
Abstract
LINGO-1 is a leucine-rich repeat and Ig domain-containing, Nogo receptor interacting protein, selectively expressed in the CNS on both oligodendrocytes and neurons. Its expression is developmentally regulated, and is upregulated in CNS diseases and injury. In animal models, LINGO-1 expression is upregulated in rat spinal cord injury, experimental autoimmune encephalomyelitis, 6-hydroxydopamine neurotoxic lesions and glaucoma models. In humans, LINGO-1 expression is increased in oligodendrocyte progenitor cells from demyelinated white matter of multiple sclerosis post-mortem samples, and in dopaminergic neurons from Parkinson’s disease brains. LINGO-1 negatively regulates oligodendrocyte differentiation and myelination, neuronal survival and axonal regeneration by activating ras homolog gene family member A (RhoA) and inhibiting protein kinase B (Akt) phosphorylation signalling pathways. Across diverse animal CNS disease models, targeted LINGO-1 inhibition promotes neuron and oligodendrocyte survival, axon regeneration, oligodendrocyte differentiation, remyelination and functional recovery. The targeted inhibition of LINGO-1 function presents a novel therapeutic approach for the treatment of CNS diseases.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
1 Introduction
CNS disorders with neurodegeneration (multiple sclerosis [MS], Parkinson’s disease [PD], glaucoma) are diseases characterized by progressive loss of function, neuronal cell death and disability [1–4]. Although there are many different causes for neuronal cell loss, the study and identification of outcome similarities at a molecular level could lead to CNS regeneration therapeutics that are effective against multiple neurodegenerative indications.
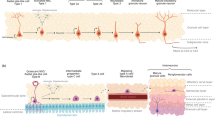
Therapeutic targets for the treatment of neural dysfunction include those that have restricted tissue distribution and critical roles in the establishment or maintenance of function of neural circuitry. Certain leucine-rich repeat (LRR) proteins play key roles in CNS biology, making them attractive development targets for the treatment of CNS disorders [5–7]. Examples include secreted ligand for the transmembrane protein (SLIT) and amphoterin-induced gene and ORF (AMIGO) that function in axon guidance, and Nogo-66 receptor (NgR1) and oligodendrocyte-myelin glycoprotein (OMgp) that regulate axon regeneration [6–9]. An effort to identify novel CNS-specific LRR proteins led to the discovery of LINGO-1 [10]. LINGO-1 is specifically expressed on oligodendrocytes and neurons of the CNS and is a potent negative regulator of oligodendrocyte differentiation, neuronal survival and axon regeneration [10, 11]. LINGO-1 expression is upregulated during CNS injury across diverse animal models and in human CNS diseases [12, 13]. The underlying mechanisms leading to the elevated LINGO-1 levels during injury and CNS diseases are unknown. LINGO-1 forms a complex with growth factor receptors to block their functions [10, 14, 15]. To date, four pathways have been elucidated (Fig. 1). In dopaminergic neurons, LINGO-1 binds to epidermal growth factor receptor (EGFR), and in retinal ganglion cells, it binds to TrkB to block survival pathways [14, 15]. In oligodendrocytes, it blocks differentiation by forming a receptor complex with erythroblastic leukaemia viral oncogene homolog 2 (ErB2) (unpublished data). LINGO-1 binds to the NgR1 complex in neurons and axons to inhibit axon regeneration by activating the RhoA pathway [10]. Many LINGO-1 antagonist modalities have been used to assess its activity. Attenuation of LINGO-1 function in vitro using interfering RNA (RNAi) to decrease LINGO-1 messenger RNA level [10, 11], dominant negative LINGO-1 with intact extracellular and transmembrane domains but lacking its cytoplasmic tail to disrupt downstream signalling [10, 11, 16], soluble LINGO-1 containing only its extracellular domain to act as a receptor antagonist [10, 11, 13, 14, 16, 17] and neutralizing anti-LINGO-1 antibodies [12–18] all promote oligodendrocyte differentiation, neuronal survival and axon regeneration. Antibodies that block LINGO-1 function in vitro promote remyelination, neuronal survival/axon regeneration and functional recovery in animal models of CNS diseases [12, 14, 15, 17–21]. Key aspects of LINGO-1 biology have also been recapitulated using LINGO-1 null mice [12, 14]. Together these studies show that LINGO-1 is an important component in pathways leading to neuronal survival and regeneration, and blocking LINGO-1 function could be an approach for developing therapeutics across diverse CNS diseases.

