
Abstract
Vedolizumab, a humanized monoclonal antibody to the α4β7 integrin, reduces lymphocyte trafficking to the intestine. This gut-selective mechanism of action offers a safer alternative to other biologics used to treat ulcerative colitis (UC) and Crohn’s disease (CD). We reviewed efficacy and safety data from randomized controlled trials (RCTs), open-label extension (OLE) and observational studies, and pooled analyses of vedolizumab therapy. In UC, RCTs demonstrate that vedolizumab is effective for induction and maintenance of remission, regardless of prior tumor necrosis factor (TNF) antagonist exposure. In CD, vedolizumab is moderately effective as an induction therapy and demonstrates efficacy as a maintenance agent. Secondary analyses indicate that prolonged induction therapy may result in greater efficacy, particularly in TNF antagonist-exposed patients. Comparative efficacy studies and network meta-analyses show similar efficacy to other biologic therapies. OLE studies in UC and CD demonstrate the durability of maintenance efficacy and low serious adverse event (SAE) rates. In an integrated safety analysis of controlled data, there was no significant difference in adverse event, SAE, infection and serious infection rates between vedolizumab and placebo. No drug-specific safety signals were identified. Immunogenicity rates were low and no cases of progressive multifocal leukoencephalopathy directly attributable to vedolizumab are reported in the literature. Vedolizumab is effective for induction and maintenance of inflammatory bowel disease with low treatment-related risks. Given the high therapeutic index of this gut-specific agent, it can be used as either a first- or second-line biologic therapy for UC and CD.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Vedolizumab is an effective first- or second-line agent for inducing and maintaining remission in UC and CD. |
The safety profile of vedolizumab is comparable with placebo. |
Future studies are needed to directly compare the efficacy and safety of vedolizumab to alternative agents and establish the role of vedolizumab in combination with other medications, and as a treatment for fistulizing CD and pouchitis. |
1 Introduction
The chronic inflammatory bowel diseases (IBDs), comprised of ulcerative colitis (UC) and Crohn’s disease (CD), are idiopathic conditions that typically affect younger people. Given that the cause of IBD is unknown, treatment is directed towards suppression of pathological inflammation in the gut. In UC, therapy consists of a step-care approach that features sequential use of aminosalicylates, corticosteroids, tumor necrosis factor (TNF)-α antagonists, and the oral small molecule JAK1/3 inhibitor tofacitinib. Alternatively, although corticosteroids remain the standard induction therapy for most patients with CD, the incremental step-care approach has fallen out of favor, with current algorithms specifying early use of highly effective treatment in high-risk patients. Specifically, this strategy involves the use of biologics such as TNF antagonists, anti-integrins or the interleukin (IL)-12/23 antagonist ustekinumab (Stelara; Janssen Biotech, Inc, Horsham, PA, USA) as monotherapy or in combination with immunosuppressants (thiopurines or methotrexate). However, up to 40% of patients experience primary non-response to TNF antagonists, and up to 46% of patients who initially respond eventually experience secondary loss of response [1]. Furthermore, corticosteroids, conventional immunosuppressants, and TNF antagonists are associated with an increased risk of serious infection. Thus, a need exists for alternative agents, such as the anti-integrins.
Natalizumab (Tysabri; Biogen, Cambridge, MA, USA), a monoclonal antibody directed to the α4 subunit shared by the α4β1- and α4β7-integrins on T cells, was the first anti-integrin therapy for CD. Although natalizumab was effective as induction and maintenance therapy for moderate to severe CD [2, 3], use was limited by the risk of progressive multifocal leukoencephalopathy (PML), a rare viral disease with a high mortality rate. In retrospect, the unique risk for PML conveyed by natalizumab was due to the non-specific effect of the drug on T cell trafficking. Specifically, inhibition of α4β1-integrin and vascular cell adhesion molecule-1 (VCAM-1) interactions by targeting α4 impairs immune surveillance in the kidney, allowing the John–Cunningham (JC) virus to replicate and become genetically diverse. Ultimately, the mutated virus invades the central nervous system, where it infects glial cells and causes PML [4,5,6]. Viral replication in the central nervous system cannot be controlled by the immune system because cytotoxic T-cell trafficking to the brain is dependent on α4β1 VCAM-1 interactions.
The mechanism of action of vedolizumab (Entyvio; Takeda Pharmaceutical Company Ltd, Japan; previous versions: LDP02, MLN02, and MLN0002) is distinct from natalizumab because the former only targets the α4β7 integrin heterodimer that governs trafficking of T lymphocytes to the gut though interaction with mucosal vascular addressin cell adhesion molecule (MAdCAM-1) [7, 8]. Vedolizumab is a humanized immunoglobulin (Ig) G1 monoclonal antibody that binds to α4β7 with no affinity for α4. Accordingly, the drug does not block α4β1-VCAM interactions nor does it affect T-cell trafficking to either the kidney or brain. Therefore, there is no theoretical risk of PML [7, 9,10,11].
Vedolizumab received approval from the European Medicines Agency (EMA) and US FDA in 2014 for the treatment of moderate to severe UC and CD. Guidelines currently recommend vedolizumab in both diseases as a first-line biologic agent or for patients who are refractory to a TNF antagonist [12, 13].
2 Benefit Evaluation
2.1 Ulcerative Colitis (UC)
2.1.1 Epidemiology
UC typically presents with symptoms of diarrhea and rectal bleeding. The incidence has been reported to be up to 23.1 per 100,000 in North America, with a prevalence of 139.8–286.3 per 100,000. The inflammatory process in UC is limited to the superficial mucosa and, in distinction to CD, which can involve any segment of the gastrointestinal tract, is restricted exclusively to the colon [14, 15]. A broad spectrum of clinical disease exists, with severe activity present in up to 25% of patients. These patients are frequently refractory to medical management and may require hospitalization with attendant risk of complications, need for colectomy, and mortality [16]. UC is also associated with a slightly increased risk of colorectal cancer [17].
2.1.2 Early Trials
In a phase Ib/IIa proof-of-concept randomized controlled trial (RCT), 29 UC patients with moderate to severe disease were randomly assigned to receive a single dose of LPD-02, a humanized monoclonal to α4β7 prepared in a Baculovirus system, in ascending doses or placebo. In the study group that received 0.5 mg/kg of LPD-02, 40% of patients had deep remission, whereas none of the placebo patients achieved this outcome [18]. No drug-related adverse events (AEs) were observed in this short-term trial.
Subsequently, a double-blind, phase II trial that evaluated the same formulation (designated MLN02; Millennium Pharmaceuticals) was performed. One hundred and eighty-one patients with active UC were randomly assigned to receive intravenous MLN02 0.5 mg/kg, 2.0 mg/kg, or placebo on days 1 and 29 [19]. Higher rates of clinical response (p = 0.002) and remission (p = 0.03) were observed at week 6 in both treatment groups compared with placebo. Endoscopic response (p = 0.001) and remission (p = 0.007) rates at week 6 were higher in the active treatment groups compared with placebo, and these patients also exhibited superior improvement in histologic inflammation, as measured by mean changes in the Riley score (p = 0.03). However, by week 8, 44% of patients who received MLN02 developed human antidrug antibodies (ADAs), including 24% with a titer greater than 1:125. Importantly, these patients had similar clinical remission rates to placebo and lower drug concentrations than patients without detectable ADAs, indicating clinically meaningful immunogenicity (Table 1).
To overcome the problem of immunogenicity, a new formulation of vedolizumab based on the MLN02 framework was developed with production in a mammalian (Chinese hamster ovary [CHO]) cell system. This formulation, which was ultimately designated vedolizumab, demonstrated similar in vitro binding and potency to MLN02 and was taken forward in further studies. In a phase II dose-ranging RCT, 46 patients with mild to moderate UC received vedolizumab (2.0, 6.0, or 10.0 mg/kg) or placebo on days 1, 15, 29, and 85, and were followed until day 253 [20]. This trial was designed to evaluate the safety, pharmacokinetics, pharmacodynamics, and immunogenicity of the new formulation. Only four patients (11%) developed ADAs and none developed infusion hypersensitivity reactions or important changes in drug pharmacokinetics. Vedolizumab was well tolerated and no treatment-related serious AEs (SAEs) occurred. Clinical response between day 29 and day 253 in the combined vedolizumab cohort was consistently > 50%, compared with a placebo response of 22–33%. In patients with active disease at baseline, clinical remission rates were 53–79% in the vedolizumab groups, compared with 25–50% in patients assigned to placebo. Vedolizumab-treated patients also had greater median reductions from baseline in fecal calprotectin concentrations compared with placebo [20]. Thirty-eight UC patients from the trial of the new formulation entered an open-label extension (OLE) study that included 56 UC and 19 CD patients [21]. In total, 88% of UC patients achieved clinical remission, and 49% had a clinical response at day 491. High rates of durable benefit were observed, indicating the potential of vedolizumab as a maintenance therapy.
