
Abstract
Background
Pregnant women and their fetuses are exposed to numerous drugs; however, they are orphan populations with respect to the safety and efficacy of drugs. Therefore, the prediction of maternal and fetal drug exposure prior to administration would be highly useful.
Methods
A physiologically-based pharmacokinetic (PBPK) model for nevirapine, which is metabolized by the cytochrome P450 (CYP) 3A4, 2B6 and 2D6 pathways, was developed to predict maternal and fetal pharmacokinetics (PK). The model was developed in both non-pregnant and pregnant women, and all physiological and enzymatic changes that could impact nevirapine PK were taken into account. Transplacental parameters estimated from ex vivo human placenta perfusion experiments were included in this PBPK model. To validate the model, observed maternal and cord blood concentrations were compared with predicted concentrations, and the impact of fetal clearance on fetal PK was investigated.
Results
By implementing physiological changes, including CYP3A4, 2D6 and 2B6 inductions, we predicted a clearance increase of 21 % in late pregnancy. The PBPK model successfully predicted the disposition for both non-pregnant and pregnant populations. Parameters obtained from the ex vivo experiments allowed the prediction of nevirapine concentrations that matched observed cord blood concentrations. The fetal-to-maternal area under the curve ratio (0–24 h interval) was 0.77, and fetal metabolism had no significant effect on fetal PK.
Conclusions
The PBPK approach is a useful tool for quantifying a priori the drug exposure of metabolized drugs during pregnancy, and can be applied to evaluate alternative dosing regimens to optimize drug therapy. This approach, including ex vivo human placental perfusion parameters, is a promising approach for predicting human fetal exposure.
Similar content being viewed by others
Avoid common mistakes on your manuscript.
Transplacental transfer parameters were estimated from the cotyledon perfusion model for névirapine. |
The approach was able to quantitatively predict maternal pharmacokinetics (PK) and drug fetal exposure by incorporating estimated transplacental transfer parameters in physiologically-based pharmacokinetic models. |
Fetal metabolism has no significant impact on fetal PK. |
1 Introduction
Pregnant women and their fetuses are orphan populations with respect to the safety and efficacy of drugs; however, they are exposed to numerous compounds [1–3]. Therefore, the prediction of maternal and fetal pharmacokinetics (PK) is highly desirable. Physiologically-based PK (PBPK) models have already been used to predict maternal PK by implementing physiological changes occurring during pregnancy [4–12]; however, few of these models have focused on the prediction of fetal exposure, and only two have validated their models by comparing predicted fetal concentrations with observed concentrations [12–16].
Physiological changes occurring during pregnancy can be associated with significant variations in PK, and PBPK models can predict these variations. Indeed, the models are based on physiology, and incorporate comprehensive information related to anatomical, physiological, and metabolic changes that can impact the PK. Numerous maternal changes can be implemented, such as increasing body weight, fat mass, cardiac output, plasma volume, glomerular filtration rate (GFR), renal plasma flow, and decreasing haematocrit, albumin and α1-acid glycoprotein (α1-AGP) [5, 6, 8, 10, 17, 18]. Moreover, enzymatic induction or inhibition can also be implemented [7, 17, 19].
Due to obvious ethical reasons, in vivo fetal risk assessment studies related to maternal drug exposure remain extremely limited. Human cord blood plasma samples are the most suitable measurements to describe transplacental transfer in late pregnancy; however, the cord blood-to-maternal concentration ratio is highly variable, according to the time elapsed between drug administration and blood sampling [20, 21]. Population PK analyses allow the area under the curve (AUC) ratio to be estimated, and should be preferred. The drawback of the population PK approach is the high number of patients who should be included in order to estimate maternal and fetal PK [22, 23]. To ensure drug safety during pregnancy, information regarding transplacental transfer prior to administration would be highly desirable. As animal studies may not be helpful in predicting human fetal PK because of interspecies differences in the structural and functional features of the placenta, other approaches have been developed. The ex vivo human placental perfusion is the gold standard and offers a better insight into the placenta drug transporters, xenobiotic metabolism and tissue binding [24, 25]. Nevertheless, this model does not take into account the variation of maternal concentration and cannot predict fetal PK profiles.