LINGO-1 signalling complexes. LINGO-1 binds to NgR, TrkB, EGFR and ErB2 receptors in the CNS and negatively regulates downstream signalling molecules involved in axon regeneration, neuronal survival and oligodendrocyte differentiation and myelination. BDNF brain-derived neurotrophic factor, EGFR epidermal growth factor receptor, ErB2 erythroblastic leukaemia viral oncogene homolog 2, NgR nogo receptor, P75 P75 neurotrophin receptor, RGC retinal ganglion cells, TrkB tyrosine kinase receptor B, Troy tnfrsf 19
2 LINGO-1 is a CNS-Specific Membrane-Associated Glycoprotein
LINGO-1 is a 614 amino acid protein encoded on chromosome 15 (15q24.3 GI:15029689) and selectively expressed in the CNS. It is a member of a new protein family that in humans comprises LINGO-1 and its three paralogs: LINGO-2 (GI:12309630, 61 % protein identity), LINGO-3 (GI:23342615, 56 % identity) and LINGO-4 (GI: 21211752, 44 % identity). LINGO-1 contains 12 LRR motifs flanked by N- and C-terminal capping domains, one Ig domain, a transmembrane domain and a short cytoplasmic tail. The cytoplasmic tail contains a canonical EGFR-like tyrosine phosphorylation site (residue 591). LINGO-1 is highly conserved evolutionarily, with human and mouse orthologues sharing 99.5 % identity. By Northern blot analysis, LINGO-1 is expressed in the brain and spinal cord, and is not detectable in non-neural tissues [10]. LINGO-1 expression occurs in all regions of the rat brain examined [10, 16, 22–24]. A rostral to caudal gradient of LINGO-1 expression was observed in the adult CNS, with highest levels found in the cortex and lowest levels in the spinal cord [10]. Taqman reverse transcription polymerase chain reaction (RT-PCR) quantitation of LINGO-1 expression from embryonic and postnatal rat brains showed expression levels peaking around postnatal day 1 (P1) and decreasing thereafter into adulthood [10]. Figure 2 shows the expression of LINGO-1 in adult rat tissues by Western blot analysis. Consistent with the Northern blot data, LINGO-1 is expressed in the brain and spinal cord, but not in other tissues. The expression of LINGO-1 in the brain was further localized by in situ hybridization, immunohistochemistry and RT-PCR. LINGO-1 is expressed in neurons and oligodendrocytes as is evident from co-localization of LINGO-1 and βIII tubulin, a specific marker of neurons, and O4, a specific marker of oligodendrocytes [10, 11, 16]. The high specificity to the CNS, developmental regulation and induction following injury and CNS diseases [20, 21] suggests important roles for LINGO-1 in regulating neuronal survival, axon regeneration and timing of oligodendrocyte and neuron interactions for myelination during development and disease.

Western blot of LINGO-1 expression in rat tissue lysates following immunoprecipitation and detection with anti-LINGO-1 monoclonal antibodies
3 Blocking LINGO-1 as a Treatment for Multiple Sclerosis
MS is an inflammatory demyelination disease of the CNS that is progressive, chronic and heterogeneous. MS is the most frequently occurring, non-traumatic neurological disease affecting young adults [25, 26]. Autoimmune mechanisms are believed to play a critical role in MS disease pathogenesis and all susceptibility genes identified to date are related to immune function rather than neurodegeneration [27]. The disease has a complex and varied pathology, but is typically characterized by acute, multi-focal, CNS autoimmune-mediated demyelination, oligodendrocyte injury and axonal loss [25, 26]. In many cases, demyelination is followed by spontaneous remyelination, a repair process in which new myelin sheaths are formed [28]. For the majority of patients, this recovery process ultimately fails, and a persistent demyelination with a subsequent loss of axons results in progressive and irreversible functional deficits and neurological disability. Currently approved and available MS treatments only modulate inflammatory components of the disease. No treatments target the regenerative process of remyelination and axonal protection.
The reasons for remyelination failure in MS are poorly understood [28–30]. The identification of undifferentiated oligodendrocyte progenitor cells (OPC) within demyelinated lesions or plaques suggests that inhibitors such as myelin debris, chondroitin sulfate proteoglycans, dysregulated Wnt signalling pathway, Notch or hyaluronan may prevent OPC differentiation and remyelination [31–36]. The OPC differentiation inhibitor, LINGO-1, is also expressed in the OPC of demyelinated MS lesions [11, 13].
The negative role of LINGO-1 in oligodendrocyte differentiation was studied by four different approaches to reduce endogenous LINGO-1 function: LINGO-1 RNAi, dominant-negative LINGO-1, soluble LINGO-1, and neutralizing anti-LINGO-1 monoclonal antibody [11]. All four approaches resulted in morphological changes characteristic of more highly differentiated mature oligodendrocytes such as increased process lengths and branching, and formation of abundant myelin sheet structures. These changes were accompanied by biochemical indicators of OPC maturation such as increased myelin basic protein (MBP) expression, a marker for oligodendrocyte differentiation. Dramatic morphological changes also seen on human OPCs following anti-LINGO-1 antibody treatment are shown in Fig. 3a. By contrast, over-expression of full-length LINGO-1 (FL-LINGO-1) inhibits OPC differentiation [16]. Consistent with the hypothesis that LINGO-1 is a negative regulator of oligodendrocyte differentiation, cultured OPCs from LINGO-1 knockout (KO) mice differentiated more rapidly than cells from wild-type (WT) littermates (Fig. 3b) [11]. Regulating the differentiation of oligodendrocytes is likely to have significant impact on myelination, and the effects of LINGO-1 antagonists on oligodendrocyte differentiation suggest its blockade might facilitate CNS myelination. This concept has been tested in vitro using oligodendrocyte/neuron co-cultures. In co-cultures of rat primary oligodendrocytes and DRG neurons, in the presence of nerve growth factor (NGF), very limited myelination was observed, consistent with findings from other laboratories [16, 37]. By contrast, addition of soluble LINGO-1, dominant negative LINGO-1 or anti-LINGO-1 antibody to the NGF-containing medium promoted myelination in a dose-dependent manner [16], which correlated with increased expression of myelin-associated glycoprotein (MAG), a myelin protein expressed early in myelination, and MBP, the major protein component of myelin. By contrast, over-expression of FL-LINGO-1 decreased the number of MBP+ cells [11]. Ultrastructure studies revealed multiple, well-formed internodes and structures that resembled nodes of Ranvier in soluble LINGO-1-treated cultures [11]. The negative role of LINGO-1 in oligodendrocyte differentiation and myelination was further verified using LINGO-1 KO mice. LINGO-1 KO mice showed early-onset CNS myelination during postnatal days 5–15, then reaching the same level of myelination as WT mice at adulthood [11]. No effects on PNS myelination were detected in the KO mice. These data support the notion that endogenous LINGO-1 negatively regulates oligodendrocyte differentiation and myelination.

