
Abstract
The treatment of patients infected with the hepatitis C virus (HCV) has been revolutionised by the development of direct-acting antiviral agents (DAAs) that target specific HCV proteins involved in viral replication. The first DAAs were associated with clinical problems such as adverse drug reactions and pharmacokinetic drug–drug interactions (DDIs). Current FDA/EMA-approved treatments are combinations of DAAs that simultaneously target the HCV N5A-protein, the HCV N5B-polymerase and the HCV NS3/4A-protease. Adverse events and DDIs are less likely with these DAA combinations but several DDIs of potential clinical significance remain. Much of the available information on the interaction of DAAs with CYP drug-metabolising enzymes and influx and efflux transporters is contained in regulatory summaries and is focused on DDIs of likely clinical importance. Important DDIs perpetrated by current DAAs include increases in the pharmacokinetic exposure to statins and dabigatran. Some mechanistic information can be deduced. Although the free concentrations of DAAs in serum are very low, a number of these DDIs are likely mediated by the inhibition of systemic influx transporters, especially OATP1B1/1B3. Other DDIs may arise by DAA-mediated inhibition of intestinal efflux transporters, which increases the systemic concentrations of some coadministered drugs. Conversely, DAAs are victims of DDIs mediated by cyclosporin, ketoconazole, omeprazole and HIV antiretroviral drug combinations, especially when boosted by ritonavir and, to a lesser extent, cobicistat. In addition, concurrent administration of inducers, such as rifampicin, carbamazepine and efavirenz, decreases exposure to some DAAs. Drug-drug interactions that increase the accumulation of HCV N3/4A-protease inhibitors like grazoprevir may exacerbate hepatic injury in HCV patients.
Plain Language Summary
Direct-acting antiviral (DAA) drugs have revolutionised the treatment of patients with hepatitis C. Compared to the earlier agents, currently-approved DAA combinations have fewer adverse effects and are less likely to be associated with pharmacokinetic drug-drug interactions (DDIs). However, adverse events and DDIs still occur when DAAs are coadministered with certain drugs. In most cases DAAs likely perpetrate DDIs by inhibiting drug transporters. However, access to more detailed information on HCV DAAs as substrates and inhibitors of drug-metabolising enzymes and transporters, and the incidence of DDIs in target populations, would enhance the understanding of the significance of the likelihood of DDIs with DAAs.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
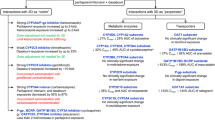
Current direct-acting antiviral agents (DAAs) for the treatment of hepatitis C produce fewer clinical problems than the earliest DAAs. |
However, current DAAs are implicated as perpetrators and victims of pharmacokinetic drug-drug interactions (DDIs) of potential clinical significance. |
Mechanisms of certain DDIs can be proposed, although the current available information on DAA biotransformation and disposition is focused primarily on major pathways of clinical importance. |
1 Introduction
1.1 Hepatitis C Infection and Treatment
The hepatitis C virus (HCV) is a major cause of chronic liver disease with up to 80 million viraemic individuals worldwide and a further 100 million who are HCV-antibody positive [1, 2]. Hepatitis C virus causes diverse hepatic pathologies, including cirrhosis and hepatocellular carcinoma, and a number of extrahepatic syndromes, including thrombocytopenia, non-insulin-dependent diabetes mellitus and cardiovascular disorders [3]. Altered drug pharmacokinetics in patients with HCV-mediated liver injury complicate drug treatment, which leads to impaired efficacy and increased drug toxicity.
In patients, a sustained virological response occurs when HCV RNA is undetectable in serum 12 weeks after the withdrawal of antiviral treatments [4]. This is associated with improved survival and decreased progression to advanced liver disease compared with untreated patients or those in whom treatment has failed [4]. For ~25 years pegylated interferon and ribavirin were the mainstays of HCV treatment but numerous side effects, suboptimal efficacy and contraindications were significant drawbacks.
1.2 New Drug Targets for HCV Treatment
The understanding of the molecular mechanisms of HCV infection and replication have increased over the last two decades. Briefly, lipoviral particles in blood attach to glycosaminoglycans and low-density lipoprotein receptors on the hepatocellular plasma membrane and are endocytosed. The HCV viral genome, which consists of a single open reading frame and two non-coding regions, is then translated by cellular ribosomes to produce a single polyprotein of ~3000 amino acids [5]. This polyprotein is cleaved by host and viral proteases to generate ten proteins, including the p7 ion channel, the HCV core protein that forms the viral capsid, the E1 and E2 envelope glycproteins that form further viral particles, and several non-structural (NS) proteins. Non-structural proteins include the viral auto-proteases (NS2/3 and NS3/4A), the RNA-binding protein NS5A, which enables assembly of the replication complex and the RNA-dependent RNA polymerase NS5B that is required for RNA replication [6]. Direct-acting antiviral agents (DAAs) that selectively target the HCV NS3/4A protease, the NS5A RNA-binding protein or the NS5B RNA-dependent RNA polymerase have been pivotal in the design of new therapeutic strategies for HCV treatment.
Hepatitus C virus is classified into six major genotypes and a number of subtypes on the basis of nucleotide sequence similarity; genotypes differ by 30–35% of nucleotides across the HCV genome [7, 8]. The NS3/4A protease inhibitors boceprevir and telaprevir were the first FDA-approved DAAs and exhibited short-term efficacy in HCV genotype 1-infected patients. However, their pharmacokinetics were suboptimal and drug-drug interactions (DDIs) due to inhibition of the transport and biotransformation of coadministered drugs were common. Furthermore, resistance to these DAAs emerged rapidly and response rates were also low in patients with hepatic cirrhosis [9].
Direct-acting antiviral agents that have been developed more recently are used in fixed-dose combinations. The 'second-wave' N3/4A protease inhibitors include glecaprevir, grazoprevir and voxilaprevir. Compared to boceprevir and telaprevir, these agents are more potent, exhibit higher barriers to resistance, have favourable pharmacokinetics and have more convenient dosage regimen, including once-daily dosing [10]. Currently used NS5A RNA binding protein inhibitors include pibrentasvir, velpatasvir, ledipasvir, elbasvir and daclatasvir, while sofosbuvir is the most important RNA-dependent RNA polymerase NS5B inhibitor in current use. The US FDA- and EMA-approved DAA combinations, their activities toward HCV genotypes and their use in patients with different grades of cirrhosis are shown in Table 1.
This article presents an overview of the absorption, distribution, biotransformation and elimination of DAAs that are currently approved by the FDA and EMA. Their clinical pharmacokinetics and the observed and potential DDIs in which the DAAs are perpetrators and victims are discussed. The available data from clinical studies with the following DAAs are included: glecaprevir, voxilaprevir and grazoprevir, pibrentasvir, velpatasvir, elbasvir, daclatasvir and ledipasvir and sofosbuvir. Direct-acting antivirals that were previously in clinical use are not discussed. Sources of information included published articles (Medline search terms: pharmacokinetics, drug interactions, toxicity, adverse events and all of the known biotransformation enzymes and transporters), the prescribing information published by the US FDA, summaries of product characteristics published by the EMA and other regulatory agencies, information from completed clinical trials (www.clinicaltrials.gov) and conference presentations (abstracts) that are available online.
1.3 Biotransformation Enzymes and Transporters in Drug Development
1.3.1 Serum HCV DAA Concentrations and Treatment Outcomes
During therapy, serum DAA concentrations are maintained at levels to suppress viral replication and prevent the emergence of resistant HCV variants. If trough concentrations of DAAs are too low, due to rapid rates of elimination, DAA-resistant viral mutations may accumulate. Conversely, if the elimination of DAAs is too slow, the drugs may elicit adverse effects. Information on the enzymes that mediate DAA biotransformation and transporters that control DAA disposition is essential to understand the mechanistic basis of DDIs in patients who receive DAA combinations.
Most information on the transporters and drug-metabolising enzymes in DAA biotransformation and disposition comes from reports submitted by pharmaceutical manufacturers to regulatory agencies. The major focus has been on the enzymes and transporters most commonly implicated in clinical drug pharmacokinetics. However, such studies are not exhaustive and alternate pathways of DAA disposition are possible.
1.3.2 Tissue Specificity of Biotransformation Enzymes and Transporters in DDIs
Multiple solute carrier (SLC) transporters facilitate the uptake of drugs, including HCV DAAs, in a tissue-specific manner [11]. Organic anion transporting polypeptide (OATP) 2B1, organic cation transporters OCT1 and OCT3 and multidrug and toxin extrusion transporter MATE1 (SLC47A1) mediate intestinal drug absorption [12]. Other SLC transporters participate in renal drug excretion [13], including OCT2, OAT1 and OAT3 which are expressed in kidney and bladder. OATP1A2, OATP1B1, OATP1B3, OATP2B1, OCT1 and OCT3 are expressed in liver [12].
Hepatic cytochrome P450 (CYP) enzymes oxidize lipophilic drugs, environmental chemicals and endobiotics to polar metabolites [14, 15]. In intestinal enterocytes CYP3A4 also oxidises a number of drugs following oral administration [16]. UDP-glucuronosyltransferases (UGTs) and other Phase II enzymes produce highly polar conjugates of some drug metabolites to enhance elimination in urine and faeces.
Intestinal ATP-binding cassette (ABC) transporters efflux drugs and decrease oral absorption [17]. In other tissues ABC transporters facilitate removal of drugs and metabolites from cells [18]. Major ABC transporters include P-glycoprotein (PgP/ABCB1), breast cancer resistance protein (BCRP/ABCG2) and the multidrug-resistance proteins MRP1 (ABCC1), MRP2 (ABCC2), MRP3 (ABCC3) and MRP4 (ABCC4). PgP, MRP1, MRP2, MRP3 and BCRP are expressed in many tissues, including liver and intestine, while MRP4 is expressed in the intestine, but is absent from liver, and bile salt export protein (BSEP/ABCB11) is expressed in hepatocytes [12, 19]. The tissue specificity of transporter and drug-metabolising enzyme expression is a critical factor in understanding DDI mechanisms.
1.3.3 Regulation of Drug-Metabolising Enzymes and Transporters
The basal expression of CYPs and transporters is regulated by tissue-specific transcription factors, such as HNF3γ and CEBPα [20]. Several of these genes are also inducible by certain drugs and chemicals that activate the pregnane X and constitutive androstane nuclear receptors (PXR and CAR, respectively) and the aryl hydrocarbon receptor (AhR) [21,22,23]. Transporters in particular are also subject to post-translational trafficking that regulates the expression of the proteins at the plasma membrane of cells [24, 25].
Pathophysiological regulation of drug-metabolising enzymes and transporters is important in drug development [26]. Recently nine CYPs and four UGTs were quantified in livers from patients with hepatitis C according to the severity of liver injury (using the Child–Pugh scale, where mild, moderate and severe impairment is graded as class A, B and C, respectively [27]). In Child–Pugh class B samples there was down-regulation of CYPs 2C19, 2E1 and 3A4, and UGTs 1A3 and 2B7. In more severe injury (Child–Pugh class C) CYP1A2 was also decreased. In contrast, expression of CYPs 1A1, 2B6, 2C8, 2C9 and 2D6 were not affected by liver injury [26]. These findings are in general accord with studies of altered CYP and transporter expression in related hepatic diseases, including cirrhosis and steatosis, and certain altered nutritional states [28,29,30,31,32,33,34,35,36,37], which may lead to impaired drug and endobiotic metabolism and transport.
The US FDA-approved prescribing information and EMA-approved summaries of product characteristics provide advice on the safe use of HCV DAAs in individuals who present with liver dysfunction of varying grade. Mavyret is contraindicated in patients with Child-Pugh class C disease and is not recommended in patients with Child-Pugh class B impairment due to the possibility of higher exposure [38]. Thus, the AUCs for glecaprevir in Child-Pugh class B and C patients are 2- and 11-fold of Child-Pugh A, respectively, while the corresponding AUCs for pibrentasvir in such patients are respectively 1.26- and 2.14-fold of Child-Pugh A [38]. Similarly, Vosevi is not recommended in Child-Pugh class B/C patients due to increased exposure [39]. Although velpatasvir exposure in patients with Child-Pugh class B/C is similar to that in Child-Pugh A, the AUCs for voxilaprevir in Child-Pugh class B and C are ~4- and ~6-fold, respectively, of Child-Pugh class A [39]. Zepatier is also contraindicated in Child-Pugh class B/C patients. Compared to non–HCV-infected subjects with normal hepatic function, grazoprevir AUC values were higher by 1.7-fold, 5-fold, and 12-fold in non–HCV-infected subjects with mild, moderate, and severe hepatic impairment, respectively, with an increased risk of alanine transaminase elevations [40]. In contrast, the exposures of daclatasvir, ledipasvir and sofosbuvir are relatively unchanged in Child-Pugh grade B/C patients and are not contraindicated [41, 42].
Pharmacogenomic factors also influence the activities of drug-metabolising enzymes and transporters [14, 43,44,45]. Single nucleotide polymorphisms (SNPs) and other gene variations can lead to impaired function or dysregulated expression. Some polymorphisms are more common in certain ethnic populations, so that altered drug-metabolising enzyme and transporter activities may be more common when HCV DAAs are used in these groups [14, 43,44,45].
1.3.4 Assessment of Biotransformation Enzymes and Transporters During Drug Development
Regulatory agencies provide guidance to the pharmaceutical industry in the development of new chemical entities (NCEs). Required information includes routes of metabolism, disposition and elimination, especially the involvement of specific drug metabolising enzymes and drug transporters. Agencies also advise on the in vitro methods to identify enzymes and transporters in NCE biotransformation and disposition. Data on the inhibition of these enzymes and transporters by NCEs are also required.
Whether the NCE is a substrate or inhibitor of efflux transporters can be determined in intestinal Caco-2 cells or in transporter over-expressing cell lines. Studies with influx transporters are conducted in over-expressing cell lines. During development of HCV DAAs the focus has usually been on the influx transporters OATP1B1/1B3, OAT1/3 and OCT2 and the efflux transporters PgP and BCRP because of their important roles in absorption, disposition and elimination.