2.1.3 Phase III Trials
The GEMINI 1 trial was an integrated phase III, double-blind, multicenter, placebo-controlled induction and maintenance study conducted in 895 UC patients (randomized cohort: n =374; open-label cohort: n =521) [22]. Approximately 40% of participants had failed previous treatment with a TNF antagonist. In the induction component of the trial, patients received intravenous vedolizumab (300 mg) or placebo at days 1 and 15 and were evaluated for response at week 6. The maintenance trial included week 6 clinical responders from the induction trial, as well as patients who responded to similarly administered open-label vedolizumab induction therapy. Responders received vedolizumab every 4 or 8 weeks, or placebo, until week 52 when the primary endpoint of clinical remission was evaluated. The induction and maintenance prespecified primary endpoints were both met. In the induction trial, vedolizumab-treated patients had higher rates of clinical response (47.1 vs. 25.5%, p < 0.001), clinical remission (16.9 vs. 5.4%, p =0.001) and mucosal healing (40.9 vs. 24.8%, p = 0.001) than placebo-treated patients. In the maintenance trial, both vedolizumab groups had higher rates of clinical remission than placebo (every 8 weeks: 41.8%, p < 0.001; every 4 weeks: 44.8%, p < 0.001; placebo: 15.9%). Mucosal healing rates and corticosteroid-free remission rates also favored vedolizumab.
2.1.4 Post Hoc Analyses
In post hoc analyses of the GEMINI 1 trial [23], vedolizumab was more effective than placebo for induction of remission in both TNF antagonist-naïve (clinical response: 53.1 vs. 26.3%; relative risk [RR] 2.0, 95% confidence interval [CI] 1.3–3.0) and exposed patients (39.0 vs. 20.6%; RR 1.9, 95% CI 1.1–3.2). Vedolizumab was also more effective than placebo for maintenance of remission in both TNF antagonist-naïve (46.9 vs. 19.0%; RR 2.5, 95% CI 1.5–4.0) and exposed patients (36.1 vs. 5.3%; RR 6.6, 95% CI 1.7–26.5). Notably, rates of any infection, serious infection and opportunistic infection in the vedolizumab groups did not differ from placebo.
2.1.5 Open-Label Extension Studies
Patients who completed GEMINI 1 had the opportunity to subsequently enroll in a long-term OLE study that analyzed the effectiveness and safety of vedolizumab [24]. Exploratory analyses were reported based on data from patients with at least 248 weeks of cumulative vedolizumab treatment (n =154). Of those who responded to induction therapy and finished the maintenance trial, 40.9% of patients completed 248 weeks of cumulative therapy. Of these patients, 98% had a clinical response and 90% achieved clinical remission. Furthermore, health-related quality-of-life improvements, based on analysis of mean reductions from baseline in the IBD Questionnaire, were observed.
2.1.6 Pooled Analyses
A systematic review and meta-analysis of the aforementioned RCTs confirmed that vedolizumab was superior to placebo for induction of clinical remission (RR 0.86, 95% CI 0.80–0.91), clinical response (RR 0.82, 95% CI 0.75–0.91), and endoscopic remission (RR 0.82, 95% CI 0.75–0.91) in patients with active UC [25]. Furthermore, maintenance therapy with vedolizumab was superior to placebo for achieving clinical remission (RR 2.73, 95% CI 1.78–4.18) and endoscopic remission (RR 2.71, 95% CI 1.88–3.93) [25].
2.1.7 Observational Studies
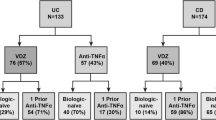
Considerable experience with vedolizumab therapy for UC has accumulated since regulatory approval in 2014. A recent analysis of pooled efficacy data from nine open-label cohorts that included 571 UC patients estimated overall week 6 response and remission rates of 43% (95% CI 37–49%) and 25% (95% CI 12–45%), respectively [26]. Remission was maintained in approximately 40% of patients after 1 year of follow-up. However, as is the case for other agents in ‘real world’ environmental studies, discontinuation rates due to loss of response, intolerance, patient or physician preference, or access considerations were approximately 40% within 1 year of treatment initiation [27, 28]. In another report of open-label vedolizumab therapy, the Groupe d’Etude Therapeutique des Affections Inflammatoires du tube Digestif (GETAID) reported that among 121 UC patients who failed TNF antagonist therapy at 41 French centers, corticosteroid-free clinical remission was achieved in 36% of patients at week 14 [29]. In a nationwide German cohort study comprising 115 UC patients in which 24.3% of patients were TNF antagonist-naïve, 11.3% were in clinical remission at week 6, and this rose to 23.5% at week 14 [30]. Consistent with the GEMINI 1 data, higher clinical remission rates were seen in TNF antagonist-naïve patients compared with those with prior exposure to these agents (39.3 vs. 18.5%, p = 0.023). In a separate study, 69% of 29 UC patients were found to have no endoscopically visible ulcerations following 52 weeks of therapy [31]. Finally, Dulai et al. reported results from the multicenter hospital-based US VICTORY (Vedolizumab for Health Outcomes in Inflammatory Bowel Disease) consortium that evaluated outcomes in 321 vedolizumab-treated UC patients, the majority of whom (71%) had failed treatment with a TNF antagonist. At 12 months, the cumulative rates of clinical and endoscopic remission were 51% and 41%, respectively [32]. Multivariable analyses demonstrated that prior TNF antagonist exposure was associated with reduced clinical (hazard ratio [HR] 0.53, 95% CI 0.38–0.75) and endoscopic remission rates (HR 0.51, 95% CI 0.29–0.88).
Collectively, these open-label studies show that while vedolizumab is likely more effective in UC patients who are naïve to TNF antagonists, a significant proportion of patients exposed to TNF antagonists can still achieve important clinical and endoscopic outcomes over time when treated with vedolizumab.
2.2 Crohn’s Disease (CD)
2.2.1 Epidemiology
The annual incidence and prevalence of CD in North America have been reported to be up to 23.8 and 318 per 100,000, respectively [33]. In distinction to UC, CD features transmural inflammation that results in structural complications such as strictures and fistulae [34]. Surgery is required to treat complications or to manage disease that is refractory to medical interventions in up to 80% of patients [35]. Clinical manifestations depend on disease phenotype. Stricturing disease, which results from both inflammation and fibrosis, is most common with ileal involvement. Intestinal obstruction typically causes crampy abdominal pain, while colonic inflammatory disease characteristically results in diarrhea with or without hematochezia. Fistulizing disease leads to perianal leakage and pain, in addition to placing patients at risk for abscess formation. Extraintestinal manifestations are more frequent in CD compared with UC, with arthralgias being the most common complaint [36].
2.2.2 Early Trials
In a phase II RCT, patients with active CD who had failed treatment with corticosteroids, aminosalicylates or immunosuppressives were randomly assigned to receive intravenous MLN0002 2.0 mg/kg, MLN0002 0.5 mg/kg, or placebo on days 1 and 29 [37]. Patients were followed for 180 days, with response (Crohn’s Disease Activity Index [CDAI] reduction of 70 points) at day 57 functioning as the primary endpoint. Higher rates of clinical response were found in the high-dose group compared with placebo at day 15, but not other timepoints (days 8, 29, 43, and 57). Likewise, time to clinical response was shorter in the high-dose group compared with placebo (17 vs. 42 days, p = 0.04). Furthermore, the CDAI response rate at day 57 (p =0.05) and clinical remission rates at days 15, 29, and 57 (p = 0.009, 0.047, and 0.049, respectively) revealed a statistically significant difference in favor of the high-dose MLN0002 group, compared with placebo. Analysis of the safety data did not identify clinical or laboratory drug-specific safety signals. Subsequent to the results of this phase II study, 19 vedolizumab-naïve CD patients participated in the previously described ‘bridging’ study of the CHO-derived formulation of vedolizumab (Table 2) [21]. Of the 10 vedolizumab-naïve CD patients with active clinical disease at baseline, 40% achieved clinical remission and 70% achieved clinical response by day 491. Again, no safety concerns were identified in this longer duration experience.
2.2.3 Phase III Trials
The GEMINI 2 trial was a phase III, double-blind, multicenter, placebo-controlled RCT conducted in patients with moderate to severe CD (induction n = 368; maintenance n = 461) [38]. The study design was analogous to that of GEMINI 1. In the induction component, patients were randomized to receive two 300 mg doses of vedolizumab at baseline and week 2. Notably, approximately 50% of patients had prior exposure to a TNF antagonist and high baseline disease activity (median CDAI score = 324). At week 6, patients assigned to vedolizumab had higher rates of clinical remission compared with placebo (14.5 vs. 6.8%, p = 0.02). However, CDAI-100 response rates, the co-primary endpoint, were similar between the vedolizumab and placebo groups (31.4 vs. 25.7%, p = 0.23). Although these results were positive from a regulatory perspective—the prespecified analysis plan required a p-value of < 0.025 for either co-primary endpoint as the criterion for statistical significance—they were considered disappointing in terms of the absolute difference in remission rates between drug and placebo, notwithstanding the relatively refractory patient population evaluated. In the maintenance trial, responders to vedolizumab following induction therapy had higher rates of remission compared with placebo when treated with either one of the two vedolizumab regimens (every 8 weeks: 39%, p < 0.001; every 4 weeks: 36.4%, p = 0.004) compared with placebo (21.6%). Furthermore, a higher proportion of patients in the vedolizumab treatment groups achieved CDAI-100 response and glucocorticoid-free remission at week 52 compared with those assigned to placebo (p = 0.005 and p = 0.04, respectively). AEs were no more common in patients who received vedolizumab, and no signal for infection, serious infection opportunistic infection or malignancy was observed.