We previously developed a PBPK model to predict fetal drug exposure [15], for which transplacental parameter estimates from the ex vivo human placental perfusion model were implemented in the model to predict fetal PK, an approach that was validated for two renally excreted drugs. The aim of the present study was to extrapolate this approach to drugs mainly metabolized via hepatic cytochrome P450 (CYP) isoenzymes. Moreover, fetal metabolism was explored, along with its impact on fetal PK.
2 Materials and Methods
Figure 1 summarizes the study workflow. First, we developed a nevirapine PBPK model for the non-pregnant population. When the model adequately described observed concentrations after oral and intravenous administration for both a single dose and at steady state, we implemented all physiological changes occurring during pregnancy. We then predicted and compared the PK profile with observed concentrations in pregnant women. Transplacental transfer parameters were estimated from the human cotyledon perfusion experiment and, after scaling to the placental size, were implemented into the PBPK model. Thus, the fetal PK profile was predicted and was compared with observed cord blood plasma concentrations. The full PBPK model used during pregnancy is depicted in Fig. 2.

Study workflow. PBPK physiologically-based pharmacokinetic, PK pharmacokinetic

Physiologically-based pharmacokinetic model representation. IV intravenously, PO orally, RB rest of the body, Amn fluid amniotic fluid, QplaM blood flow from the mother to the placental tissue, QPlaF blood flow from the fetus to the placental tissue, QrbF blood flow to the fetal body, k SW swallowing constant, CLr F fetal urinary excretion, M maternal, F fetal, k1 and k2 transplacental transfer constants, k L lung excretion, k INT intramembranous pathway
2.1 Physiologically-Based Pharmacokinetic (PBPK) Modelling in the Non-Pregnant Population
A full PBPK model was built for nevirapine using R software. Physiological parameters obtained from Price et al., in which organ volumes and blood flows are scaled relative to individual characteristics, are summarized in Table 1 [26]. The cardiac output (CO) was obtained by summing all tissue perfusion rates. In Table 1, the body mass index (BMI) of both men and women was simulated to match the population of Ibarra et al. [28], and the pregnant women had a BMI that matched the population included in the study by Benaboud et al. [41]. Drug-dependent parameters are summarized in Table 2. Partition coefficients were estimated using the Rodgers and Roland method [27], and absorption was described using a first-order rate constant (ka) [28].
The nevirapine clearance fraction for each metabolite, as well as urinary excretion, were obtained from the recovery of urine/faeces and in vitro studies [29, 30]. Based on different metabolite profiles between single and multiple doses, we estimated enzymatic induction and thus clearance fraction [30, 31]. Interindividual variability parameters were fixed to 40 % for clearance, 70 % for ka and 10 % for bioavailability (F). These values are in agreement with clinical data [28, 32].
2.2 Ex Vivo Model
2.2.1 Placental Tissue Collections
Placenta from normal pregnancies were obtained from the Port Royal Hospital (Paris, France) after uncomplicated vaginal delivery or caesarean section. All mothers were sero-negative for HIV infection, were not infected by hepatitis B or C viruses, and took no medication other than oxytocin or epidural anaesthesia during labour. Written informed consents were obtained from all participants in the study.