Recombinant enzymes and hepatic microsomes are used to identify biotransformation pathways. For HCV DAAs, the focus of preclinical investigations has been on the major CYPs 1A2, 2C8, 2C9, 2C19, 2D6 and 3A4. The capacity of NCEs to inhibit these enzymes is evaluated in the same systems. In addition, preincubation of NCEs with NADPH-supplemented CYPs and microsomal fractions is used to assess time-dependent inhibition that is due to the formation of potent inhibitory metabolites. Preincubation of some NCEs with transporters may also increase the extent of inhibition of drug transport, although the mechanism is currently unclear [46].
The capacity of NCEs to induce CYPs and transporters is also assessed during preclinical development using cryopreserved hepatocytes and positive control drugs which are established PXR, CAR and AhR inducers [21,22,23]. If the NCE shows induction potential, a small clinical study may be justified to further evaluate the significance in patients. Several current HCV DAAs have been evaluated as potential inducers.
2 NS3/4A Protease Inhibitors in Clinical DAA Combinations
Three US FDA- and EMA-approved second-generation NS3/4A protease inhibitors are in use in HCV therapy: glecaprevir, voxilaprevir and grazoprevir. These agents are used in combination with other DAA drugs.
2.1 Absorption and Elimination
The NS3/4A protease inhibitors are hydrophobic (clog P range 4.10–6.79; Table 2), rapidly absorbed after oral dosage and are eliminated primarily in faeces, which is consistent with biliary excretion. Glecaprevir, grazoprevir and voxilaprevir are excreted in faeces as both parent drug and metabolites [38, 39, 47, 48].
There is limited detail on the interactions of these agents with drug transporters. Glecaprevir and grazoprevir are substrates of the influx transporters OATP1B1 (Km 0.098 µM and 0.43 µM, respectively) and OATP1B3 (Km 0.19 µM and 0.18 µM, respectively) and the efflux transporter PgP; glecaprevir is also transported by BCRP [38, 47]. Voxilaprevir is a substrate for OATP1B1, OATP1B3, PgP and BCRP [49].
2.2 CYP-Dependent Metabolism
Cytochrome P450s oxidize NS3/4A protease inhibitors to multiple metabolites. cDNA-expressed CYP3A4 and CYP2D6 catalysed glecaprevir oxidation [47]. In human liver microsomes, voxilaprevir formed nine metabolites [49] with CYP3A4 being the most important enzyme and lesser roles for CYP2C8 and CYP1A2 [39]. CYP3A4 was also the major catalyst of grazoprevir oxidation [48].
2.3 Pharmacokinetics
Across three studies, the half-life of glecaprevir varied between 5.86–8.69 h (Table 2). A single clinical dose (300 mg coadministered with pibrentasvir 120 mg) produced mean Cmax and AUC values in 23 patients of 294 ng/mL and 1150 ng.h/mL, respectively (Table 2; [47]). Glecaprevir exposure was influenced by diet: thus, a medium fat diet increased AUC and Cmax to ~2.6-fold and ~3.2-fold of fasted control, respectively (Table 2; [47]). After multiple doses (300 mg daily for 14 days) glecaprevir exposure was similar in Caucasian, Chinese and Japanese patients (Cmax ranges were 1150–1390 ng/mL and AUC ranges were 3630–4500 ng.h/mL) (Table 2; [50]). Exposure was greater than dose-proportional over the dose range 25–800 mg [51, 52].
There is less information on voxilaprevir pharmacokinetics. The median half-life was 8.51 h (Table 2 [53]). Pharmacokinetics were linear over the dose range 50–300 mg but the AUC increase was greater than dose-proportional over the range 100–900 mg (>100-fold); nonlinearity could be due to inhibition of intestinal PgP or BCRP by the drug [54]. Food also affected voxilaprevir exposure. When administered as Vosevi, light, moderate and high fat meals increased the AUC of voxilaprevir by 2-fold, ~3-fold and ~5-fold, respectively and Cmax values by 2.5-fold, 3.5-fold and ~8-fold, respectively [54].
In fasting individuals, grazoprevir (100 mg) exposure (Cmax and AUC) was 40.8 ng/mL and 595 ng.h/mL, respectively (Table 2; [48]). After food, exposure increased to 1.8- and 1.5-fold of corresponding fasting levels. The pharmacokinetics of grazoprevir was assessed in single dose (2–1600 mg) and multiple dose studies (100–1000 mg daily over 10 days). Exposure increased dose-proportionately to 200 mg (single dose) and multiple doses between 100 and 400 mg, but was non-linear at higher doses [55, 56]. The half-life of grazoprevir across four studies was 27–39.8 h (Table 2).
3 NS5A RNA-binding Protein Inhibitors in Clinical DAA Combinations
There are five current NS5A inhibitors: pibrentasvir, velpatasvir, ledipasvir, elbasvir and daclatasvir.
3.1 Absorption and Elimination
Like the NS3/4A protease inhibitors, NS5A RNA-binding protein inhibitors are highly hydrophobic (clog P 4.72–7.92; Table 2), are rapidly absorbed from the intestine and are eliminated primarily in faeces. With pibrentasvir ~97% of a dose was excreted unchanged in faeces by 144 h [47]. Similarly, 77% and 70% of velpatasvir and ledipasvir doses were excreted unchanged in faeces [52, 57]. Over 94% of a dose of elbasvir and its major metabolites appeared in faeces [58] and around 88% of an oral daclatasvir dose appeared in faeces by 10 days, of which ~59% was unchanged drug [59]. These findings suggest that biliary excretion is also the predominant route of elimination of NS5A RNA-binding protein inhibitors.
Velpatasvir and ledipasvir, but not pibrentasvir or elbasvir, were substrates for the influx transporters OATP1B1 and OATP1B3 [38,39,40, 60]. Ledipasvir was also a substrate for OCT1 [57], while daclatasvir was not a substrate for OATP1B1, OATP1B3, OATP2B1 or OCT1 [61]. With regard to efflux transporters, velpatasvir and ledipasvir were substrates for PgP and BCRP, pibrentasvir was a substrate for PgP and/or BCRP, and elbasvir and daclatasvir were substrates for PgP [38, 52, 57].
3.2 CYP-dependent Metabolism
Pibrentasvir and ledipasvir biotransformation was minimal and was CYP-independent [47, 57]. Although minor, velpatasvir oxidation in vitro was mediated by CYPs 2B6, 2C8 and 3A4. Elbasvir oxidation was mediated by CYP3A4, but not other CYPs [62]. Daclatasvir oxidation was mediated by CYPs 3A4, 3A5 and 2C8 [63]. Daclatasvir was also found to be a time-dependent CYP inhibitor, which is consistent with the formation of a reactive metabolite during biotransformation, possibly by ∂-oxidation within the pyrrolidine ring to an amino-aldehyde intermediate [63]. This potential pathway of daclatasvir activation is distinct from other time-dependent (or mechanism-based) inhibitors in which oxidation of specific chemical moieties generates reactive metabolites that elicit CYP destruction or sequestration in a catalytically inactive state. These moieties include alkylamines [64, 65], benzodioxoles [66, 67], aldehydes [68], isocyanates [69] and unsaturated hydrocarbons [70]. Daclatasvir and several of its metabolites also contain two substituted imidazole systems, which are associated with potent CYP inhibition [71].
3.3 Pharmacokinetics
The pharmacokinetics of pibrentasvir has been studied: the half-life was in the range 13–28.1 h (Table 2). In 18 Caucasian subjects, the clinical regimen (120 mg daily for 14 days) produced Cmax and AUC values of 244 ng/mL and 1900 ng.h/mL, respectively, and were similar in Chinese and Japanese subjects (Table 2; [50]). Exposure was dose-independent [50] and was increased by a medium-fat diet to ~1.4-fold (AUC) and ~1.9-fold (Cmax) of fasted control [38].
A single dose of velpatasvir (100 mg) produced Cmax values in the range 569–764 ng/mL and AUC values in the range 4610–5652 ng.h/mL (Table 2; [72, 73]), that were increased by a multiple dose regimen (100 mg for 28 days) [53]. Pharmacokinetics were linear over the dose range 30–300 mg when administered by single- and multiple-dose regimen [74] and the half-life was 11.59–19.6 h [53, 72, 75].
Pharmacokinetic exposure to ledipasvir was linear across the dose range 3–100 mg [76] and the half-life was in the range 35.4–54.7 h [42, 75, 77]. A single 90-mg dose produced Cmax values between 197–342 ng/mL and AUC between 7616–12875 ng.h/mL (Table 2; [42, 77]).
Elbasvir (50 mg single dose), with or without grazoprevir (100 mg), produced Cmax values between 95–168 ng/mL and AUC values between 1330–2276 ng.h/mL (Table 2; [40, 78,79,80]). AUC and Cmax were dose-dependent to 100 mg (single dose) and to 200 mg (multiple doses over 10 days) [40]; this has been attributed to the possible saturation of CYP3A4-dependent biotransformation or OATP1B1-mediated influx into tissues. The half-life ranged between 18.2–25.02 h [40, 78, 79].
In 11 subjects a single 60-mg dose of daclatasvir produced Cmax and AUC values of 1110 ng/mL and 11220 ng.h/mL, respectively (Table 2; [81]). Multiple doses produced Cmax between 1335–1582 ng/mL and AUC between 12677–15666 ng.h/mL [82]. Daclatasvir pharmacokinetics were dose-dependent to 100 mg when administered according to single or multiple dose regimen [82]. The half-life of daclatasvir across three studies was in the range 12–15 h [81,82,83,84].
4 Sofosbuvir
4.1 Absorption and Elimination
Sofosbuvir is more hydrophilic than other HCV DAAs (clog P 0.84; Table 2) and urinary excretion is the primary elimination route [85]. Biotransformation is CYP-independent and the pharmacologically active GS-461203 and inactive GS-331007 metabolites are formed by cathepsin A or carboxylesterase 1 [85]. The latter metabolite accounts for over 90% of the systemic exposure of sofosbuvir [85]. Sofosbuvir is a substrate for BCRP and PgP, but not major SLC influx transporters [85].
4.2 Pharmacokinetics
A single dose of sofosbuvir (400 mg) produced Cmax and AUC values of 622 ng/mL and 629 ng.h/mL, respectively (Table 2; [85]). Similar exposures occurred in some studies after multiple doses [57, 86], although other studies reported higher exposures [53, 75, 77, 87]. Sofosbuvir pharmacokinetics was dose-dependent over the range 200–1200 mg [85] and the half-life was 0.4–0.68 h [57, 75, 77, 86, 88].
5 Direct-acting Antiviral Combinations
Direct-acting antiviral combinations are used to treat multiple HCV genotypes (Table 1). Mavyret, Epclusa and Vosevi are administered once daily for the treatment of patients infected with HCV genotype 1–6 without cirrhosis or who have compensated cirrhosis (Child-Pugh class A) [38, 39]. Grazoprevir and elbasvir (Zepatier), with or without ribavirin, is used to treat adult patients with HCV genotypes 1a, 1b, or 4 [40]. Sofosbuvir and ledipasvir (Harvoni) are indicated for HCV genotype 1, either without cirrhosis or with compensated cirrhosis [42]. Daclatasvir (Daklinza) is used with sofosbuvir for HCV genotypes 1 or 3, in patients without cirrhosis or who have Child-Pugh class A impairment [41]. Daclatasvir and sofosbuvir may be used with ribavirin for patients with decompensated liver disease (Child-Pugh class B or C) or in patients post-transplant [41]. Sofosbuvir is also used with ribavirin, with or without pegylated interferon-α, for patients infected with HCV genotypes 1–4.
Current HCV DAAs appear less likely than the earliest DAAs to elicit pharmacokinetic DDIs [89] but, as outlined in the following sections, DDIs may still occur. Most information on DAA pharmacokinetics and DDIs as perpetrator or victim drugs has emerged from small-scale clinical studies with approved DAA combinations; few studies have evaluated DDIs with individual agents. Drug-drug interaction mechanisms have usually been inferred from altered DAA pharmacokinetics and the CYPs and transporters that mediate their disposition.
6 HCV DAAs as Perpetrators and Victims of DDIs
6.1 Prediction of the DDI Potential of NCEs
Regulatory agencies recommend the use of pharmacokinetic modelling to assess the DDI potential of NCEs and that may be used to justify a targeted clinical study [90]. As mentioned, the tissue specific expression of drug transporters and drug-metabolising enzymes is an important consideration in assessing potential DDI mechanisms.
6.1.1 DDIs in the Intestine
The initial criterion is that an IC50 or Ki value of 10 μM is the upper limit for an inhibitory interaction involving a biotransformation enzyme or transporter for potential clinical significance [91]. The ratio Igut/IC50 or Igut/Ki, is then calculated, where IC50 and Ki are inhibition constants for the drug of interest, and Igut is the maximum clinical dose of the drug divided by 250 mL. This is the theoretical maximal intestinal concentration of the agent after oral administration. The cut-off value of the Igut/IC50 or Igut/Ki ratio for intestinal DDIs of potential significance is taken as ≥10 [91]. However, it is noteworthy that the 10 μM cut-off concentration is often exceeded by intestinal Igut concentrations for DAAs that have been calculated (Table 3).
6.1.2 Systemic DDIs
After oral administration, a drug enters the liver via the hepatic artery if it is already in the systemic circulation, or via the portal vein immediately after absorption from the gut. The equation Imax = Cmax + A describes the maximal concentration of an inhibitory drug in the portal vein (Imax), where Cmax is its maximal systemic concentration and A is the portal concentration of inhibitory drug after absorption, as calculated using the expression (ka.D/Qh.Fa). Here, ka is the first order absorption rate constant, D is the oral dose of the drug, Qh is the hepatic blood flow (~1.5 L/h) and Fa is the fraction of drug absorbed from the gastrointestinal tract [92]. The absorption term A has been found to contribute significantly to the value of Imax for some drugs. Thus, inclusion of A may improve DDI predictions over those that consider systemic inhibitory drug concentrations alone (Cmax) [93].
The unbound fraction (fu) is considered to be the pharmacologically active form of a drug, and is also used to assess DDI potential. The ratios R = 1+(fu.Imax/IC50) or R = 1+(fu.Imax/Ki) have been derived to assess the propensity for DDIs involving inhibition of CYPs [93] and SLC influx transporters by drugs in the systemic circulation [94]. The recommended cut-off for systemic R values differs between regulatory agencies and is R ≥ 1.1 for the FDA and R ≥ 1.05 for the EMA [95]; R ≥ 1.25 is the recommended cut-off value for time-dependent inhibition [90]. When the cut-off value is exceeded it is recommended that an in vivo clinical study could be conducted.
