
Abstract
Afamelanotide, the first α-melanocyte-stimulating hormone (MSH) analogue, synthesized in 1980, was broadly investigated in all aspects of pigmentation because its activity and stability were higher than the natural hormone. Afamelanotide binds to the melanocortin-1 receptor (MC1R), and MC1R signaling increases melanin synthesis, induces antioxidant activities, enhances DNA repair processes and modulates inflammation. The loss-of-function variants of the MC1R present in fair-skinned Caucasians are less effectively activated by the natural hormone. Afamelanotide was the first α-MSH analogue to be applied to human volunteers. Ten daily doses of between 0.08 and 0.21 mg/kg in saline injected subcutaneously resulted in long-lasting skin pigmentation and enabled basic pharmacokinetics. Subcutaneous application had full bioavailability, but neither oral nor transdermal application resulted in measurable plasma concentrations or pigmentation response. Two trials in human volunteers showed that neither MC1R variants nor fair skin reduced the afamelanotide-induced increase in skin pigmentation. A controlled-release formulation optimizes administration in man and is effective at a lower dose than the daily saline injections. Promising therapeutic results were published in polymorphic light eruption, erythropoietic protoporphyria (EPP), solar urticaria, Hailey–Hailey disease and vitiligo. In 2014, afamelanotide was approved by the European Medicines Agency for the prevention of phototoxicity in adult patients with EPP. No late effects were reported in volunteers 25 years after the first exposure or after continuous long-term application of up to 8 years in EPP patients, and an immunogenic potential has been excluded. Generally, adverse effects were benign in all trials.
Similar content being viewed by others
Avoid common mistakes on your manuscript.
Afamelanotide is the first α-melanocyte-stimulating hormone (MSH) analogue applied to humans. |
We overview the fate of afamelanotide in the human body and its characteristics of α-MSH as a therapeutic drug (including side effects) in the treatment of different skin disorders. |
1 Introduction
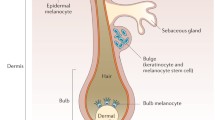
The skin, as the surface of an organism, protects the organism from the influence of environmental factors and helps to conserve homoeostasis within the body. Some of these environmental factors, particularly ultraviolet (UV) radiation, have the potential to damage the integrity of the skin, which is counteracted by specific cellular reactivity of skin cells. One of these protecting processes conserved from invertebrates to humans relates to the activation of the proopiomelanocortin (POMC) gene [1]. Its expression is stimulated by TP53, which, in turn, is upregulated in response to UV radiation-induced DNA damage in keratinocytes in the epidermis [2]. The product of the POMC gene is cleaved by proconvertases into different active peptides, including adrenocorticotropin (ACTH), α- and β-melanocyte-stimulating hormones (MSHs) and β-lipotropin. Following light-induced damage, the α-MSH released acts as a paracrine factor on the epidermal pigment cells (melanocytes) and autocrine within melanocytes. After its agonistic binding to the melanocortin-1 receptor (MC1R), α-MSH activates the synthesis of (eu)-melanin (Fig. 1) by increasing the expression of the rate-limiting enzyme tyrosinase, as well as tyrosinase-related proteins 1 and 2. The latter two induce a shift from yellow-red pheomelanin to brown–black eumelanin production, with eumelanin being more protective against UV radiation than pheomelanin [3, 4]. MC1R signaling also stimulates the transcription and folding of tyrosinase within the Golgi network, and ensures that the newly produced melanin is incorporated into small vesicles (melanosomes) and then distributed by the dendrites of the melanocytes to the surrounding keratinocytes. Keratinocytes concentrate the melanosomes above their cell nucleus, and these form a cap to protect the organelle most sensitive to UV insult [5]. α-MSH-induced MC1R signaling is shown to support differentiation and maintains the function of mature melanocytes, while melanocyte stem cells do not express the MC1R and are therefore unresponsive to α-MSH or its analogues [6, 7].

Schematic of the supposed multiple effects of the α-MSH analog afamelanotide on a melanocyte (left side) and a keratinocyte (right side). Upon binding to the MC1R at melanocytes, transcription, translation, proper folding, and transport into melanosomes of tyrosinase and tyrosinase-related proteins is stimulated, the outgrowth of dendrites propagated, and melanosomes are distributed to adjacent keratinocytes. In addition, MC1R signaling promotes DNA repair and synthesis of antioxidant enzymes. MC1R melanocortin-1 receptor, ER endoplasmatic reticulum, TGN trans-Golgi network, IL interleukin, UV ultraviolet, α-MSH α-melanocyte-stimulating hormone
In melanocytes and other dermal cells, including fibroblasts and endothelial cells, which express the MC1R and therefore react on MC1R signaling, other protective processes, such as an increase in antioxidant activities, stimulation of DNA repair mechanism, and secretion of the immunomodulator interleukin (IL)-10, are promoted by α-MSH [8–11].
In premalignant melanocytes, α-MSH-dependent MC1R signaling induces senescence and therefore prevents proliferation and melanoma formation [12]. Anchorage-independent, clonogenic growth of freshly explanted human melanoma cells were not enhanced; rather, afamelanotide inhibited the proliferation and induced differentiation [13, 14]. In addition, α-MSH reduces migration and invasion of melanoma cells, which may reduce or suppress metastasis of melanoma [15, 16]. Afamelanotide 2 and 10 mg/kg did not enhance the number and size of human melanomas implanted into the severe combined immunodeficiency mouse model, and afamelanotide did not induce malignant transformation of normal human melanocytes [13]. Both pigment enhancement and melanoma induction have a common denominator, namely UV radiation, leading to an unjustified assumption on α-MSH and its analogues being involved in melanoma induction.