Given the equivocal induction results obtained in GEMINI 2, the GEMINI 3 trial was highly important because the design featured a 10-week induction period. A total of 416 patients with moderate to severe CD were treated with vedolizumab 300 mg or placebo at weeks 0, 2, and 6 [39]. The primary analysis, which evaluated remission at week 6, was performed in 315 patients who failed or were intolerant to TNF antagonists, with a secondary analysis restricted to TNF antagonist-naïve patients. While the week 6 clinical remission rate was similar between vedolizumab-treated (15.2%) and placebo-treated (12.1%) patients (p = 0.433), vedolizumab was statistically superior to placebo (26.6% vs. 12.1%, p = 0.001) at week 10. Furthermore, CDAI-100 response rates were higher at both week 6 (39.2 vs. 22.3%, p = 0.001) and week 10 (46.8 vs. 24.8%, p < 0.0001) in the vedolizumab group. A subgroup analysis performed using week 10 data demonstrated that vedolizumab was more effective than placebo for induction of remission in TNF antagonist-naïve patients (35.3 vs. 16.0%, p = 0.025).
2.2.4 Pooled Analyses
An integrated analysis of data from 516 TNF antagonist-naïve and 960 TNF antagonist-exposed patients enrolled in GEMINI 2 and GEMINI 3 confirmed a significant difference in favor of vedolizumab when clinical remission rates among TNF antagonist-naïve patients at week 6 (22.7% vs. 10.6%, 95% CI 3.7–21.4) and week 10 (26.6 vs. 15.4%, 95% CI 1.5–21.1) were calculated [40]. At week 52, vedolizumab-treated patients who were naïve to TNF antagonist therapy had higher rates of clinical remission compared with the placebo group (48.9% vs. 26.8%, 95% CI 8.9–35.4). In patients who had previously failed TNF antagonist therapy, vedolizumab treatment had similar clinical remission rates to placebo at week 6 (13.3 vs. 9.7%, 95% CI−1.6–9.8); however, significantly greater clinical remission rates were seen at week 10 (21.8 vs. 11.0%, 95% CI 4.5–18.6). During maintenance therapy, clinical remission rates were higher for vedolizumab-treated patients with prior TNF antagonist failure compared with placebo at week 52 (27.7 vs. 12.8%, 95% CI 4.7–25.0). Collectively, these data indicate that patients who fail TNF antagonist therapy are more refractory to induction therapy and a relatively longer treatment period is required to demonstrate a benefit. In distinction, responders to vedolizumab have a durable benefit regardless of prior TNF antagonist exposure. The greater efficacy observed at 10 weeks was central to decisions by regulatory authorities to specify evaluation of the benefits and risks of continued vedolizumab therapy at 10 weeks (EMA) and 14 weeks (FDA).
2.2.5 Observational Studies
The GEMINI 2 OLE study [41] allowed clinical responders from the randomized trial who completed therapy until week 52 to continue treatment for a prolonged period. An initial report of this experience evaluated 61 of 146 patients who had received 248 weeks of cumulative vedolizumab therapy. Of these patients, 95 and 89% were in clinical response and remission, respectively, with a consistent benefit observed between weeks 52 and 248.
Multiple ‘real-world’ experiences with vedolizumab therapy for CD have been reported in studies originating primarily from academic tertiary care centers. A pooled analysis based on data from 994 participants [26] reported week 6 clinical response and remission rates of 54% (95% CI 41–66%) and 22% (95% CI 13–35%), respectively. Similar rates were reported at week 14. Remission was maintained in 32% (95% CI 12–62%) of patients at week 52. The VICTORY consortium [42] included 212 patients with moderate-to-severe CD. At 12-months, clinical remission, mucosal healing, and deep remission were achieved in 35, 63, and 26% of participants, respectively. Furthermore, improvement in radiographic disease has been demonstrated with vedolizumab in a large, single-center, retrospective cohort [43].
Several of these observational studies evaluated predictors of response. Baseline clinical disease activity, previous TNF antagonist exposure, and inflammatory burden as measured by serum or fecal biomarkers were the most common factors associated with clinical outcomes [44]. Of these predictors, it is notable that previous failure with a TNF antagonist was most frequently associated with poorer clinical response [28, 42, 45]. For example, in the VICTORY consortium, CD patients with prior TNF antagonist exposure had a HR of 0.40 (95% CI 0.20–0.81) for response (defined as a ≥ 50% reduction in CD-related symptom activity or severity on Physician Global Assessment) in comparison with TNF antagonist-naïve patients [30, 45,46,47]. In this data set, concomitant immunosuppressive therapy was not associated with greater vedolizumab efficacy [29]. Furthermore, cumulative mucosal healing rates at 6 and 12 months were 20 and 63%, respectively. Although the results of these open-label, uncontrolled studies should be interpreted cautiously, they are informative and reassuring to clinicians as they demonstrate a similar benefit of vedolizumab in difficult-to-treat patients to what was observed in the GEMINI 2 trial.
3 Rapidity of Onset
A post hoc analysis of outcomes at weeks 2, 4, and 6 of treatment based on patient-level data from 374 UC and 784 CD patients in the GEMINI trials was conducted [48]. TNF antagonist-naïve patients comprised 55% of the UC patients and 36.5% of the CD patients. In UC, higher rates of clinical remission, defined as a rectal bleeding score of 0 and a stool frequency score of < 2, were observed in the active treatment groups compared with placebo at weeks 2 (19.1 vs. 10.1%, difference-adjusted percentage change: 9.0%, 95% CI 2.0–16.1), week 4 (28 vs. 14.8%, difference-adjusted percentage change: 13.2%, 95% CI 5.1–21.4), and week 6 (33.8% vs. 16.8%, difference-adjusted percentage change: 17.0%, 95% CI 8.4–25.6). In CD, higher rates of achieving both an abdominal pain score of < 2 and liquid stool frequency score of > 4 was seen in the treatment groups compared with placebo at week 4 (14.7 vs. 9.3%, difference-adjusted percentage change: 5.4%, 95% CI 0.8–9.9), but not at weeks 2 or 6. While subgroup analyses failed to demonstrate significant differences between treatment and placebo among TNF antagonist-exposed patients at weeks 2, 4, or 6, TNF antagonist-naïve patients had higher remission rates at weeks 2 (15 vs. 7.9%, difference-adjusted percentage change: 7.4%, 95% CI 0.1–14.6) and week 4 (23.8% vs. 10.3%, difference-adjusted percentage change: 13.6%, 95% CI 5.1–22.2).
Collectively, these data, which are based on validated measures of clinical disease activity [49, 50], demonstrate that improvement following initiation of vedolizumab therapy can be observed as early as week 2 for UC patients and week 4 for TNF antagonist-naïve CD patients.
4 Fistulizing CD
Exploratory analyses of patient-level data from the GEMINI 2 trial were conducted in responders to induction therapy with draining fistula [51]. Of the 57 patients with active fistulizing disease at baseline, patients treated with vedolizumab had numerically, but not significantly, higher week 52 fistula closure rates (31% vs. 11%, absolute risk reduction [ARR] 19.7%, 95% CI − 8.9 to 46.2). Similarly, vedolizumab-treated patients had a more rapid time to fistula closure (HR 2.54, 95% CI 0.54–11.96), although this difference was not statistically significant. In a postmarketing study of 35 CD patients with active perianal disease at baseline, 12 patients achieved complete perianal remission after 54 weeks of follow-up [52].
5 Extraintestinal Manifestations
In a post hoc analysis of the GEMINI studies, CD patients receiving vedolizumab were less likely to develop new or worsening arthritis or arthralgia compared with those assigned to placebo (HR 0.63, 95% CI 0.44–0.89). In UC, similar rates of new or worsening arthritis or arthralgia were observed between the treatment and placebo groups. In corticosteroid-free CD patients, lower arthritis or arthralgia rates were seen with vedolizumab (HR 0.14, 95% CI 0.05–0.35). These rates were similar between vedolizumab and placebo in UC patients [53]. A postmarketing study from a claims database demonstrated similar rates of extraintestinal manifestations between vedolizumab and TNF antagonist-treated UC patients, and higher rates in vedolizumab-treated CD patients (adjusted incident rate ratio [IRR] 1.28, 95% CI 1.02–1.62) relative to TNF antagonist-treated patients [54]. However, this estimate should be interpreted with caution given that no adjustment was made for underlying disease activity or intensity of prior treatment. In another postmarketing study that followed 173 CD and 121 UC patients for 54 weeks, 50 of the patients with available data (17.2%) presented with extraintestinal manifestations at baseline. At week 54, 45.7% (n =46) of patients with arthropathy and 60% (n =5) of patients with skin manifestations at baseline were in complete remission of their extraintestinal manifestations. Development of new arthropathies was observed in 15.8% (n =32) of patients, and skin manifestations developed in 4.8% (n =14) of patients [52]. Finally, case reports have identified vedolizumab as a potentially effective treatment for several extraintestinal manifestations, including pyoderma gangrenosum, peripheral arthralgia/arthritis, axial arthropathies, erythema nodosum, and uveitis [55].