6.2 Capacity of HCV DAAs to Inhibit Drug Metabolising Enzymes and Transporters
6.2.1 NS3/4A Protease Inhibitors
Glecaprevir is a potent inhibitor of the transporters OATP1B1, OATP1B3, BSEP, PgP and BCRP (Table 3; [47, 96, 97]). Voxilaprevir also inhibited OATP1B1, OATP1B3, and BCRP [49]. Grazoprevir inhibits OATP1B1 and OATP1B3 and, to a lesser extent, BCRP, MRP2, MRP3 and MRP4 (Table 3; [48]).
The inhibition of major drug-metabolising enzymes by NS3/4A protease inhibitors has been investigated. In human liver microsomes, glecaprevir inhibited CYP2C8 and the Phase II enzymes UGT1A1 and UGT1A4 (Kis ~6.2–7.5 μM)[98]. In contrast, voxilaprevir and grazoprevir were relatively non-potent inhibitors of CYPs and UGT1A1; CYP2C8 was relatively susceptible to inhibition by grazoprevir (IC50 6.1 μM, Table 3; [54]).
6.2.2 NS5A RNA-binding Protein Inhibitors
The influx transporter OATP1B1 is inhibited by pibrentasvir, velpatasvir, daclatasvir and, to a lesser extent, ledipasvir (Table 3; [54, 59, 62, 96, 97]). OATP1B3 is susceptible to inhibition by velpatasvir and elbasvir, while daclatasvir, ledipasvir and pibrentasvir are considerably less potent (Table 3; [54, 59, 62, 96, 97]). In the case of elbasvir, IC50s against OATP1B1, OAT1, OAT3 and OCT1 reportedly exceeded 0.5 µM, but were not fully estimated (Table 3; [62]). Daclatasvir is also an inhibitor of OCT1 and, to a lesser extent, OCT2 (Table 3; [59]). In vitro daclatasvir is a time-dependent inhibitor of OCT1 and OCT2, although its potency was low [99]. The efflux transporter PgP is inhibited potently by pibrentasvir and elbasvir (IC50s <1 µM) and BCRP is susceptible to inhibition by velpatasvir, elbasvir and daclatasvir, while velpatasvir and daclatasvir are non-potent PgP inhibitors and pibrentasvir was a non-potent inhibitor of BCRP (Table 3; [54, 59, 62, 96, 97]).
Pibrentasvir is a moderately effective inhibitor of biotransformation by CYP2C8, UGT1A1 and UGT1A4 [98]. Similarly, daclatasvir inhibits UGT1A1 and CYP3A4-mediated midazolam 1’-hydroxylation and testosterone hydroxylation [59]. In contrast, elbasvir does not inhibit CYP3A4, six other CYPs or UGT1A1 [62] and ledipasvir is non-potent against UGT1A1 and CYP3A [57]. Preincubation of daclatasvir with NADPH-supplemented liver microsomes decreases the IC50 of midazolam 1’-hydroxylation around two-fold, consistent with time-dependent inhibition [63]. The mechanism of this intensification of inhibition is presently unclear because the structure of daclatasvir lacks any of the characteristic chemical moieties that have been shown to undergo biotransformation to inhibitory metabolites, such as alkylamine, thionosulfur, benzodioxole, olefinic, acetylenic and isocyano substituents [64,65,66,67,68,69]. This suggests that daclatasvir contains a different structural feature that may be converted to a metabolite that inhibits CYP3A4. Indeed, it has been proposed that an amino-aldehyde intermediate is formed during daclatasvir biotransformation and could chemically modify lysine and histidine residues in CYP3A4 [63, 100]. If these residues were catalytically important this mechanism could account for time-dependent inhibition, although this possibility remains to be established.
6.2.3 NS5B Polymerase Inhibitors
Sofosbuvir did not inhibit the influx transporters OATP1B1/1B3, OAT1/3, OCT1/2 and MATE, CYP enzymes or UGT1A1, or the efflux transporters BCRP and PgP [57].
7 Clinical Evidence for DDIs Produced by Approved HCV DAAs
Small-scale clinical studies have been used to assess whether NCEs alter pharmacokinetic parameters, such as AUC and Cmax, that could indicate DDI potential. Agents that increase AUC ratios to ≥ 5-fold, or 2- to 5-fold, of control are considered to be potential inhibitors of high or moderate potency, respectively [90, 97, 101]. Similarly, drugs that decrease AUC ratios to 0.2 or to 0.2- to 0.5-fold of control are considered to be strong or moderate potential inducers, respectively [90, 97, 101]. The following sections focus on potential DDIs associated with HCV DAAs that are of moderate to high potency, based on the magnitude of AUC changes outlined above. Interactions where potential victim and perpetrator drugs elicited AUC changes that were <2-fold are not discussed in great detail, although it is acknowledged that information on smaller changes in AUC ratios may also facilitate the understanding of DDI mechanisms.
7.1 DAAs as Perpetrators of DDIs
The Igut/IC50 and R values for DAAs as inhibitors of transporters and drug-metabolising enzymes were calculated as described in section 6. The calculated Igut/IC50 values suggest that intestinal PgP may be subject to inhibition by glecaprevir, pibrentasvir, elbasvir and daclatasvir, while intestinal BCRP may be inhibited by glecaprevir, velpatasvir, elbasvir and daclatasvir (Table 3). Similarly, intestinal MRP2, MRP3 and OAT1 may also be susceptible to inhibition by grazoprevir and OCT1 by daclatasvir (Table 3).
In considering potential systemic DDIs, it is noted that the fu values for HCV DAAs are low. However, some of the calculated and reported R values against transporters and drug-metabolising enzymes exceed the FDA cut-off of R ≥ 1.1, which suggests certain inhibitory interactions mediated by DDAs may have potential clinical significance (Table 3). Thus, OATP1B1 was inhibited by glecaprevir, voxilaprevir, grazoprevir, pibrentasvir and, possibly, elbasvir (Table 3). OATP1B3 was inhibited by glecaprevir, voxilaprevir, grazoprevir, velpatasvir and elbasvir (Table 3). Literature R values for glecaprevir and voxilaprevir against OATP1B1/1B3 [96, 102] were in good agreement with those in Table 3. At this stage further information is required to assess whether elbasvir is a significant inhibitor of OAT1, OAT3 and OCT1 (Table 3). From the calculated systemic R values PgP might also be inhibited by glecaprevir, pibrentasvir and elbasvir, BCRP could be inhibited by velpatasvir and elbasvir, grazoprevir could inhibit BSEP and pibrentasvir may inhibit UGT1A4 (Table 3). The potential inhibition of these transporters and/or enzymes by DAAs has led to several Phase I clinical studies in healthy subjects that have evaluated potential DDIs with the PgP substrates digoxin and dabigatran etexilate, the BCRP substrate rosuvastatin and the OATP1B1/3 substrates pravastatin and rosuvastatin [96].
In the following sections, the focus is on potential DDIs with HCV DAAs where AUC ratios for victim drugs increase to at least 2- to 5-fold of those in the absence of an inhibitory DAA or where AUC ratios decrease to at least 0.2-fold of those in the absence of a DAA inducer. Drugs whose AUC values did not change to this extent were excluded, with the exception of digoxin that has a narrow therapeutic index.
7.1.1 Statins
Statins are important victim drugs in DAA-mediated DDIs. Once-daily administration of glecaprevir (non-clinical dose of 400 mg) and pibrentasvir (120 mg) to 11 subjects increased exposure to atorvastatin (10 mg). Thus, the Cmax and AUC parameters were increased to 22.0- and 8.3-fold of respective control (Table 4) [55]. Administration of glecaprevir (non-clinical dose of 400 mg) and pibrentasvir (120 mg once daily) increased the AUC and Cmax for rosuvastatin (5 mg) to 2.2- and 5.6-fold, and pravastatin (10 mg) to 2.3- and 2.2-fold of respective control values [47, 96]. Administration of Mavret (glecaprevir/pibrentasvir 300/120 mg) once daily in 12 patients slightly increased exposure to lovastatin (10 mg once daily; AUC 1.7-fold of control), although exposure to the active metabolite lovastatin acid was increased markedly (Cmax and AUC to 5.73- and 4.10-fold of respective control; [38]). Similarly, although simvastatin (5 mg once daily) exposure was increased only slightly by Mavyret (Cmax and AUC to 1.99- and 2.32-fold of respective control), that of the active metabolite simvastatin acid was markedly increased (Cmax and AUC) to 10.7- and 4.48-fold of control) [38, 96]. Similarly, Vosevi (sofosbuvir/velpatasvir/voxilaprevir 400/100/200 mg) increased the Cmax and AUC of rosuvastatin (10 mg single dose) to 18.9- and 7.4-fold and the Cmax and AUC of pravastatin to 1.89- and 2.16-fold of respective control [39]. Zepatier administration also increased the Cmax and AUC ratios of atorvastatin (10 mg single-dose) to 4.34- and 1.94-fold of control and those of rosuvastatin (10 mg single-dose) to 5.49- and 2.26-fold of control [40].
According to the US FDA prescribing information, Mavyret is not recommended in patients who also receive atorvastatin, lovastatin or simvastatin. Dose reductions or restrictions are recommended when pravastatin, fluvastatin and pitavastatin are coadministered because there may be an increased risk of myopathy due to increased serum statin concentrations. Similarly, Vosevi is not recommended with coadministered rosuvastatin and pitavastatin and dose restrictions are recommended with other statins [39]. Dose restrictions are recommended with coadministered statins and Zepatier [40] and monitoring for myopathy is recommended when Daklinza and statins are coadministered [41]. The coadministration of Harvoni and rosuvastatin is not recommended [42].
Most statins are transporter substrates. Rosuvastatin is transported by OATP1A2, OATP2B1 and OCT3, and PgP and BCRP [103, 104]. Pravastatin is transported by OATP1B1, OATP1B3, OATP2B1 and MRP2, but not by PgP [105,106,107,108]. Atorvastatin is a substrate for OATP1B1, OATP2B1 and BCRP and, to a lesser extent, PgP [106, 109]. Lovastatin is a substrate for PgP, but not BCRP [110,111,112,113], whereas simvastatin is not a major substrate for intestinal drug transporters [111, 112, 114]. The available information suggests that the active metabolites of lovastatin and simvastatin (lovastatin and simvastatin acids) do not interact significantly with PgP [106, 114]. Clinical and in vitro data suggest that statins that modulate PgP have a higher probability for eliciting DDIs. Taken together, because the calculated R values for several HCV DAAs against OATP1B1, OATP1B3, PgP and/or BCRP exceed the FDA cut-off value of R≥1.1 (Table 3), transporter inhibition is a likely mechanism underlying DDIs with statins.
7.1.2 Dabigatran
Dabigatran etexilate is a prodrug that undergoes carboxylesterase-mediated hydrolysis to the active oral anticoagulant dabigatran [115]. The prodrug is a PgP substrate whereas dabigatran itself is a relatively poor substrate for either PgP or BCRP [116]. In addition, Udomnilobol et al, reported very recently that sub-therapeutic doses of dabigatran etexilate may also undergo biotransformation by CYP3A4 [117]. Some coadministered HCV DAAs have been found to increase the serum accumulation of dabigatran and the risk of bleeding. Thus, in 54 subjects, Mavyret increased dabigatran Cmax and AUC (150 mg single dose) to 2.04- and 2.38-fold of control, respectively [47, 96]. Similarly, Vosevi also enhanced dabigatran exposure (75 mg one dose), as shown by increases in the Cmax and AUC to 2.9- and 2.6-fold of respective control, most likely due to inhibition by voxilaprevir of PgP-mediated dabigatran etexilate efflux [39, 96] Although the coadministration of dabigatran and daclatasvir or elbasvir has not been studied directly, both HCV DAAs inhibit PgP, so that increased serum dabigatran concentrations may be anticipated [40, 41]. At present dose modifications for dabigatran etexilate are recommended when the drug is coadministered with Mavyret and Vosevi [38, 39]. The potential increases in dabigatran concentrations has led to suggestions that serum monitoring could be considered [118, 119].
7.1.3 Digoxin
Digoxin is a PgP substrate that exhibits variable bioavailability and is predominantly eliminated by renal excretion [120]. The AUC of digoxin (0.25 mg single dose) is increased to 1.48- and 1.34-fold of respective control when Mavyret and Vosevi are coadministered, which suggests that digoxin efflux by intestinal PgP is inhibited [39, 96]; therapeutic monitoring and potential dose modifications of digoxin are recommended [38, 39]. Drug-drug interactions with digoxin that are mediated by PgP inhibitors have high potential clinical relevance because of the narrow therapeutic index of the drug [96]. The AUC of digoxin (0.125 mg once daily) was also increased by daclatasvir (60 mg once daily) to 1.27-fold of control [41].
7.1.4 Amiodarone
The coadministration of sofosbuvir-containing DAA combinations and amiodarone is contraindicated due to potential life-threatening arrhythmias [121]. Initially attributed to PgP inhibition and altered amiodarone pharmacokinetics it now appears that the mechanism involves disrupted intracellular calcium handling and cellular electrophysiology in stem cell-derived cardiomyocytes [122]. Thus, the DDI may be due to altered signalling by second messengers, membrane depolarisation or sarcoplasmic calcium release. Because of the long half-life of amiodarone, close monitoring is required in patients who receive sofosbuvir, even after treatment has been discontinued [123].
7.1.5 Mutual Interactions Between HCV DAAs
During Mavyret administration, glecaprevir increased pibrentasvir exposure in a dose-dependent fashion [124]. At clinical doses the Cmax and AUC of pibrentasvir were respectively increased by glecaprevir to ~2.9- and ~3.5-fold of pibrentasvir alone, which was likely due to inhibition of intestinal pibrentasvir efflux by PgP and/or BCRP [124]; the reverse interaction was not significant.
7.2 DAAs as Victims
The mechanisms by which HCV DAAs are victim drugs in DDIs are difficult to define because the available information is somewhat limited. In most cases, the pharmacokinetic information is restricted to altered exposure (Cmax and AUC) of the HCV DAAs produced by coadministered agents. However, the mechanism of increased exposure could be due to inhibition of influx transporters that impairs DAA uptake from blood into tissues, by impaired DAA biotransformation, or to decreased DAA efflux from tissues due to inhibition of systemic efflux transporters. Increased HCV DAA exposure could also occur if coadministered perpetrator drugs inhibit intestinal drug efflux transporters. Potential DDIs in which HCV DAAs are victims again focus on interactions where AUC ratios for DAAs were increased to at least 2- to 5-fold of control by an inhibitory perpetrator drug, or were decreased to at least 0.2-fold of control after induction by a perpetrator drug, respectively.