In addition to the MC1R, α-MSH and its analogues also bind to melanocortin receptors 3–5; these receptors have certain effects on feeding and body weight control, thermogenesis, and sexual functions [17]. As the K i of afamelanotide for the MC3R is approximately five times higher than that for the MC1R, and those for the MC4R and MC5R are approximately 50 times higher than that for the MC1R [18], it is questionable as to whether they are activated by the pharmacological application of afamelanotide. Moreover, the MC4R is localized in the brain only and is most likely inaccessible to systemically applied afamelanotide [18].
2 Preclinical Pharmacology
2.1 Physiochemical Properties and Biological Effectiveness
The 13-amino acid peptide afamelanotide, first synthesized in 1980 at the University of Arizona as an analogue of α-MSH [19], shows an exchange of amino acids at positions 4 (methionine to norleucine) and 7 (l-phenylalanine to d-phenylalanine) compared with the physiological hormone. The exchange of amino acids increases both the affinity to the MC1R and the biological half-life, resulting in a strong biological effectiveness [20]. Because of these characteristics, afamelanotide, under the name NDP-MSH, was widely used in studies on the biological effects of α-MSH, and the effects of NDP-MSH were considered equivalent to those of the native hormone. Since then, the development of NDP-MSH as a drug for human use required optimization in synthesis processes, with effects on structure and configuration. These processes have not been reported in detail but, nonetheless, they likely positively influence the reproducibility of effectiveness and minimize adverse effects.
2.2 Preclinical Toxicology
The intraperitoneal dose used in early toxicological studies in mice and rats was more than 200 times above that currently applied to humans. A study of four male Yucatan pigs, using a dose of 0.16 mg/kg subcutaneously for 30 days, did not show any significant toxicological signs [13, 21]. Moreover, the intraperitoneal and intrauterine application of afamelanotide in pregnant rodents failed to produce an effect on the sex ratio, weight, morphology or histology of the developing fetuses, and there was no evidence of congenital defects, change in maturation, growth, premature parturition or pigmentation changes in the fetuses [22].
2.3 Metabolism and Elimination
The natural hormone α-MSH is rapidly degraded by unspecific proteases in frog or rat serum, or in vitro by trypsin or chymotrypsin, whereas afamelanotide is resistant to all these enzymes [23]. The biological activity of afamelanotide on frog skin pigmentation is preserved after prolonged in vitro incubation for up to 5 h, and is resistant to elimination by rinsing. Therefore, an irreversible binding or sequestration of the compound within the melanocytes, or even in the melanosomes, was assumed [23, 24]. Details regarding the metabolic fate of afamelanotide within the human organism have not been published.
3 Pharmacodynamic Properties
3.1 Pigmentation Response
Early studies have shown that repeated intramuscular application of α-MSH, β-MSH or ACTH to dark-skinned humans at 3–8 mg for 10 days induced fast skin pigmentation within the first few days, and the effect of the pigmentation resolved at approximately 6 weeks after the end of the application [25]. About the same temporal effect was found in healthy volunteers of skin type I–IV after 10 subcutaneous applications of afamelanotide 0.08 mg/kg solubilized in saline [26]. When afamelanotide treatment was combined with solar UV exposure, melanogenesis was amplified and the epidermal pigmentation was reported to last considerably longer than after sunlight exposure only [27].
3.2 MC1R Variants and Pigmentation Response
The MC1R shows numerous allelic variants in Caucasians, some of which were loss-of-function variants [28]. Carriers of these loss-of-function variants have fair skin and red hair (known as Fitzpatrick skin type I or II) and are at increased risk for skin cancer. In contrast to the physiological hormone, afamelanotide binds to genetically modified MC1Rs in vitro [29]. However, these artificially induced mutations do not exist in human MC1Rs and the effect found in vitro may not be extensible to the natural variants. Therefore, the effect of afamelanotide was investigated in human volunteers with skin type I or II and compared with those with skin type III and IV. Furthermore, the effect of afamelanotide was tested in volunteers who carried different MC1R variants [30, 31]. In fact, after application of afamelanotide, the melanin density of skin increased more dramatically in fair-skinned persons and persons carrying an MC1R variant compared with dark-skinned persons and persons carrying the wild-type MC1R.
3.3 Additional Effects of Afamelanotide
Afamelanotide enhanced the DNA repair process after UV damage in keratinocytes in vitro and this effect was reproduced in human volunteers in vivo [11, 30]. Other effects of afamelanotide found in vitro or in animal studies, including inhibition of inflammation, improvement of experimental colitis or reduction of reperfusion injury, have, to our knowledge, not been studied in humans [32–34].
4 Pharmacokinetics
4.1 Transdermal Delivery
Afamelanotide was transdermally effective in pigmentation induction in mice, but not in rats. Its penetration through artificial human skin models was demonstrated [35], but apparently it was not effective in the intact organism [13].
