus rituximab. The most frequent serious adverse reactions reported for patients treated with idelalisib plus rituximab were pneumonia (23%), diarrhoea (10%), pyrexia (9%), sepsis (8%), and febrile neutropenia (5%). Adverse reactions that led to discontinuation of idelalisib occurred in 19 (17%) patients. The most common adverse reactions that led to treatment discontinuations were hepatotoxicity and diarrhoea/colitis. 42 (38%) patients had dose interruptions and 16 (15%) patients had dose reductions due to adverse reactions or laboratory abnormalities. The most common reasons for dose interruptions or reductions were pneumonia, diarrhoea, colitis, rash and elevated transaminases.
In pre-approval clinical trials, serious adverse reactions were reported in 44 (26%) patients treated with copanlisib. Adverse reactions resulted in dose reduction in 36 (21%) and discontinuation in 27 (16%) patients receiving copanlisib. The most common reasons for drug discontinuation were pneumonitis (2%) and hyperglycaemia (2%).
Considering that idelalisib is a selective inhibitor of PI3Kδ and copanlisib is a selective inhibitor of PI3Kδ and PI3Kα isoforms, serious adverse effects induced by these two PI3K inhibitors can be conveniently divided into those that are likely class-related to PI3Kδ inhibition and those that are more drug-specific. These are discussed in Sects. 3.1 and 3.2 as reported in association with the two currently approved PI3K inhibitors.
3.1 Class-Related Effects
As a class, both idelalisib and copanlisib are associated with serious dermatological, myelosuppressive, metabolic, gastrointestinal and respiratory adverse effects. Unlike p110α and p110β, which are ubiquitously expressed, p110δ is mainly expressed in leukocytes and therefore, the PI3Kδ pathway and its inhibitors have attracted much attention for their likely involvement in immune disorders. As stated in Sect. 1, inhibition of PI3Kδ leads to activation of immune response [16, 17], which is believed to account for some of the adverse reactions to these agents, especially the PI3Kδ-selective idelalisib [12]. Idelalisib-induced colitis, hepatitis and pneumonitis are believed to be immune-mediated effects [12, 17, 38, 40, 41]. These effects are more frequently observed in first-line patients who are more immunocompetent, are associated with T cell infiltrates and can often be treated by administration of corticosteroids. One review reported data showing higher incidences of idelalisib-induced diarrhoea, pneumonitis and raised hepatic transaminases in previously untreated patients than in those who were previously treated [42], the former group being immunocompetent. Neither is it surprising that the pattern of idelalisib-induced immune-mediated toxicity is similar to that of checkpoint-blocking antibodies [43, 44]. The FDA-approved label of idelalisib includes a detailed black box warning concerning its potential for “Fatal and Serious toxicities: Hepatic, severe diarrhoea, colitis, pneumonitis, infections and intestinal perforations”.
3.1.1 Dermatological Reactions
Fatal cases of Stevens–ohnson syndrome and toxic epidermal necrolysis have occurred in patients treated with idelalisib. If either of these effects is confirmed to be due to idelalisib, treatment with idelalisib should be permanently discontinued. Other severe or life-threatening (grade ≥ 3) cutaneous reactions have been reported in 25% of idelalisib-treated patients and have included exfoliative dermatitis, erythematous rash, generalized rash, macular rash, maculopapular rash, pruritic rash and exfoliative rash.
Grades 3 and 4 cutaneous reactions occurred in 2.8% and 0.6%, respectively, of 317 patients treated with copanlisib monotherapy. Serious cutaneous adverse events were reported in 0.9%. The reported events included exfoliative dermatitis, exfoliative rash, pruritus and rash (including maculopapular rash).
3.1.2 Myelosuppression and Infections
Treatment-emergent grade 3 or 4 neutropenia occurred in 25% of patients treated with idelalisib monotherapy and 58% of patients treated with idelalisib in combination with rituximab or with unapproved combination therapies. Thrombocytopenia and anaemia have also been reported as laboratory abnormalities in about 27% (all grades) of patients treated with idelalisib. Fatal and/or serious infections occurred in 21% of patients treated with idelalisib monotherapy and 48% of patients treated with idelalisib in combination with rituximab or with unapproved combination therapies. The most common infections were pneumonia, sepsis and febrile neutropenia. Upper respiratory tract infections were reported in 12% of the 146 patients receiving idelalisib monotherapy.
Following copanlisib monotherapy, grade ≥ 3 neutropenia occurred in 24% of 317 patients. Serious neutropenic events occurred in 1.3%. In 168 patients treated with copanlisib, leukopenia, neutropenia (including febrile neutropenia) and thrombocytopenia of any grade were reported in 36, 32 and 22%, respectively. The corresponding figures for grade ≥ 3 events were 27, 25 and 8%, respectively. Serious, including fatal, infections occurred in 19% of 317 patients treated with copanlisib monotherapy. The most common serious infection was pneumonia.
3.1.3 Metabolic Effects
Both idelalisib and copanlisib are reported to cause hyperglycaemia and hypertriglyceridaemia, although copanlisib is by far the more potent in this respect.
Idelalisib plus rituximab was associated with hyperglycaemia (all grade) in 54% of 110 patients compared with 46% of 108 patients who received placebo plus rituximab. This small difference between the frequencies in the two arms suggests that idelalisib has a low potential for inducing hyperglycaemia. The corresponding frequencies for hypertriglyceridaemia in the two treatment arms were 56% and 34%, respectively.
Due to the central role of PI3Kα in regulating glucose homeostasis, PI3K inhibition in patients often gives rise to hyperglycaemia and/or hyperinsulinaemia. Not surprisingly, grade 3 or 4 hyperglycaemia occurred in 41% of 317 patients treated with copanlisib monotherapy. Serious hyperglycaemic events occurred in 2.8% of patients. Blood glucose levels typically peaked 5–8 h post-infusion and subsequently declined to baseline levels in majority of the patients; however, blood glucose levels had remained elevated in 17.7% of patients 1 day after copanlisib infusion. Of the 155 patients with baseline glycosylated haemoglobin (HbA1c) < 5.7%, 16 (10%) patients had HbA1c > 6.5% at the end of treatment. Of the 20 patients with diabetes mellitus (treated with copanlisib in one of the studies), seven developed grade 4 hyperglycaemia and two discontinued treatment as a result. Therefore, it is recommended that patients with diabetes mellitus should only be treated with copanlisib following adequate glucose control and should be monitored closely. In 168 patients receiving copanlisib monotherapy, hypertriglyceridaemia was reported in 58% (any grade) and 5% (grade ≥ 3).
3.1.4 Gastrointestinal Reactions
Among the serious gastrointestinal reactions, the most troublesome clinically are diarrhoea, colitis and stomatitis.
In 146 patients treated with idelalisib monotherapy, diarrhoea was reported in 47% (any grade). Severe diarrhoea or colitis (grade 3 or higher) occurred in 14% of patients treated with idelalisib monotherapy and 20% of patients treated with idelalisib in combination with rituximab or with unapproved combination therapies. Diarrhoea can occur at any time and responds poorly to antimotility agents. Among idelalisib-treated patients who reported diarrhoea or colitis, the median time to onset of any grade diarrhoea or colitis was 1.9 months (range 0.0–29.8 months), of grade 1 or 2 was 1.5 months (range 0.0–15.2 months) and of grades 3 or 4 was 7.1 months (range 0.5–29.8 months). Median time to resolution ranged between 1 week and 1 month across trials following interruption of idelalisib therapy and, in some instances, use of corticosteroids. Stomatitis was reported in 6% of 110 patients treated with idelalisib plus rituximab and 1% of 108 patients treated with placebo plus rituximab. The FDA-approved label carries a black box warning explicitly warning that “fatal and/or serious and severe diarrhoea or colitis occurred in 14% to 20% of idelalisib-treated patients”.