7.2.1 Cyclosporin
A low dose of cyclosporin (100 mg) had a limited effect on glecaprevir or pibrentasvir exposure [125]. However, in 12 subjects who received glecaprevir/pibrentasvir (300 mg/120 mg), a single dose of cyclosporin (400 mg) substantially increased glecaprevir exposure (Cmax and AUC to 4.5- and 5.1-fold of control, respectively); pibrentasvir AUC was increased to 1.93-fold of control (Table 5; [47]). The US FDA-approved prescribing information indicates that Mavyret is not recommended in patients who require ongoing cyclosporin therapy [38]. Glecaprevir is a substrate for OATP1B1, OATP1B3, CYP3A4, PgP and BCRP, while pibrentasvir is transported by PgP, but not by OATP1B1, OATP1B1B3 or BCRP, and is minimally metabolised by CYP3A4 [47]. Cyclosporin inhibits OATP1B1/1B3, CYP3A4, Pgp and MRP2 [126]. Thus, it is feasible that cyclosporin could inhibit OATP1B1/1B3 to increase serum glecaprevir exposure without altering serum pibrentasvir exposure, but modulation of CYP3A4 activity or other as-yet unidentified transporters cannot be ruled out.
Increased systemic exposure to HCV NS3/4A protease inhibitors also occurred when cyclosporin was coadministered with other DAA combinations. In 14 patients who received Zepatier, grazoprevir exposure (Cmax and AUC) was increased markedly by cyclosporin (400 mg single dose) to 15- and 17-fold of respective control, while elbasvir exposure was moderately increased to 2.0-fold of control [48, 62]. The increase in grazoprevir exposure was also associated with an increase in the alanine transaminase/aspartate transaminase ratio to >5-fold of normal upper limits after 4 weeks of treatment, suggesting an increased propensity for liver injury [127]. Thus, cyclosporin is contraindicated in HCV patients who receive Zepatier. Similarly, in individuals who received Vosevi, cyclosporin coadministration increased systemic exposures (Cmax and AUC) of voxilaprevir (100 mg dose) to 19.0- and 9.4-fold of respective control and sofosbuvir (400 mg dose) to 2.5- and 4.5-fold of respective control, while exposure to velpatasvir (100 mg single dose) was minimally altered (Table 5; [39]). The FDA-approved prescribing information recommends that Vosevi and cyclosporin are not coadministered [39].
Cytochrome P450-dependent biotransformation of NS3/4A protease inhibitors appears more important than for other classes of DAAs. An exception is the NS5A inhibitor daclatasvir, which undergoes biotransformation by CYP3A4. However, DDIs between daclatasvir and cyclosporin appeared to be relatively unimportant in HCV-infected patients after liver transplantation and dose adjustments are not required [128]. Taken together, it seems likely that the observed cyclosporin-mediated DDIs are more likely due to altered transport of HCV DAAs rather than to CYP inhibition.
7.2.2 Rifampicin
Administration of multiple doses of rifampicin activates PXR-inducible genes, including CYPs 3A, PgP and several other drug-metabolising enzymes and transporters [21, 22]. In contrast, treatment with single doses of rifampicin produces short-term inhibition of CYP3A4 and several transporters. Such inhibitory interactions are potentially significant during the initiation phase of treatment with rifampicin. The interplay of multiple interactions with transporters and enzymes is a complex clinical issue.
Rifampicin is a substrate for OATP1B1 [129] and PgP [130] and is also an OATP1B1/1B3 inhibitor [131]. In 12 subjects who received Mavyret, rifampicin (600 mg single dose) increased exposure to glecaprevir (Cmax and AUC to 6.5- and 8.6-fold of respective control), but not pibrentasvir (Table 5; [47]). Similarly, single-dose rifampicin strongly increased exposure to voxilaprevir (Cmax and AUC) to 7.9- and 11-fold of control, but not velpatasvir (1.46- and 1.28-fold of control) (Table 5; [39]). Single-dose rifampicin (600 mg orally) did not markedly affect exposure to elbasvir (Cmax and AUC were 1.29- and 1.29-fold of control, respectively) but strongly increased grazoprevir exposure (Cmax and AUC to 6.52- and 8.35-fold of control) (Table 5; [40]). It is possible that increased exposure could be due to inhibition by rifampicin of influx transporters, such as OATP1B1, that would decrease DAA uptake from serum into tissues. Alternately, rifampicin could inhibit intestinal efflux transporters and promote absorption. However, rifampicin was found to be a relatively non-potent inhibitor of efflux transporters so this possibility may not be clinically important [132, 133]. Further information on the relative affinities of individual HCV DAAs for transporters would facilitate a mechanistic understanding of rifampicin-mediated effects on DAA exposure.
The administration of multiple doses of rifampicin activates the PXR gene battery. In 12 patients rifampicin (600 mg daily) markedly decreased the Cmax and AUC of glecaprevir to 0.14- and 0.12-fold of control, respectively, and the Cmax and AUC for pibrentasvir to 0.17- and 0.13-fold of respective control (Table 5; [47]). Multiple-dose rifampicin (600 mg daily) also decreased exposure to voxilaprevir (Cmax and AUC to 0.27- and 0.91-fold of control), velpatasvir (to 0.18- and 0.29-fold of control), daclatasvir (to 0.44- and 0.21-fold of control), and ledipasvir (90 mg single dose) to 0.65- and 0.41-fold of control, respectively (Table 5; [39, 41, 42]). These findings are consistent with induction of CYP3A4, PgP and/or BCRP, which would be expected to enhance oxidation and efflux of the DAAs [134]. Rifampicin (600 mg for 10 days) also strongly decreased exposure (Cmax and AUC) to sofosbuvir (400 mg single dose) to 0.23- and 0.28-fold of respective control, which is probably due to PgP induction because CYP3A4 does not mediate sofosbuvir biotransformation (Table 5; [85]). In contrast, multiple-dose rifampicin (600 mg daily) did not significantly alter exposure to grazoprevir (Cmax and AUC to 1.16- and 0.93-fold of control) (Table 5; [40]). Differences between the effects of induction on exposure to different HCV DAAs are likely due to the relative importance of PXR-inducible genes in their disposition. However, even though CYP3A4 has a minor role in the basal biotransformation of several DAAs, after induction, this pathway could assume greater quantitative significance. This may well contribute to the marked decreases in exposure to several NS5A inhibitors after multiple doses of rifampicin. The coadministration of rifampicin with Mavyret, Vosevi, Zepatier and Daklinza is contraindicated, and not recommended with Harvoni and Epclusa due to the potential loss of therapeutic effect of the HCV DAAs [38,39,40,41,42, 60].
Consistent with these findings, other agents that activate the PXR also decreased the systemic exposure to some DAAs. In 10 patients who received carbamazepine (200 mg twice daily) exposure to glecaprevir/pibrentasvir (300/120 single dose) was decreased. Thus, Cmax and AUC values (fold of control) for glecaprevir were 0.33 and 0.34, respectively, and for pibrentasvir were 0.49 and 0.50, respectively (Table 5; [47]). Similarly, PXR activation by efavirenz, when administered in combination with emtricitabine and tenofovir, decreased the Cmax and AUC of velpatasvir to 0.50 and 0.47-fold of control, respectively, most likely due to induction of PgP and possibly CYP3A4 (Table 5; [39]). Accordingly, coadministration of Vosevi with efavirenz-containing antiretroviral regimen is not recommended. Clinical studies have shown that rifampicin also decreases the AUC of the PgP substrates digoxin and dabigatran etexilate [135]. Despite these changes, the regimen were well tolerated and did not elicit major adverse events.
7.2.3 Antivirals
Over 2.3 million individuals worldwide are co-infected with HCV and the human immunodeficiency virus (HIV) [136]. Because multiple drugs are administered in these patients there is a significant potential for DDIs that may complicate therapy.
Ritonavir and cobicistat are used as boosters to prolong the duration of action of HIV protease inhibitors by decreasing their elimination. Ritonavir elicits a wide range of DDIs by inhibiting multiple influx and efflux transporters and CYPs [137,138,139]. Cobicistat is a structural analogue of ritonavir that was introduced because it has a lower DDI potential [140,141,142]. However, despite a narrower range of targets, pharmacokinetic data indicate that the elimination of HCV DAAs may still be impaired by cobicistat-containing regimen.
Ritonavir-boosted HIV drug regimen disrupt Mavyret therapy [143]. Thus, in 12 subjects, coadministration of atazanavir/ritonavir (300/100 mg) increased exposure to glecaprevir (Cmax and AUC to 4.1- and 6.5-fold of respective control), but not pibrentasvir (Table 5; [47]). Similarly, lopinavir/ritonavir (400/100 mg twice daily) also increased glecaprevir exposure (Cmax and AUC to 2.56- and 4.4-fold of control)[38], as did darunavir/ritonavir (800/100 mg once daily) to 3.1- and 5.0-fold of control (Table 5; [47]).
Ritonavir-containing regimen also increased exposure to grazoprevir and elbasvir, when administered as Zepatier. Thus, atazanavir/ritonavir (300 mg/100 mg once daily) increased grazoprevir (200 mg once daily) exposure (Cmax and AUC to 10.6 and 6.2-fold of control) in 12 subjects and elbasvir (50 mg once daily) exposure in 10 subjects (Cmax and AUC to 4.8- and 4.2-fold of control; Table 5) [48, 62]. Similarly, in 10 subjects, lopinavir/ritonavir (400/100 mg twice daily) increased elbasvir (50 mg once daily) exposure (Cmax and AUC were altered to 3.7- and 2.9-fold of respective control) and exposure to grazoprevir (200 mg once daily) in 13 patients (Cmax and AUC to 12.9- and 7.3-fold of respective control) [48, 62]. Ritonavir (100 mg) with darunavir (600 mg twice daily) also increased grazoprevir (200 mg once daily) exposure in 13 patients (Cmax and AUC) to 7.5- and 5.3-fold of control, but increases in exposure to elbasvir (50 mg once daily) in 10 patients were much less pronounced [48, 62]. Glecaprevir and grazoprevir are substrates of CYP3A4, OATP1B1/1B3 and PgP, while pibrentasvir and elbasvir are PgP substrates, but are minimally metabolised by CYP3A4. Thus, it is feasible that potential DDIs could be due to inhibition of OATP1B1/3, PgP or CYP3A4 by ritonavir.
Ritonavir in combination with other antiretrovirals also increased HCV DAA exposure. Thus, the coadministration of ritonavir/darunavir/emtricitabine/tenofovir disoproxil fumarate (100/800/200/300 mg) or atazanavir/ritonavir (300/100 mg) with Vosevi (single dose) increased the AUC of voxilaprevir to 2.4- and 4.3-fold of control, respectively, which could be due to inhibition of OATP1B1/1B3, PgP, and/or CYP3A. In combination with atazanavir/emtricitabine/tenofovir disoproxil fumarate (300/200/300 mg once daily), ritonavir (100 mg) moderately increased velpatasvir (100 mg daily) exposure (Cmax and AUC) to 1.6- and 2.4-fold of control, respectively (Table 5; [39]). This mechanism likely involves transporter inhibition because velpatasvir is a substrate of OATP1B1/1B3, PgP and BCRP and both atazanavir and ritonavir are established inhibitors of these transporters [144]. Ritonavir-containing regimen also increased sofosbuvir and GS-331007 exposure, which is probably due to inhibition of BCRP and/or PgP [86]. It was suggested that coadministration of sofosbuvir with tipranavir/ritonavir would likely decrease the serum concentrations of sofosbuvir, leading to reduced therapeutic effect; the use of this combination is not recommended [145].
Cobicistat, in combination with elvitegravir, emtricitabine and tenofovir, increased the Cmax and AUC of glecaprevir (300 mg once daily) to 2.5- and 3.1-fold of respective control, while pibrentasvir (120 mg once daily) exposure was unchanged [47]. Similarly, in the case of Zepatier, grazoprevir exposure (Cmax and AUC) was increased markedly by the elvitegravir, emtricitabine and tenofovir combination to 5.4- and 4.6-fold of control, while elbasvir exposure was only slightly increased (Cmax and AUC to 2.2- and 1.9-fold of control; Table 5; [48, 62]). Increased grazoprevir exposure could be related to inhibition by cobicistat of OATP1B-mediated influx of the DAA into tissues [144].
Cobicistat/elvitegravir/emtricitabine/tenofovir alafenamide also increased exposure (Cmax and AUC) to the NS3/4A protease inhibitor voxilaprevir (100 mg once daily) to 1.9- and 2.7-fold of control (Table 5; [39]). Voxilaprevir is a substrate for OATP1B1/1B3, PgP and BCRP, while elvitegravir, like cobicistat, is an inhibitor of PgP and, to a lesser extent, BCRP [146]. Emtricitabine is a substrate for several transporters including BCRP [147] and is an inhibitor of OCT2 [148] and MRPs 1/2/3 [149]. Thus, cobicistat, emtricitabine or elvitegravir could contribute to increased exposure produced by the antiretroviral combination by competing for multiple transporters.
Ritonavir is also an inducer when used in multiple-dose regimen because it activates the PXR pathway [150]. The combination of ritonavir and lopinavir (100/400 mg twice daily) decreased daclatasvir exposure (Cmax and AUC) to 0.34- and 0.58-fold of respective control (Table 5; [41]). Similarly, darunavir/ritonavir (800 mg/100 mg daily) also decreased daclatasvir exposure (Cmax to 0.38- and AUC to 0.70-fold of control; [41]). To date, there is no evidence that cobicistat activates the PXR pathway.
Liver dysfunction may occur in some patients who receive Mavyret or Zepatier [151]. The US FDA-approved prescribing information recommends caution when these drugs are combined with certain HIV antiretroviral agents because of altered serum concentrations of the HCV DAAs. Thus, coadministration with atazanavir is contraindicated. Similarly, coadministration of Mavyret with darunavir, lopinavir, ritonavir and efavirenz is not recommended [38] and the concurrent use of Zepatier with darunavir, lopinavir, saquinavir and tipranavir is contraindicated due to possible increases in alanine transaminase consistent with liver dysfunction [40]. While coadministration of Vosevi with atazanavir, lopinavir, tipranavir, ritonavir or efavirenz is also not recommended, its combination with tenofovir disoproxil fumarate should be accompanied by monitoring for serum tenofovir concentrations [39].