LINGO-1 antagonists promote oligodendrocyte differentiation detected with anti-MBP staining. a Human OPC treated with an anti-LINGO-1 antibody or an isotype control antibody. b OPC from wild-type (WT) or LINGO-1 knockout (KO) mice. MBP myelin basic protein, OPC oligodendrocyte progenitor cell
The ability of LINGO-1 antagonists to promote remyelination was tested in lysolecithin (LPC)-induced and cuprizone demyelination models [13]. In the LPC model, demyelination occurs within 1 day of LPC treatment. LPC was injected into the dorsal columns of adult rats on day 0 and intraperitoneal administration of anti-LINGO-1 antibody occurred on day 2 (3 mg/kg every third day for an additional 7 days). Remyelination was determined by Luxol fast blue (LFB) staining of lesion tissue sections; myelinated white matter appeared dark blue and demyelinated lesions were pale blue or white. Sections from control antibody-treated animals showed large lesions with extensive areas of demyelination, whereas much smaller lesions were apparent in the anti-LINGO-1 antibody-treated group, 9 days after LPC injection (Fig. 4). Consistent with the LFB staining, anti-LINGO-1 antibody-treated animals also showed increased MBP staining in the lesions with morphology typical of myelinated axons when compared with controls. To determine whether the differences in LFB staining represented enhanced remyelination, anti-LINGO-1 antibody and control treated lesions were examined by light and electron microscopy. Quantitation of putative myelinated axons in 1-μm sections stained with toluidine blue demonstrated a threefold increase in myelinated fibres in anti-LINGO-1 antibody-treated animals compared with controls. Electron microscopy confirmed a threefold increase in myelinated axons from anti-LINGO-1 antibody-treated animals compared with control-treated animals [13]. With the goal of developing a non-invasive method of analysis that could be applied to humans, remyelination was also examined by magnetization transfer ratio (MTR), a non-conventional MRI technology. In these studies, LPC was stereotactically injected into the corpus callosum in rat brains to induce demyelination and anti-LINGO-1 antibody was administered by intraperitoneal delivery 2 days after LPC treatment. A significantly higher MTR signal was obtained in the anti-LINGO-1 antibody treatment group, which correlated with increased LFB staining for remyelination in corresponding tissue sections from the same brains (Fig. 5) [38]. The cuprizone CNS demyelination model is an alternative to the LPC model. Blocking LINGO-1 with anti-LINGO-1 antibody treatment similarly led to an increase in remyelination in the brain of mice that were fed with cuprizone [13].

LINGO-1 antagonist treatment promotes remyelination. Remyelination by an anti-LINGO-1 antibody and isotype control were evaluated histochemically in a rat LPC-induced spinal cord demyelination model following systemic administration. Top panel LFB staining for detecting myelination in the LPC lesion. Bottom panel MBP immunohistochemical staining remyelinated axons in LPC lesion. LFB luxol fast blue, LPC lysolecithin, MBP myelin basic protein

Anti-LINGO-1 antibody treatment promotes remyelination. Remyelination by an anti-LINGO-1 antibody and isotype control were evaluated using MTR imaging in a rat LPC-induced brain demyelination model following systemic administration. Arrow in top panel denotes the higher MTR signal in an anti-LINGO-1 antibody-treated brain lesion. Arrow in bottom panel denotes the stronger LFB staining in a rat corpus callosum lesion following anti-LINGO-1 antibody treatment. LFB luxol fast blue, LPC lysolecithin, MTR magnetization transfer ratio
Experimental autoimmune encephalomyelitis (EAE) is a widely accepted model for studying the clinical and pathological features of MS. The clinical manifestations of myelin oligodendrocyte glycoprotein (MOG)-induced EAE, mirroring MS, comprise an autoimmune inflammatory component and a neurological component, including demyelination and axonal loss [39]. Two approaches were used to demonstrate that blocking LINGO-1 promoted remyelination in EAE [12]. First, LINGO-1 knockout mice exhibit greater resistance to the development of MOG-induced EAE. EAE scores, which quantify the disease progression by measuring motor dysfunction, were used to monitor the functional improvement. Both WT and LINGO-1 KO mice developed motor impairment; however, EAE scores were significantly lower in LINGO-1 KO mice [12]. Damaged myelin sheaths, often displaying loose and separated layers and degraded sheath structures were present in the WT animals [12]. In contrast, LINGO-1 KO EAE animals showed an abundance of newly formed myelin sheaths that were notably thinner (Fig. 6). Twofold more myelinated axons were present in LINGO-1 KO EAE mice compared with their WT counterparts. In a second approach, an anti-LINGO-1 antibody was tested in the rat MOG-EAE model, a more clinically relevant assessment of the therapeutic potential of LINGO-1 antagonism. 