In summary, evidence from RCTs and postmarketing studies does not support the notion that vedolizumab is associated with worsening extraintestinal manifestations. Vedolizumab is likely an effective therapy for IBD-associated arthritis or arthralgia. Further research on this topic is needed.
6 Risks
6.1 Biologic Basis for the Safety of Vedolizumab
PML is a serious opportunistic infection of the central nervous system that classically occurs in the setting of intensive chemotherapy or advanced infection with HIV. Thus, it was unexpected when cases were reported in patients treated with natalizumab for multiple sclerosis [56] and CD [57]. The drug was subsequently confirmed as a major risk factor for PML. Basic and clinical studies have determined that natalizumab therapy impairs viral surveillance by T cells based on blockade of interactions between the α4β1-integrin and VCAM-1. Reduced immune surveillance in a renal reservoir and the central nervous system allows the JC virus to become neurotropic and target glial cells, which ultimately causes the disease [4,5,6]. Vedolizumab specifically targets the α4β7 integrin, without binding to α4β1 specifically. Given that the α4β7 integrin heterodimer specifically traffics leukocytes to gut endothelium expressing MAdCAM-1 [7, 8], inhibition of α4β7 causes selective leukocyte trafficking blockade in the gut [7]. Vedolizumab does not inhibit the α4β1–VCAM-1 interaction, and therefore should not put patients at risk of developing PML [7, 9,10,11]. Furthermore, selective α4β7 blockade does not reduce effector T-cell function in the central nervous system of primates [58], nor does it alter CD4 to CD8 ratios in humans [59]. However, safety concerns necessitated a comprehensive evaluation of PML risk during the conduct of the vedolizumab development program.
6.2 Safety in UC
The primary safety advantage of vedolizumab over other IBD therapies is a gut-selective mechanism of action that avoids systemic immunosuppression. Experience with the initial formulations (LPD-02, MLN02) [18] showed it to be well-tolerated as an intravenous infusion. In a phase II trial of MLN02 (n =181) [19], similar AE rates were observed across treatment and placebo groups. However, by week 8, 44% of patients had developed ADAs, including 24% with high titers. Importantly, these patients had reduced serum drug concentrations and similar clinical remission rates to placebo, indicating a clinically relevant immunogenicity problem. This issue and the need for a more efficient manufacturing process led to re-formulation of the drug with production in CHO cells. An open-label ‘bridging’ study of the new vedolizumab formulation was performed in UC and CD patients [20], with a mean exposure between 440 and 859 days [21]. This study demonstrated a safety profile consistent with the original product. Most AEs were mild to moderate, and no deaths occurred. In total, 7% (5/72) of participants developed treatment-related SAEs (e.g. salmonella sepsis, furuncle, blurred vision, a spontaneous abortion, and one infusion-related reaction). No opportunistic infections were observed.
Considerable safety experience was accrued during the conduct of the GEMINI trials. In GEMINI 1, the rate of AEs and SAEs did not differ between the treatment and placebo groups. One patient died from acute coronary syndrome 14 days after the first vedolizumab dose. No significant differences were identified in laboratory test results between active drug- and placebo-treated patients. In the OLE study, 154 of 247 patients in GEMINI 1 who responded to vedolizumab induction at week 6 and received vedolizumab maintenance therapy were enrolled. Of these, 137 patients experienced AEs, which led to discontinuation of treatment in 17 patients. Forty-four SAEs were reported, with seven of these events considered by the investigator to be drug-related [24].
A systematic review and meta-analysis of RCTs that included 1122 UC patients demonstrated similar rates of SAEs between the vedolizumab and placebo-treated groups (12% [97/775] vs. 12% [43/347]; RR 1.02, 95% CI 0.73–1.42) [25].
6.3 Safety in CD
In the GEMINI 2 trial [38], infections and serious infections were more frequent in patients treated with vedolizumab compared with those assigned to placebo. SAEs occurred more often (24.4%) in vedolizumab-treated patients compared with placebo (15.3%); however, these rates were not exposure-adjusted. Four deaths occurred in the vedolizumab group compared with one in the placebo group. One case of breast cancer occurred during vedolizumab induction therapy. In the maintenance component of the trial, one case of the following conditions was observed: tuberculosis, appendiceal carcinoid tumor, squamous cell carcinoma, and basal cell skin carcinoma. In the short-term GEMINI 3 trial [39], similar rates of AEs were seen in vedolizumab (n =209) and placebo (n =207) patients (56 vs. 60%, respectively). In both groups, < 1% of patients experienced SAEs and no deaths occurred.
In the safety population of the GEMINI OLE study in CD [41], AEs occurred in 134 patients and 41 patients experienced SAEs, with three of the latter deemed drug-related. After 248 weeks of follow-up, 15 patients had discontinued treatment due to an AE. One non-drug-related death occurred due to a motor vehicle accident.
6.4 Pooled Analyses in UC and CD
Colombel et al. compared the safety of vedolizumab with placebo in a pooled analysis of six RCTs that collectively evaluated 2830 UC and CD patients with 4811 patient-years of vedolizumab exposure [60]. Notably, exposure-adjusted incidence rates for AEs and SAEs were lower in the vedolizumab groups compared with placebo. The overall AE rates were 247.8 per 100 patient-years (95% CI 229.8–265.8) and 419.4 per 100 patient-years (95% CI 359.3–479.5) for the vedolizumab and placebo groups, respectively. The corresponding SAE rates were 20.0 per 100 patient-years (95% CI 18.5–1.5) and 28.3 per 100 patient-years (95% CI 20.6–35.9), respectively. Prolonged exposure to the drug did not increase the AE and SAE rates. The risk of developing any infection or serious infection was not higher in vedolizumab-treated patients compared with placebo. Serious infections (such as Clostridium difficile infections, sepsis, and Mycobacterium tuberculosis) were infrequent, occurring in < 0.6% of patients. Furthermore, prolonged exposure did not increase gastrointestinal SAEs or serious infections. Congestive heart failure and demyelinating diseases were not observed. In UC, prior TNF antagonist failure (HR 1.99, 95% CI 1.16–3.42; p = 0.0122) and narcotic use (HR 2.68, 95% CI 1.57–4.58; p = 0.0003) were identified as risk factors for serious infection. In CD, younger age (HR 0.97, 95% CI 0.95–0.98; p < 0.0001), corticosteroid use (HR 1.88, 95% CI 1.35–2.63; p = 0.0002) and narcotic use (HR 2.72, 95% CI 1.90–3.89; p < 0.0001) were associated with these events. Among CD patients, the rate of fistula development was lower in vedolizumab-treated patients (4.6 per 100 patient-years, 95% CI 3.8–5.5) compared with those who received placebo (10.0 per 100 patient-years, 95% CI 4.5–15.5). Eighteen malignancies occurred in vedolizumab-treated patients (< 1%). The rates of colorectal cancer and non-melanoma skin cancer were similar between groups and were consistent or lower than rates in the general IBD population. Thirteen deaths occurred in vedolizumab-treated patients.
6.5 Immunogenicity
In the open-label ‘bridging’ study of the new formulation, ADAs were detected in 4% (3/72) of patients, and one of these patients experienced an infusion reaction [21]. The relatively low immunogenicity of the new formulation was confirmed in the GEMINI trials. In GEMINI 1, ADAs were detected in 3.7% of patient samples, with 1.0% of patients testing positive at two or more consecutive time points. Concomitant immunosuppressive therapy was associated with decreased immunogenicity. Three patients experienced clinically important infusion reactions that resulted in drug discontinuation, and two of these patients tested positive for ADAs. In GEMINI 2, ADAs were detected in 4.1% (33/814) of patients and in 0.4% (3/814) of patients with two or more positive samples. Concomitant immunosuppressive therapy was associated with decreased immunogenicity. In GEMINI 3, ADAs were detected in 1% (3/209) of patients, none of which were persistently positive. Furthermore, pooled data from the GEMINI 1 and 2 trials demonstrated reduced immunogenicity in groups receiving concomitant immunosuppressive therapy (3% [1/32] vs. 18% [44/247]) compared with vedolizumab monotherapy. The pooled analysis of six RCTs by Colombel et al. determined that ADAs were detected in 4% (56/1434) of patients from the initial GEMINI 1 and 2 trials. Furthermore, infusion reactions unrelated to ADA formation occurred in 4% (87/2243) of patients in the GEMINI long-term studies [60]. However, interpretation of the immunogenicity data from the GEMINI program requires some caution since the assay used to detect ADAs was not drug-tolerant and the patient populations were heavily exposed to vedolizumab throughout the dosing interval. In keeping with this caveat, ADAs were detected in 10% of the 320 patients who were evaluated during a final safety visit 16 weeks after the last dose, with monotherapy and intermittent exposure identified as important risks for sensitization.