4.2 Afamelanotide in Saline Solutions
In a phase I trial, three routes of application were compared: oral (0.16 mg/kg), intravenous (0.16 mg/kg) and subcutaneous (0.08–0.21 mg/kg) [36]. Each application was administered once daily, ten times within 2 weeks. In addition to the afamelanotide blood levels, pigmentation response was assessed by skin reflectometry. The oral application resulted in undetectable blood concentration and no pigmentation occurred, while the subcutaneous application showed complete bioavailability compared with the intravenous application and resulted in the highest pigmentation response. The plasma half-life following subcutaneous dosing ranged between 0.07 and 0.79 h during the absorption phase and between 0.7 and 1.7 h (mean 1.30 ± 0.46) during the β-phase. No detectable drug was present in blood after 9.5 h and, consequently, no accumulation was detected by repeated dosing every 24 h. After intravenous dosing, the area under the curve was between 297 and 519 ng/(ml h), the mean half-life of the α-phase was between 0.14 and 0.52 h, and the mean half-life of the β-phase was 1.07 ± 0.46 h in the three tested volunteers. In addition, the mean systemic clearance was 0.41 ± 0.13 L/(h kg). The average apparent volume of distribution after intravenous dosing was 0.54 L/kg, which indicates that afamelanotide is distributed in a space exceeding the blood volume. As afamelanotide is water soluble, it may be speculated that it is rapidly bound to specific receptors and potentially sequestered into melanocytes (Sect. 2.3).
Only minimal amounts of native drug were recovered in urine, indicating an extensive metabolism of afamelanotide. Although no studies have been published on the site of metabolism, afamelanotide is most likely degraded by unspecific proteases in the serum, the same as the physiological hormone α-MSH but at a slower rate.
4.3 Slow-Release Formulations
The lack of bioavailability after oral application, the fast elimination after subcutaneous application, and the requirement for repeated applications in order to achieve pigmentation necessitated the development of a slow-release formulation. A biodegradable polymer, poly(d,l-lactide-co-glycolide), with 50% lactide and 50% glycolide, was the matrix for the formulation [37–39]. It contained variable additives such as poly(vinylpyrrolidone), methylcellulose, and hydroxypropyl methylcellulose, which influences the release of the peptide hormone as well as the dissolution profiles. The biological activity of the peptide in the biodegradable polymer, as measured by the frog skin assay, was not affected by gamma irradiation up to 2.5 Mrad for sterilization, but was increased after 3.5 Mrad. Hairless and haired guinea pigs were treated with 4 mg slow-release implants, and melanogenesis was measured by skin reflectometry and by the eumelanin content in biopsies of the epidermis. Hair color darkened in the haired guinea pigs and skin darkening was observed in the hairless guinea pigs. The epidermis of hairless guinea pigs showed an increased melanin content of epidermal cells, whereas in the haired guinea pigs more melanin was contained in the melanocytes of the hair follicle. The effects reached their maximum 1 month after implantation and lasted for at least 3 months.
The elution profile of the 16 or 20 mg slow-release formulation applied to humans in the trials of afamelanotide in erythropoietic protoporphyria (EPP) [40, 41], polymorphic light eruption (PLE) [42], solar urticaria (SU) [43], Hailey–Hailey disease (HHD) [44], acne vulgaris [45] or vitiligo [46] has not been published. It is important to note that the slow-release formulation contains only approximately one-seventh to one-eighth of the active compound required for pigmentation induction in the saline preparations, which indicates that the slow-release formulation most likely has better therapeutic margins than the saline preparations.
4.4 Pharmacokinetics in Special Populations
Studies on bioavailability have not been published neither for renal disease nor for hepatic disease. As only trace amounts of unchanged drug was recovered in urine, it is probable that renal impairment has no influence on bioavailability. Afamelanotide is degraded intracellularly, either by endocytosis after binding to its receptors or by unspecific proteases [23, 47]. Consequently, hepatic impairment will most probably also have no effect on bioavailability.
5 Trials on the Efficacy of Afamelanotide in Skin Diseases
5.1 Effects of Afamelanotide on Light-Induced Skin Diseases
5.1.1 Polymorphic Light Eruption
PLE is an acquired light-induced immunological disorder manifested by itchy, papulovesicular lesions on sun-exposed skin in spring and summer. A high lifetime prevalence was found in European populations, with a preponderance in females. Therapeutic sun protection by clothing, broad spectrum sunscreens, local or systemic corticosteroids, and phototherapy are applied, which may be replaced by hydroxychloroquine, azathioprine or cyclosporine in therapeutically resistant cases [48]. A pilot trial on 36 PLE patients with 20 mg slow-release afamelanotide showed a trend in reduction of symptom severity compared with placebo [49]. A phase III trial using afamelanotide 16 mg was conducted and, to our knowledge, the data have only been published on the company (Clinuvel Pharmaceuticals, Melbourne) website.
5.1.2 Solar Urticaria
SU is also an acquired, immunologically elicited photodermatosis, with acute swelling of sun-exposed skin within minutes of irradiation and, in some cases, angioedema of the face. SU may be due to an immunoglobulin (Ig)E-mediated reaction to photo-induced allergens. As a treatment, antihistamines and sunlight protection have been proposed and, in severe disease, omalizumab, an anti-IgE antibody, was applied to single cases, with significant improvement in light tolerance. Because of limited effectiveness, the need for a new therapy was expressed [43]. Slow-release afamelanotide 16 mg was applied to five SU patients, and their minimal urticaria dose increased in tendency (p = 0.058) at day 30 after the implant [43].
5.1.3 Erythropoietic Protoporphyria
EPP is a rare inherited disorder of heme biosynthesis leading to accumulation of protoporphyrin IX (PPIX) due to a deficiency of the enzyme ferrochelatase or by an overactive erythropoietic aminolevulinate synthase (ALAS2), the rate-limiting enzyme in heme biosynthesis [50]. Upon skin radiation by visible light, the accumulated protoporphyrin disseminates the gathered light energy to oxygen, which is converted to several reactive oxygen species [51, 52]. The resulting tissue damage affects mostly mast cells and endothelial cells of the subpapillary capillaries [53, 54]. Clinically, patients experience acute, severely painful phototoxicity that may last up to 2 weeks, and the associated intolerable pain renders patients incapable of sleeping or performing daily activities. Although β-carotene, cysteine, and a number of other therapeutic attempts have been made, effectiveness has not been proven [55].