15 days post-MOG immunization, rats with EAE scores of 1.0 were treated either with an anti-LINGO-1 antibody or an isotype-matched antibody delivered systemically by intraperitoneal injections. Similar to the studies with the LINGO-1 KO mice, the anti-LINGO-1 antibody significantly mitigated disease severity across all stages of disease progression based on rat EAE scores, when compared with an isotype antibody control or a no-treatment control. After a 2-week treatment, the anti-LINGO-1 antibody group showed significantly lower EAE scores than the control group. Most impressively, disease progression appeared stabilized with a perceptible downtrend [12]. The functional improvement correlated with increased axonal remyelination in anti-LINGO-1 antibody-treated rats when compared with control animals. Animals treated with a control antibody showed severe white matter demyelination with a predominance of naked axons while anti-LINGO-1 antibody-treated animals showed significantly increased numbers of thinly myelinated axons in the demyelinated area, suggesting that anti-LINGO-1 antibody treatment promoted axonal remyelination. Diffusion tensor imaging (DTI) was used to assess the integrity of myelinated axons in the EAE spinal cords. The F value (DTI signal) from the anti-LINGO-1 antibody-treated group was significantly higher compared with the control group and correlated well with the remyelination area seen in toluidine blue-stained sections [12]. Axonal integrity was also confirmed by immunohistochemistry staining for β amyloid precursor protein (APP), a marker for axonal damage elevated in MS lesions. Fewer APP-positive axons were observed in the white matter of the anti-LINGO-1 antibody-treated EAE spinal cords compared with the controls [12]. These studies suggest that antagonism of LINGO-1 is a viable approach for the treatment of CNS demyelinating diseases, such as MS.

Electron microscopy to visualize remyelinated axons in EAE spinal cords from WT and KO LINGO-1 mice (red arrows denote some remyelinated axons). Scale bar 5μm. EAE experimental autoimmune encephalomyelitis, KO knockout, WT wild-type
The pharmacokinetic/pharmacodynamic relationships following treatment with anti-LINGO-1 antibody were investigated in normal and MOG-induced EAE rats by measuring antibody levels in blood and the CNS and correlating these with dose-efficacy responses [40]. Antibody levels in the brain and spinal cord followed an exponential disposition that paralleled levels in blood, with calculated area under the curve values of 0.15 % of blood levels. CNS antibody levels in normal and MOG-EAE animals were similar. To correlate antibody levels that promoted remyelination in the LPC model with receptor occupancy due to LINGO-1 binding, the mg/kg doses were converted to levels of antibody in the spinal cord and these data were directly compared with in vitro LINGO-1 binding data. The overlay of the binding and efficacy data sets revealed a remarkable correlation between binding and extent of remyelination. The direct dependence of dose on the extent of remyelination in the LPC model provides clear evidence that remyelination is directly linked to binding.
4 Blocking LINGO-1 Promotes Axonal Regeneration and Functional Recovery Following Spinal Cord Injury
LINGO-1 is a component of the NgR1 complex involved in RhoA activation and axon degeneration. The potential roles of LINGO-1 were first revealed by demonstrating its physical interaction with the NgR1 receptor complex, using either protein interaction or cell-binding assays [10]. NgR1 forms a receptor complex with LINGO-1 and P75/Troy that activates RhoA and blocks neurite outgrowth upon binding to inhibitory molecules such as MAG, OMgp and NogoA [9, 41–43]. Blocking the LINGO-1/NgR1 interaction by soluble LINGO-1 or dominant-negative LINGO-1 promoted neurite outgrowth and RhoA inactivation in vitro. To explore the physiological role of LINGO-1 in vivo, soluble LINGO-1 was tested for its ability to promote axon regeneration after spinal cord injury (SCI) induced by dorsal hemi section at thoracic vertebra 6/7. The dorsal and dorsolateral components of the corticospinal tract (CST) were completely interrupted, with the ventral portion of the CST left intact. The day after SCI, all animals exhibited near complete hind limb paralysis. Continuous intrathecal infusion of soluble LINGO-1 for 4 weeks after spinal cord transection resulted in 60 % of treated animals showing significant improvements in mobility [19]. To determine if treatment with soluble LINGO-1 improved axonal regeneration, 2 weeks prior to termination of the study, CST axons were anterogradely traced down the spinal cord by injecting biotin dextran amine (BDA) into the sensory-motor cortex to label regenerated axons. Significantly more BDA labelled axons were observed in soluble LINGO-1-treated rats compared with controls. The number of BDA-labelled regenerated axons that physically contacted motor neurons in the lumbar motor neuron pool, approximately 15 mm caudal to the site of transection, was greater in soluble LINGO-1-treated animals than in controls [19]. Degenerated axons were quantified after spinal cord transection as the mean distance between the closest BDA-labelled axon terminal and the lesion edge [41]. Impressively, axon retraction was reduced by approximately 50 % in soluble LINGO-1-treated animals and this was directly correlated with reduced RhoA activation [19]. In addition, significantly more surviving neurons and oligodendrocytes were observed in the lesion sites of spinal cord tissue in soluble LINGO-1-treated animals compared with controls [19]. Treatment with soluble LINGO-1 therefore improved hind limb function, and this correlated directly with enhanced axonal regeneration, less axon retraction, reduced RhoA activation, and increased neuron and oligodendrocyte survival adjacent to the lesion. In a similar study, a neutralizing LINGO-1 antibody also significantly decreased RhoA activation and increased neuronal survival, with recovery of certain hind limb motor functions [44]. These studies suggest that antagonism of LINGO-1 is a viable approach for the treatment of SCI.