2.2.2 Placental Perfusion
Placenta were perfused in a recirculating (closed-closed) circuit according to the method adapted from Schneider et al. and Forestier et al. [33, 34]. Perfusion experiments started within 30 min after delivery. After visual examination for lack of evident lesions on the chorionic plate, a truncal branch of the chorionic artery and the associated vein were cannulated. Fetal circulation was established at a flow rate of 6 mL/min (Q f), and the perfused area progressively whitened and allowed visualization of the selected cotyledon. After confirmation of the absence of vascular leakage, the perfusion was subsequently initiated by insertion of two catheters into the intervillous space on the maternal side. Maternal circulation was established at a flow rate of 12 mL/min (Q m), and the pHs of maternal and fetal solutions were adjusted to 7.4 ± 0.1 and 7.2 ± 0.1, respectively. These solutions were prepared with earle medium containing 30 and 40 g/L of human serum albumin, respectively. Antipyrine, an inert molecule that freely diffuses through the placenta, was added at 20 mg/L as a marker to validate the viability of the cotyledon through the experiment. Nevirapine and antipyrine were perfused into the maternal reservoir. The maternal reservoir volume (V m) was 250 mL, and the fetal reservoir volumes (V f) were 150, 200 and 250 ml for the first, second and other placentas, respectively. Samples were collected every 10 min during the first half-hour and then every 30 min until 150 min, from both the fetal and maternal sides, to determine the concentrations (C f and C m, respectively). Samples were then stored at −20 °C until analysis.
2.2.3 Sample Analysis
Nevirapine concentrations were determined using high-performance liquid chromatography (HPLC) with ultraviolet (UV) detection. The mobile phase was made of 0.025 mol/L of phosphate buffer + pentanesulfonate and acetonitrile (75/25 [vol/vol]). The intraday variability was <10 % and the relative standard error was <12 %, while the intraday variability was <12 % and the relative standard error was <14 % at the limit of quantification of 0.125 mg/L. Antipyrine concentrations were determined using HPLC with UV detection at 290 nm [35].
The maternal-to-fetal transfer was described using the fetal transfer rate (FTR) [Eq. 1]:
where C f and C m are the drug concentrations in fetal and maternal perfusates, and V f and V m are the fetal and maternal perfusate volumes. An antipyrine FTR >20 % was required to validate each experiment. The clearance index (CLI) was calculated as the ratio of nevirapine FTR to that of antipyrine.
2.2.4 Transplacental Parameter Estimation
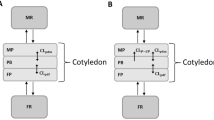
Placentas with obvious leakage were set aside by looking at the return fetal flow rate and measuring the antipyrine FTR. Drug transfer across the placenta was modelled as a cotyledon split into maternal and fetal compartments (Fig. 3). The cotyledon volume averaged 60 mL, and the maternal cotyledon volume (V mp) was assumed to be 9 mL.

Human cotyledon perfusion experiment. Q flow rate (L/h), V volume (L), mp maternal placenta, fp fetal placenta, m maternal, f fetal, k1 and k2 transplacental parameter constants (h−1), Vm maternal volume, Qm maternal flow, Vf fetal volume, Qf fetal flow
Data were analysed using a non-linear mixed-effect modelling approach, using Monolix version 4.2. Parameters were estimated by computing the maximum likelihood estimator of the parameters, using the stochastic approximation expectation maximization (SAEM) algorithm combined with a Markov Chain Monte Carlo (MCMC) procedure. The number of MCMC chains was fixed to five for all estimations. Several transplacental transfer models were investigated, i.e. simple diffusion, linear transfer, saturable transfer, combination of diffusion and saturable transfer, and addition of placental elimination rate. For one parameter change, a model was kept if the objective function value (OFV) was decreased by at least 3.84 (Chi-square test with 1 degree of freedom). Simulated concentrations in the maternal and fetal reservoirs were compared with concentrations from the ex vivo experiment previously described. Equations 2 (maternal reservoir), 3 (maternal cotyledon), 4 (fetal cotyledon) and 5 (fetal reservoir) were used to describe the ex vivo experiment:
where C denotes concentration (mg/ L), Q denotes flow rate (L/h) and V denotes volume (L). Subscripted m, f, mp, and fp denote mother, fetus, maternal placenta and fetal placenta, respectively. Kppl, k1cot and k2cot denote the placental coefficient partition (dimensionless) and transplacental parameters (L/h) estimated for a cotyledon.