6.6 Risk of Progressive Multifocal Leukoencephalopathy
As summarized in the study by Colombel et al., no cases of PML were reported in any controlled trials or OLE studies. As of May 2018, exposure to vedolizumab in the postmarketing setting was approximately 208,000 patient-years, with over 150,000 patients exposed, and no cases of PML have been reported. In July 2018, an HIV-positive patient with CD receiving vedolizumab developed PML. An independent adjudication committee of PML experts concluded that vedolizumab was not the cause. Instead, they attributed the cause to the patient’s HIV status, along with prolonged immunosuppressant medication use (Takeda Pharmaceuticals, data on file).
6.7 Vedolizumab Safety in Observational Studies
The safety experience of vedolizumab reported in observational studies resembles the data collected in RCTs. A recently published systematic review of safety data summarized findings from published postmarketing open-label cohort studies [61]. In six studies that enrolled 1049 patients (403 patient-years of exposure), the total non-infectious AE rate was 15.8%, with arthralgia (3.1%), arthritis (1.3%), and headache (1.8%) most commonly reported. The combined infectious complication rate in these ‘real-world’ cohorts is approximately 7.8%.
6.8 Clostridium Difficile Infection
The pooled analysis of six RCTs by Colombel et al. reported that C. difficile infection rates were 0.7 per 100 patient-years (95% CI 0.5–1.0), compared with placebo rates of 0.0 per 100 patient-years (95% CI 0.0–1.4). To place these observations in context, these rates are lower than the rate derived from the Health Core Integrated Research Database data (3.1 per 1000 patient-years, 95% CI 2.1–4.5), one of the largest commercial insurance databases in the US [60]. The systematic review of safety data from postmarketing open-label studies found a C. difficile infection rate of 1.2% (13/1049) [61]. In the VICTORY consortium, C. difficile infection occurred in 3.3% of treated patients (5 cases per 100 patient-years) [42], however lower rates have been observed by others (< 1%) [62]. In summary, C. difficile infection rates in RCTs and postmarketing studies were similar between vedolizumab- and placebo-treated patients, with significant overlap in CIs, and lower than those recorded in large health population databases.
6.9 Vedolizumab and the Risk of Postoperative Complications
Several retrospective observational studies have suggested an increased risk of postoperative complications in vedolizumab-treated patients, including surgical site infections [63,64,65,66]. However, a meta-analysis of observational studies after intestinal surgery does not support the presence of this association [67]. In the GEMINI 1 and 2 trials, similar IBD-related surgical rates (vedolizumab: 3.6%, 51/1434; placebo: 2.4%, 7/297), postoperative complications (vedolizumab: 5.9%, 3/51; placebo: 14.3%, 1/7) and serious postoperative complications (vedolizumab: 2.0%, 1/51; placebo: 14.3%, 1/7) were observed [68]. The difference in risk estimates between the findings of the controlled trials and some of the observational studies that suggested an increase in risk is likely explained by the inability of the former to adequately control for confounders—notably disease activity and corticosteroid exposure—that are important risk factors for the development of perioperative complications.
7 Comparison with Other Therapies
No RCTs comparing vedolizumab with other therapies have been reported. In a recent publication, the VICTORY consortium used a propensity score-adjusted model to indirectly compare the efficacy of vedolizumab with TNF antagonist therapy. In UC, vedolizumab-treated patients had higher clinical remission and endoscopic healing rates, regardless of prior TNF antagonist exposure [69]. In CD, vedolizumab-treated patients had higher endoscopic healing rates and higher clinical remission rates, specifically in patients with colonic disease [70]. In UC and CD, overall SAEs and rates of serious infection were lower in vedolizumab-treated patients [71].
A network meta-analysis of RCTs in UC determined that vedolizumab was highly ranked for induction of clinical remission and mucosal healing in biologic-naïve patients. In biologic-experienced patients, low-quality evidence supported the use of vedolizumab [72]. Maintenance outcomes could only be compared between vedolizumab, golimumab, and tofacitinib. All were superior to placebo for clinical remission and mucosal healing, and maintenance outcomes were similar for the active treatments. In contrast to the other agents evaluated, vedolizumab was not associated with an increased risk of infection [72]. In a network analysis of RCTs in CD, vedolizumab performed similarly to other biologics as an induction agent. As a maintenance therapy, vedolizumab had similar efficacy to most other therapies, yet it was estimated to be inferior to adalimumab and combination therapy using infliximab and azathioprine. Vedolizumab had lower withdrawal rates due to AEs than immunosuppressives, infliximab, and combination therapy [73]. Evidence from network meta-analyses of RCTs comparing vedolizumab with ustekinumab in CD is conflicting [74,75,76].
In summary, the results of indirect comparisons are consistent with the notion that vedolizumab has a similar efficacy profile to other IBD treatments with a reduced risk of infectious complications. However, it is important to recognize that such indirect comparison is highly susceptible to bias and cannot adequately control for the effects of important confounding variables that may explain differences between the efficacy of vedolizumab and other therapies. Thus, randomized comparisons are essential.
8 Benefit–Risk Evaluation
8.1 Summary of Benefits
In UC, vedolizumab is effective for induction of clinical and endoscopic remission at week 6. Prolonged treatment with 300 mg administered every 4 or 8 weeks is effective regardless of TNF antagonist exposure status, although more effective in biologic-naïve patients. In CD, the GEMINI 3 study demonstrated that vedolizumab was most effective as an induction therapy at week 10. Continued treatment in induction responders was an effective maintenance therapy for both TNF antagonist-naïve and antagonist-failure patients. In OLE studies of both UC and CD, clinical efficacy rates > 90% at 5 years were observed for patients who had responded to 52 weeks of therapy. Observational studies showed similar induction and maintenance outcomes to those reported in RCTs. Comparative studies and network meta-analyses suggest similar efficacy between vedolizumab and TNF antagonists or tofacitinib, while efficacy data are conflicting for comparisons to ustekinumab.
Data regarding the cost effectiveness of vedolizumab compared with TNF antagonists are conflicting. A literature review conducted by Schneider et al. found that while evidence supports vedolizumab as a cost effective first- and second-line therapy for moderate to severe UC, there is no evidence that vedolizumab is the most cost effective first-line therapy for moderate to severe CD [77]. However, these conclusions should be interpreted with caution, given that direct comparisons of vedolizumab versus TNF antagonists are lacking. It may be possible to directly compare these agents in the near future as several head-to-head trials are currently underway (NCT02497469, NCT03679546).
8.2 Summary of Main Risks
In UC and CD, individual trials and OLE studies did not reveal any specific safety signals for vedolizumab. In a pooled analysis using data from six RCTs and OLEs, lower exposure-adjusted AE and SAE rates were detected with vedolizumab use compared with placebo. Serious infections, upper respiratory tract infections, and lower respiratory tract infections also occurred less frequently in vedolizumab-treated patients. Similar malignancy rates existed compared with placebo and the general IBD population. ADA formation (4%) and infusion reactions (4%) were uncommon. Similar findings have been documented in ‘real world’ observational studies. A single case of PML has occurred in an HIV-infected patient. An increased risk of perioperative complications is not supported by the totality of the data. Comparative ‘real world’ studies and network meta-analyses of RCTs show superior safety profiles to TNF antagonists and tofacitinib.
9 Conclusions
Vedolizumab is effective for the induction and maintenance of remission in both UC and CD, either as a first-line therapy or after failure of other agents. Given that vedolizumab has a comparable safety profile to placebo, it likely has a superior therapeutic index relative to other advanced treatments for IBD. However, future studies are needed to directly compare the efficacy and safety of vedolizumab to alternative agents. In addition, high-quality data regarding the role of vedolizumab in combination with other biologic therapies, and as a treatment for fistulizing CD and pouchitis, are lacking.
References
Ben-Horin S, Kopylov U, Chowers Y. Optimizing anti-TNF treatments in inflammatory bowel disease. Autoimmun Rev. 2014;13:24–30.
Targan SR, Feagan BG, Fedorak RN, Lashner BA, Panaccione R, Present DH, et al. Natalizumab for the treatment of active Crohn’s disease: results of the ENCORE Trial. Gastroenterology. 2007;132(5):1672–83.
Sandborn WJ, Colombel JF, Enns R, Feagan BG, Hanauer SB, Lawrance IC, et al. Natalizumab induction and maintenance therapy for Crohn’s disease. N Engl J Med. 2005;353(18):1912–25.
Kuehn BM. Rare neurological condition linked to newer monoclonal antibody biologics. JAMA. 2009;301(14):1423–4.
Chen Y, Bord E, Tompkins T, Miller J, Tan CS, Kinkel RP, et al. Asymptomatic reactivation of JC virus in patients treated with natalizumab. N Engl J Med. 2009;361(11):1067–74.
Hunt D, Giovannoni G. Natalizumab-associated progressive multifocal leucoencephalopathy: a practical approach to risk profiling and monitoring. Pract Neurol. 2012;12(1):25–35.
Soler D, Chapman T, Yang LL, Wyant T, Egan R, Fedyk ER. The binding specificity and selective antagonism of vedolizumab, an anti-alpha4beta7 integrin therapeutic antibody in development for inflammatory bowel diseases. J Pharmacol Exp Ther. 2009;330(3):864–75.