5 Blocking LINGO-1 Promotes Neuronal Survival in Parkinson’s Disease Models
PD is the second most prevalent adult neurodegenerative disorder and is characterized by differential degeneration of dopamine (DA)-producing cells in the substantia nigra and other midbrain structures. LINGO-1 is expressed in DA neurons and it is upregulated in PD [14]. The role of LINGO-1 in the survival of dopaminergic neurons has been assessed in cell culture and animal models [14]. Blocking LINGO-1 function by dominant-negative LINGO-1, soluble LINGO-1 or anti-LINGO-1 antibody led to increases in DA neurite length, and survival in a MPP+ neurotoxicity paradigm in vitro. Increased DA neuron survival and neurite outgrowth by LINGO-1 antagonism were also observed in the 6-hydroxydopamine-induced cell degeneration model in vivo. Consistent with the LINGO-1 antagonist studies, increased DA neuron survival and reduced motor asymmetry were observed in LINGO-1 KO mice in a progressive 6-hydroxydopamine model [14].
The mechanism by which LINGO-1 affects DA neuron survival is being elucidated. In animal models of PD, upregulation of LINGO-1 coincided with decreased EGFR protein levels, suggesting that endogenous LINGO-1 may negatively regulate the EGFR/Akt signalling pathway [14]. EGFR treatment protected DA neurons in primary ventral motor (VM) cultures and animal models of PD [14, 45–48]. Intracellular signalling of EGFR is mediated by the phosphoinositide 3-kinase (PI3 kinase) pathway [14], which increases phosphorylation and activation of Akt [49]. Growth factor-activated PI3-kinase/Akt signalling pathways enhance neuronal survival and axonal regeneration. Consistent with the functional link between LINGO-1 and EGFR signalling, blocking LINGO-1 function increased EGFR and phosphorylated-Akt levels in the DA neurons coincident with its ability to protect DA neurons from injury-induced cellular damage [14]. The clear effects of LINGO-1 antagonists on DA neuron survival in vitro and in vivo suggest that blocking LINGO-1 function could provide therapeutic benefit for the treatment of PD.
6 Blocking LINGO-1 Promotes Neuronal Survival in Glaucoma Disease Models
Glaucoma is a neurodegenerative disease characterized by structural damage to the optic nerve and slow progressive death of retinal ganglion cells (RGCs) [50]. Glaucomatous damage leads to an elevation of intraocular pressure (IOP). Current therapies, surgery or medications lower the IOP to delay disease progression, but do not prevent RGC loss and axon degeneration. Therefore promoting RGC survival is a great unmet need for the treatment of glaucoma.
LINGO-1 is expressed and upregulated in injured RGCs where it complexes with and negatively regulates TrkB activation, a brain-derived neurotrophic factor (BDNF) receptor [15, 17, 18]. Anti-LINGO-1 antibody and soluble LINGO-1 treatment promoted TrkB phosphorylation by BDNF and significantly reduced RGC loss when administered intravitreously in a rat experimental ocular hypertension model [15, 17, 18]. Interestingly, BDNF-alone treatment had only a modest neuroprotective effect; while combination treatment with BDNF and soluble LINGO-1 significantly prevented RGC death [17]. These results indicate that LINGO-1 antagonists could protect RGCs from degeneration and death that occur in glaucoma.
7 Status of the Anti-LINGO-1 Antibody (BIIB033) Clinical Trial
BIIB033 is the first anti-LINGO-1 antibody to enter clinical development. It is a fully human, IgG1 monoclonal antibody that binds human LINGO-1 with high affinity and specificity. BIIB033 has been engineered to have reduced Fc gamma (Fcγ) and complement effector functions compared with WT IgG1. A phase I, single ascending-dose study in healthy human volunteers and a phase I multiple-dose study in subjects with relapsing remitting or secondary progressive MS have been completed. These studies examined the safety, tolerability and pharmacodynamic properties of BIIB033 (ClinicalTrials.gov Identifier NCT01244139). The following is a summary of the key findings from the two phase I studies: [1] following a single intravenous and subcutaneous dose in healthy volunteers and two repeated intravenous doses up to 100 mg/kg in MS subjects, BIIB033 appeared to be safe and well tolerated; [2] BIIB033 pharmacokinetics was linear with small volume of distribution, minimal target-mediated clearance and a terminal elimination half-life of approximately 2–3 weeks; [3] although brain penetration of BIIB033 in humans is expected to be low due to the blood–brain barrier, intravenous infusion of doses ≥3 mg/kg or ≥10 mg/kg in MS subjects resulted in BIIB033 concentrations in CSF comparable to the half maximal effective concentration or greater or ≥90 % effective concentration, respectively, based on dose-efficacy data in the rat LPC demyelination spinal cord model [51].