Histologically, the colon biopsies in all 11 cases reported by Louie et al. [45] showed some degree of apoptosis within crypts, with five cases showing moderate to severe apoptosis involving the majority of the crypts with loss of goblet cells. In another study of 50 patients, 23 (46%) patients experienced diarrhoea during treatment with idelalisib, including eight with severe symptoms (≥ 7 stools/day above baseline and/or requiring hospitalization) [46]. 14 patients underwent colonoscopic examination with mucosal biopsy. 12 (86%) of these had colitis characterised by intraepithelial lymphocytosis, crypt cell apoptosis and neutrophilic infiltration of crypt epithelium. Eleven patients had symptoms severe enough to warrant drug withdrawal, including nine who were also treated with corticosteroids. Hammami et al. [47] have reported the case of a 56-year-old male who developed severe diarrhoea with a skin eruption mimicking graft-versus-host disease 6 months after starting idelalisib. Colonoscopy revealed normal colon and terminal ileum and biopsies showed mild active ileitis, colitis and proctitis with frequent epithelial apoptosis. Recently, Yeung et al. [48] reported evidence of T cell dysregulation and a substantial infectious component in association with idelalisib-related diarrhoea/colitis.
With regard to idelalisib and the toxic effects listed in the black box of the FDA label, an expert panel has provided guidance on management of these toxic effects [43]. Interestingly, this panel concluded that there are two types of diarrhoea observed in idelalisib clinical trials. The first type of diarrhoea tended to be self-limiting. This type generally occurs within the first 8 weeks and is typically mild or moderate (grades 1–2) and responsive to common antidiarrheal agents. The second type of diarrhoea tends to occur relatively late (although a few cases happened early) and responds poorly to antidiarrheal or empiric antimicrobial therapy. This second type of diarrhoea is most likely related to idelalisib-induced colitis. Recently, de Weerdt et al. [42] have also provided recommendations for the management of idelalisib-induced adverse respiratory and hepatic effects.
In 168 patients treated with copanlisib monotherapy, diarrhoea was reported in 36% (any grade) and 5% (grade ≥ 3) of patients. The corresponding frequencies for stomatitis were 14% and 2%, respectively. Colitis does not seem to have been a problem associated with copanlisib during clinical trials.
3.1.5 Pneumonitis
Fatal and serious pneumonitis has been reported in patients treated with idelalisib. Pneumonia (including non-infectious pneumonitis) was reported in 25% (any grade) and 16% (grade ≥ 3) of 146 patients treated with idelalisib monotherapy. In randomised clinical trials of combination therapies, pneumonitis (manifested by interstitial infiltrates and organising pneumonia) occurred in 4% of patients treated with idelalisib compared with 1% on the comparator arms. Time to onset of pneumonitis ranged from < 1 to 15 months. Patients with pneumonitis thought to be caused by idelalisib have been treated by discontinuing idelalisib and administration of corticosteroids.
Haustraete et al. [49] have reported five patients with idelalisib-induced pneumonitis. All patients complained of cough, dyspnoea and fever. Four had progression of their clinical symptoms over 3–9 weeks, whereas one had progression over 5 days. Four patients had crackles on physical examination and one had a normal examination. A lung computed tomography scan showed diffuse ground-glass opacities (n = 3), consolidations (n = 2), diffuse micronodules (n = 1) and pleural effusions (n = 2). Three of the patients had a bronchoalveolar lavage. Lymphocytic alveolitis was found in two patients (mean lymphocyte proportion 71 ± 5%), whereas one patient had neutrophilic alveolitis (77% neutrophils, 1% lymphocytes). Broad-spectrum antibiotics were ineffective in all five patients. Idelalisib was stopped in all patients and four received corticosteroids. Among patients who received corticosteroids, three had a resolution of pneumonitis and one patient died 12 days after admission due to multi-organ failure. The patient who was not treated with corticosteroids had a favourable outcome after the interruption of idelalisib.
Non-infectious pneumonitis occurred in 5% of 317 patients treated with copanlisib monotherapy. Its features included pulmonary symptoms such as cough, dyspnoea, hypoxia or interstitial infiltrates on radiologic examination. These patients have been managed by withholding copanlisib and administration of systemic corticosteroids.
3.2 Agent-Specific Adverse Effects
Each of the two PI3K inhibitors currently approved is also associated with specific additional adverse effects that are not shared by the other member in the class.
3.2.1 Hepatotoxicity
Idelalisib is reported to induce anaphylaxis, hepatotoxicity, intestinal perforation and hyponatraemia. Idelalisib is contraindicated in patients who have a history of serious allergic reactions including anaphylaxis and toxic epidermal necrolysis. Fatal and/or serious hepatotoxicity occurred in 18% of patients treated with idelalisib monotherapy and 16% of patients treated with idelalisib in combination with rituximab or with unapproved combination therapies. Elevations in ALT or AST greater than 5 times the upper limit of normal were reported and these findings were generally observed within the first 12 weeks of treatment and were reversible with dose interruption. After resumption of treatment at a lower dose, 26% of patients had recurrence of ALT and AST elevations. Therefore, it is recommended that patients should be monitored for hepatic function prior to and during treatment. Rates of recurrent toxicity were lower in patients taking corticosteroids when idelalisib was re-initiated. The advice is to discontinue idelalisib for recurrent hepatic injury or hepatotoxicity. The reader is referred to a review of idelalisib-induced hepatotoxicity for further details of the mechanisms and histological changes involved [41]. Fatal and serious intestinal perforation occurred in idelalisib-treated patients. At the time of perforation, some patients had moderate to severe diarrhoea. As stated earlier, the FDA-approved label of idelalisib includes a black box warning on its potential for hepatotoxicity and intestinal perforation.
In clinical trials with copanlisib, 20–30% of the patients experienced elevations of serum transaminases but there were no cases of clinically relevant hepatic injury [50].
3.2.2 Hypertension
Copanlisib has some activity at PI3Ky, an isoform which has been implicated in blood pressure (BP) homeostasis [12, 15]. Not surprisingly, copanlisib monotherapy induced grade 3 hypertension in 26% of 317 patients treated. Serious hypertensive events occurred in 0.9% of 317 patients. The mean change of systolic and diastolic BP from baseline to 2 h post-infusion on day 1 of cycle 1 was 16.8 and 7.8 mmHg, respectively. The mean BP started decreasing approximately 2 h post-infusion; BP remained elevated for 6–8 h after the start of infusion. It is recommended that optimal BP control should be achieved before starting each infusion and BP should be monitored pre- and during infusion. Depending on the severity and persistence of hypertension, therapy with copanlisib should be withheld, the dose reduced or treatment discontinued. Raised serum levels of lipase and uric acid and hypophosphataemia were reported as laboratory abnormalities in 21, 25 and 44%, respectively, of 168 patients treated with copanlisib monotherapy.
3.2.3 Cardiac Effects Including QT Interval Prolongation
Across clinical trials with copanlisib, there were other cardiac adverse effects, including atrial fibrillation (1.8%), tachycardia, bradycardia and arrhythmia (0.06%). There was one patient with an event of syncope, but was not associated with QT interval prolongation.