In 2006, a pilot study of slow-release formulated afamelanotide 20 mg was undertaken in five EPP patients, showing an improved tolerance to artificial white light [40]. A phase III, placebo-controlled, double-blinded, crossover trial in 100 European patients confirmed the efficacy of afamelanotide in EPP, enabling patients to prolong their spontaneous exposure to sunlight, as well as reducing their EPP-related pain [56]. Another phase II trial in the US was followed by two randomized, double-blinded, placebo-controlled, phase III trials—one in Europe and one in the US [41, 57]. In both phase III trials, patients showed a prolonged spontaneous exposure to sunlight during the active treatment. The clinical significance of these study results was underlined by an exceptionally high adherence of Italian and Swiss EPP patients to afamelanotide treatment during prolonged compassionate use and the special access scheme program, of up to 8 years [58].
5.2 Effects of Afamelanotide on Other Skin Diseases
5.2.1 Vitiligo
Vitiligo is an acquired skin disorder due to the loss of functioning melanocytes in the skin or hair causing patchy areas of depigmentation. Vitiligo may be stigmatizing, especially in dark-skinned individuals. The estimated prevalence worldwide is 1% but may vary strongly among different populations [59]. Vitiligo exists in both a segmental and a generalized form, with the latter being more common. Several guidelines for optimal treatment have been published, including topical treatments (corticosteroids and calcineurin inhibitors), phototherapy (narrowband UVB [NB-UVB] and psoralen plus ultraviolet A [PUVA]), systemic corticosteroids, surgical grafting techniques, and depigmenting treatments in extended vitiligo [59].
The leading hypothesis of vitiligo causation is that of an autoimmune or autoinflammatory process, potentially due to a specific genetic background [59]. This theory is based on its association with several other autoimmune disorders, including thyroiditis. Reactive oxygen species, and therefore melanocyte-intrinsic abnormalities, are possible inducers of the whole inflammatory cascade. Afamelanotide, by its multiple effects on melanogenesis, inflammation and antioxidant activity (Sect. 3.3), has the potential to arrest such a process in vitiligo as long as melanocytes are present in the skin. In vitiligo, resting and undifferentiated melanoblasts are preserved in the hair bulbs; UVB irradiation can activate and differentiate these to form mature cells that carry an MC1R. These cells will then be accessible to the agonistic upregulation by afamelanotide.
Both a pilot study and a multicenter study combined the treatment of UVB with afamelanotide [46, 60]. The multicenter trial enrolled adult patients with Fitzpatrick skin phototypes III–VI and stable, generalized vitiligo. Fifty-six patients were randomized to the combination therapy of UVB and afamelanotide versus UVB monotherapy. After 1 month of UVB phototherapy, afamelanotide 16 mg was administered subcutaneously monthly to the combination therapy group for 4 months, while the monotherapy was continued for the same period in the control group. The authors reported a superior response of the combination therapy group compared with the NB–UVB monotherapy group at day 56, especially in the face and on the upper extremities. In a subgroup analysis, patients with skin type IV–VI responded well, but not those with skin type III.
5.2.2 Hailey–Hailey Disease
HHD, also known as familial benign chronic pemphigus, belongs to the autosomal dominant genodermatoses. Deficiency of ATP2C1 in keratinocytes from HHD patients is associated with alterations in proliferation and differentiation, as well as increased oxidative stress [61, 62]. Affected patients develop recurrent blisters, erosions and crusts in the intertriginous areas. In addition, heat and rubbing promote lesions, which can be complicated by superinfection. HHD has a substantial negative effect on the quality of life (QOL) of patients. The onset of lesions is generally after adolescence, showing a peak at around the age of 30–40 years, but the symptoms can develop at any age. The most commonly used therapies include corticosteroids, antifungals and antibiotics, administered either topically or systemically. These therapies are aimed to control the underlying inflammatory immune response in order to achieve remission; however, these existing treatments do not provide a long-lasting positive therapeutic result. α-MSH was shown to increase the expression of nuclear factor (erythroid-derived 2)-like 2 (Nrf2), a transcription factor coordinating the expression of antioxidative and Nrf-dependent phase II detoxifying enzymes in primary keratinocytes [9]. This finding suggests the potential role of α-MSH beyond pigment induction as a guarantor of epidermal homeostasis and oxidative stress balance [44]. In an investigator-initiated, phase II, open-label, pilot study, afamelanotide 16 mg controlled release implant was administered subcutaneously to two patients with HHD who had a number of long-standing skin lesions. Both patients had 100% clearance of HHD lesions 60 days after the first injection, independently of the lesion location [44].
5.2.3 Facial Acne Vulgaris
Acne vulgaris, a frequent inflammatory skin disease in Caucasian adolescents, is caused multifactorially, including increased sebum production, proliferation of propionibacterium acnes, and hyperkeratinization of the pilosebaceous duct. The open-label study by Bohm et al. reported on three male patients with chronic stable disease who were treated with two to three implants of afamelanotide, in 3- to 4-week intervals [45]. The number of acne lesions decreased from 68 ± 27.6 to 30 ± 19.7 between days 0 and 56, and life quality, as measured by the Dermatological Life Quality Index, improved.