Phase II studies for BIIB033 are planned for 2013. As the first drug candidate to enter the clinic for CNS repair, BIIB033 presents several drug development challenges. These include the need to further evaluate its safety, tolerability and efficacy using clinical endpoints (Table 1) and non-invasive methods to assess two potentially beneficial mechanisms: (i) CNS remyelination and (ii) axonal protection/repair. Although several non-invasive techniques (evoked potentials, MTR, diffusion tensor imaging, myelin water imaging, optical coherence tomography) are available for the measurement of CNS protection and remyelination/repair, their sensitivity and specificity to these processes remain incompletely understood. Much work remains to better understand these various potential clinical measurements for CNS remyelination and protection/repair. There is also a great need for developing biomarkers to identify responders and non-responders early into the treatment. The lessons we learn in evaluating efficacy readouts for BIIB033 should provide valuable insights in the design of trials for new drug candidates that target CNS remyelination and repair.
8 Conclusion
Many CNS diseases including MS, PD, glaucoma and SCI are CNS neurodegeneration disorders with adverse consequences that include neuronal death, axon degeneration, gliosis and demyelination. Regeneration and repair of CNS damage is complex, involving multiple cell types, inflammatory and neurological components, and the presence of inhibitory factors. It is reasonable to assume that treatments that reduce neuronal degeneration and maintain or restore neuronal pathways and physiological circuits in CNS neurodegenerative diseases would be of therapeutic benefit. Neurotrophins and growth factors such as BDNF, NGF, ciliary neurotrophic factor, platelet-derived growth factor and glioma-derived growth factor have been clinically tested without success due to their lack of tissue specificity and associated non-target tissue toxicity [52–61]. Identifying the inhibitors of regeneration and blocking their function comprises a relatively unexplored strategy for CNS regeneration. LINGO-1 is a particularly compelling target because it is a CNS-specific protein and has negative regulatory functions in axonal regeneration, neuronal survival, and oligodendrocyte differentiation and myelination. The elevated levels of LINGO-1 in neuropathologies such as in rat models of spinal cord injury, EAE and glaucoma and in human MS and PD suggest that LINGO-1 and associated pathways may inhibit functional repair in these conditions. Preclinical experiments with LINGO-1 antagonists have validated the hypothesis that antagonism of LINGO-1 is a viable approach for the treatment of neurodegenerative diseases. The ongoing clinical studies with BIIB033 should, for the first time, allow evaluation of the impact of drugs that target CNS repair on MS.
References
Meissner WG, Frasier M, Gasser T, et al. Priorities in Parkinson’s disease research. Nat Rev Drug Discov. 2011;10(5):377–93.
Miller RG, Mitchell JD, Moore DH. Riluzole for amyotrophic lateral sclerosis (ALS)/motor neuron disease (MND). Cochrane Database Syst Rev. 2012;(3):CD001447.
Schmier JK, Halpern MT, Jones ML. The economic implications of glaucoma: a literature review. Pharmacoeconomics. 2007;25(4):287–308.
Dutta R, Trapp BD. Mechanisms of neuronal dysfunction and degeneration in multiple sclerosis. Prog Neurobiol. 2011;93(1):1–12.
Chen Y, Aulia S, Li L, et al. AMIGO and friends: an emerging family of brain-enriched, neuronal growth modulating, type I transmembrane proteins with leucine-rich repeats (LRR) and cell adhesion molecule motifs. Brain Res Rev. 2006;51(2):265–74.
Kuja-Panula J, Kiiltomaki M, Yamashiro T, et al. AMIGO, a transmembrane protein implicated in axon tract development, defines a novel protein family with leucine-rich repeats. J Cell Biol. 2003;160(6):963–73.
Nguyen-Ba-Charvet KT, Picard-Riera N, Tessier-Lavigne M, et al. Multiple roles for slits in the control of cell migration in the rostral migratory stream. J Neurosci. 2004;24(6):1497–506.
Fournier AE, GrandPre T, Strittmatter SM. Identification of a receptor mediating Nogo-66 inhibition of axonal regeneration. Nature. 2001;409(6818):341–6.
McGee AW, Strittmatter SM. The Nogo-66 receptor: focusing myelin inhibition of axon regeneration. Trends Neurosci. 2003;26(4):193–8.
Mi S, Lee X, Shao Z, et al. LINGO-1 is a component of the Nogo-66 receptor/p75 signaling complex. Nat Neurosci. 2004;7(3):221–8.
Mi S, Miller RH, Lee X, et al. LINGO-1 negatively regulates myelination by oligodendrocytes. Nat Neurosci. 2005;8(6):745–51.
Mi S, Hu B, Hahm K, et al. LINGO-1 antagonist promotes spinal cord remyelination and axonal integrity in MOG-induced experimental autoimmune encephalomyelitis. Nat Med. 2007;13(10):1228–33.