In patients with indolent non-Hodgkin’s lymphoma, no patients had a QTcB (QT correction with Bazett formula) or QTcF (QT correction with Fridericia formula) > 500 ms. At subsequent visits during treatment, four of 137 patients (2.9%) and two of 118 patients (1.7%) had a QTcB and QTcF > 500 ms, respectively. Further, nine of 137 patients (6.6%) and two of 118 (1.7%) had a QTcB and QTcF > 480–500 ms, respectively. Three patients (2.2%) experienced a QTcB increase ≥ 60 ms and five patients (4.2%) had a QTcF ≥ 60 ms. It should be noted that copanlisib inhibits the PI3Kα isoform and PI3Kα inhibition is potentially arrhythmogenic [51]. The FDA has therefore required a post-marketing commitment from the sponsor to complete and submit results of a study to determine the effect of copanlisib on QT/corrected QT (QTc) interval in subjects with advanced solid tumours and non-Hodgkin’s lymphoma.
In a partially blinded, randomised, placebo- and positive-controlled crossover study, conducted in 46 healthy volunteers to evaluate the effect of idelalisib on the QT/QTc interval, no significant QTc interval prolongation was observed at idelalisib doses of 150 or 400 mg. Neither was there any concentration–QT response relationship.
4 Post-Marketing Safety of Idelalisib
Table 1 provides a summary of selected adverse events associated with idelalisib contained in EMA’s EudraVigilance Database of spontaneous reports. It is stressed that in compiling this table, these reports have not been validated or evaluated for causality but, nevertheless, the table provides a perspective of the safety of idelalisib in the real world.
There were only handful of reports of hyponatraemia (n = 5), intestinal perforation (n = 3), anaphylaxis (n = 1), hyperglycaemia (n = 1) and hypertriglyceridaemia (n = 1). It appears from these spontaneous reports that the safety observed in clinical trials correlates and predicts the safety during routine clinical use of the drug.
Reports of atrial fibrillation in association with idelalisib were unexpected and their significance is unclear. Interestingly, ibrutinib (another small-molecule agent used to treat B cell lymphomas and leukaemias) and idelalisib share a number of common safety issues [42]. Ibrutinib has been associated with high rates of atrial fibrillation. As of July 2018, there were a total of 7344 reports of adverse events associated with ibrutinib in the EMA’s EudraVigilance Database and, of these, 699 (9.5%) were of atrial fibrillation. In a pooled safety analysis of four randomised trials involving 756 ibrutinib-treated and 749 comparator-treated patients, O’Brien et al. [52] found that atrial fibrillation was reported in 6% of ibrutinib-treated versus 2% of comparator-treated patients; grade 3/4 atrial fibrillation occurred in 3% versus < 1%, respectively. In an earlier analysis by the same investigators, the incidence of ibrutinib-induced atrial fibrillation was 9% with a median time on ibrutinib of 46 months [53]. Thus, this potentially serious arrhythmia is unlikely to be identified during short-term clinical trials.
There are other aspects of idelalisib safety that merit consideration. Furman et al. [54] reported very early on that the combination of idelalisib and rituximab, as compared with placebo and rituximab, significantly improved progression-free survival (PFS), response rate and overall survival among patients with relapsed CLL who were less able to undergo chemotherapy. Various reviews have also concluded that patients treated with single-agent therapies with PI3K inhibitors have lower efficacy outcomes than those treated with combination therapies [8, 38, 55]. For example, an international, multicentre, double-blind, placebo-controlled trial [56] in 416 adult patients with relapsed/refractory CLL requiring treatment for their disease randomised 207 patients to idelalisib and 209 to the placebo arm; both in combination with bendamustine and rituximab. The median PFS was 20.8 months in the idelalisib arm and 11.1 months in the placebo arm [56]. However, in March 2016, the FDA alerted healthcare professionals to reports of an increased rate of adverse events, including deaths, in clinical trials with idelalisib in combination with other cancer medicines [57]. The sponsor confirmed that they were stopping six clinical trials in patients with CLL, small lymphocytic lymphoma and indolent non-Hodgkin lymphomas. At the same time, the FDA emphasised that idelalisib is not approved for previously untreated CLL. In order to further mitigate the risks, the FDA recently required the sponsor of idelalisib to publish in six designated professional journals an information piece that included the risks of fatal and/or serious hepatotoxicity, fatal and/or serious and severe diarrhoea or colitis, fatal and serious pneumonitis, and serious intestinal perforation associated with idelalisib treatment [58].
Since idelalisib-induced PI3Kδ inhibition leads to activation of immune response [16, 17], it is interesting to note that Novak et al. [59]. have reported a case of a heavily pre-treated CLL patient with an atypically high immunoglobulin production who developed clinically significant immunoglobulin flare following idelalisib treatment. Interestingly, Castillo et al. [60] have also reported high incidence of idelalisib-induced hepatotoxicity in patients with Waldenström macroglobulinaemia. A recent study has also highlighted the potential of idelalisib to increase the risk of adverse effects of radiotherapy (strong grade 2 radiodermatitis and grade 3 mucositis) [61]. In December 2017, a safety review by Health Canada identified nine international reports of progressive multifocal leukoencephalopathy (PML) in patients treated with idelalisib [62]. In eight of the nine reports, the link between PML and the use of idelalisib was considered to be possible. The review of the literature suggested that a possible link may be explained by the actions of idelalisib on the immune system. However, other factors such as the type of cancer the patient had and the use of other medications at the same time or in the recent past may have also played a role.
5 Later Generations of PI3K Inhibitors in the Pipeline
PI3K inhibitors have the potential to be a major therapeutic class of drugs in the treatment of various cancers, especially haematological malignancies. They have the potential to reverse resistance to a variety of anticancer therapies, including chemotherapy, radiotherapy, hormone therapy and targeted agents. Not surprisingly, there are no less than 50 PI3K inhibitors have been synthesised and are under investigation. Newer agents are more potent and have greater isoform-specific selective activity (second-generation inhibitors).
Among the many candidates either in the pipeline or discontinued are alpelisib (BYL719), apitolisib (GDC-0980), bimiralisib (PQR309), buparlisib (BKM120), dactolisib (BEZ235), dezapelisib (INCB040093), duvelisib (IPI-145), gedatolisib (PF-5212384), nemiralisib (GSK-2269557), omipalisib (GSK2126458), pictilisib (GDC-0941), pilaralisib (XL147 or SAR245408), serabelisib (INK-1117), sonolisib (PX-866), taselisib (GDC-0032), tenalisib (RP6530), umbralisib (TGR-1202) and voxtalisib (XL765 or SAR245409). These compounds display a range of activities, as shown in Table 2. The IC50 values for many of these novel inhibitors have been listed by other publications [14, 22, 34]. These agents are being investigated as monotherapy as well as in combination with other therapies.
These newer agents do not appear to be free from the major class-related serious adverse effects typically associated with currently available PI3K inhibitors. Table 3 summarises the overall adverse effects profiles, gathered from various sources, of some of these newer PI3K inhibitors. Many of these agents have been studied in early phase first-in-human studies and phase I/II clinical trials investigating their maximum tolerated doses, dose-limiting toxicities and early evidence of efficacy, often in combination with other chemotherapeutic agents. The study samples are often small and, therefore, the data on their safety profile should be considered only indicative of potential risks. Nevertheless, it is evident that these newer agents do not appear to have a particularly more favourable safety profile than their older counterparts; indeed, some of them appear to have a risk of unexpected adverse effects, such as renal failure and peripheral neuropathy, hitherto not associated with the two currently approved PI3K inhibitors (idelalisib or copanlisib). Consequently, the development of many of these new agents has been discontinued [33].