Erle DJ, Briskin MJ, Butcher EC, Garcia-Pardo A, Lazarovits AI, Tidswell M. Expression and function of the MAdCAM-1 receptor, integrin alpha 4 beta 7, on human leukocytes. J Immunol. 1994;153(2):517–28.
Fedyk ER, Wyant T, Yang L-L, Csizmadia V, Burke K, Yang H, et al. Exclusive antagonism of the α4β7 integrin by vedolizumab confirms the gut-selectivity of this pathway in primates. Inflamm Bowel Dis. 2012;18(11):2107–19.
Haanstra KG, Hofman SO, Estêvão DML, Blezer EL, Bauer J, Yang L-L, et al. Antagonizing the α4β1 integrin, but not α4β7, inhibits leukocytic infiltration of the central nervous system in rhesus monkey experimental autoimmune encephalomyelitis. J Immunol. 2013;190(5):1961–73.
Milch C, Wyant T, Xu J, Kent W, Berger J, Fox I. Vedolizumab does not reduce the CD4 + : CD8 + ratio in the CSF of healthy volunteers: P-136. Inflamm Bowel Dis. 2011;17:S54.
Bressler B, Marshall JK, Bernstein CN, Bitton A, Jones J, Leontiadis GI, et al. Clinical practice guidelines for the medical management of nonhospitalized ulcerative colitis: the Toronto consensus. Gastroenterology. 2015;148(5):1035–1058.e3.
Dassopoulos T, Cohen RD, Scherl EJ, Schwartz RM, Kosinski L, Regueiro MD. Ulcerative colitis care pathway. Gastroenterology. 2015;149(1):238–45.
Danese S, Fiocchi C. Ulcerative colitis. N Engl J Med. 2011;365(18):1713–25.
Ananthakrishnan AN. Epidemiology and risk factors for IBD. Nat Reviews Gastroenterol Hepatol. 2015;12(4):205–17.
Bitton A, Buie D, Enns R, Feagan BG, Jones JL, Marshall JK, et al. Treatment of hospitalized adult patients with severe ulcerative colitis: toronto consensus statements. Am J Gastroenterol. 2012;107(2):179–94.
Lutgens MW, van Oijen MG, van der Heijden GJ, Vleggaar FP, Siersema PD, Oldenburg B. Declining risk of colorectal cancer in inflammatory bowel disease: an updated meta-analysis of population-based cohort studies. Inflamm Bowel Dis. 2013;19(4):789–99.
Feagan BG, McDonald J, Greenberg G, Wild G, Pare P, Fedorak RN, et al. An ascending dose trial of a humanized A4 B7 antibody in ulcerative colitis (UC). Gastroenterology. 2000;118(4):A874.
Feagan BG, Greenberg GR, Wild G, Fedorak RN, Pare P, McDonald JW, et al. Treatment of ulcerative colitis with a humanized antibody to the alpha4beta7 integrin. N Engl J Med. 2005;352(24):2499–507.
Parikh A, Leach T, Wyant T, Scholz C, Sankoh S, Mould DR, et al. Vedolizumab for the treatment of active ulcerative colitis: a randomized controlled phase 2 dose-ranging study. Inflamm Bowel Dis. 2012;18(8):1470–9.
Parikh A, Fox I, Leach T, Xu J, Scholz C, Patella M, et al. Long-term clinical experience with vedolizumab in patients with inflammatory bowel disease. Inflamm Bowel Dis. 2013;19(8):1691–9.
Feagan BG, Rutgeerts P, Sands BE, Hanauer S, Colombel JF, Sandborn WJ, et al. Vedolizumab as induction and maintenance therapy for ulcerative colitis. N Engl J Med. 2013;369(8):699–710.
Feagan BG, Rubin DT, Danese S, Vermeire S, Abhyankar B, Sankoh S, et al. Efficacy of vedolizumab induction and maintenance therapy in patients with ulcerative colitis, regardless of prior exposure to tumor necrosis factor antagonists. Clin Gastroenterol Hepatol. 2017;15(2):229–39.e5.
Loftus EV, Colombel JF, Feagan BG, Vermeire S, Sandborn WJ, Sands BE, et al. Long-term effectiveness and safety of vedolizumab in patients with ulcerative colitis: 5-year cumulative exposure of GEMINI 1 completers rolling into the GEMINI open-label extension study. Gastroenterology. 2017;152(5):S602.
Mosli MH, MacDonald JK, Bickston SJ, Behm BW, Tsoulis DJ, Cheng J, et al. Vedolizumab for induction and maintenance of remission in ulcerative colitis: a Cochrane systematic review and meta-analysis. Inflamm Bowel Dis. 2015;21(5):1151–9.
Engel T, Ungar B, Yung DE, Ben-Horin S, Eliakim R, Kopylov U. Vedolizumab in IBD-lessons from real-world experience; a systematic review and pooled analysis. J Crohns Colitis. 2018;12(2):245–57.
Allegretti JR, Barnes EL, Stevens B, Storm M, Ananthakrishnan A, Yajnik V, et al. Predictors of clinical response and remission at 1 year among a multicenter cohort of patients with inflammatory bowel disease treated with vedolizumab. Dig Dis Sci. 2017;62(6):1590–6.
Eriksson C, Marsal J, Bergemalm D, Vigren L, Bjork J, Eberhardson M, et al. Long-term effectiveness of vedolizumab in inflammatory bowel disease: a national study based on the Swedish National Quality Registry for Inflammatory Bowel Disease (SWIBREG). Scand J Gastroenterol. 2017;52(6–7):722–9.
Amiot A, Grimaud JC, Peyrin-Biroulet L, Filippi J, Pariente B, Roblin X, et al. Effectiveness and safety of vedolizumab induction therapy for patients with inflammatory bowel disease. Clin Gastroenterol Hepatol. 2016;14(11):1593.e2–1601.e2.
Baumgart DC, Bokemeyer B, Drabik A, Stallmach A, Schreiber S, et al. Vedolizumab induction therapy for inflammatory bowel disease in clinical practice–a nationwide consecutive German cohort study. Aliment Pharmacol Ther. 2016;43(10):1090–102.
Vivio EE, Kanuri N, Gilbertsen JJ, Monroe K, Dey N, Chen CH, et al. Vedolizumab effectiveness and safety over the first year of use in an IBD clinical practice. J Crohns Colitis. 2016;10(4):402–9.
Narula N, Peerani F, Meserve J, Kochhar G, Chaudrey K, Hartke J, et al. Vedolizumab for ulcerative colitis: treatment outcomes from the VICTORY consortium. Am J Gastroenterol. 2018;113(9):1345–54.
Ng SC, Shi HY, Hamidi N, Underwood FE, Tang W, Benchimol EI, et al. Worldwide incidence and prevalence of inflammatory bowel disease in the 21st century: a systematic review of population-based studies. Lancet. 2018;390(10114):2769–78.
Cosnes J, Cattan S, Blain A, Beaugerie L, Carbonnel F, Parc R, et al. Long-term evolution of disease behavior of Crohn’s disease. Inflamm Bowel Dis. 2002;8(4):244–50.
Munkholm P, Langholz E, Davidsen M, Binder V. Disease activity courses in a regional cohort of Crohn’s disease patients. Scand J Gastroenterol. 1995;30(7):699–706.
Baumgart DC, Sandborn WJ. Crohn’s disease. Lancet. 2012;380(9853):1590–605.
Feagan BG, Greenberg GR, Wild G, Fedorak RN, Paré P, McDonald JWD, et al. Treatment of active Crohn’s disease with MLN0002, a humanized antibody to the alpha4beta7 integrin. Clin Gastroenterol Hepatol. 2008;6(12):1370–7.
Sandborn WJ, Feagan BG, Rutgeerts P, Hanauer S, Colombel JF, Sands BE, et al. Vedolizumab as induction and maintenance therapy for Crohn’s disease. N Engl J Med. 2013;369(8):711–21.
Sands BE, Feagan BG, Rutgeerts P, Colombel JF, Sandborn WJ, Sy R, et al. Effects of vedolizumab induction therapy for patients with Crohn’s disease in whom tumor necrosis factor antagonist treatment had failed. Gastroenterology. 2014;147(3):618–627 e3.
Sands BE, Sandborn WJ, Van Assche G, Lukas M, Xu J, James A, et al. Vedolizumab as induction and maintenance therapy for Crohn’s disease in patients naïve to or who have failed tumor necrosis factor antagonist therapy. Inflamm Bowel Dis. 2017;23(1):97–106.
Vermeire S, Loftus EV, Colombel JF, Feagan BG, Sandborn WJ, Sands BE, et al. Long-term effectiveness and safety of vedolizumab in patients with Crohn’s disease: 5-year cumulative exposure of GEMINI 2 completers rolling into the GEMINI open-label extension study. Gastroenterology. 2017;152(5):S601.
Dulai PS, Singh S, Jiang X, Peerani F, Narula N, Chaudrey K, et al. The real-world effectiveness and safety of vedolizumab for moderate-severe Crohn’s disease: results from the US VICTORY consortium. Am J Gastroenterol. 2016;111(8):1147–55.