7.2.4 Ketoconazole and Omeprazole
CYP3A4 inhibitors may increase exposure to HCV DAAs that undergo biotransformation by the enzyme (Table 5). Thus, ketoconazole (400 mg once daily) increased the AUCs of grazoprevir (100 mg single dose) and daclatasvir (10 mg) to 3-fold of control [41, 152, 153]. In contrast, clinically significant interactions were not reported with other HCV DAAs. Daclatasvir plasma concentrations were also reportedly increased by other CYP3A inhibitors, such as erythromycin and itraconazole, which may require possible dose adjustments [118]. Indeed, it has been suggested that, to prevent increased exposure, the dose of daclatasvir should be halved when potent inhibitors of CYP3A4 are coadministered [41].
Omeprazole decreases exposure to glecaprevir (Cmax to 0.36- and AUC to 0.49-fold of control) [47], and velpatasvir to 0.49- and 0.49-fold of control, respectively (Table 5; [39]). The precise mechanism is unclear but, because the time-frame over which this interaction occurred is short, it is unlikely to reflect induction. It is feasible that omeprazole alters DAA absorption after oral administration. However, no clinically relevant changes were reported with other HCV DAAs.
7.3 Drugs that do not Mediate Clinically Significant Interactions with HCV DAAs
During the course of HCV DAA drug development and clinical evaluation, a large number of other agents have been evaluated for the propensity to elicit pharmacokinetic DDIs. Based on such studies conducted with Vosevi or its components, no DDIs of potential clinical significance were observed with darunavir, elvitegravir, emtricitabine, ethinyl estradiol/norgestimate, gemfibrozil, rilpivirine, tenofovir alafenamide or voriconazole (Vosevi), dolutegravir, ketoconazole or raltegravir (Epclusa) and darunavir/ritonavir, efavirenz, emtricitabine, methadone, oral contraceptives, raltegravir, rilpivirine, tenofovir disoproxil fumarate, methadone or tacrolimus (Sovaldi) [39, 60, 145].
Dosage adjustments are not required when Mavyret is coadministered with abacavir, amlodipine, buprenorphine, caffeine, dextromethorphan, dolutegravir, emtricitabine, felodipine, lamivudine, lamotrigine, losartan, methadone, midazolam, naloxone, norethindrone or other progestin-only contraceptives, omeprazole, raltegravir, rilpivirine, tacrolimus, tenofovir alafenamide, tenofovir disoproxil fumarate, tolbutamide or valsartan [38]. Similarly, dose adjustments are not required when Zepatier is used with proton pump inhibitors, H2-receptor antagonists, antacids, buprenorphine/naloxone, digoxin, dolutegravir, methadone, mycophenolate mofetil, oral contraceptives, phosphate binders, pitavastatin, pravastatin, prednisone, raltegravir, ribavirin, rilpivirine or tenofovir disoproxil fumarate [40]. Clinically relevant DDIs are not expected when Zepatier is co-administered with abacavir, emtricitabine, entecavir, and lamivudine.
In the case of Daklinza, clinically relevant interactions were not anticipated with peginterferon-α, ribavirin, rilpivirine or antacids. Clinically relevant alterations in daclatasvir exposure were not observed with cyclosporin, darunavir/ritonavir, dolutegravir, escitalopram, ethinyl estradiol/norgestimate, lopinavir/ritonavir, methadone, midazolam, tacrolimus or tenofovir. Dosage modifications with daclatasvir are not required with coadministered darunavir/cobicistat or moderate CYP3A inhibitors, including atazanavir, fosamprenavir, ciprofloxacin, diltiazem, erythromycin, fluconazole, or verapamil [41].
In the case of Harvoni, DDIs of potential clinical significance did not occur with abacavir, cyclosporin, efavirenz, emtricitabine, lamivudine, methadone, oral contraceptives, pravastatin, raltegravir, rilpivirine, tacrolimus, tenofovir disoproxil fumarate, or verapamil [42].
8 Summary and Conclusions
Current HCV DAAs elicit fewer DDIs and appear safer than the original DAA drugs. Nevertheless, there is evidence that the agents currently approved by the US FDA and EMA also have the potential to elicit serious DDIs and toxicities. An understanding of the mechanisms of these DDIs is critical in avoiding the use of potentially hazardous drug combinations in patients. Thus, detailed information on the drug-metabolising enzymes and transporters that determine HCV DAA pharmacokinetics is essential. Most of the currently accessible information is contained in documents approved by regulatory agencies, based on data provided by pharmaceutical manufacturers. These documents focus on major potential preclinical DDI mechanisms and also provide important follow-up assessments of clinical relevance in healthy volunteers. However, additional evaluations of whether DAAs are substrates for a wider range of drug-metabolising enzymes and transporters are now warranted, as are more detailed estimations of inhibitory data (e.g., IC50 values) against other CYPs and transporters.
Agents that are potential inhibitors or inducers of drug metabolising enzymes or transporters may alter the systemic exposure to coadministered drugs [90, 97, 101]. Regulatory agencies recommend the use of pharmacokinetic modelling to assess the DDI potential of new drugs. As stated in the US FDA guidance to industry, modelling approaches can help translate in vitro observations into in vivo predictions of potential clinical DDIs [90]. Unbound serum concentrations of HCV DAAs are extremely low relative to their apparent inhibitory potencies against enzymes and transporters. However, using the US FDA-recommended cut-off of R≥1.1, a number of potential systemic DDIs could be clinically significant (Table 3). Some DDIs could also be mediated in the intestine where the local concentrations of HCV DAAs after oral administration may be high. Indeed, calculated Igut/IC50 values greatly exceeded the proposed cut-off value of 10 for a number of DAAs (Table 3).
As suggested by Kiang, additional pharmacokinetic studies analysed statistically would also be helpful to more fully evaluate the DDI potential of DAAs in target populations [58]. More information would assist the interpretation of the mechanisms by which HCV DAAs are victim drugs in DDIs. In most cases, the only available information is altered pharmacokinetic exposure (AUC and Cmax). Thus, the provision of more detailed pharmacokinetic information, including clearance and half-life, could provide greater insight into whether clinical DDI mechanisms involve inhibition or induction of drug-metabolising enzymes or transporters in vivo. While it has previously been difficult to discriminate between altered bioavailability and altered clearance, Sodhi and Benet recently proposed a method by which this might be achieved [154]. It was noted that such an approach could assist DDI interpretation during drug development and regulatory evaluation.
Even though the available data are valuable for predictions of clinically relevant scenarios, pharmacokinetic-pharmacodynamic relationships for HCV DAAs have not always been evaluated. Potential DDIs have rarely been studied in target populations. Indeed, pathophysiological regulation of drug-metabolising enzymes and transporters is an important consideration for the safety and use of many drugs, including HCV DAAs. Thus, it is established that cirrhosis and other forms of chronic liver disease may be characterised by fibrosis, chronic lipid infiltration, portosystemic shunting and increased activity of proinflammatory mediators that dysregulates multiple CYPs and transporters, which impairs drug metabolism and transport activity [28,29,30,31,32]. Altered nutrition and diet are other factors that may alter the expression of drug-metabolising enzymes and transporters [35,36,37]. Because HCV DAAs are used in individuals with chronic liver injury, who may also have impaired nutritional status, it is now important to assess how disease alters the metabolism and transport of the drugs. Moreover, increased exposure with DAAs like Mavyret or Zepatier could exacerbate the hepatic impairment that is already present [40, 151].
Comorbidities are also potentially important for the propensity of DDIs. Many individuals worldwide are coinfected with HCV and HIV [136]. Because multiple drugs are used in these patients the potential for DDIs that may complicate therapy is high. The US FDA-approved prescribing information recommends caution with certain HIV antiretroviral agents because of increased serum concentrations of HCV agents. Coadministration of Mavyret with darunavir, lopinavir, ritonavir and efavirenz or coadministration of Vosevi with atazanavir, lopinavir, efavirenz, tipranavir and ritonavir is not recommended [38, 39]. Coadministration of Harvoni with tipranavir/ritonavir is not recommended due to decreases in the concentration of ledipasvir and sofosbuvir, leading to decreased therapeutic effect [42]. Because of potential toxicity, serum monitoring of tenofovir is recommended when the drug is used in combination with Vosevi [39] and possibly Harvoni [42]. The use of alternate HCV DAAs or antiretroviral agents may avoid potential increases in tenofovir exposures.
Individual patient factors may also influence the incidence of DDIs and associated drug safety. Single nucleotide polymorphisms (SNPs) and other polymorphisms are prevalent in the genes that encode and regulate many drug-metabolising enzymes and transporters [14, 43,44,45]. Some polymorphisms are more common in certain ethnic populations, so that altered metabolism and transport may be more common when HCV DAAs are used in these groups [14, 43,44,45]. How pharmacogenomic factors influence DDIs with HCV DAAs is an underexplored area that should be evaluated more fully in future.
References
Petruzziello A, Marigliano S, Loquercio G, Cozzolino A, Cacciapuoti C. Global epidemiology of hepatitis C virus infection: An up-date of the distribution and circulation of hepatitis C virus genotypes. World J Gastroenterol. 2016;22:7824–40. https://doi.org/10.3748/wjg.v22.i34.7824.
Lanini S, Easterbrook PJ, Zumla A, Ippolito G. Hepatitis C: global epidemiology and strategies for control. Clin Microbiol Infect. 2016;22:833–8. https://doi.org/10.1016/j.cmi.2016.07.035.
Cacoub P, Sadoun D. Extrahepatic manifestations of chronic HCV infection. N Engl J Med. 2021;384:1038–52. https://doi.org/10.1056/NEJMra2033539.
Zappulo E, Scotto R, Buonomo AR, Maraolo AE, Pinchera B, Gentile I. Efficacy and safety of a fixed dose combination tablet of asunaprevir + beclabuvir + daclatasvir for the treatment of Hepatitis C. Exp Opin Pharmacother. 2020;21:261–73. https://doi.org/10.1080/14656566.2019.1697674.
Dubuisson J. Hepatitis C virus proteins. World J Gastroenterol. 2007;13:2406–15. https://doi.org/10.3748/wjg.v13.i17.2406.
Gouklani H, Bull RA, Beyer C, Coulibaly F, Gowans EJ, Drummer HE, Netter HJ, White PA, Haqshenas G. Hepatitis C virus nonstructural protein 5B is involved in virus morphogenesis. J Virol. 2012;86:5080–8. https://doi.org/10.1128/JVI.07089-11.
Simmonds P, Bukh J, Combet C, Deléage G, Enomoto N, Feinstone S, Halfon P, Inchauspé G, Kuiken C, Maertens G, Mizokami M, Murphy DG, Okamoto H, Pawlotsky JM, Penin F, Sablon E, Shin-I T, Stuyver LJ, Thiel HJ, Viazov S, Weiner AJ, Widell A. Consensus proposals for a unified system of nomenclature of hepatitis C virus genotypes. Hepatology. 2005;42:962–73. https://doi.org/10.1002/hep.20819.
Simmonds P. Genetic diversity and evolution of hepatitis C virus–15 years on. J Gen Virol. 2004;85:3173–88. https://doi.org/10.1099/vir.0.80401-0.
Zeuzem S, Andreone P, Pol S, Lawitz E, Diago M, Roberts S, Focaccia R, Younossi Z, Foster GR, Horban A, Ferenci P, Nevens F, Müllhaupt B, Pockros P, Terg R, Shouval D, van Hoek B, Weiland O, Van Heeswijk R, De Meyer S, Luo D, Boogaerts G, Polo R, Picchio G, Beumont M. Telaprevir for retreatment of HCV infection. N Engl J Med. 2011;364:2417–28. https://doi.org/10.1056/NEJMoa1013086.
Ciesek S, von Hahn T, Manns MP. Second-wave protease inhibitors: choosing an heir. Clin Liver Dis. 2011;15:597–609. https://doi.org/10.1016/j.cld.2011.05.014.
Nakanishi T, Tamai I. Solute carrier transporters as targets for drug delivery and pharmacological intervention for chemotherapy. J Pharm Sci. 2011;100:3731–50. https://doi.org/10.1002/jps.22576.
https://www.proteinatlas.org (Accessed 18 April 2023).
Robertson EE, Rankin GO. Human renal organic anion transporters: characteristics and contributions to drug and drug metabolite excretion. Pharmacol Ther. 2006;109:399–412. https://doi.org/10.1016/j.pharmthera.2005.07.005.
Zanger UM, Schwab M. Cytochrome P450 enzymes in drug metabolism: regulation of gene expression, enzyme activities, and impact of genetic variation. Pharmacol Ther. 2013;138:103–41. https://doi.org/10.1016/j.pharmthera.2012.12.007.
Rendic S, Guengerich FP. Survey of human oxidoreductases and cytochrome P450 enzymes involved in the metabolism of xenobiotic and natural chemicals. Chem Res Toxicol. 2015;28:38–42. https://doi.org/10.1021/tx500444e.
Kato M. Intestinal first-pass metabolism of CYP3A4 substrates. Drug Metab Pharmacokinet. 2008;23:87–94. https://doi.org/10.2133/dmpk.23.87.
Szakács G, Váradi A, Ozvegy-Laczka C, Sarkadi B. The role of ABC transporters in drug absorption, distribution, metabolism, excretion and toxicity (ADME-Tox). Drug Discov Today. 2008;13:379–93. https://doi.org/10.1016/j.drudis.2007.12.010.
Schinkel AH, Jonker JW. Mammalian drug efflux transporters of the ATP binding cassette (ABC) family: an overview. Adv Drug Deliv Rev. 2003;55:3–29. https://doi.org/10.1016/s0169-409x(02)00169-2.
Vander Borght S, Libbrecht L, Katoonizadeh A, van Pelt J, Cassiman D, Nevens F, Van Lommel A, Peterson BE, Fevery J, Jansen PL, Roskams TA. Breast cancer resistance protein (BCRP/ABCG2) is expressed by progenitor cells/reactive ductules and hepatocytes and its expression pattern is influenced by disease etiology and species type: possible functional consequences. J Histochem Cytochem. 2006;54:1051–9. https://doi.org/10.1369/jhc.5A6912.2006.