6 Future Challenges in the Development of PI3K Inhibitors
Given the efficacy and the pattern of serious adverse effects associated with PI3K inhibitors, both those approved and those in development, four clinical settings can be envisioned for their clinical use, with each setting requiring a careful assessment of the risks involved and potential benefits. When it comes to selecting an agent that targets the PI3K-mediated pathway, there are complex pharmacological considerations [22, 23].
Firstly, PI3K inhibitors could be used as monotherapy. However, although PI3K signalling is commonly activated in tumour cells, PI3K inhibitors have shown only modest therapeutic efficacy as monotherapy in solid tumours. Janku [9] has summarised efficacy and safety data on various PI3K inhibitors that have been investigated in haematological as well as solid tumours and concluded that challenges to the therapeutic effectiveness of some PI3K inhibitors include the absence of reliable and effective biomarkers, their limited efficacy as single agents, insufficient development of rational therapeutic combinations, the use of schedules with a broad spectrum of associated off-target adverse effects and suboptimal therapeutic exposures. Similarly, Lampson and Brown [38] have also reviewed efficacy and safety data on a number of PI3K inhibitors, focusing on agents with significant selectivity for PI3Kα and PI3Kδ that have entered clinical trials for the treatment of lymphoma. They concluded that while PI3K inhibitors, particularly those that target p110δ, have robust efficacy, their clinical use is limited by their infectious and autoimmune toxicities.
Secondly, these drugs can assist in managing resistance to anticancer therapies. Activation or dysregulation of the class 1A PI3K signalling pathway has been found to be a major contributor to resistance against a diverse range of anticancer agents, including chemotherapy, radiotherapy, hormone therapy and targeted agents [25, 28,29,30, 89, 90]. Loss of PTEN is known to promote resistance to T cell-mediated immunotherapy [91]. PI3K inhibitors have the potential to restore sensitivity to other modalities of treatments when administered as part of combination regimens and, therefore, PI3K inhibition may be positioned as another important combination strategy [30, 31]. Cancer cells are very effective at resisting PI3K inhibition and can activate the PI3K pathway in many ways, thus limiting the power of specific PI3K pathway mutations in predicting drug sensitivity [23]. Also, inhibition of one isoform can be compensated by activation of another [92, 93]. Pan–class I PI3K inhibitors are less likely than isoform-selective class I PI3K inhibitors to allow compensation by other PI3K isoforms. Therefore, they may be expected to be more effective than isoform-selective inhibitors. However, because of their wider off-target effects, they are associated with serious adverse effects, precluding their long-term use. Early indications are that isoform-selective PI3Kα inhibitors may have a more favourable safety profile than pan–class I PI3K inhibitors but it is difficult to administer them in high enough doses for the desired efficacy. All in all, the relative merits of pan–class I versus isoform-selective class I PI3K inhibitors in the clinical setting remain unclear. As inhibition of PI3K upregulates or activates a number of tyrosine kinases, combination of targeted tyrosine kinase inhibitors with PI3K inhibitors is potentially another strategy to optimise antitumour effect. The challenge is to identify the most rational combinations with acceptable safety and tolerability.
PI3K inhibitors are also being studied in combination with other anticancer drugs from different pharmacologic classes, and a large number of these studies are in progress [26]. Breast cancer is an example where multiple mechanisms of resistance to endocrine therapy has led to investigations of combinations of a wide range of drugs [94]. Li et al. [95] have reported a meta-analysis of 46 randomised controlled trials which suggested that the addition of the PI3K pathway inhibitors to the therapy regiment for advanced solid tumours significantly improves PFS. Fulvestrant illustrates well the added-value of combination therapy. It is a selective estrogen receptor antagonist first approved by the FDA in April 2002 for the treatment of hormone receptor (HR)-positive metastatic breast cancer in postmenopausal women with disease progression following antiestrogen therapy. Subsequently, its indication was extended to include its combination with palbociclib or abemaciclib following clinical trials demonstrating much improved efficacy of the combinations. More importantly, approximately 40% of HR-positive (HR+) advanced breast cancer patients have PIK3CA mutations, and the PI3K pathway is the most commonly mutated pathway associated with tumour progression in HR+ advanced breast cancer. Interestingly, Hoste et al. [96] recently reported a postmenopausal patient who was successfully treated with fulvestrant and alpelisib (a PI3Kα-selective agent with an IC50 of 5 nmol/L) following six lines of therapy. The tumour showed two uncommon PIK3CA mutations, and with the combination of alpelisib and fulvestrant the patient went from Eastern Cooperative Oncology Group (ECOG) grade 3 before the start of this therapy to ECOG grade 1 during treatment until progressive disease after 6 months. This combination has been studied in a large phase III (SOLAR-1) trial [97] and recently announced results from this trial have indicated that the combination of fulvestrant and alpelisib almost doubled the median PFS in patients with PIK3CA-mutated HR+/HER2-advanced breast cancer compared with fulvestrant alone [98]. Whilst these efficacy results demonstrating the value of combination therapy are welcome, it is noted that in the same study, the combination was associated with much higher incidences of hyperglycaemia, diarrhoea and skin reactions as well as higher incidence of treatment discontinuation. PI3Kα-specific inhibitors, including alpelisib, are associated with high frequency of diarrhoea and hyperglycaemia [71, 99] and PI3Kα inhibition is potentially arrhythmogenic [51]. SANDPIPER was a similar randomised, double-blind phase III study [100] investigating fulvestrant plus placebo versus the combination of fulvestrant with taselisib (a PI3K inhibitor with IC50 values of 0.29/0.12/0.97 nmol/L for PI3Kα/δ/γ). In this study, after randomisation of 516 patients, efficacy was modest but the most common grade ≥ 3 adverse events in the combination arm were diarrhoea (12%), hyperglycaemia (10%), colitis (3%) and stomatitis (2%). Adverse events led to more taselisib discontinuations (17% vs. 2%) and dose reductions (37% vs. 2%) than placebo [100]. The development of this combination has been discontinued [101].
Finally, in terms of improving clinical response and increasing overall survival, new immunotherapy agents that target the cytotoxic T lymphocyte-associated antigen (CTLA) 4 or programmed cell death (PD) 1 inhibitory receptors on T cells, or PD1 ligands on tumour cells have been very effective [102,103,104]. However, so far, only select patient populations benefit from this form of immunotherapy. Recent findings indicate that PI3K inhibitors could also be used to target the immune system as well as the cancer stroma (non-cancerous cells that also form the tumour mass), in particular the vasculature. Available data indicate that PI3Kδ inhibitors, such as idelalisib, have the potential to be used as cancer immunotherapeutic agents. It seems unlikely that monotherapy with PI3Kδ inhibitors will be sufficient as effective immunotherapy in order to eliminate the cancer. When used together with other immunotherapies or conventional therapies, PI3Kδ inhibitors may prove to be highly effective in promoting anticancer immune responses [17, 26, 105]. While combining PI3Kδ inhibitors with immunotherapy also seems attractive, recent non-clinical studies have demonstrated that that the immune response to different tumours can be highly distinct, which dictates their response to each therapeutic strategy [106]. The mechanisms by which PI3Kδ inhibition promotes antitumour immunity is distinct from that mediated by immune checkpoint blockade and this needs to be considered as PI3Kδ inhibitors are being developed for immunotherapy [106].