Mi S, Miller RH, Tang W, et al. Promotion of central nervous system remyelination by induced differentiation of oligodendrocyte precursor cells. Ann Neurol. 2009;65(3):304–15.
Inoue H, Lin L, Lee X, et al. Inhibition of the leucine-rich repeat protein LINGO-1 enhances survival, structure, and function of dopaminergic neurons in Parkinson’s disease models. Proc Natl Acad Sci USA. 2007;104(36):14430–5.
Fu QL, Hu B, Wu W, et al. Blocking LINGO-1 function promotes retinal ganglion cell survival following ocular hypertension and optic nerve transection. Invest Ophthalmol Vis Sci. 2008;49(3):975–85.
Lee X, Yang Z, Shao Z, et al. NGF regulates the expression of axonal LINGO-1 to inhibit oligodendrocyte differentiation and myelination. J Neurosci. 2007;27(1):220–5.
Fu QL, Hu B, Li X, et al. LINGO-1 negatively regulates TrkB phosphorylation after ocular hypertension. Eur J Neurosci. 2010;31(6):1091–7.
Fu QL, Li X, Yip HK, et al. Combined effect of brain-derived neurotrophic factor and LINGO-1 fusion protein on long-term survival of retinal ganglion cells in chronic glaucoma. Neuroscience. 2009;162(2):375–82.
Ji B, Li M, Wu WT, et al. LINGO-1 antagonist promotes functional recovery and axonal sprouting after spinal cord injury. Mol Cell Neurosci. 2006;33(3):311–20.
Mi S, Sandrock A, Miller RH. LINGO-1 and its role in CNS repair. Int J Biochem Cell Biol. 2008;40(10):1971–8.
Rudick RA, Mi S, Sandrock AW Jr. LINGO-1 antagonists as therapy for multiple sclerosis: in vitro and in vivo evidence. Expert Opin Biol Ther. 2008;8(10):1561–70.
Barrette B, Vallieres N, Dube M, et al. Expression profile of receptors for myelin-associated inhibitors of axonal regeneration in the intact and injured mouse central nervous system. Mol Cell Neurosci. 2007;34(4):519–38.
Carim-Todd L, Escarceller M, Estivill X, et al. LRRN6A/LERN1 (leucine-rich repeat neuronal protein 1), a novel gene with enriched expression in limbic system and neocortex. Eur J Neurosci. 2003;18(12):3167–82.
Okafuji T, Tanaka H. Expression pattern of LINGO-1 in the developing nervous system of the chick embryo. Gene Expr Patterns. 2005;6(1):57–62.
Compston A, Coles A. Multiple sclerosis. Lancet. 2002;359(9313):1221–31.
Hauser SL, Oksenberg JR. The neurobiology of multiple sclerosis: genes, inflammation, and neurodegeneration. Neuron. 2006;52(1):61–76.
Sawcer S, Hellenthal G, Pirinen M, et al. Genetic risk and a primary role for cell-mediated immune mechanisms in multiple sclerosis. Nature. 2011;476(7359):214–9.
Franklin RJ. Why does remyelination fail in multiple sclerosis? Nat Rev Neurosci. 2002;3(9):705–14.
Franklin RJ. Remyelination of the demyelinated CNS: the case for and against transplantation of central, peripheral and olfactory glia. Brain Res Bull. 2002;57(6):827–32.
Franklin RJ, Hinks GL. Understanding CNS remyelination: clues from developmental and regeneration biology. J Neurosci Res. 1999;58(2):207–13.
Chang A, Tourtellotte WW, Rudick R, et al. Premyelinating oligodendrocytes in chronic lesions of multiple sclerosis. N Engl J Med. 2002;346(3):165–73.
Lau LW, Keough MB, Haylock-Jacobs S, et al. Chondroitin sulfate proteoglycans in demyelinated lesions impair remyelination. Ann Neurol. 2012;72(3):419–32.
Kotter MR, Li WW, Zhao C, et al. Myelin impairs CNS remyelination by inhibiting oligodendrocyte precursor cell differentiation. J Neurosci. 2006;26(1):328–32.
Fancy SP, Baranzini SE, Zhao C, et al. Dysregulation of the Wnt pathway inhibits timely myelination and remyelination in the mammalian CNS. Genes Dev. 2009;23(13):1571–85.
Back SA, Tuohy TM, Chen H, et al. Hyaluronan accumulates in demyelinated lesions and inhibits oligodendrocyte progenitor maturation. Nat Med. 2005;11(9):966–72.
Zhang Y, Argaw AT, Gurfein BT, et al. Notch1 signaling plays a role in regulating precursor differentiation during CNS remyelination. Proc Natl Acad Sci USA. 2009;106(45):19162–7.
Chan JR, Watkins TA, Cosgaya JM, et al. NGF controls axonal receptivity to myelination by Schwann cells or oligodendrocytes. Neuron. 2004;43(2):183–91.
Mi S, Cadavid D. A novel CNS remyelination and repair therapy by blocking LINGO-1 pathway. In: 26th congress of european committee for treatment and research in multiple sclerosis, Gothenburg, 12–16 Oct 2010, p. P731.
Papadopoulos D, Pham-Dinh D, Reynolds R. Axon loss is responsible for chronic neurological deficit following inflammatory demyelination in the rat. Exp Neurol. 2006;197(2):373–85.