The goodness-of-fit was evaluated via the following graphs: observed versus predicted concentrations, weighted residuals versus time, and weighted residuals versus predictions. To validate the final model, a visual predictive check (VPC) was performed. The final model was used to simulate 1000 nevirapine concentrations at each sampling time, and the simulated and observed data were compared.
Transplacental transfer parameters depend on the surface of exchange, therefore we scaled these parameters on placental volume, as shown in Eq. 6 (parameter adjustment). Once scaled, the parameters were implemented in the PBPK model:
where k1pl denotes the placental transplacental transfer, k1cot denotes the cotyledon transplacental transfer, V pl denotes the placental volume, and V cot denotes the cotyledon volume.
2.3 PBPK Modelling in Pregnant Women
Changes in haematocrit, GFR, adipose tissue, or plasma volume were implemented according to the equations developed by Abduljalil et al. [17]. The physiological parameters are summarized in Table 1. CYP3A4, 2B6 and 2D6 induction was also taken into account in the model and resulted in an increase in clearance (Table 2) [8, 17]. The increase in clearance during pregnancy was deduced from the normal adult clearance and the induction rate(s) of the metabolic pathway(s). For example, if pregnancy induces a 90 % CYP2B6 increase, then Cl2B6,pregnant = Cl2B6,non-pregnant × 1.9. The free fraction was assumed to be unchanged during pregnancy (Table 2), and the fetal free fraction values were assumed to be equal to maternal free fraction values. Figure 2 shows the PBPK model. A structure with distinct placental, fetal and amniotic fluid compartments was chosen. Numerous exchanges can impact fetal and amniotic fluid PK, and parameter values of these exchanges are summarized in Table 3 [36–40].
Equations 7 (in vivo maternal placenta), 8 (in vivo fetal placenta), 9 (fetal blood), 10 (fetal body) and 11 (amniotic fluid) describe the feto-placental unit of the pregnancy PBPK models:
where Q denotes blood flow (L/h) and V denotes tissue volume (L). The tissue subscripts ab, plaM, plaF, bloodF, rbF and amf denote maternal blood, maternal placenta, fetal placenta, fetal blood, rest of the fetal body (fetal body–fetal blood) and amniotic fluid, respectively. B/P denotes the blood-to-plasma concentration ratio, KpRB denotes the fetal coefficient partition, Kppl denotes the placental coefficient partition, k1pl and k2pl denote the transplacental transfer parameters (L/h), GFR denotes the glomerular filtration rate (L/h), GFRf denotes the fetal GFR (L/h), ClR denotes the maternal renal clearance (L/h), fu denotes the free drug fraction, k INT denotes the intramembranous constant (L/h), k SW denotes the swallowing constant (L/h), and k L denotes the oral, nasal, tracheal and pulmonary secretion constant (L/h).
A sensitivity analysis was performed on the physiological fetal parameters, as well as the transplacental transfer parameters, to evaluate their impact on fetal PK. As elimination pathways are not well known for fetuses, we evaluated whether fetal metabolism could have an impact on fetal PK. To do so, we have assumed that fetal metabolism in late pregnancy was equal to newborn clearance, and compared fetal AUC with or without the inclusion of fetal metabolism.
To validate our model we compared nevirapine predicted concentrations with observed concentrations in different populations. A description of the populations used for the observed data are summarized in Table 4.
3 Results
3.1 Non-Pregnant Population
Our model was able to correctly describe nevirapine PK following a 15 mg intravenous administration, a single 200 mg oral dose, and multiple 200 mg oral doses (Fig. 4). Using the Chi-square test, the distribution of observed data outside the 90 % prediction interval was tested, and no significant difference was observed for the distribution of concentrations. For a single dose, 11 % of the observations fall outside the 90 % prediction interval, while after multiple doses, this rate rose to 14 %.