7 Conclusions
Knowledge of the PI3K isoforms and their distribution in tissue may help clinicians to anticipate toxicities. Notably, novel isoform-selective PI3K inhibitors have the potential for therapeutic efficacy with improved toxicity profiles compared with non-isoform-selective agents. However, there is no evidence at present to indicate that newer or more targeted PI3K inhibitors have any better safety profile. In addition, there are challenges to the therapeutic use and effectiveness of some PI3K inhibitors. Among others, these challenges include satisfactory management of a variety of serious adverse effects. More importantly, there are no validated biomarkers to identify patients who may respond satisfactorily to, or those who may be troubled by serious adverse effects of, PI3K inhibitors. Neither is there any current evidence of improved safety or efficacy as a result of two targets being combined in dual PI3K/mTOR inhibitors. Monotherapy with most PI3K inhibitors has now been shown to be of limited efficacy compared to their combination with other therapeutic modalities. Data from the SOLAR-1 trial, referred to in Sect. 6, provided evidence of greater benefit from alpelisib when combined with fulvestrant in molecularly selected metastatic breast cancer patients harbouring PI3KCa mutations on primary or metastatic tumours. However, combination of taselisib with fulvestrant was associated with unacceptable risks. Furthermore, reports of an increased rate of adverse events, including deaths, in clinical trials of idelalisib in combination with other cancer medicines (as alerted by the FDA in March 2016) are a matter of concern since they suggest that even combinations of PI3K inhibitors with other therapies may not be free of unacceptable hazards.
The clinical development of many PI3K inhibitors has been discontinued due to insufficient efficacy, problematic toxicities and the absence of biomarkers that correlate with clinical activity. There remain many questions on their optimal use, including the choice of the most appropriate drug in specific cancer, the dosing schedules and possible rational combination strategies. The next step in the clinical development of PI3K inhibitors might be to focus on exploration of new dosing schedules that are better tolerated and more efficacious, the investigation of new rational combinations that overcome PI3K resistance or act in synergy, and the identification of predictive biomarkers of clinical activity. Arising from the toxicity profiles of novel PI3K inhibitors, it is also clear that while guidelines have been established for dealing with toxicities associated with idelalisib and copanlisib (that target PI3Kδ), there will arise a need for the development and wider dissemination of formal guidelines for identification and management of (additional) toxicities associated with these agents with different target selectivity and/or their novel combinations with agents in other pharmacologic classes.
References
Porta C, Paglino C, Mosca A. Targeting PI3K/Akt/mTOR signaling in cancer. Front Oncol. 2014;4:64.
Markman B, Atzori F, Pérez-García J, Tabernero J, Baselga J. Status of PI3K inhibition and biomarker development in cancer therapeutics. Ann Oncol. 2010;21:683–91.
Fruman DA, Chiu H, Hopkins BD, Bagrodia S, Cantley LC, Abraham RT. The PI3K pathway in human disease. Cell. 2017;170:605–35.
Yip PY. Phosphatidylinositol 3-kinase-AKT-mammalian target of rapamycin (PI3K-Akt-mTOR) signaling pathway in non-small cell lung cancer. Transl Lung Cancer Res. 2015;4:165–76.
Fry MJ. Phosphoinositide 3-kinase signalling in breast cancer: how big a role might it play? Breast Cancer Res. 2001;3:304–12.
Engelman JA, Luo J, Cantley LC. The evolution of phosphatidylinositol 3-kinases as regulators of growth and metabolism. Nat Rev Genet. 2006;7:606–19.
Curran E, Smith SM. Phosphoinositide 3-kinase inhibitors in lymphoma. Curr Opin Oncol. 2014;26:469–75.
Wang X, Ding J, Meng LH. PI3K isoform-selective inhibitors: next-generation targeted cancer therapies. Acta Pharmacol Sin. 2015;36:1170–6.
Janku F. Phosphoinositide 3-kinase (PI3K) pathway inhibitors in solid tumors: from laboratory to patients. Cancer Treat Rev. 2017;59:93–101.
Tzenaki N, Papakonstanti EA. p110δ PI3Kinase pathway: emerging roles in cancer. Front Oncol. 2013;3:40.
Stark AK, Sriskantharajah S, Hessel EM, Okkenhaug K. PI3K inhibitors in inflammation, autoimmunity and cancer. Curr Opin Pharmacol. 2015;23:82–91.
Greenwell IB, Ip A, Cohen JB. PI3K inhibitors: understanding toxicity mechanisms and management. Oncology (Williston Park). 2017;31:821–8.
Baselga J. Targeting the phosphoinositide-3 (PI3) kinase pathway in breast cancer. Oncologist. 2011;16(Suppl 1):12–9.
Zhao W, Qiu Y, Kong D. Class I phosphatidylinositol 3-kinase inhibitors for cancer therapy. Acta Pharm Sin B. 2017;7:27–37.
Perrotta M, Lembo G, Carnevale D. The multifaceted roles of PI3Kγ in hypertension, vascular biology, and inflammation. Int J Mol Sci. 2016;17:1858.
Ali K, Soond DR, Pineiro R, Hagemann T, Pearce W, Lim EL, et al. Inactivation of PI(3)K p110δ breaks regulatory T-cell-mediated immune tolerance to cancer. Nature. 2014;510:407–11.
Okkenhaug K, Graupera M, Vanhaesebroeck B. Targeting PI3K in cancer: impact on tumor cells, their protective stroma, angiogenesis, and immunotherapy. Cancer Discov. 2016;6:1090–105.
Vivanco I, Sawyers CL. The phosphatidylinositol 3-kinase AKT pathway in human cancer. Nat Rev Cancer. 2002;2:489–501.
Brader S, Eccles SA. Phosphoinositide 3-kinase signalling pathways in tumor progression, invasion and angiogenesis. Tumori. 2004;90:2–8.
Massacesi C, Di Tomaso E, Urban P, Germa C, Quadt C, Trandafir L, et al. PI3K inhibitors as new cancer therapeutics: implications for clinical trial design. Onco Targets Ther. 2016;9:203–10.
Janku F, Yap TA, Meric-Bernstam F. Targeting the PI3K pathway in cancer: are we making headway? Nat Rev Clin Oncol. 2018;15:273–91.
Dienstmann R, Rodon J, Serra V, Tabernero J. Picking the point of inhibition: a comparative review of PI3K/AKT/mTOR pathway inhibitors. Mol Cancer Ther. 2014;13:1021–31.
Thorpe LM, Yuzugullu H, Zhao JJ. PI3K in cancer: divergent roles of isoforms, modes of activation and therapeutic targeting. Nat Rev Cancer. 2015;15:7–24.
Millis SZ, Ikeda S, Reddy S, Gatalica Z, Kurzrock R. Landscape of phosphatidylinositol-3-kinase pathway alterations across 19784 diverse solid tumors. JAMA Oncol. 2016;2:1565–73.
LoRusso PM. Inhibition of the PI3K/AKT/mTOR pathway in solid tumors. J Clin Oncol. 2016;34:3803–15.
O’Donnell JS, Massi D, Teng MWL, Mandala M. PI3K-AKT-mTOR inhibition in cancer immunotherapy, redux. Semin Cancer Biol. 2018;48:91–103.
Yehia L, Eng C. 65 years of the double helix: one gene, many endocrine and metabolic syndromes: PTEN-opathies and precision medicine. Endocr Relat Cancer. 2018;25:T121–40.
Engelman JA. The role of phosphoinositide 3-kinase pathway inhibitors in the treatment of lung cancer. Clin Cancer Res. 2007;13(15 Pt 2):s4637–40.
Lux MP, Fasching PA, Schrauder MG, Hein A, Jud SM, Rauh C, et al. The PI3K pathway: background and treatment approaches. Breast Care (Basel). 2016;11:398–404.
Keegan NM, Gleeson JP, Hennessy BT, Morris PG. PI3K inhibition to overcome endocrine resistance in breast cancer. Expert Opin Investig Drugs. 2018;27:1–15.