Kotze PG, Ma C, Almutairdi A, Al-Darmaki A, Devlin SM, Kaplan Gilaad G, et al. Real-world clinical, endoscopic and radiographic efficacy of vedolizumab for the treatment of inflammatory bowel disease. Aliment Pharmacol Ther. 2018;48(6):626–37.
Barre A, Colombel JF, Ungaro R. Review article: predictors of response to vedolizumab and ustekinumab in inflammatory bowel disease. Aliment Pharmacol Ther. 2018;47(7):896–905.
Stallmach A, Langbein C, Atreya R, Bruns T, Dignass A, Ende K, et al. Vedolizumab provides clinical benefit over 1 year in patients with active inflammatory bowel disease: a prospective multicenter observational study. Aliment Pharmacol Ther. 2016;44(11–12):1199–212.
Kopylov U, Ron Y, Avni-Biron I, Koslowsky B, Waterman M, Daher S, et al. Efficacy and safety of vedolizumab for induction of remission in inflammatory bowel disease-the Israeli real-world experience. Inflamm Bowel Dis. 2017;23(3):404–8.
Shelton E, Allegretti JR, Stevens B, Lucci M, Khalili H, Nguyen DD, et al. Efficacy of vedolizumab as induction therapy in refractory IBD patients: a multicenter cohort. Inflamm Bowel Dis. 2015;21(12):2879–85.
Feagan BG, Lasch K, Lissoos T, Cao C, Wojtowicz AM, Khalid JM, et al. Rapid response to vedolizumab therapy in biologic-naïve patients with inflammatory bowel diseases. Clin Gastroenterol Hepatol. 2019;17(1):130–138.e7.
Jairath V, Khanna R, Zou GY, Stitt L, Mosli M, Vandervoort MK, et al. Development of interim patient-reported outcome measures for the assessment of ulcerative colitis disease activity in clinical trials. Aliment Pharmacol Ther. 2015;42(10):1200–10.
Khanna R, Zou G, D’Haens G, Feagan BG, Sandborn WJ, Vandervoort MK, et al. A retrospective analysis: the development of patient reported outcome measures for the assessment of Crohn’s disease activity. Aliment Pharmacol Ther. 2015;41(1):77–86.
Feagan BG, Schwartz D, Danese S, Rubin DT, Lissoos TW, Xu J, et al. Efficacy of vedolizumab in fistulising Crohn’s disease: exploratory analyses of data from GEMINI 2. J Crohns Colitis. 2018;12(5):621–6.
Tadbiri S, Grimaud J-C, Peyrin-Biroulet L, Filippi J, Pariente B, Roblin X, et al. Efficacy of vedolizumab on extraintestinal manifestation in patients with inflammatory bowel diseases: a post hoc analysis of the observ-IBD cohort of the Getaid. Gastroenterology. 2017;152(5):S396.
Feagan BG, Sandborn WJ, Colombel JF, O’Byrne S, Khalid JM, Kempf C, et al. Incidence of arthritis/arthralgia in inflammatory bowel disease with long-term vedolizumab treatment: post hoc analyses of the GEMINI trials. J Crohns Colitis. 2019;13(1):50–7.
Dubinsky MC, Cross RK, Sandborn WJ, Long M, Song X, Shi N, et al. Extraintestinal manifestations in vedolizumab and anti-TNF-treated patients with inflammatory bowel disease. Inflamm Bowel Dis. 2018. https://doi.org/10.1093/ibd/izy065 (Epub 13 Apr 2018).
Fleisher M, Marsal J, Lee SD, Frado LE, Parian A, Korelitz BI, et al. Effects of Vedolizumab therapy on extraintestinal manifestations in inflammatory bowel disease. Dig Dis Sci. 2018;63(4):825–33.
Bloomgren G, Richman S, Hotermans C, Subramanyam M, Goelz S, Natarajan A, et al. Risk of natalizumab-associated progressive multifocal leukoencephalopathy. N Engl J Med. 2012;366(20):1870–80.
Van Assche G, Van Ranst M, Sciot R, Dubois B, Vermeire S, Noman M, et al. Progressive multifocal leukoencephalopathy after natalizumab therapy for Crohn’s disease. N Engl J Med. 2005;353(4):362–8.
Haanstra KG, Hofman SO, Estêvão DML, Blezer EL, Bauer J, Yang L-L, et al. Antagonizing the α4β1 integrin, but not α4β7, inhibits leukocytic infiltration of the central nervous system in rhesus monkey experimental autoimmune encephalomyelitis. J Immunol. 2013;190(5):1961–73.
Fuchs F, Schillinger D, Atreya R, Hirschmann S, Fischer S, Neufert C, et al. clinical response to vedolizumab in ulcerative colitis patients is associated with changes in integrin expression profiles. Front Immunol. 2017;8:764.
Colombel JF, Sands BE, Rutgeerts P, Sandborn W, Danese S, D’Haens G, et al. The safety of vedolizumab for ulcerative colitis and Crohn’s disease. Gut. 2017;66(5):839–51.
Bye WA, Jairath V, Travis SPL. Systematic review: the safety of vedolizumab for the treatment of inflammatory bowel disease. Aliment Pharmacol Ther. 2017;46(1):3–15.
Ladd AHM, Scott FI, Grace R, Bownik H, Lichtenstein GR. Safety of vedolizumab in inflammatory bowel disease patients: real world experience from a large university practice. Gastroenterology. 2016;150(4):S977.
Kotze P, Mckenna N, Ma C, Almutairdi A, Raffals L, Loftus E Jr, et al. Vedolizumab and early postoperative complications in non-intestinal surgery: a case-matched analysis. J Crohns Colitis. 2018;12(Suppl 1):S290.
Lightner AL, McKenna NP, Moncrief S, Pemberton JH, Raffals LE, Mathis KL. Surgical outcomes in vedolizumab-treated patients with ulcerative colitis. Inflamm Bowel Dis. 2017;23(12):2197–201.
Lightner A, McKenna N, Tse C, Raffals L, Loftus E Jr, Mathis K. Postoperative outcomes in vedolizumab-treated Crohn’s disease patients undergoing major abdominal operations. Aliment Pharmacol Ther. 2018;47(5):573–80.
Lightner AL, Mathis KL, Tse CS, Pemberton JH, Shen B, Kochlar G, et al. Postoperative outcomes in vedolizumab-treated patients undergoing major abdominal operations for inflammatory bowel disease: retrospective multicenter cohort study. Inflamm Bowel Dis. 2018;24(4):871–6.
Law CCY, Narula A, Lightner AL, McKenna NP, Colombel J-F, Narula N. Systematic review and meta-analysis: preoperative vedolizumab treatment and postoperative complications in patients with inflammatory bowel disease. J Crohns Colitis. 2018;12(5):538–45.
Shen B, Blake A, Lasch K, Palo W, Smyth M, Bhayat F. Vedolizumab use in patients with IBD undergoing surgery: a summary from clinical trials and post-marketing experience. Inflamm Bowel Dis. 2017;23(Suppl 1):S47.
Faleck D, Shashi P, Meserve J, Rahal M, Kadire S, Tran G, et al. Comparative effectiveness of vedolizumab and TNF-antagonist therapy in ulcerative colitis: a multicentre consortium propensity score-matched analysis. J Crohns Colitis. 2018;12(Suppl 1):S019.
Bohm M, Sagi SV, Fischer M, Kadire S, Tran G, Rahal M, et al. Comparative effectiveness of vedolizumab and tumour necrosis factor-antagonist therapy in Crohn’s disease: a multicentre consortium propensity score-matched analysis. J Crohns Colitis. 2018;12(Suppl 1):18.
Lukin D, Weiss A, Aniwan S, Kadire S, Tran G, Rahal M, et al. Comparative safety profile of vedolizumab and tumour necrosis factor–antagonist therapy for inflammatory bowel disease: a multicentre consortium propensity score-matched analysis. J Crohns Colitis. 2018;12(Suppl 1):S036.
Singh S, Fumery M, Sandborn WJ, Murad MH. Systematic review with network meta-analysis: first- and second-line pharmacotherapy for moderate-severe ulcerative colitis. Aliment Pharmacol Ther. 2018;47(2):162–75.
Hazlewood GS, Rezaie A, Borman M, Panaccione R, Ghosh S, Seow CH, et al. Comparative effectiveness of immunosuppressants and biologics for inducing and maintaining remission in Crohn’s disease: a network meta-analysis. Gastroenterology. 2015;148(2):344–54.e5.
Hather G, Curtis R, Minda K, Zouraq IA, Khalid JM. Indirect comparison of two novel biologics for the treatment of Crohn’s disease: network-meta analysis of ustekinumab vs vedolizumab. J Crohns Colitis. 2017;11(Suppl 1):S232–3.
Singh S, Fumery M, Dulai P, Murad M, Sandborn W. Comparative efficacy of pharmacological agents for moderate-severe Crohn’s disease in patients with prior exposure to anti-TNF agents: a network meta-analysis. Inflamm Bowel Dis. 2017;23(Suppl 1):44.