Rodríguez-Antona C, Bort R, Jover R, Tindberg N, Ingelman-Sundberg M, Gómez-Lechón MJ, Castell JV. Transcriptional regulation of human CYP3A4 basal expression by CCAAT enhancer-binding protein-α and hepatocyte nuclear factor-3β. Mol Pharmacol. 2003;63:1180–9. https://doi.org/10.1124/mol.63.5.1180.
Waxman DJ. P450 gene induction by structurally diverse xenochemicals: central role of nuclear receptors CAR, PXR, and PPAR. Arch Biochem Biophys. 1999;369:11–23. https://doi.org/10.1006/abbi.1999.1351.
Maglich JM, Stoltz CM, Goodwin B, Hawkins-Brown D, Moore JT, Kliewer SA. Nuclear pregnane X receptor and constitutive androstane receptor regulate overlapping but distinct sets of genes involved in xenobiotic detoxification. Mol Pharmacol. 2002;62:638–46. https://doi.org/10.1124/mol.62.3.638.
Roberts EA, Johnson KC, Harper PA, Okey AB. Characterization of the Ah receptor mediating aryl hydrocarbon hydroxylase induction in the human liver cell line Hep G2. Arch Biochem Biophys. 1990;276:442–50. https://doi.org/10.1016/0003-9861(90)90743-i.
Kipp H, Arias IM. Trafficking of canalicular ABC transporters in hepatocytes. Annu Rev Physiol. 2002;64:595–608. https://doi.org/10.1146/annurev.physiol.64.081501.155793.
Murray M, Zhou F. Trafficking and other regulatory mechanisms for Organic anion transporting polypeptides (OATPs) and Organic Anion transporters (OATs) that modulate cellular drug and xenobiotic influx and that are dysregulated in disease. Br J Pharmacol. 2017;174:1908–24. https://doi.org/10.1111/bph.13785.
Drozdzik M, Lapczuk-Romanska J, Wenzel C, Skalski L, Szelag-Pieniek S, Post M, Parus A, Syczewska M, Kurzawski M, Oswald S. Protein abundance of drug metabolizing enzymes in human hepatitis C livers. Int J Mol Sci. 2023;24:4543. https://doi.org/10.3390/ijms24054543.
Pugh RN, Murray-Lyon IM, Dawson JL, Pietroni MC, Williams R. Transection of the oesophagus for bleeding oesophageal varices. Br J Surg. 1973;60:646–9. https://doi.org/10.1002/bjs.1800600817.
Guengerich FP, Turvy CG. Comparison of levels of several human microsomal cytochrome P-450 enzymes and epoxide hydrolase in normal and disease states using immunochemical analysis of surgical liver samples. J Pharmacol Exp Ther. 1991;256:1189–94.
George J, Murray M, Byth K, Farrell GC. Differential alterations of cytochrome P450 proteins in livers from patients with severe chronic liver disease. Hepatology. 1995;21:120–8. https://doi.org/10.1016/0270-9139(95)90418-2.
Hardwick RN, Fisher CD, Canet MJ, Scheffer GL, Cherrington NL. Variations in ATP-binding cassette transporter regulation during the progression of human nonalcoholic fatty liver disease. Drug Metab Dispos. 2011;39:2395–402. https://doi.org/10.1124/dmd.111.041012.
Merrell MD, Cherrington NJ. Drug metabolism alterations in nonalcoholic fatty liver disease. Drug Metab Rev. 2011;43:317–34. https://doi.org/10.3109/03602532.2011.577781.
Thakkar N, Slizgi JR, Brouwer KLR. Effect of liver disease on hepatic transporter expression and function. J Pharm Sci. 2017;106:2282–94. https://doi.org/10.1016/j.xphs.2017.04.053.
Murray M, Zaluzny L, Dannan GA, Guengerich FP, Farrell GC. Altered regulation of cytochrome P-450 enzymes in choline-deficient cirrhotic male rat liver: Impaired regulation and activity of the male-specific androst-4-ene-17-dione 16α-hydroxylase, cytochrome P-450UT-A, in hepatic cirrhosis. Mol Pharmacol. 1987;31:117–21.
Murray M, Zaluzny L, Farrell GC. Impaired androgen 16α-hydroxylation in hepatic microsomes from carbon tetrachloride-cirrhotic male rats. Gastroenterology. 1987;93:141–7. https://doi.org/10.1016/0016-5085(87)90326-x.
Ioannides C. Effect of diet and nutrition on the expression of cytochromes P450. Xenobiotica. 1999;29:109–54. https://doi.org/10.1080/004982599238704.
Alvarez AI, Real R, Pérez M, Mendoza G, Prieto JG, Merino G. Modulation of the activity of ABC transporters (P-glycoprotein, MRP2, BCRP) by flavonoids and drug response. J Pharm Sci. 2010;99:598–617. https://doi.org/10.1002/jps.21851.
Guo Y, Cui JY, Lu H, Klaassen CD. Effect of nine diets on xenobiotic transporters in livers of mice. Xenobiotica. 2015;45:634–41. https://doi.org/10.3109/00498254.2014.1001009.
US Food and Drug Administration. Highlights of prescribing information: Mavyret. 2023. https://www.accessdata.fda.gov/drugsatfda_docs/label/2017/209394s000lbl.pdf. (Accessed 22 April 2023).
US Food and Drug Administration. Highlights of prescribing information: Vosevi. 2023. https://www.accessdata.fda.gov/drugsatfda_docs/label/2017/209195s000lbl.pdf. (Accessed 22 April 2023).
US Food and Drug Administration. Highlights of prescribing information: Zepatier. 2023. https://www.accessdata.fda.gov/drugsatfda_docs/label/2017/208261s002lbl.pdf. (Accessed 22 April 2023).
US Food and Drug Administration. Highlights of prescribing information: Daklinza. 2023. https://www.accessdata.fda.gov/drugsatfda_docs/label/2017/206843s006lbl.pdf. (Accessed 22 April 2023).
US Food and Drug Administration. Highlights of prescribing information: Harvoni. 2023. https://www.accessdata.fda.gov/drugsatfda_docs/label/2017/205834s017lbl.pdf. (Accessed 22 April 2023).
Bertilsson L. Geographical/interracial differences in polymorphic drug oxidation. Current state of knowledge of cytochromes P450 (CYP) 2D6 and 2C19. Clin Pharmacokinet. 1995;29:192–209. https://doi.org/10.2165/00003088-199529030-00005.
Ito S, Ieiri I, Tanabe M, Suzuki A, Higuchi S, Otsubo K. Polymorphism of the ABC transporter genes, MDR1, MRP1 and MRP2/cMOAT, in healthy Japanese subjects. Pharmacogenetics. 2001;11:175–84. https://doi.org/10.1097/00008571-200103000-00008.
Zhou F, Zhu L, Wang K, Murray M. Recent advance in the pharmacogenomics of human Solute Carrier Transporters (SLCs) in drug disposition. Adv Drug Deliv Rev. 2017;116:21–36. https://doi.org/10.1016/j.addr.2016.06.004.
Nozaki Y, Izumi S. Preincubation time-dependent, long-lasting inhibition of drug transporters and impact on the prediction of drug-drug interactions. Drug Metab Dispos. 2023. https://doi.org/10.1124/dmd.122.000970.
Pharmaceuticals and Medical Devices Agency (Japan). Review Report: Maviret. 2023. https://pmda.go.jp/files/000230308.pdf. (Accessed 20 April 2023).
Pharmaceuticals and Medical Devices Agency (Japan). Review Report: Grazoprevir. 2023. https://pmda.go.jp/files/000224902.pdf. (Accessed 20 April 2023).
https://www.tga.gov.au/sites/default/files/auspar-sofosbuvir-velpatasvir-voxilaprevir-190304.pdf (Accessed 18 April 2023).
Lin CW, Dutta S, Ding B, Wang T, Zadeikis N, Asatryan A, Kort J, Campbell A, Podsadecki T, Liu W. Pharmacokinetics, safety, and tolerability of glecaprevir and pibrentasvir in healthy White, Chinese, and Japanese adult subjects. J Clin Pharmacol. 2017;57:1616–24. https://doi.org/10.1002/jcph.959.
Lin CW, Dutta S, Asatryan A, Chiu YL, Wang H, Clifton J 2nd, Campbell A, Liu W. Pharmacokinetics, safety, and tolerability of single and multiple doses of ABT-493: a first-in-human study. J Pharm Sci. 2017;106:645–51. https://doi.org/10.1016/j.xphs.2016.10.007.
Smolders EJ, Jansen AME, Ter Horst PGJ, Rockstroh J, Back DJ, Burger DM. Viral hepatitis C therapy: pharmacokinetic and pharmacodynamic considerations—a 2019 update. Clin Pharmacokinet. 2019;58:1237–63. https://doi.org/10.1007/s40262-019-00774-0.
https://clinicaltrials.gov/ct2/show/NCT02533427 (Accessed 17 April 2023).
European Medicines Agency. Vosevi: assessment report. 2023. https://www.ema.europa.eu/en/documents/assessment-report/voxevi-epar-public-assessment-report_en.pdf. (Accessed 23 April 2023).
Brainard DM, Petry A, Anderson MS, Mitselos A, Laethem T, Heirman I, Caro L, Stone JA, Sun P, Panorchan P, Van Bortel LM, Iwamoto M, Wagner JA. Safety, tolerability, and pharmacokinetics after single and multiple doses of MK-5172, a novel HCV NS3/4A protease inhibitor with potent activity against known resistance mutants, in healthy subjects. Hepatology. 2010;52:1216A-A1217.
Summa V, Ludmerer SW, McCauley JA, Fandozzi C, Burlein C, Claudio G, Coleman PJ, Dimuzio JM, Ferrara M, Di Filippo M, Gates AT, Graham DJ, Harper S, Hazuda DJ, Huang Q, McHale C, Monteagudo E, Pucci V, Rowley M, Rudd MT, Soriano A, Stahlhut MW, Vacca JP, Olsen DB, Liverton NJ, Carroll SS. MK-5172, a selective inhibitor of hepatitis C virus NS3/4A protease with broad activity across genotypes and resistant variants. Antimicrob Agents Chemother. 2012;56:4161–7. https://doi.org/10.1128/AAC.00324-12.
Pharmaceuticals and Medical Devices Agency (Japan). Review Report: Harvoni. 2023. https://pmda.go.jp/files/000215262.pdf. (Accessed 20 April 2023).
Kiang TKL. Clinical pharmacokinetics and drug-drug interactions of elbasvir/grazoprevir. Eur J Drug Metab Pharmacokinet. 2018;43:509–31. https://doi.org/10.1007/s13318-018-0471-0.
Pharmaceuticals and Medical Devices Agency (Japan). Review Report: Daklinza. 2023. https://pmda.go.jp/files/000209021.pdf. (Accessed 20 April 2023).
US Food and Drug Administration. Highlights of prescribing information: Epclusa. 2016. https://www.accessdata.fda.gov/drugsatfda_docs/label/2016/208341s000lbl.pdf. (Accessed 9 July 2023).
Eley T, Han YH, Huang SP, He B, Li W, Bedford W, Stonier M, Gardiner D, Sims K, Rodrigues AD, Bertz RJ. Organic anion transporting polypeptide-mediated transport of, and inhibition by, asunaprevir, an inhibitor of hepatitis C virus NS3 protease. Clin Pharmacol Ther. 2015;97:159–66. https://doi.org/10.1002/cpt.4.
Pharmaceuticals and Medical Devices Agency (Japan). Review Report: Elbasvir. 2023. https://pmda.go.jp/files/000224903.pdf. (Accessed 20 April 2023).
Li W, Zhao W, Liu X, Huang X, Lopez OD, Leet JE, Fancher RM, Nguyen V, Goodrich J, Easter J, Hong Y, Caceres-Cortes J, Chang SY, Ma L, Belema M, Hamann LG, Gao M, Zhu M, Shu YZ, Humphreys WG, Johnson BM. Biotransformation of daclatasvir in vitro and in nonclinical species: Formation of the main metabolite by pyrrolidine δ-oxidation and rearrangement. Drug Metab Dispos. 2016;44:809–20. https://doi.org/10.1124/dmd.115.068866.
Murray M. Complexation of cytochrome P-450 isoenzymes in hepatic microsomes from SKF 525-A-induced rats. Arch Biochem Biophys. 1988;262:381–8. https://doi.org/10.1016/0003-9861(88)90202-0.
Murray M, Field SL. Inhibition and metabolite complexation of rat hepatic microsomal cytochrome P450 by tricyclic antidepressants. Biochem Pharmacol. 1992;43:2065–71. https://doi.org/10.1016/0006-2952(92)90163-d.
Murray M, Wilkinson CF, Marcus C, Dubé CE. Structure-activity relationships in the interactions of alkoxymethylenedioxybenzene derivatives with rat hepatic microsomal mixed-function oxidases in vivo. Mol Pharmacol. 1983;24:129–36.
Marcus CB, Murray M, Wilkinson CF. Spectral and inhibitory interactions of methylenedioxyphenyl and related compounds with purified isozymes of cytochrome P-450. Xenobiotica. 1985;15:351–62. https://doi.org/10.3109/00498258509045370.
Murray M, Butler AM, Stupans I. Competitive inhibition of human liver microsomal P450 3A-dependent steroid 6β-hydroxylation activity by cyclophosphamide and ifosfamide in vitro. J Pharmacol Exp Ther. 1994;270:645–9.
Moreno RL, Kent UM, Hodge K, Hollenberg PF. Inactivation of cytochrome P450 2E1 by benzyl isothiocyanate. Chem Res Toxicol. 1999;12:582–7. https://doi.org/10.1021/tx9900019.
Ortiz de Montellano PR, Kunze KL, Beilan HS, Wheeler C. Destruction of cytochrome P-450 by vinyl fluoride, fluroxene, and acetylene. Evidence for a radical intermediate in olefin oxidation. Biochemistry. 1982;21:1331-9. doi: https://doi.org/10.1021/bi00535a035.
Wilkinson CF, Hetnarski K, Yellin TO. Imidazole derivatives–a new class of microsomal enzyme inhibitors. Biochem Pharmacol. 1972;21:3187–92. https://doi.org/10.1016/0006-2952(72)90147-5.