6 Safety
6.1 Adverse Effects
The adverse effects of afamelanotide have been reported to be mild [41, 58], and, until now, no serious drug-related adverse effects have been reported [41]. Mild adverse effects include nausea, headache and pigmentation at the insertion site. Treatment-related events, such as bruises or hematomas, and redness or itching at the implant insertion site, have been reported. The summary of product characteristics published by the European Medicines Agency [63] lists additional potential adverse effects, most of which are either due to the pigmentation effect of afamelanotide, such as freckles and skin darkening, or unspecific or rare, such as migraine, back pain, abdominal pain, diarrhea, vomiting, decreased appetite, fatigue, dizziness, drowsiness and weakness, hot flushes and upper respiratory tract infections.
The observation time for adverse effects extends back to early 1990s, when the first doses of afamelanotide were applied to healthy volunteers [36]. Although treatment duration was limited at these early time periods (see Sect. 4.2), the amount of drug to which these volunteers were exposed was extremely high compared with the currently applied commercial slow-release product; namely, a volunteer was exposed to up to 10 doses of afamelanotide 0.21 mg/kg. Assuming the volunteer’s body weight to be 70 kg, the exposure reached 147 mg of afamelanotide within the 2 weeks of administration. To reach this same amount of exposure with the current commercial slow-release formulation, more than nine implantations are required; these nine implants are currently applied within a period of 1.5–3 years [58]. Despite this early high exposure to afamelanotide in saline, no long-term negative sequelae in these volunteers have been reported until now.
In addition, a long-term observational study of the now commercialized controlled-release implant of afamelanotide in EPP patients, which covers up to 8 years of continuous exposure, showed no serious drug-related adverse event [58], and the frequency and nature of observed adverse events were similar to those in the clinical trials. Since this report, our center administered another 400 afamelanotide implants to EPP patients and our experience now extends to a maximum of 10 years of continuous application of the controlled-release formulation, with no new afamelanotide-related adverse events.
As a caveat, illegal internet-distributed chemical agents are sold for lifestyle and tanning purposes under false pretences using the false name of afamelanotide, or its previous names and, in the medical literature, serious conditions associated with the use of these compounds have been described [64–70]. As we did not observe any of the pathologies reported from these illegally-distributed compounds, and after having implanted more than 1000 implants of the genuine afamelanotide drug, we assume that the reported adverse effects of the illegal compounds are elicited by toxic additives or impurities that may be contained in those preparations, besides some kind of α-MSH-like peptides.
In conclusion, all current clinical observations and reports support a very benign safety profile of afamelanotide.
6.2 Immunogenicity
In recent years, it has been observed that biological drugs could elicit an immunogenic reaction in a treated human. The 13-amino acid peptide afamelanotide has a very low molecular weight of only 1647 Da compared with other biological drugs. This low weight reduces the risk of eliciting antibodies, as long as no aggregates are released into the organism [71, 72]. On the other hand, the repeated administration of afamelanotide can potentially increase the risk of provoking an immunogenic response. The existence of both antidrug antibodies and neutralizing antibodies were tested during long-term treatment of EPP patients [73, 74]. Pre-existing immunoreactivity was found in some patients and this immunoreactivity was seen against both afamelanotide and the physiological hormone α-MSH. A similar reactivity of some human sera against α-MSH has been previously described [75]. The reactivity in those EPP patients with pre-existing immunoreactivity did not increase during the repeated application of afamelanotide. Moreover, none of these sera had a neutralizing effect against the activity of afamelanotide in mouse melanoma cells transfected with the human MC1R. These data exclude the fact that afamelanotide induces immunogenic reactions during prolonged application.
7 Conclusion
In a slow-release formulation, afamelanotide, a highly active α-MSH analogue, has proven to be effective in a number of skin disorders, most likely due to its combined effects on skin pigmentation, immunomodulation, enhancement of antioxidant activity and DNA repair. Adverse effects were mild and no late sequelae after early high doses or prolonged application were encountered.
References
Malagoli D, Accorsi A, Ottaviani E. The evolution of pro-opiomelanocortin: looking for the invertebrate fingerprints. Peptides. 2011;32:2137–40.
Cui R, Widlund HR, Feige E, Lin JY, Wilensky DL, Igras VE, et al. Central role of p53 in the suntan response and pathologic hyperpigmentation. Cell. 2007;128:853–64.
Aroca P, Urabe K, Kobayashi T, Tsukamoto K, Hearing VJ. Melanin biosynthesis patterns following hormonal stimulation. J Biol Chem. 1993;268:25650–5.
del Marmol V, Beermann F. Tyrosinase and related proteins in mammalian pigmentation. FEBS Lett. 1996;381:165–8.
Gibbs S, Murli S, De BG, Mulder A, Mommaas AM, Ponec M. Melanosome capping of keratinocytes in pigmented reconstructed epidermis: effect of ultraviolet radiation and 3-isobutyl-1-methyl-xanthine on melanogenesis. Pigment Cell Res. 2000;13:458–66.
Kauser S, Thody AJ, Schallreuter KU, Gummer CL, Tobin DJ. A fully functional proopiomelanocortin/melanocortin-1 receptor system regulates the differentiation of human scalp hair follicle melanocytes. Endocrinology. 2005;146:532–43.
Osawa M, Egawa G, Mak SS, Moriyama M, Freter R, Yonetani S, et al. Molecular characterization of melanocyte stem cells in their niche. Development. 2005;132:5589–99.
Brzoska T, Luger TA, Maaser C, Abels C, Bohm M. Alpha-melanocyte-stimulating hormone and related tripeptides: biochemistry, antiinflammatory and protective effects in vitro and in vivo, and future perspectives for the treatment of immune-mediated inflammatory diseases. Endocr Rev. 2008;29:581–602.