Pharmacokinetics in the non-pregnant population. Median predicted concentrations (solid lines) and 90 % prediction intervals (dashed lines) compared with observed mean concentrations from Lamson et al. [32] (open circle), and individuals concentrations from Ibarra et al. [28] (+) and Moltó et al. [61] (triangle). bid twice daily
3.2 Ex Vivo Model
A total of 13 placentas were successfully perfused and 12 experiments were validated for integrity of the placental membrane and adequate conditions of perfusion. A total of 109 maternal and 103 fetal reservoir samples were available for the analysis. The mean antipyrine FTR at the end of the experiment was 40.7 % (±standard deviation [SD] 9.07 %), and the mean nevirapine FTR and CLI (60–180 min) were 26 ± 7 % and 0.73 ± 0.15, respectively. The CLI reflects that nevirapine has a high transplacental transfer.
The Kppl, k1cot and k2cot were estimated. Residual variability was best described as proportional for the maternal concentrations, and additive for fetal concentrations. Additionally, interindividual variability was estimated for Kppl and k1cot. Table 5 summarizes the final parameters and shows that parameters were well estimated, with small residual standard errors (RSEs).
The VPC of the final model (Fig. 5) confirmed that the average prediction matched the observed concentrations and that variability was well estimated.

Visual predictive check. Concentrations observed for the fetal side (open circle) and maternal side (+) compared with estimated median profiles (solid lines) and 90 % prediction interval (dashed lines) for the fetal side (green) and maternal side (maroon). VPC visual predictive check
3.3 Pregnant Population
As a result of CYP3A4, 2D6 and 2B6 inductions, we predicted a clearance increase of 21 and 38 % in late pregnancy after a single-dose administration and at steady state, respectively. As shown in Fig. 6, the model was able to describe the maternal PK profile. Assuming a bioavailability of 0.93 for pregnant women [32], the apparent clearances were 1.65 and 3.55 L/h for a single dose and at steady state, respectively, for a gestational age (GA) of 39 weeks. These results are in accordance with the reported 1.42 and 3.57 L/h for a single dose and at steady state, respectively [41, 42].

Maternal and fetal pharmacokinetic profiles. Median predicted concentrations (solid lines) and 90 % prediction interval (dashed lines) compared with individual observed concentrations [41]
The parameters estimated ex vivo (Kppl, k1 and k2) were implemented in the PBPK model. PK profiles during labour were simulated and were compared with observed data [41]. Figure 6 shows that simulated fetal PK profiles using our PBPK model were in agreement with observed cord blood concentrations [41]. At delivery, the fetal-to-maternal AUC 024 ratio has been predicted to be 77 % for a single dose of nevirapine, while the predicted AUC 0∞ ratio for a single dose was 84 %. Accordingly, Fig. 6 shows that fetal concentrations were generally lower than maternal concentrations, reflecting this AUC ratio. Using the Chi-square test, the distribution of observed data outside the 90 % prediction interval was tested. For pregnant women, the variability was underestimated, i.e. 22 % of concentrations were outside the 90 % prediction interval (confidence interval: 17–27 %). No significant difference was observed for fetal prediction; 8 % of the observations fall outside the 90 % prediction interval.
Some physiological parameters are not well documented and are not easy to access. Sensitivity analyses have been conducted to ensure that if the true values are, to some extent, higher or lower than those used in the models, the effect on fetal predictions would not be significant. On the other hand, we wanted to ensure that placental transfer was highly dependent on parameters estimated ex vivo. Figure 7 shows that the fetal renal excretion (CLRfetus), intramembranous pathway and swallowing constant had no significant impact on the fetal PK profile. A small impact on fetal PK was shown when the fetal blood flow was divided by 5. Therefore, uncertainties on these physiological constants were not a major issue. The transplacental transfer parameters estimated from the human cotyledon experiments have been shown to be key parameters in predicting the fetal PK profile; if the k1 value was divided by two, the fetal maximal concentrations were also approximately divided by two.