Mogalian E, German P, Kearney BP, Yang CY, Brainard D, McNally J, Moorehead L, Mathias A. Use of multiple probes to assess transporter- and cytochrome P450-mediated drug-drug interaction potential of the pangenotypic HCV NS5A inhibitor velpatasvir. Clinical Pharmacokinet. 2016;55:605–13. https://doi.org/10.1007/s40262-015-0334-7.
https://clinicaltrials.gov/ct2/show/NCT02002767 (Accessed 17 April 2023).
Soriano V, Benítez-Gutiérrez L, Arias A, Carrasco I, Barreiro P, Peña JM, de Mendoza C. Evaluation of sofosbuvir, velpatasvir plus voxilaprevir as fixed-dose co-formulation for treating hepatitis C. Expert Opin Drug Metab Toxicol. 2017;13:1015–22. https://doi.org/10.1080/17425255.2017.1359254.
Li C, Li X, Zhu X, Zhang H, Shen G, Kersey K, Ding Y. Pharmacokinetics, safety, and tolerability of ledipasvir/sofosbuvir and sofosbuvir/velpatasvir in healthy Chinese subjects. Clin Ther. 2020;42:448–57. https://doi.org/10.1016/j.clinthera.2020.01.013.
German P, Mathias A, Brainard DM, Kearney BP. Drug-drug interaction profile of the fixed-dose combination tablet regimen ledipasvir/sofosbuvir. Clin Pharmacokinet. 2018;57:1369–83. https://doi.org/10.1007/s40262-018-0654-5.
Ankrom W, Sanchez RI, Yee KL, Fan L, Mitra P, Wolford D, Triantafyllou I, Sterling LM, Stoch SA, Iwamoto M, Khalilieh S. Investigation of pharmacokinetic interactions between doravirine and elbasvir-grazoprevir and ledipasvir-sofosbuvir. Antimicrob Agents Chemother. 2019;63:e02491-e2518. https://doi.org/10.1128/AAC.02491-18.
https://clinicaltrials.gov/ct2/show/NCT01937975 (Accessed 17 April 2023).
https://clinicaltrials.gov/ct2/show/NCT01797536 (Accessed 17 April 2023).
Cheung TT, Chiu JWY, Yuen MF, Lam KSL, Cheung BMY, Feng HP, Yeh WW, Wang J, Li W, Zhao XM, Wang Z, Mu S. A Phase I, single- and multiple-dose study to evaluate the pharmacokinetics of elbasvir and grazoprevir in healthy Chinese participants. Clin Ther. 2018;40:719-732.e1. https://doi.org/10.1016/j.clinthera.2018.03.014.
https://clinicaltrials.gov/ct2/show/NCT01830205 (Accessed 17 April 2023).
Esposito I, Marciano S, Trinks J. Pharmacokinetic and pharmacodynamic evaluation of daclatasvir, asunaprevir plus beclabuvir as a fixed-dose co-formulation for the treatment of hepatitis C. Expert Opin Drug Metab Toxicol. 2018;14:649–57. https://doi.org/10.1080/17425255.2018.1483336.
Pharmaceuticals and Medical Devices Agency (Japan). Review Report: Ximency. 2023. https://pmda.go.jp/files/000226231.pdf. (Accessed 20 April 2023).
https://clinicaltrials.gov/ct2/show/NCT00859053 (Accessed 17 April 2023).
Kirby BJ, Symonds WT, Kearney BP, Mathias AA. Pharmacokinetic, pharmacodynamic, and drug-interaction profile of the hepatitis C virus NS5B polymerase inhibitor sofosbuvir. Clin Pharmacokinet. 2015;54:677–90. https://doi.org/10.1007/s40262-015-0261-7.
King JR, Dutta S, Cohen D, Podsadecki TJ, Ding B, Awni WM, Menon RM. Drug-Drug Interactions between sofosbuvir and ombitasvir-paritaprevir-ritonavir with or without dasabuvir. Antimicrob Agents Chemother. 2015;60:855–61. https://doi.org/10.1128/AAC.01913-15.
Indolfi G, Kelly D, Nebbia G, Iorio R, Mania A, Giacomet V, Szenborn L, Shao J, Sang Yue M, Hsueh CH, Parhy B, Kersey K, Mangia A, Pawlowska M, Bansal S. Sofosbuvir-velpatasvir-voxilaprevir in adolescents 12 to 17 years old with HCV infection. Hepatology. 2022;76:445–55. https://doi.org/10.1002/hep.32393.
Cuenca-Lopez F, Rivero A, Rivero-Juárez A. Pharmacokinetics and pharmacodynamics of sofosbuvir and ledipasvir for the treatment of hepatitis C. Expert Opin Drug Metab Toxicol. 2017;13:105–12. https://doi.org/10.1080/17425255.2017.1255725.
Kiser JJ, Burton JR, Anderson PL, Everson GT. Review and management of drug interactions with boceprevir and telaprevir. Hepatology. 2012;55:1620–8. https://doi.org/10.1002/hep.25653.
US Food and Drug Administration. https://www.fda.gov/regulatory-information/search-fda-guidance-documents/clinical-drug-interaction-studies-cytochrome-p450-enzyme-and-transporter-mediated-drug-interactions. 2020. (Accessed 17 July 2023).
Yu J, Zhou Z, Tay-Sontheimer J, Levy RH, Ragueneau-Majlessi I. Intestinal drug interactions mediated by OATPs: a systematic review of preclinical and clinical findings. J Pharm Sci. 2017;106:2312–25. https://doi.org/10.1016/j.xphs.2017.04.004.
Ito K, Chiba K, Horikawa M, Ishigami M, Mizuno N, Aoki J, Gotoh Y, Iwatsubo T, Kanamitsu SI, Kato M, Kawahara I, Niinuma K, Nishino A, Sato N, Tsukamoto Y, Ueda K, Itoh T, Sugiyama Y. Which concentration of the inhibitor should be used to predict in vivo drug interactions from in vitro data? AAPS PharmSci. 2002;4:E25. https://doi.org/10.1208/ps040425.
Ito K, Iwatsubo T, Kanamitsu S, Ueda K, Suzuki H, Sugiyama Y. Prediction of pharmacokinetic alterations caused by drug-drug interactions: metabolic interaction in the liver. Pharmacol Rev. 1998;50:387–412.
Giacomini KM, Huang SM, Tweedie DJ, Benet LZ, Brouwer KLR, Chu X, Dahlin A, Evers R, Fischer V, Hillgren KM, Hoffmaster KA, Ishikawa T, Keppler D, Kim RB, Lee CA, Niemi M, Polli JW, Sugiyama Y, Swaan PW, Ware JA, Wright SH, Yee SW, Zamek-Gliszczynski MJ, Zhang L. Membrane transporters in drug development. Nat Rev Drug Discov. 2010;9:215–36. https://doi.org/10.1038/nrd3028.
Parkinson A. Regulatory recommendations for calculating the unbound maximum hepatic inlet concentration: a complicated story with a surprising and happy ending. Drug Metab Dispos. 2019;47:779–84. https://doi.org/10.1124/dmd.119.086496.
Kosloski MP, Bow DAJ, Kikuchi R, Wang H, Kim EJ, Marsh K, Mensa F, Kort J, Liu W. Translation of in vitro transport inhibition studies to clinical drug-drug interactions for glecaprevir and pibrentasvir. J Pharmacol Exp Ther. 2019;370:278–87. https://doi.org/10.1124/jpet.119.256966.
Yu J, Petrie ID, Levy RH, Ragueneau-Majlessi I. Mechanisms and clinical significance of pharmacokinetic-based drug-drug interactions with drugs approved by the US Food and Drug Administration in 2017. Drug Metab Dispos. 2019;47:135–44. https://doi.org/10.1124/dmd.118.084905.
European Medicines Agency. Maviret:assessment report. 2023. https://www.ema.europa.eu/en/documents/assessment-report/maviret-epar-public-assessment-report_en.pdf. (Accessed 23 April 2023).
Gandhi Y, Eley T, Fura A, Li W, Bertz RJ, Garimella T. Daclatasvir: A review of preclinical and clinical pharmacokinetics. Clinical Pharmacokinet. 2018;57:911–28. https://doi.org/10.1007/s40262-017-0624-3.
Kalgutkar AS, Soglia JR. Minimising the potential for metabolic activation in drug discovery. Expert Opin Drug Metab Toxicol. 2005;1:91–142. https://doi.org/10.1517/17425255.1.1.91.
Yu J, Zhou Z, Tay-Sontheimer J, Levy RH, Ragueneau-Majlessi I. Risk of clinically relevant pharmacokinetic-based drug-drug interactions with drugs approved by the US Food and Drug Administration between 2013 and 2016. Drug Metab Dispos. 2018;46:835–45. https://doi.org/10.1124/dmd.117.078691.
US Food and Drug Administration. Clinical Pharmacology and Biopharmaceutics Review: Vosevi. 2023. https://www.accessdata.fda.gov/drugsatfda_docs/nda/2017/209195Orig1s000ClinPharmR.pdf. (Accessed 12 July 2023).
Ho RH, Tirona RG, Leake BF, Glaeser H, Lee W, Lemke CJ, Wang Y, Kim RB. Drug and bile acid transporters in rosuvastatin hepatic uptake: function, expression, and pharmacogenetics. Gastroenterology. 2006;130:1793–806. https://doi.org/10.1053/j.gastro.2006.02.034.
Wang Q, Zheng M, Leil T. Investigating transporter-mediated drug-drug interactions using a physiologically based pharmacokinetic model of rosuvastatin. CPT Pharmacometrics Syst Pharmacol. 2017;6:228–38. https://doi.org/10.1002/psp4.12168.
Kivistö KT, Niemi M. Influence of drug transporter polymorphisms on pravastatin pharmacokinetics in humans. Pharm Res. 2007;24:239–47. https://doi.org/10.1007/s11095-006-9159-2.
Bogman K, Peyer AK, Török M, Küsters E, Drewe J. HMG-CoA reductase inhibitors and P-glycoprotein modulation. Br J Pharmacol. 2001;132:1183–92. https://doi.org/10.1038/sj.bjp.0703920.
Afrouzian M, Al-Lahham R, Patrikeeva S, Xu M, Fokina V, Fischer WG, Abdel-Rahman SZ, Costantine M, Ahmed MS, Nanovskaya T. Role of the efflux transporters BCRP and MRP1 in human placental bio-disposition of pravastatin. Biochem Pharmacol. 2018;156:467–78. https://doi.org/10.1016/j.bcp.2018.09.012.
Seithel A, Eberl S, Singer K, Auge D, Heinkele G, Wolf NB, Dörie F, Fromm MF, König J. The influence of macrolide antibiotics on the uptake of organic anions and drugs mediated by OATP1B1 and OATP1B3. Drug Metab Dispos. 2007;35:779–86. https://doi.org/10.1124/dmd.106.014407.
Vildhede A, Karlgren M, Svedberg EK, Wisniewski JR, Lai Y, Norén A, Artursson P. Hepatic uptake of atorvastatin: influence of variability in transporter expression on uptake clearance and drug-drug interactions. Drug Metab Dispos. 2014;42:1210–8. https://doi.org/10.1124/dmd.113.056309.
Kalliokoski A, Niemi M. Impact of OATP transporters on pharmacokinetics. Br J Pharmacol. 2009;158:693–705. https://doi.org/10.1111/j.1476-5381.2009.00430.x.
Goard CA, Mather RG, Vinepal B, Clendening JW, Martirosyan A, Boutros PC, Sharom FJ, Penn LZ. Differential interactions between statins and P-glycoprotein: implications for exploiting statins as anticancer agents. Int J Cancer. 2010;127:2936–48. https://doi.org/10.1002/ijc.25295.
Li J, Volpe DA, Wang Y, Zhang W, Bode C, Owen A, Hidalgo IJ. Use of transporter knockdown Caco-2 cells to investigate the in vitro efflux of statin drugs. Drug Metab Dispos. 2011;39:1196–202. https://doi.org/10.1124/dmd.111.038075.
Tornio A, Vakkilainen J, Neuvonen M, Backman JT, Neuvonen PJ, Niemi M. SLCO1B1 polymorphism markedly affects the pharmacokinetics of lovastatin acid. Pharmacogenet Genomics. 2015;25:382–7. https://doi.org/10.1097/FPC.0000000000000148.
Hochman JH, Pudvah N, Qiu J, Yamazaki M, Tang C, Lin JH, Prueksaritanont T. Interactions of human P-glycoprotein with simvastatin, simvastatin acid, and atorvastatin. Pharm Res. 2004;21:1686–91. https://doi.org/10.1023/b:pham.0000041466.84653.8c.
Ishiguro N, Kishimoto W, Volz A, Ludwig-Schwellinger E, Ebner T, Schaefer O. Impact of endogenous esterase activity on in vitro P-glycoprotein profiling of dabigatran etexilate in Caco-2 monolayers. Drug Metab Dispos. 2014;42:250–6. https://doi.org/10.1124/dmd.113.053561.
Shen H, Yao M, Sinz M, Marathe P, Rodrigues AD, Zhu M. Renal excretion of dabigatran: the potential role of multidrug and toxin extrusion (MATE) proteins. Mol Pharm. 2019;16:4065–76. https://doi.org/10.1021/acs.molpharmaceut.9b00472.
Udomnilobol U, Jianmongkol S, Prueksaritanont T. The potentially significant role of CYP3A-mediated oxidative metabolism of dabigatran etexilate and its intermediate metabolites in drug-drug interaction assessments using microdose dabigatran etexilate. Drug Metab Dispos. 2023. https://doi.org/10.1124/dmd.123.001353.
Garimella T, You X, Wang R, Huang SP, Kandoussi H, Bifano M, Bertz R, Eley T. A Review of daclatasvir drug-drug interactions. Adv Ther. 2016;33:1867–84. https://doi.org/10.1007/s12325-016-0407-5.
https://www.ema.europa.eu/en/documents/product-information/zepatier-epar-product-information_en.pdf (accessed July 12, 2023)/
Fromm MF, Kim RB, Stein CM, Wilkinson GR, Roden DM. Inhibition of P-glycoprotein-mediated drug transport: a unifying mechanism to explain the interaction between digoxin and quinidine. Circulation. 1999;99:552–7. https://doi.org/10.1161/01.cir.99.4.552.