Kokot A, Metze D, Mouchet N, Galibert MD, Schiller M, Luger TA, et al. Alpha-melanocyte-stimulating hormone counteracts the suppressive effect of UVB on Nrf2 and Nrf-dependent gene expression in human skin. Endocrinology. 2009;150:3197–206.
Bohm M, Luger TA, Tobin DJ, Garcia-Borron JC. Melanocortin receptor ligands: new horizons for skin biology and clinical dermatology. J Invest Dermatol. 2006;126:1966–75.
Abdel-Malek ZA, Ruwe A, Kavanagh-Starner R, Kadekaro AL, Swope V, Haskell-Luevano C, et al. alpha-MSH tripeptide analogs activate the melanocortin 1 receptor and reduce UV-induced DNA damage in human melanocytes. Pigment Cell Melanoma Res. 2009;22:635–44.
Bennett DC, Medrano EE. Molecular regulation of melanocyte senescence. Pigment Cell Res. 2002;15:242–50.
Hadley ME, Dorr RT. Melanocortin peptide therapeutics: historical milestones, clinical studies and commercialization. Peptides. 2006;27:921–30.
Jiang J, Sharma SD, Nakamura S, Lai JY, Fink JL, Hruby VJ, et al. The melanotropic peptide, [Nle4, D-Phe7] alpha-MSH, stimulates human melanoma tyrosinase activity and inhibits cell proliferation. Pigment Cell Res. 1995;8:314–23.
Eves P, Haycock J, Layton C, Wagner M, Kemp H, Szabo M, et al. Anti-inflammatory and anti-invasive effects of alpha-melanocyte-stimulating hormone in human melanoma cells. Br J Cancer. 2003;89:2004–15.
Zhu N, Eves PC, Katerinaki E, Szabo M, Morandini R, Ghanem G, et al. Melanoma cell attachment, invasion, and integrin expression is upregulated by tumor necrosis factor alpha and suppressed by alpha melanocyte stimulating hormone. J Invest Dermatol. 2002;119:1165–71.
Cone RD. Studies on the physiological functions of the melanocortin system. Endocr Rev. 2006;27:736–49.
Minder EI. Afamelanotide, an agonistic analog of alpha-melanocyte-stimulating hormone, in dermal phototoxicity of erythropoietic protoporphyria. Expert Opin Investig Drugs. 2010;19:1591–602.
Sawyer TK, Sanfilippo PJ, Hruby VJ, Engel MH, Heward CB, Burnett JB, et al. 4-Norleucine, 7-d-phenylalanine-alpha-melanocyte-stimulating hormone: a highly potent alpha-melanotropin with ultralong biological activity. Proc Natl Acad Sci USA. 1980;77:5754–8.
Hadley ME, Heward CB, Hruby VJ, Sawyer TK, Yang YC. Biological actions of melanocyte-stimulating hormone. Ciba Found Symp. 1981;81:244–62.
Dorr RT, Dawson BV, al-Obeidi F, Hadley ME, Levine N, Hruby VJ. Toxicologic studies of a superpotent alpha-melanotropin, [Nle4, D-Phe7]alpha-MSH. Invest New Drugs. 1988;6:251–8.
Dawson BV, Ford CA, Holloway H, Dorr RT, Johnson P. Administration of melanotropic peptides during gestation in the rodent. Toxicology. 1993;77:91–101.
Castrucci AM, Hadley ME, Sawyer TK, Hruby VJ. Enzymological studies of melanotropins. Comp Biochem Physiol B. 1984;78:519–24.
Peters EM, Tobin DJ, Seidah NG, Schallreuter KU. Pro-opiomelanocortin-related peptides, prohormone convertases 1 and 2 and the regulatory peptide 7B2 are present in melanosomes of human melanocytes. J Invest Dermatol. 2000;114:430–7.
Lerner AB, McGuire JS. Effect of alpha- and betamelanocyte stimulating hormones on the skin colour of man. Nature. 1961;189:176–9.
Levine N, Sheftel SN, Eytan T, Dorr RT, Hadley ME, Weinrach JC, et al. Induction of skin tanning by subcutaneous administration of a potent synthetic melanotropin. JAMA. 1991;266:2730–6.
Dorr RT, Ertl G, Levine N, Brooks C, Bangert JL, Powell MB, et al. Effects of a superpotent melanotropic peptide in combination with solar UV radiation on tanning of the skin in human volunteers. Arch Dermatol. 2004;140:827–35.
Newton RA, Smit SE, Barnes CC, Pedley J, Parsons PG, Sturm RA. Activation of the cAMP pathway by variant human MC1R alleles expressed in HEK and in melanoma cells1. Peptides. 2005;26:1818–24.
Frandberg PA, Muceniece R, Prusis P, Wikberg J, Chhajlani V. Evidence for alternate points of attachment for alpha-MSH and its stereoisomer [Nle4, D-Phe7]-alpha-MSH at the melanocortin-1 receptor. Biochem Biophys Res Commun. 1994;202:1266–71.
Barnetson RS, Ooi TK, Zhuang L, Halliday GM, Reid CM, Walker PC, et al. [Nle4-D-Phe7]-alpha-melanocyte-stimulating hormone significantly increased pigmentation and decreased UV damage in fair-skinned Caucasian volunteers. J Invest Dermatol. 2006;126:1869–78.
Fitzgerald LM, Fryer JL, Dwyer T, Humphrey SM. Effect of MELANOTAN, [Nle(4), D-Phe(7)]-alpha-MSH, on melanin synthesis in humans with MC1R variant alleles. Peptides. 2006;27:388–94.