Varu A, Palimaka S, Hutton B, Wilson FR, Dyrda P, Naessens D, et al. Treatment sequence network-meta analysis in Crohn’s disease. Can J Gastroenterol Hepatol. 2018;1(Suppl 1):262–3.
Schneider Y, Saumoy M, Cohen-Mekelburg S, Steinlauf AF, Scherl EJ. The cost-effectiveness of vedolizumab for inflammatory bowel disease: a review of the current literature. Gastroenterol Hepatol. 2016;12(10):617–21.
Schreiber S, Dignass A, Peyrin-Biroulet L, et al. Systematic review with meta-analysis: real-world effectiveness and safety of vedolizumab in patients with inflammatory bowel disease. J Gastroenterol. 2018;53:1048–64.
Chaparro M, Sierra-Ausin M, Mesonero F, Maroto N, Fernandez de Castro C, García-Sánchez V, et al. Effectiveness and safety of vedolizumab for the induction of remission in inflammatory bowel disease. Inflamm Bowel Dis. 2016;10(Suppl 1):S416–7.
Chaudrey K, Whitehead D, Dulai PS, Peerani F, Narula N, Hudesmanet D, et al. Safety of vedolizumab in inflammatory bowel disease in a multi-center real world consortium. Gastroenterology. 2016;150(4 suppl 1):S974.
Christensen B, Goeppinger SR, Colman R, Siddiqui D, Yarur A, Bochenek AA. Post-marketing experience of vedolizumab for IBD: The University of Chicago experience. J Crohns Colitis. 2015;9(Suppl 1):S388–9.
Dulai P, Meserve J, Hartke J, Chilukuri P, Chaudrey K, Koliani-Pace JL, et al. Predictors of clinical and endoscopic response with vedolizumab for the treatment of moderately-severely active ulcerative colitis: results from the US VICTORY consortium. J Crohns Colitis. 2017;11(suppl 1):S40–1.
Hoog C, Eberhardson M, Almer S. P419. Efficacy of vedoli-zumab in patients with inflammatory bowel disease and failure of anti-TNF-antibodies. Presented at: European Crohn’s and Colitis Organisation Congress, March 16–19, 2016, Amsterdam, The Netherlands.
Lenti MV, Levison S, Eliadou E, et al. P525. Effectiveness and safety of vedolizumab in IBD patients: a multicentre experience of ‘‘real world data’’ from the UK. J Crohns Colitis. 2017;11(suppl 1):S347.
Mankongpaisarnrung C, Mattar M, Charabaty A. Single-center experience: vedolizumab in patients with Crohn’s disease and ulcerative colitis at Georgetown University Hospital. Inflamm Bowel Dis. 2016;22(suppl 1):S32.
Pauwels RWM, De Vries AC, Van der Woude CJ. P447. Vedolizumab induces significantly higher endoscopic remission rates at week 16 in ulcerative colitis as compared to Crohn’s disease. J Crohns Colitis. 2017;11(suppl 1):S305.
Samaan MA, Pavlidis P, Johnston E, et al. Vedolizumab: early experience and medium-term outcomes from two UK tertiary IBD centres. Frontline Gastroenterol. 2017;8(3):196–202.
Shivashankar R, Mendoza Ladd AH, Grace R, et al. Effect of vedolizumab dose escalation on recapturing response in patients with inflammatory bowel disease. Gastroenterology. 2017;152(5 suppl 1):S77.
Ungar B, Kopylov U, Waterman M, et al. Early vedolizumab drug levels and induction success in patients with inflammatory bowel disease. United European Gastroenterol J. 2016;4(5 suppl):A3.
Wright AP, Fontana RJ, Stidham RW. Vedolizumab is safe and effective in moderate-to-severe inflammatory bowel disease following liver transplantation. Liver Transpl. 2017;23(7):968–71.
Zezos P, Kabakchiev B, Weizman AV, et al. P502. Ulcerative colitis patients on vedolizumab lacking response at induction phase continue to improve over the first 6 months of treatment. J Crohns Colitis. 2017;11(suppl 1):S334–5.
Abramowitz M, Dale M, Saumoy M, et al. Harvey-Bradshaw index captures clinical efficacy of vedolizumab induction therapy for active Crohn’s disease. Inflamm Bowel Dis. 2016;22(suppl 1):S27.
Blum A, Gregory N, Keyur P, et al. Vedolizumab for the treatment of Crohn’s disease: experience in an inflammatory bowel disease clinical practice. Am J Gastroenterol. 2016;111(suppl 1):S276.
Gils A, Dreesen E, Compernolle G, et al. Vedolizumab exposure correlates with clinical, biological and endoscopic outcome in patients with inflammatory bowel disease. United Eur Gas-troenterol J. 2016;4(5 suppl):A3.
Glover S, Edminster T, Garcia L, et al. Efficacy of vedolizumab in Crohn’s disease after at least 14 weeks of therapy as measured by HBI scores. Am J Gastroenterol. 2015;110(suppl 1):S826.
De Vos M, Dhooghe B, Vermeire S, et al. P0868. Open label observational study evaluating the effect of induction therapy with vedolizumab in patients with moderate to severe inflam-matory bowel disease intolerant/resistant to at least two bio-logicals. United Eur Gastroenterol J. 2016;4(5 suppl):A452.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Funding
This narrative review was not funded by an external source.
Conflict of interest
Robert Battat has no conflicts to declare. Christopher Ma is supported by a Clinician Fellowship from the Canadian Association of Gastroenterology and the Canadian Institutes of Health Research. Vipul Jairath has received consulting fees from AbbVie, Eli Lilly, GlaxoSmithKline, Arena Pharmaceuticals, Genetech, Pendopharm, Sandoz, Merck, Takeda, Janssen, Robarts Clinical Trials, Topivert and Celltrion, and speaker’s fees from Takeda, Janssen, Shire, Ferring, Abbvie and Pfizer. Reena Khanna has received consulting fees from AbbVie, Janssen, Pfizer, Takeda, and Robarts Clinical Trials Inc., and speaker’s fees from AbbVie, Janssen, Shire, and Takeda. Brian G. Feagan has received grant/research support from AbbVie Inc., Amgen Inc., AstraZeneca/MedImmune Ltd, Atlantic Pharmaceuticals Ltd, Boehringer-Ingelheim, Celgene Corporation, Celltech, Genentech Inc./Hoffmann-La Roche Ltd, Gilead Sciences Inc., GlaxoSmithKline (GSK), Janssen Research & Development LLC, Pfizer Inc., Receptos Inc./Celgene International, Sanofi, Santarus Inc., Takeda Development Center Americas Inc., Tillotts Pharma AG and UCB; consulting fees from Abbott/AbbVie, Ablynx, Akebia Therapeutics, Allergan, Amgen, Applied Molecular Transport Inc., Aptevo Therapeutics, Astra Zeneca, Atlantic Pharma, Avir Pharma, Baxter Healthcare Corp., Biogen Idec, Boehringer-Ingelheim, Bristol-Myers Squibb, Calypso Biotech, Celgene, Elan/Biogen, EnGene, Ferring Pharma, Roche/Genentech, Galapagos, GiCare Pharma, Gilead, Given Imaging Inc., GSK, Inception IBD Inc., Ironwood Pharma, Janssen Biotech (Centocor), JnJ/Janssen, Kyowa Kakko Kirin Co Ltd, Lexicon, Lilly, Lycera BioTech, Merck, Mesoblast Pharma, Millennium, Nektar, Nestles, Nextbiotix, Novonordisk, Pfizer, Prometheus Therapeutics and Diagnostics, Progenity, Protagonist, Receptos, Roche/Genentech, Salix Pharma, Serano, Shire, Sigmoid Pharma, Sterna Biologicals, Synergy Pharma Inc., Takeda, Teva Pharma, TiGenix, Tillotts, UCB Pharma, Vertex Pharma, Vivelix Pharma, VHsquared Ltd, Warner-Chilcott, Wyeth, Zealand and Zyngenia; speakers bureau fees from Abbott/AbbVie, JnJ/Janssen, Lilly, Takeda, Tillotts, and UCB Pharma; advisory board fees from Abbott/AbbVie, Allergan, Amgen, Astra Zeneca, Atlantic Pharma, Avaxia Biologics Inc., Boehringer-Ingelheim, Bristol-Myers Squibb, Celgene, Centocor Inc., Elan/Biogen, Ferring, Galapagos, Genentech/Roche, JnJ/Janssen, Merck, Nestles, Novartis, Novonordisk, Pfizer, Prometheus Laboratories, Protagonist, Salix Pharma, Sterna Biologicals, Takeda, Teva, TiGenix, Tillotts Pharma AG and UCB Pharma; and is a consultant (Senior Scientific Officer) for Robarts Clinical Trials, Inc.
Rights and permissions
About this article

Cite this article
Battat, R., Ma, C., Jairath, V. et al. Benefit–Risk Assessment of Vedolizumab in the Treatment of Crohn’s Disease and Ulcerative Colitis. Drug Saf 42, 617–632 (2019). https://doi.org/10.1007/s40264-018-00783-1
Published:
Issue Date:
DOI: https://doi.org/10.1007/s40264-018-00783-1