Burris HA 3rd. Overcoming acquired resistance to anticancer therapy: focus on the PI3K/AKT/mTOR pathway. Cancer Chemother Pharmacol. 2013;71:829–42.
Fruman DA, Rommel C. PI3Kδ inhibitors in cancer: rationale and serendipity merge in the clinic. Cancer Discov. 2011;1:562–72.
Rodon J, Tabernero J. Improving the armamentarium of PI3K inhibitors with isoform-selective agents: a new light in the darkness. Cancer Discov. 2017;7:666–9.
Liu Y, Wan WZ, Li Y, Zhou GL, Liu XG. Recent development of ATP-competitive small molecule phosphatidylinostitol-3-kinase inhibitors as anticancer agents. Oncotarget. 2017;8:7181–200.
Bergholz JS, Roberts TM, Zhao JJ. Isoform-selective phosphatidylinositol 3-kinase inhibition in cancer. J Clin Oncol. 2018;36:1339–42.
Food and Drug Administration. Drug-specific reviews on Drugs@FDA; 2018. https://www.accessdata.fda.gov/scripts/cder/daf/index.cfm.
European Medicines Agency. Drug-specific assessment reports and labels; 2018. http://www.ema.europa.eu/ema/index.jsp?curl=pages/medicines/landing/epar_search.jsp&mid=WC0b01ac058001d124.
Lampson BL, Brown JR. PI3Kδ-selective and PI3Kα/δ-combinatorial inhibitors in clinical development for B-cell non-Hodgkin lymphoma. Expert Opin Investig Drugs. 2017;26:1267–79.
Bossaer JB, Chakraborty K. Drug interaction between idelalisib and diazepam resulting in altered mental status and respiratory failure. J Oncol Pharm Pract. 2017;23:470–2.
Pleyer C, Wiestner A, Sun C. Immunological changes with kinase inhibitor therapy for chronic lymphocytic leukemia. Leuk Lymphoma. 2018. https://doi.org/10.1080/10428194.2018.1457147 (Epub 2018 May 15).
Lampson BL, Kasar SN, Matos TR, Morgan EA, Rassenti L, Davids MS, et al. Idelalisib given front-line for treatment of chronic lymphocytic leukemia causes frequent immune-mediated hepatotoxicity. Blood. 2016;128:195–203.
de Weerdt I, Koopmans SM, Kater AP, van Gelder M. Incidence and management of toxicity associated with ibrutinib and idelalisib: a practical approach. Haematologica. 2017;102:1629–39.
Coutre SE, Barrientos JC, Brown JR, de Vos S, Furman RR, Keating MJ, et al. Management of adverse events associated with idelalisib treatment: expert panel opinion. Leuk Lymphoma. 2015;56:2779–86.
Callahan MK, Postow MA, Wolchok JD. Targeting T cell co-receptors for cancer therapy. Immunity. 2016;44:1069–78.
Louie CY, DiMaio MA, Matsukuma KE, Coutre SE, Berry GJ, Longacre TA. Idelalisib-associated enterocolitis: clinicopathologic features and distinction from other enterocolitides. Am J Surg Pathol. 2015;39:1653–60.
Weidner AS, Panarelli NC, Geyer JT, Bhavsar EB, Furman RR, Leonard JP, et al. Idelalisib-associated colitis: histologic findings in 14 patients. Am J Surg Pathol. 2015;39:1661–7.
Hammami MB, Al-Taee A, Meeks M, Fesler M, Hurley MY, Cao D, et al. Idelalisib-induced colitis and skin eruption mimicking graft-versus-host disease. Clin J Gastroenterol. 2017;10:142–6.
Yeung CC, Hockenbery DM, Westerhoff M, Coutre SE, Sedlak RH, Dubowy RL, et al. Pathological assessment of gastrointestinal biopsies from patients with idelalisib-associated diarrhea and colitis. Future Oncol. 2018;14:2265–77.
Haustraete E, Obert J, Diab S, Abbes S, Zini JM, Valade S, et al. Idelalisib-related pneumonitis. Eur Respir J. 2016;47:1280–3.
Dreyling M, Santoro A, Mollica L, Leppä S, Follows GA, Lenz G, et al. Phosphatidylinositol 3-kinase inhibition by copanlisib in relapsed or refractory indolent lymphoma. J Clin Oncol. 2017;35:3898–905.
Yang T, Meoli DF, Moslehi J, Roden DM. Inhibition of the α-subunit of phosphoinositide 3-kinase in heart increases late sodium current and is arrhythmogenic. J Pharmacol Exp Ther. 2018;365:460–6.
O’Brien S, Hillmen P, Coutre S, Barr PM, Fraser G, Tedeschi A, et al. Safety analysis of four randomized controlled studies of ibrutinib in patients with chronic lymphocytic leukemia/small lymphocytic lymphoma or mantle cell lymphoma. Clin Lymphoma Myeloma Leuk. 2018;18(10):648.e15–657.e15.
O’Brien SM, Furman RR, Coutre SE, Flinn IW, Burger J, Blum K, et al. Five-year experience with single-agent ibrutinib in patients with previously untreated and relapsed/refractory chronic lymphocytic leukemia/small lymphocytic leukemia [abstract]. Presented at: 58th American Society of Hematology Annual Meeting; San Diego, CA; December 3–6, 2016. Blood. 2016;128:233.
Furman RR, Sharman JP, Coutre SE, Cheson BD, Pagel JM, Hillmen P, et al. Idelalisib and rituximab in relapsed chronic lymphocytic leukemia. N Engl J Med. 2014;370:997–1007.
Janku F, Hong DS, Fu S, Piha-Paul SA, Naing A, Falchook GS, et al. Assessing PIK3CA and PTEN in early-phase trials with PI3K/AKT/mTOR inhibitors. Cell Rep. 2014;6:377–87.
Zelenetz AD, Barrientos JC, Brown JR, Coiffier B, Delgado J, Egyed M, et al. Idelalisib or placebo in combination with bendamustine and rituximab in patients with relapsed or refractory chronic lymphocytic leukaemia: interim results from a phase 3, randomised, double-blind, placebo-controlled trial. Lancet Oncol. 2017;18:297–311.
Food and Drug Administration. FDA alerts healthcare professionals about clinical trials with Zydelig (idelalisib) in combination with other cancer medicines. 14 Mar 2016; 2018. https://www.fda.gov/drugs/drugsafety/ucm490618.htm.
Food and Drug Administration. Zydelig® risk evaluation and mitigation strategy (modified on 26 January 2018); 2018. https://www.accessdata.fda.gov/drugsatfda_docs/rems/Zydelig_2018_03_22_REMS_Full.pdf.
Novak J, Havrda M, Gaherova L, Spicka J, Kozak T. Clinical case: idelalisib-induced immunoglobulin flare. Immunopharmacol Immunotoxicol. 2017;39:251–2.
Castillo JJ, Gustine JN, Meid K, Dubeau T, Yang G, Xu L, et al. Idelalisib in Waldenström macroglobulinemia: high incidence of hepatotoxicity. Leuk Lymphoma. 2017;58:1002–4.
Gryc T, Putz F, Goerig N, Ziegler S, Fietkau R, Distel LV, et al. Idelalisib may have the potential to increase radiotherapy side effects. Radiat Oncol. 2017;12:109.
Health Canada. Summary safety review—ZYDELIG (idelalisib)—assessing the potential risk of a rare brain infection (progressive multifocal leukoencephalopathy). 15 Dec 2017; 2017. https://www.canada.ca/en/health-canada/services/drugs-health-products/medeffect-canada/safety-reviews/zydelig-assessing-potential-risk-rare-brain-infection.html.