Luger TA, Scholzen T, Grabbe S. The role of alpha-melanocyte-stimulating hormone in cutaneous biology. J Investig Dermatol Symp Proc. 1997;2:87–93.
Chiao H, Kohda Y, McLeroy P, Craig L, Housini I, Star RA. Alpha-melanocyte-stimulating hormone protects against renal injury after ischemia in mice and rats. J Clin Invest. 1997;99:1165–72.
Rajora N, Boccoli G, Catania A, Lipton JM. alpha-MSH modulates experimental inflammatory bowel disease. Peptides. 1997;18:381–5.
Dawson BV, Hadley ME, Levine N, Kreutzfeld KL, Don S, Eytan T, et al. In vitro transdermal delivery of a melanotropic peptide through human skin. J Invest Dermatol. 1990;94:432–5.
Ugwu SO, Blanchard J, Dorr RT, Levine N, Brooks C, Hadley ME, et al. Skin pigmentation and pharmacokinetics of melanotan-I in humans. Biopharm Drug Dispos. 1997;18:259–69.
Bhardwaj R, Blanchard J. Controlled-release delivery system for the alpha-MSH analog melanotan-I using poloxamer 407. J Pharm Sci. 1996;85:915–9.
Bhardwaj R, Hadley ME, Dorr RT, Dvorakova K, Brooks C, Blanchard J. Pharmacologic response of a controlled-release PLGA formulation for the alpha-melanocyte stimulating hormone analog, melanotan-I. Pharm Res. 2000;17:593–9.
Bhardwaj R, Blanchard J. In vitro characterization and in vivo release profile of a poly (d, l-lactide-co-glycolide)-based implant delivery system for the alpha-MSH analog, melanotan-I. Int J Pharm. 1998;170:109–17.
Harms J, Lautenschlager S, Minder CE, Minder EI. An alpha-melanocyte-stimulating hormone analogue in erythropoietic protoporphyria. N Engl J Med. 2009;360:306–7.
Langendonk JG, Balwani M, Anderson KE, Bonkovsky HL, Anstey AV, Bissell DM, et al. Afamelanotide for erythropoietic protoporphyria. N Engl J Med. 2015;373:48–59.
Fabrikant J, Touloei K, Brown SM. A review and update on melanocyte stimulating hormone therapy: afamelanotide. J Drugs Dermatol. 2013;12:775–9.
Haylett AK, Nie Z, Brownrigg M, Taylor R, Rhodes LE. Systemic photoprotection in solar urticaria with alpha-melanocyte-stimulating hormone analogue [Nle4-D-Phe7]-alpha-MSH. Br J Dermatol. 2011;164:407–14.
Biolcati G, Aurizi C, Barbieri L, Cialfi S, Screpanti I, Talora C. Efficacy of the melanocortin analogue Nle4-D-Phe7-alpha-melanocyte-stimulating hormone in the treatment of patients with Hailey–Hailey disease. Clin Exp Dermatol. 2014;39:168–75.
Bohm M, Ehrchen J, Luger TA. Beneficial effects of the melanocortin analogue Nle4-D-Phe7-alpha-MSH in acne vulgaris. J Eur Acad Dermatol Venereol. 2014;28:108–11.
Grimes PE, Hamzavi I, Lebwohl M, Ortonne JP, Lim HW. The efficacy of afamelanotide and narrowband UV-B phototherapy for repigmentation of vitiligo. JAMA Dermatol. 2013;149:68–73.
Jiang J. Microscopic visualization of melancyte/melanoma melanotropic receptors. Tucson: University of Arizona; 1993.
Smith E, Kiss F, Porter RM, Anstey AV. A review of UVA-mediated photosensitivity disorders. Photochem Photobiol Sci. 2012;11:199–206.
Clinuvel Pharmaceuticals Limited. Clinuvel anounces PLE phase III preliminary results. Clinuvel Pharmaceuticals Ltd; 2009. http://www.clinuvel.com/2009-announcements/item/140-clinuvel-announces-ple-phase-iii-preliminary-results. Accessed 3 Jan 2017.
Schneider-Yin X, Minder EI. Erythropoietic protoporphyria and X-linked dominant protoporphyria. In: Ferreira GC, editor. Porphyrias and sideroblastic anemias. Vol. 29 of the handbook of porphyrin science: 299–328. Series edited by Kadish KM, Simth KM, Guilard R. Singapore: World Scientific Publishing Company; 2013.
Afonso SG, Enriquez DS, Batlle A. Photodynamic and light independent action of 8 to 2 carboxylic free porphyrins on some haem-enzymes. Int J Biochem Cell Biol. 2001;33:1208–14.
Menon IA, Becker MA, Persad SD, Haberman HF. Quantitation of hydrogen peroxide formed during UV-visible irradiation of protoporphyrin, coproporphyrin and uroporphyrin. Clin Chim Acta. 1990;186:375–81.
Lim HW. Mechanisms of phototoxicity in porphyria cutanea tarda and erythropoietic protoporphyria. Immunol Ser. 1989;46:671–85.
Timonen K, Kariniemi AL, Niemi KM, Teppo AM, Tenhunen R, Kauppinen R. Vascular changes in erythropoietic protoporphyria: histopathologic and immunohistochemical study. J Am Acad Dermatol. 2000;43:489–97.
Minder EI, Schneider-Yin X, Steuer J, Bachmann LM. A systematic review of treatment options for dermal photosensitivity in erythropoietic protoporphyria. Cell Mol Biol (Noisy-le-grand). 2009;55:84–97.