Wessler JD, Grip LT, Mendell J, Giugliano RP. The P-glycoprotein transport system and cardiovascular drugs. J Am Coll Cardiol. 2013;61:2495–502. https://doi.org/10.1016/j.jacc.2013.02.058.
Millard DC, Strock CJ, Carlson CB, Aoyama N, Juhasz K, Goetze TA, Stoelzle-Feix S, Becker N, Fertig N, January CT, Anson BD, Ross JD. Identification of drug-drug interactions in vitro: A case study evaluating the effects of sofosbuvir and amiodarone on hiPSC-derived cardiomyocytes. Toxicol Sci. 2016;154:174–82. https://doi.org/10.1093/toxsci/kfw153.
https://www.hep-druginteractions.org/drug_queries/21504/drug_query_interactions. (Accessed 17 April 2023).
Lin CW, Dutta S, Zhao W, Asatryan A, Campbell A, Liu W. Pharmacokinetic interactions and safety of coadministration of glecaprevir and pibrentasvir in healthy volunteers. Eur J Drug Metab Pharmacokinet. 2018;43:81–90. https://doi.org/10.1007/s13318-017-0428-8.
Kosloski M, Li H, Wang S, Mensa F, Kort J, Liu W. Characterizing complex and competing drug-drug interactions between the antiviral regimen of glecaprevir and pibrentasvir with rifampin or carbamazepine. Clin Transl Sci. 2023;16:593–605. https://doi.org/10.1111/cts.13471.
Yang Y, Li P, Zhang Z, Wang Z, Liu L, Liu X. Prediction of cyclosporin-mediated drug interaction using physiologically based pharmacokinetic model characterizing interplay of drug transporters and enzymes. Int J Mol Sci. 2020;21:7023. https://doi.org/10.3390/ijms21197023.
Feng HP, Caro L, Fandozzi C, Chu X, Guo Z, Talaty J, Panebianco D, Dunnington K, Du L, Hanley WD, Fraser IP, Mitselos A, Denef JF, De Lepeleire I, de Hoon JN, Vandermeulen C, Marshall WL, Jumes P, Huang X, Martinho M, Valesky R, Butterton JR, Iwamoto M, Yeh WW. Pharmacokinetic interactions between the hepatitis C virus inhibitors elbasvir and grazoprevir and HIV protease inhibitors ritonavir, atazanavir, lopinavir, and darunavir in healthy volunteers. Antimicrob Agents Chemother. 2019;63:e02142-e2218. https://doi.org/10.1128/AAC.02142-18.
Bifano M, Adamczyk R, Hwang C, Kandoussi H, Marion A. Bertz RJ An open-label investigation into drug-drug interactions between multiple doses of daclatasvir and single-dose cyclosporine or tacrolimus in healthy subjects. Clinical Drug Invest. 2015;35:281–9. https://doi.org/10.1007/s40261-015-0279-5.
Tirona RG, Leake BF, Wolkoff AW, Kim RB. Human organic anion transporting polypeptide-C (SLC21A6) is a major determinant of rifampin-mediated pregnane X receptor activation. J Pharmacol Exp Ther. 2003;304:223–8. https://doi.org/10.1124/jpet.102.043026.
Kirby BJ, Collier AC, Kharasch ED, Whittington D, Thummel KE, Unadkat JD. Complex drug interactions of the HIV protease inhibitors 3: Effect of simultaneous or staggered dosing of digoxin and ritonavir, nelfinavir, rifampin, or bupropion. Drug Metab Dispos. 2012;40:610–6. https://doi.org/10.1124/dmd.111.042705.
Vavricka SR, Van Montfoort J, Ha HR, Meier PJ, Fattinger K. Interactions of rifamycin SV and rifampicin with organic anion uptake systems of human liver. Hepatology. 2002;36:164–72. https://doi.org/10.1053/jhep.2002.34133.
Karlgren M, Vildhede A, Norinder U, Wisniewski JR, Kimoto E, Lai Y, Haglund U, Artursson P. Classification of inhibitors of hepatic organic anion transporting polypeptides (OATPs): influence of protein expression on drug-drug interactions. J Med Chem. 2012;55:4740–63. https://doi.org/10.1021/jm300212s.
Te Brake LHM, Russel FGM, van den Heuvel JJMW, de Knegt GJ, de Steenwinkel JE, Burger DM, Aarnoutse RE, Koenderink JB. Inhibitory potential of tuberculosis drugs on ATP-binding cassette drug transporters. Tuberculosis. 2016;96:150–7. https://doi.org/10.1016/j.tube.2015.08.004.
Creamer BA, Sloan SNB, Dennis JF, Rogers R, Spencer S, McCuen A, Persaud P, Staudinger JL. Associations between pregnane X receptor and breast cancer growth and progression. Cells. 2020;9:2295. https://doi.org/10.3390/cells9102295.
Kosloski M, Zhao W, Li H, Pugatch D, Asatryan A, Kort J, Mensa FJ, Liu W. Drug-drug interactions of tacrolimus or cyclosporine with glecaprevir and pibrentasvir in healthy subjects. Clin Pharmacol Drug Dev. 2019;8:779–89. https://doi.org/10.1002/cpdd.671.
Gobran ST, Ancuta P, Shoukry NH. A tale of two viruses: immunological insights into HCV/HIV coinfection. Front Immunol. 2021;12: 726419. https://doi.org/10.3389/fimmu.2021.726419.
Bierman WFW, Scheffer GL, Schoonderwoerd A, Jansen G, van Agtmael MA, Danner SA, Scheper RJ. Protease inhibitors atazanavir, lopinavir and ritonavir are potent blockers, but poor substrates, of ABC transporters in a broad panel of ABC transporter-overexpressing cell lines. J Antimicrob Chemother. 2010;65:1672–80. https://doi.org/10.1093/jac/dkq209.
Shitara Y, Takeuchi K, Horie T. Long-lasting inhibitory effects of saquinavir and ritonavir on OATP1B1-mediated uptake. J Pharm Sci. 2013;102:3427–35. https://doi.org/10.1002/jps.23477.
Eagling VA, Back DJ, Barry MG. Differential inhibition of cytochrome P450 isoforms by the protease inhibitors, ritonavir, saquinavir and indinavir. Br J Clin Pharmacol. 1997;44:190–4. https://doi.org/10.1046/j.1365-2125.1997.00644.x.
Lepist EI, Zhang X, Hao J, Huang J, Kosaka A, Birkus G, Murray BP, Bannister R, Cihlar T, Huang Y, Ray AS. Contribution of the organic anion transporter OAT2 to the renal active tubular secretion of creatinine and mechanism for serum creatinine elevations caused by cobicistat. Kidney Int. 2014;86:350–7. https://doi.org/10.1038/ki.2014.66.
https://liverpool-hiv-hep.s3.amazonaws.com/fact_sheets/pdfs/000/000/118/original/HIV_FactSheet_Cobi_2016_Mar.pdf?1458130953 (Accessed 17 April 2023).
Marzolini C, Gibbons S, Khoo S, Back D. Cobicistat versus ritonavir boosting and differences in the drug–drug interaction profiles with co-medications. J Antimicrob Chemother. 2016;71:1755–8. https://doi.org/10.1093/jac/dkw032.
Kosloski MP, Oberoi R, Wang S, Viani RM, Asatryan A, Hu B, Ding B, Qi X, Kim EJ, Mensa F, Kort J, Liu W. Drug-drug interactions of glecaprevir and pibrentasvir coadministered with Human Immunodeficiency Virus antiretrovirals. J Infect Dis. 2020;221:223–31. https://doi.org/10.1093/infdis/jiz439.
Mogalian E, Stamm LM, Osinusi A, Brainard DM, Shen G, Ling KHJ, Mathias A. Drug-drug interaction studies between hepatitis C virus antivirals sofosbuvir/velpatasvir and boosted and unboosted Human Immunodeficiency Virus antiretroviral regimens in healthy volunteers. Clin Infect Dis. 2018;67:934–40. https://doi.org/10.1093/cid/ciy201.
US Food and Drug Administration. Highlights of prescribing information: Solvaldi. 2013. https://www.accessdata.fda.gov/drugsatfda_docs/label/2013/204671s000lbl.pdf. (Accessed 22 April 2023).
Feng HP, Guo Z, Fandozzi C, Panebianco D, Caro L, Wolford D, Dreyer DP, Valesky R, Martinho M, Rizk ML, Iwamoto M, Yeh WW. Pharmacokinetic interactions between the fixed-dose combinations of elvitegravir/cobicistat/tenofovir disoproxil fumarate/emtricitabine and elbasvir/grazoprevir in healthy adult participants. Clin Pharmacol Drug Developm. 2019;8:952–61. https://doi.org/10.1002/cpdd.702.
Zeng Q, Bai M, Li C, Lu S, Ma Z, Zhao Y, Zhou H, Jiang H, Sun D, Zheng C. Multiple drug transporters contribute to the placental transfer of emtricitabine. Antimicrob Agents Chemother. 2019;63:e00199-e219. https://doi.org/10.1128/AAC.00199-19.
Minuesa G, Volk C, Molina-Arcas M, Gorboulev V, Erkizia I, Arndt P, Clotet B, Pastor-Anglada M, Koepsell H, Martinez-Picado J. Transport of lamivudine [(-)-beta-L-2’,3’-dideoxy-3’-thiacytidine] and high-affinity interaction of nucleoside reverse transcriptase inhibitors with human organic cation transporters 1, 2, and 3. J Pharmacol Exp Ther. 2009;329:252–61. https://doi.org/10.1124/jpet.108.146225.
Weiss J, Theile D, Ketabi-Kiyanvash N, Lindenmaier H, Haefeli WE. Inhibition of MRP1/ABCC1, MRP2/ABCC2, and MRP3/ABCC3 by nucleoside, nucleotide, and non-nucleoside reverse transcriptase inhibitors. Drug Metab Dispos. 2007;35:340–4. https://doi.org/10.1124/dmd.106.012765.
Dussault I, Lin M, Hollister K, Wang EH, Synold TW, Forman BM. Peptide mimetic HIV protease inhibitors are ligands for the orphan receptor SXR. J Biol Chem. 2001;276:33309–12. https://doi.org/10.1074/jbc.C100375200.
Tamai H, Okamura J. Risk factors of glecaprevir/pibrentasvir-induced liver injury and efficacy of ursodeoxycholic acid. Viruses. 2023;15:489. https://doi.org/10.3390/v15020489.
Sulejmani N, Jafri SM, Gordon SC. Pharmacodynamics and pharmacokinetics of elbasvir and grazoprevir in the treatment of hepatitis C. Expert Opin Drug Metab Toxicol. 2016;12:353–61. https://doi.org/10.1517/17425255.2016.1148685.
Garrison KL, German P, Mogalian E, Mathias A. The drug-drug interaction potential of antiviral agents for the treatment of chronic hepatitis C infection. Drug Metab Dispos. 2018;46:1212–25. https://doi.org/10.1124/dmd.117.079038.
Sodhi JK, Benet LZ. A simple methodology to differentiate changes in bioavailability from changes in clearance following oral dosing of metabolized drugs. Clin Pharmacol Ther. 2020;108:306–15. https://doi.org/10.1002/cpt.1828.
https://clinicaltrials.gov/ct2/show/NCT03067129 (Accessed 17 April 2023).
https://clinicaltrials.gov/ct2/show/NCT02402452 (Accessed 17 April 2023).
Caro L, Wenning L, Guo Z, Fraser IP, Fandozzi C, Talaty J, Panebianco D, Ho M, Uemura N, Reitmann C, Angus P, Gane E, Marbury T, Smith WB, Iwamoto M, Butterton JR, Yeh WW. Effect of hepatic impairment on the pharmacokinetics of grazoprevir, a hepatitis C virus protease inhibitor. Antimicrob Agents Chemother. 2017;61:e00813-e817. https://doi.org/10.1128/AAC.00813-17.
https://clinicaltrials.gov/ct2/show/NCT02159352 (Accessed 17 April 2023).
Furihata T, Matsumoto S, Fu Z, Tsubota A, Sun Y, Matsumoto S, Kobayashi K, Chiba K. Different interaction profiles of direct-acting anti-hepatitis C virus agents with human organic anion transporting polypeptides. Antimicrob Agents Chemother. 2014;58:4555–64. https://doi.org/10.1128/AAC.02724-14.
Al-Nahari MM, Abbassi MM, Ebeid FS, Hassany M, El-Sayed MH, Farid SF. Pharmacokinetics of daclatasvir in Egyptian adolescents with genotype-4 HCV infection. Antiviral Ther. 2020;25:101–10. https://doi.org/10.3851/IMP3357.
Ramsden D, Fullenwider CL. Characterization of correction factors to enable assessment of clinical risk from in vitro CYP3A4 induction data and basic drug–drug interaction models. Eur J Drug Metab Pharmacokinet. 2022;47:467–82. https://doi.org/10.1007/s13318-022-00763-y.
https://www.tga.gov.au/sites/default/files/auspar-grazoprevir-170515.pdf. (Accessed 18 April 2023).
https://www.tga.gov.au/sites/default/files/auspar-sofosbuvir-velpatasvir-171108-pi.pdf. (Accessed 18 April 2023).
https://www.tga.gov.au/sites/default/files/auspar-ledipasvir-sofosbuvir-171020-pi.pdf. (Accessed 18 April 2023).
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Author Contributions
Conceptualisation, Methodology, Data curation, formal analysis, investigation, Visualisation and Writing: M.M.
Funding
The author received no funding for this work.
Conflict of interests
The author declares no competing interests for this work.
Ethics approval
Not applicable.
Consent to participate
Not applicable.
Consent for publication
Not applicable.
Data availability
Not applicable.
Code availability
Not applicable.
Rights and permissions
Springer Nature or its licensor (e.g. a society or other partner) holds exclusive rights to this article under a publishing agreement with the author(s) or other rightsholder(s); author self-archiving of the accepted manuscript version of this article is solely governed by the terms of such publishing agreement and applicable law.
About this article

Cite this article
Murray, M. Mechanisms and Clinical Significance of Pharmacokinetic Drug Interactions Mediated by FDA and EMA-approved Hepatitis C Direct-Acting Antiviral Agents. Clin Pharmacokinet 62, 1365–1392 (2023). https://doi.org/10.1007/s40262-023-01302-x
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s40262-023-01302-x