Martín M, Chan A, Dirix L, O’Shaughnessy J, Hegg R, Manikhas A, et al. A randomized adaptive phase II/III study of buparlisib, a pan-class I PI3K inhibitor, combined with paclitaxel for the treatment of HER2-advanced breast cancer (BELLE-4). Ann Oncol. 2017;28:313–20.
Younes A, Salles G, Martinelli G, Bociek RG, Barrigon DC, Barca EG, et al. Pan-phosphatidylinositol 3-kinase inhibition with buparlisib in patients with relapsed or refractory non-Hodgkin lymphoma. Haematologica. 2017;102:2104–12.
Heudel PE, Fabbro M, Roemer-Becuwe C, Kaminsky MC, Arnaud A, Joly F, et al. Phase II study of the PI3K inhibitor BKM120 in patients with advanced or recurrent endometrial carcinoma: a stratified type I-type II study from the GINECO group. Br J Cancer. 2017;116:303–9.
Sarker D, Ang JE, Baird R, Kristeleit R, Shah K, Moreno V, et al. First-in-human phase I study of pictilisib (GDC-0941), a potent pan-class I phosphatidylinositol-3-kinase (PI3K) inhibitor, in patients with advanced solid tumors. Clin Cancer Res. 2015;21:77–86.
Shapiro GI, Rodon J, Bedell C, Kwak EL, Baselga J, Braña I, et al. Phase I safety, pharmacokinetic, and pharmacodynamic study of SAR245408 (XL147), an oral pan-class I PI3K inhibitor, in patients with advanced solid tumors. Clin Cancer Res. 2014;20:233–45.
Matulonis U, Vergote I, Backes F, Martin LP, McMeekin S, Birrer M, et al. Phase II study of the PI3K inhibitor pilaralisib (SAR245408; XL147) in patients with advanced or recurrent endometrial carcinoma. Gynecol Oncol. 2015;136:246–53.
Edelman G, Rodon J, Lager J, Castell C, Jiang J, Van Allen EM, et al. Phase I trial of a tablet formulation of pilaralisib, a pan-class I PI3K inhibitor, in patients with advanced solid tumors. Oncologist. 2018;23:401-e38.
Bowles DW, Kochenderfer M, Cohn A, Sideris L, Nguyen N, Cline-Burkhardt V, et al. A randomized, phase II trial of cetuximab with or without PX-866, an irreversible oral phosphatidylinositol 3-kinase inhibitor, in patients with metastatic colorectal carcinoma. Clin Colorectal Cancer. 2016;15(337–44):e2.
Mayer IA, Abramson VG, Formisano L, Balko JM, Estrada MV, Sanders ME, et al. A phase Ib study of alpelisib (BYL719), a PI3Kα-specific inhibitor, with letrozole in ER+/HER2- metastatic breast cancer. Clin Cancer Res. 2017;23:26–34.
Juric D, Rodon J, Tabernero J, Janku F, Burris HA, Schellens JHM, et al. Phosphatidylinositol 3-kinase α-selective inhibition with alpelisib (BYL719) in PIK3CA-altered solid tumors: results from the first-in-human study. J Clin Oncol. 2018;36:1291–9.
Flinn IW, O’Brien S, Kahl B, Patel M, Oki Y, Foss FF, et al. Duvelisib, a novel oral dual inhibitor of PI3K-δ, γ, is clinically active in advanced hematologic malignancies. Blood. 2018;131:877–87.
Horwitz SM, Koch R, Porcu P, Oki Y, Moskowitz A, Perez M, et al. Activity of the PI3K-δ, γ inhibitor duvelisib in a phase 1 trial and preclinical models of T-cell lymphoma. Blood. 2018;131:888–98.
Flinn IW, Patel M, Oki Y, Horwitz S, Foss FF, Allen K, et al. Duvelisib, an oral dual PI3K-δ, γ inhibitor, shows clinical activity in indolent non-Hodgkin lymphoma in a phase 1 study. Am J Hematol. 2018;93:1311–7.
Juric D, Krop I, Ramanathan RK, Wilson TR, Ware JA, Sanabria Bohorquez SM, et al. Phase I dose-escalation study of taselisib, an oral PI3K inhibitor, in patients with advanced solid tumors. Cancer Discov. 2017;7:704–15.
Tamura K, Kodaira M, Shimizu C, Yonemori K, Yunokawa M, Shimomura A, et al. Phase I study of taselisib in Japanese patients with advanced solid tumors or hormone receptor-positive advanced breast cancer. Cancer Sci. 2018;109:1592–601.
Dolly SO, Wagner AJ, Bendell JC, Kindler HL, Krug LM, Seiwert TY, et al. Phase I study of apitolisib (GDC-0980), dual phosphatidylinositol-3-kinase and mammalian target of rapamycin kinase inhibitor, in patients with advanced solid tumors. Clin Cancer Res. 2016;22:2874–84.
Powles T, Lackner MR, Oudard S, Escudier B, Ralph C, Brown JE, et al. Randomized open-label phase II trial of apitolisib (GDC-0980), a novel inhibitor of the PI3K/mammalian target of rapamycin pathway, versus everolimus in patients with metastatic renal cell carcinoma. J Clin Oncol. 2016;34:1660–8.
Wicki A, Brown N, Xyrafas A, Bize V, Hawle H, Berardi S, et al. First-in human, phase 1, dose-escalation pharmacokinetic and pharmacodynamic study of the oral dual PI3K and mTORC1/2 inhibitor PQR309 in patients with advanced solid tumors (SAKK 67/13). Eur J Cancer. 2018;96:6–16.
Carlo MI, Molina AM, Lakhman Y, Patil S, Woo K, DeLuca J, et al. A phase Ib study of BEZ235, a dual inhibitor of phosphatidylinositol 3-kinase (PI3K) and mammalian target of rapamycin (mTOR), in patients with advanced renal cell carcinoma. Oncologist. 2016;21:787–8.
Seront E, Rottey S, Filleul B, Glorieux P, Goeminne JC, Verschaeve V, et al. Phase II study of dual phosphoinositol-3-kinase (PI3K) and mammalian target of rapamycin (mTOR) inhibitor BEZ235 in patients with locally advanced or metastatic transitional cell carcinoma. BJU Int. 2016;118:408–15.
Wise-Draper TM, Moorthy G, Salkeni MA, Karim NA, Thomas HE, Mercer CA, et al. A phase Ib study of the dual PI3K/mTOR inhibitor dactolisib (BEZ235) combined with everolimus in patients with advanced solid malignancies. Target Oncol. 2017;12:323–32.
Shapiro GI, Bell-McGuinn KM, Molina JR, Bendell J, Spicer J, Kwak EL, et al. First-in-human study of PF-05212384 (PKI-587), a small-molecule, intravenous, dual inhibitor of PI3K and mTOR in patients with advanced cancer. Clin Cancer Res. 2015;21:1888–95.
Del Campo JM, Birrer M, Davis C, Fujiwara K, Gollerkeri A, Gore M, et al. A randomized phase II non-comparative study of PF-04691502 and gedatolisib (PF-05212384) in patients with recurrent endometrial cancer. Gynecol Oncol. 2016;142:62–9.
Papadopoulos KP, Tabernero J, Markman B, Patnaik A, Tolcher AW, Baselga J, et al. Phase I safety, pharmacokinetic, and pharmacodynamic study of SAR245409 (XL765), a novel, orally administered PI3K/mTOR inhibitor in patients with advanced solid tumors. Clin Cancer Res. 2014;20:2445–56.