Sensitivity analyses. Predicted fetal concentrations when changing a parameter value. Q FO fetal blood flow, k SW fetal swallowing constant, k int intramembranous pathway constant, CL Rfetus renal clearance excretion, k1 and k2 transplacental transfer parameters, kp placental coefficient partition
The implementation of fetal metabolism based on newborn clearance did not change fetal AUC. Even if this elimination was multiplied by 2, there was a less than 0.1 % change in fetal AUC. Compared with feto-maternal exchanges, fetal metabolism did not significantly influence fetal PK.
4 Discussion
The PBPK model developed was able to quantitatively predict maternal and fetal PK after nevirapine intake in late pregnancy. By implementing physiological changes, including CYP3A4, 2D6 and 2B6 inductions, we predicted a clearance increase of 21 and 38 % in late pregnancy after a single-dose administration and at steady state, respectively. However, the concentrations 24 h post-dose are still at least 100 times the IC50, and dosing adjustment is not mandatory [41]. The mean fetal-to-maternal AUC 024 and AUC 0∞ ratios were 77 and 84 %, respectively. Maternal and fetal PK profiles predicted by the model were in accordance with observed concentrations [41].
To our knowledge, few PBPK models have predicted pregnancy-induced changes in clearance for drugs metabolized by several CYP pathways [5, 7, 9]. Alqahtani and Kaddoumi studied indomethacin, which is metabolized by CYP2C9 and uridine diphosphate glucuronosyltransferase (UGT) 2B7, and Ke et al. studied phenytoin, metabolized by CYP2C9 and 2C19, and glyburide, metabolized by CYP3A, 2C9 and 2C19 [7, 9]. Our model demonstrates that PBPK modelling can predict these changes. We previously developed this approach for two renally excreted drugs, and another study has used a similar approach, but without any human data, to validate their predictions [15, 16, 43]. This is the first PBPK model that predicts fetal PK using transplacental transfer parameters from ex vivo experiments, validated by observed concentrations for a drug metabolized by several CYP pathways.
At delivery, the fetal-to-maternal AUC 024 ratio has been estimated, using a population PK model, to be 75 % for a single dose of nevirapine, which is in agreement with our model prediction of 77 % [41]. At steady state, observed cord blood-to-maternal concentration ratios were 1.02 (n = 21, range 0.59–2.08), 0.91 (n = 9, SD 0.9), 0.6 (n = 5, SD 0.19) and 0.59 (n = 1) [44–47]. These observed ratios were similar to our mean predicted fetal-to-maternal concentration ratio of 0.91 (95 % confidence interval 0.5–1.5, range 0.3–2.2). In our model, we assumed that pregnancy had no effect on nevirapine autoinduction. Observed clearances were similar to the predicted clearances in all cases.
Some CYP450 s are active in the fetal liver [48–52], therefore we investigated whether fetal metabolism could significantly impact the fetal PK. When the fetal metabolism rate is significant, this parameter is mandatory for predicting the fetal PK using our approach. In our model, whether or not fetal metabolism was added did not change the fetal exposure, thus confirming its negligible effect on fetal PK. Moreover, fetal metabolism is much smaller than exchanges between the mother and the fetus, as well as maternal clearance.
Protein binding can lead to misleading results with the placental ex vivo model, therefore we added albumin to the media to ensure the same protein binding and thus good estimation of transplacental parameters. Nevirapine is 60 % plasma bound, thus protein binding should influence the transplacental transfer. Two options are available to decrease the risk of misleading transplacental parameter estimation: (i) the ex vivo free fraction must be the same as the in vivo free fraction; or (ii) the transplacental transfer must be corrected by the in vivo free fraction. The first option was chosen, and physiological concentrations of albumin were added in the solutions.
Plasma protein binding can decrease during pregnancy due to a decrease in albumin for highly bound drugs [18]. For example, phenytoin free fraction has been shown to increase by 30 %; however, as nevirapine is moderately plasma bound it is unlikely that a significant change in free fraction would occur. We assumed that CYP2D6 activity increased, according to Abduljalil et al. [17]. However, other studies have reported a bigger increase [8]. In our model, a 100 % increase led to an overestimation of clearance when compared with observed clearances [41, 42]. The variability in the pregnant population was underestimated, which could be explained by variability in CYP induction that was not implemented in the model. This approach has been validated for three drugs. Further validation should be undertaken to ensure the reliability of the approach, especially for drugs that are highly plasma bound.