Minder EI, Harms J, Lautenschlager S, Schneider-Yin X, Deybach JC, Minder CE. A double-blind, randomized, controlled phase III trial of afamelanotide (an alpha-MSH analogue) in erythropoietic protoporphyria (EPP): preliminary data on a Swiss cohort of patients and a model to determine efficacy in EPP [abstract]. Berzelius Symposium 81: Porphyrins and Porphyrias. Stockholm: 2009.
Langendonk J, Karstens F, SiJbrands E, Hanneken S, Anstey A, Deybach J, et al. Afamelanotide implants effectively reduce pain and prolong sun-tolerance in patients with erythropoietic protoporphyria; results of a phase III, multicenter, double-blind, randomized, placebo-controlled trial. Clin Chem Lab Med. 2013;51:eA12.
Biolcati G, Marchesini E, Sorge F, Barbieri L, Schneider-Yin X, Minder EI. Long-term observational study of afamelanotide in 115 patients with erythropoietic protoporphyria. Br J Dermatol. 2015;172:1601–12.
Ezzedine K, Eleftheriadou V, Whitton M, van Geel N. Vitiligo. Lancet. 2015;386:74–84.
Lim HW, Grimes PE, Lebwohl M. Indications and limitations of afamelanotide for treating vitiligo-reply. JAMA Dermatol. 2015;151:350.
Cialfi S, Oliviero C, Ceccarelli S, Marchese C, Barbieri L, Biolcati G, et al. Complex multipathways alterations and oxidative stress are associated with Hailey–Hailey disease. Br J Dermatol. 2010;162:518–26.
Manca S, Magrelli A, Cialfi S, Lefort K, Ambra R, Alimandi M, et al. Oxidative stress activation of miR-125b is part of the molecular switch for Hailey–Hailey disease manifestation. Exp Dermatol. 2011;20:932–7.
Summary of product characteristics (afamelanotide/Scenesse). 2015.
Ong S, Bowling J. Melanotan-associated melanoma in situ. Australas J Dermatol. 2012;53:301–2.
Hjuler KF, Lorentzen HF. Melanoma associated with the use of melanotan-II. Dermatology. 2014;228:34–6.
Reid C, Fitzgerald T, Fabre A, Kirby B. Atypical melanocytic naevi following melanotan injection. Ir Med J. 2013;106:148–9.
Ellis RA, Kirkham N, Seukeran D. Malignant melanoma in a user of melanotan I. BMJ. 2009;338:b566.
Cardones AR, Grichnik JM. Alpha-melanocyte-stimulating hormone-induced eruptive nevi. Arch Dermatol. 2009;145:441–4.
Cousen P, Colver G, Helbling I. Eruptive melanocytic naevi following melanotan injection. Br J Dermatol. 2009;161:707–8.
Langan EA, Ramlogan D, Jamieson LA, Rhodes LE. Change in moles linked to use of unlicensed “sun tan jab”. BMJ. 2009;338:b277.
Sela M. Antigenicity: some molecular aspects. Science. 1969;166:1365–74.
Mariani M, Bracci L, Presentini R, Nucci D, Neri P, Antoni G. Immunogenicity of a free synthetic peptide: carrier-conjugation enhances antibody affinity for the native protein. Mol Immunol. 1987;24:297–303.
Spichty R, Balimann M, Barman J, Minder EI. A bioassay for detection of neutralizing antibodies against the alpha-melanocyte stimulating hormone analogue afamelanotide in patients with erythropoietic protoporphyria. J Pharm Biomed Analy. 2013;75:192–8.
Lengweiler S, Kreim S, Barman-Aksozen J, Maurer M, Minder EI. Evaluation of the immunogenicity of the synthetic alpha-melanocyte-stimulating hormone (alpha-MSH) analogue afamelanotide ([Nle4-D-Phe7]-alpha-MSH, Scenesse®) in erythropoietic protoporphyria patients by ELISA detecting both anti-afamelanotide and anti-alpha-MSH antibodies. Skin Pharmacol Physiol. 2015;28:103–13.
Fetissov SO, Harro J, Jaanisk M, Jarv A, Podar I, Allik J, et al. Autoantibodies against neuropeptides are associated with psychological traits in eating disorders. Proc Natl Acad Sci USA. 2005;102:14865–70.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Funding
Elisabeth I. Minder, Jasmin Barman-Aksoezen and Xiaoye Schneider-Yin were partly supported by grants from the Foundation for Scientific Research of Triemli Hospital, the Foundation for Scientific Research of the University of Zurich, the Hartmann–Müller Foundation, and the Velux Foundation. The immunogenicity studies were partly supported by a grant from Clinuvel Pharmaceutical, Melbourne, VIC, Australia.
Conflict of interest
Elisabeth Minder was the principal investigator of two trials of afamelanotide by Clinuvel Pharmaceutical, Melbourne, VIC, Australia. Jasmin Barman-Aksoezen and Xiaoye Schneider-Yin declare that they have no conflicts of interest that might be relevant to the contents of this article.
Rights and permissions
About this article

Cite this article
Minder, E.I., Barman-Aksoezen, J. & Schneider-Yin, X. Pharmacokinetics and Pharmacodynamics of Afamelanotide and its Clinical Use in Treating Dermatologic Disorders. Clin Pharmacokinet 56, 815–823 (2017). https://doi.org/10.1007/s40262-016-0501-5
Published:
Issue Date:
DOI: https://doi.org/10.1007/s40262-016-0501-5