Wen PY, Omuro A, Ahluwalia MS, Fathallah-Shaykh HM, Mohile N, Lager JJ, et al. Phase I dose-escalation study of the PI3K/mTOR inhibitor voxtalisib (SAR245409, XL765) plus temozolomide with or without radiotherapy in patients with high-grade glioma. Neuro Oncol. 2015;17:1275–83.
Brown JR, Hamadani M, Hayslip J, Janssens A, Wagner-Johnston N, Ottmann O, et al. Voxtalisib (XL765) in patients with relapsed or refractory non-Hodgkin lymphoma or chronic lymphocytic leukaemia: an open-label, phase 2 trial. Lancet Haematol. 2018;5:e170–80.
Brown KK, Toker A. The phosphoinositide 3-kinase pathway and therapy resistance in cancer. F1000Prime Rep. 2015;7:13.
Wang Q, Liu P, Spangle JM, Von T, Roberts TM, Lin NU, et al. PI3K-p110α mediates resistance to HER2-targeted therapy in HER2+, PTEN-deficient breast cancers. Oncogene. 2016;35:3607–12.
Peng W, Chen JQ, Liu C, Malu S, Creasy C, Tetzlaff MT, et al. Loss of PTEN promotes resistance to T cell-mediated immunotherapy. Cancer Discov. 2016;6:202–16.
Costa C, Ebi H, Martini M, Beausoleil SA, Faber AC, Jakubik CT, et al. Measurement of PIP3 levels reveals an unexpected role for p110β in early adaptive responses to p110α-specific inhibitors in luminal breast cancer. Cancer Cell. 2015;27:97–108.
Schwartz S, Wongvipat J, Trigwell CB, Hancox U, Carver BS, Rodrik-Outmezguine V, et al. Feedback suppression of PI3Kα signaling in PTEN-mutated tumors is relieved by selective inhibition of PI3Kβ. Cancer Cell. 2015;27:109–22.
Mayer IA, Dent R, Tan T, Savas P, Loi S. Novel targeted agents and immunotherapy in breast cancer. Am Soc Clin Oncol Educ Book. 2017;37:65–75.
Li X, Dai D, Chen B, Tang H, Xie X, Wei W. Efficacy of PI3K/AKT/mTOR pathway inhibitors for the treatment of advanced solid cancers: a literature-based meta-analysis of 46 randomised control trials. PLoS One. 2018;13:e0192464.
Hoste G, Slembrouck L, Jongen L, Punie K, Matton T, Vander Borght S, et al. Unexpected benefit from alpelisib and fulvestrant in a woman with highly pre-treated ER-positive, HER2-negative PIK3CA mutant metastatic breast cancer. Clin Drug Investig. 2018;38:1071–5.
Andre F, Kaufman B, Juric D, Ciruelos EM, Iwata H, Mayer IA, et al. SOLAR-1: a phase III study of alpelisib and fulvestrant in men and postmenopausal women with hormone receptor-positive (HR+), human epidermal growth factor receptor 2-negative (HER2–) advanced breast cancer (BC) progressing on or after aromatase inhibitor (AI) therapy [abstract no. OT2-01-04]. Cancer Res. 2017;77(4 Suppl):OT2-01-04. https://doi.org/10.1158/1538-7445.SABCS16-OT2-01-04.
André F, Ciruelos EM, Rubovszky G, Campone M, Loibl S, Rugo HS, et al. Alpelisib (ALP) + fulvestrant (FUL) for advanced breast cancer (ABC): results of the phase III SOLAR-1 trial. Ann Oncol. 2018;29(suppl_8):mdy424.010 (Abstract LBA3-PR).
Rodon J, Curigliano G, Delord JP, Harb W, Azaro A, Han Y, et al. A phase Ib, open-label, dose-finding study of alpelisib in combination with paclitaxel in patients with advanced solid tumors. Oncotarget. 2018;9:31709–18.
Baselga J, Dent SF, Cortés J, Im Y-H, Diéras V, Harbeck N, et al. Phase III study of taselisib (GDC-0032) + fulvestrant (FULV) v FULV in patients (pts) with estrogen receptor (ER)-positive, PIK3CA-mutant (MUT), locally advanced or metastatic breast cancer (MBC): primary analysis from SANDPIPER [abstract no. LBA1006]. J Clin Oncol. 2018;36(suppl):LBA1006.
Carroll J. Roche dumps its PhIII PI3K effort on taselisib after researchers track poor survival edge, harsh side effects for breast cancer. Chicago, 3 Jun 2018; 2018. https://endpts.com/roche-dumps-its-phiii-pi3k-effort-on-taselisib-after-researchers-track-poor-survival-edge-harsh-side-effects-for-breast-cancer/.
Larkin J, Hodi FS, Wolchok JD. Combined nivolumab and ipilimumab or monotherapy in untreated melanoma. N Engl J Med. 2015;373:1270–1.
Hao C, Tian J, Liu H, Li F, Niu H, Zhu B. Efficacy and safety of anti-PD-1 and anti-PD-1 combined with anti-CTLA-4 immunotherapy to advanced melanoma: a systematic review and meta-analysis of randomized controlled trials. Medicine (Baltimore). 2017;96:e7325.
Hodi FS, Chiarion-Sileni V, Gonzalez R, Grob JJ, Rutkowski P, Cowey CL, et al. Nivolumab plus ipilimumab or nivolumab alone versus ipilimumab alone in advanced melanoma (CheckMate 067): 4-year outcomes of a multicentre, randomised, phase 3 trial. Lancet Oncol. 2018;19(11):1480–92.
Collins DC, Chenard-Poirier M, Lopez JS. The PI3K pathway at the crossroads of cancer and the immune system: strategies for next generation immunotherapy combinations. Curr Cancer Drug Targets. 2018;18:355–64.
Lim EL, Cugliandolo FM, Rosner DR, Gyori D, Roychoudhuri R, Okkenhaug K. Phosphoinositide 3-kinase δ inhibition promotes antitumor responses but antagonizes checkpoint inhibitors. JCI Insight. https://doi.org/10.1172/jci.insight.120626 (Epub 2018 Jun 7).
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Ethical Standards
This is a review of data in the public domain and the authors declare compliance with all ethical standards.
Funding
No sources of funding were used to assist in the preparation of this review.
Conflict of Interest
Rashmi Shah (RS) was formerly a Senior Clinical Assessor at the Medicines and Healthcare products Regulatory Agency (MHRA), London, UK, and now provides expert consultancy services to a number of pharmaceutical companies. Giuseppe Curigliano (GC) has received consulting fees from Pfizer, Novartis, Eli Lilly and Roche; travel support from Roche; and lecture payments from Pfizer. RS and GC have no other conflicts of interest that are relevant to the content of this review.
Contributions for Authorship
GC and RS conceived the topic and RS assisted with literature search. RS prepared the first draft and GC revised the first and subsequent drafts. Both GC and RS approved the final version.
Additional information
Part of a theme issue on “Safety of Novel Anticancer Therapies: Future Perspectives”. Guest Editors: Rashmi R. Shah, Giuseppe Curigliano.
Rights and permissions
About this article

Cite this article
Curigliano, G., Shah, R.R. Safety and Tolerability of Phosphatidylinositol-3-Kinase (PI3K) Inhibitors in Oncology. Drug Saf 42, 247–262 (2019). https://doi.org/10.1007/s40264-018-0778-4
Published:
Issue Date:
DOI: https://doi.org/10.1007/s40264-018-0778-4