Pepinsky RB, Shao Z, Ji B, et al. Exposure levels of anti-LINGO-1 Li81 antibody in the central nervous system and dose-efficacy relationships in rat spinal cord remyelination models after systemic administration. J Pharmacol Exp Ther. 2011;339(2):519–29.
Pallini R. Anatomy of “regenerating axons”. Science. 1998;280(5361):181–2.
Park JB, Yiu G, Kaneko S, et al. A TNF receptor family member, TROY, is a coreceptor with Nogo receptor in mediating the inhibitory activity of myelin inhibitors. Neuron. 2005;45(3):345–51.
Shao Z, Browning JL, Lee X, et al. TAJ/TROY, an orphan TNF receptor family member, binds Nogo-66 receptor 1 and regulates axonal regeneration. Neuron. 2005;45(3):353–9.
Lv J, Xu RX, Jiang XD, et al. Passive immunization with LINGO-1 polyclonal antiserum afforded neuroprotection and promoted functional recovery in a rat model of spinal cord injury. Neuroimmunomodulation. 2010;17(4):270–8.
Ericson C, Georgievska B, Lundberg C. Ex vivo gene delivery of GDNF using primary astrocytes transduced with a lentiviral vector provides neuroprotection in a rat model of Parkinson’s disease. Eur J Neurosci. 2005;22(11):2755–64.
Hanke M, Farkas LM, Jakob M, et al. Heparin-binding epidermal growth factor-like growth factor: a component in chromaffin granules which promotes the survival of nigrostriatal dopaminergic neurones in vitro and in vivo. Neuroscience. 2004;124(4):757–66.
Iwakura Y, Piao YS, Mizuno M, et al. Influences of dopaminergic lesion on epidermal growth factor-ErbB signals in Parkinson’s disease and its model: neurotrophic implication in nigrostriatal neurons. J Neurochem. 2005;93(4):974–83.
Seroogy KB, Numan S, Gall CM, et al. Expression of EGF receptor mRNA in rat nigrostriatal system. Neuroreport. 1994;6(1):105–8.
Wang X, McCullough KD, Franke TF, et al. Epidermal growth factor receptor-dependent Akt activation by oxidative stress enhances cell survival. J Biol Chem. 2000;275(19):14624–31.
Quigley HA, Nickells RW, Kerrigan LA, et al. Retinal ganglion cell death in experimental glaucoma and after axotomy occurs by apoptosis. Invest Ophthalmol Vis Sci. 1995;36(5):774–86.
Tran J. BIIB033 phase I study. In: Annual meeting of the American Academy of Neurology, New Orleans. 21–28 2012, p. P02–021.
Bongioanni P, Reali C, Sogos V. Ciliary neurotrophic factor (CNTF) for amyotrophic lateral sclerosis/motor neuron disease. Cochrane Database Syst Rev. 2004;(3):CD004302.
Lavail MM. Survival factors for treatment of retinal degenerative disorders: preclinical gains and issues for translation into clinical studies. Retina. 2005;25(8 Suppl.):S25–6.
Sahenk Z. Neurotrophins and peripheral neuropathies. Brain Pathol. 2006;16(4):311–9.
Sarchielli P, Gallai V. Nerve growth factor and chronic daily headache: a potential implication for therapy. Expert Rev Neurother. 2004;4(1):115–27.
Sherer TB, Fiske BK, Svendsen CN, et al. Crossroads in GDNF therapy for Parkinson’s disease. Mov Disord. 2006;21(2):136–41.
Tuszynski MH. Nerve growth factor gene therapy in Alzheimer disease. Alzheimer Dis Assoc Disord. 2007;21(2):179–89.
Williams BJ, Eriksdotter-Jonhagen M, Granholm AC. Nerve growth factor in treatment and pathogenesis of Alzheimer’s disease. Prog Neurobiol. 2006;80(3):114–28.
Woodruff RH, Fruttiger M, Richardson WD, et al. Platelet-derived growth factor regulates oligodendrocyte progenitor numbers in adult CNS and their response following CNS demyelination. Mol Cell Neurosci. 2004;25(2):252–62.
Zazpe A, Del Rio J. Neurotrophins. II: therapeutic potential. Rev Med Univ Navarra. 1997;41(3):180–4.
Zhang J, Lineaweaver WC, Oswald T, et al. Ciliary neurotrophic factor for acceleration of peripheral nerve regeneration: an experimental study. J Reconstr Microsurg. 2004;20(4):323–7.
Acknowledgments
No sources of funding were received to prepare this article. We thank Zhaohui Shao, Xinhua Lee, Benxiu Ji, Bruce Jenkins and Guangron Huang for their contributions to these studies. Sha Mi, R. Blake Pepinsky and Diego Cadavid are employees of Biogen Idec Inc. Biogen Idec Inc. markets two products for the treatment of multiple sclerosis.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Mi, S., Blake Pepinsky, R. & Cadavid, D. Blocking LINGO-1 as a Therapy to Promote CNS Repair: From Concept to the Clinic. CNS Drugs 27, 493–503 (2013). https://doi.org/10.1007/s40263-013-0068-8
Published:
Issue Date:
DOI: https://doi.org/10.1007/s40263-013-0068-8