Another limitation is the prediction of placental transfer at the end of pregnancy only. Indeed, since the placenta structure changes throughout gestation, this approach cannot be extrapolated to early pregnancy. During the gestational period, the surface area available for exchange, and blood flow perfusing the placenta, increase and membrane thickness decreases, which could lead to an increase in passive diffusion with GA. Thus, transplacental transfer should be less important before full-term [25, 53]. Taking this into account, our model should reflect the maximal transplacental transfer; however, since transporter activities during pregnancy are not well known and could influence transplacental transfer, no conclusions for transplacental transfer before full-term can be deduced from our approach [53].
5 Conclusions
Transplacental parameters obtained from the human cotyledon perfusion experiments allowed for good predictions of fetal PK profiles, and no random scaling factors were needed. Moreover, the sensitivity analyses performed on these parameters showed that any modification significantly impacted fetal exposure. Thus, the ex vivo parameters integrated into the PBPK model seem to be a sensitive and accurate method for predicting fetal exposure at full-term pregnancy for metabolized drugs.
These populations are orphan populations with respect to the safety and efficacy of drugs. Indeed, clinical trials are difficult in this context and the modelling approach can provide a better insight into maternal and fetal PK profiles in late pregnancy, and therefore avoid some overexposure or ineffective exposure.
References
Daw JR, Hanley GE, Greyson DL, Morgan SG. Prescription drug use during pregnancy in developed countries: a systematic review. Pharmacoepidemiol Drug Saf. 2011;20(9):895–902.
Mitchell AA, Gilboa SM, Werler MM, Kelley KE, Louik C, Hernández-Díaz S. Medication use during pregnancy, with particular focus on prescription drugs: 1976–2008. Am J Obstet Gynecol. 2011;205(1):51.e1–51.e8.
Zaki NM, Albarraq AA. Use, attitudes and knowledge of medications among pregnant women: a Saudi study. Saudi Pharm J. 2014;22(5):419–28.
Eyal S, Easterling TR, Carr D, Umans JG, Miodovnik M, Hankins GDV, et al. Pharmacokinetics of metformin during pregnancy. Drug Metab Dispos. 2010;38(5):833–40.
Gaohua L, Abduljalil K, Jamei M, Johnson TN, Rostami-Hodjegan A. A pregnancy physiologically based pharmacokinetic (p-PBPK) model for disposition of drugs metabolized by CYP1A2, CYP2D6 and CYP3A4. Br J Clin Pharmacol. 2012;74(5):873–85.
Ke AB, Nallani SC, Zhao P, Rostami-Hodjegan A, Unadkat JD. A PBPK model to predict disposition of CYP3A-metabolized drugs in pregnant women: verification and discerning the site of CYP3A induction. CPT Pharmacomet Syst Pharmacol. 2012;1(9):e3.
Ke AB, Nallani SC, Zhao P, Rostami-Hodjegan A, Unadkat JD. Expansion of a PBPK model to predict disposition in pregnant women of drugs cleared via multiple CYP enzymes, including CYP2B6, CYP2C9 and CYP2C19. Br J Clin Pharmacol. 2014;77(3):554–70.
Ke AB, Nallani SC, Zhao P, Rostami-Hodjegan A, Isoherranen N, Unadkat JD. A physiologically based pharmacokinetic model to predict disposition of CYP2D6 and CYP1A2 metabolized drugs in pregnant women. Drug Metab Dispos Biol Fate Chem. 2013;41(4):801–13.
Alqahtani S, Kaddoumi A. Development of physiologically based pharmacokinetic/pharmacodynamic model for indomethacin disposition in pregnancy. PLoS One. 2015;10(10):e0139762.
Xia B, Heimbach T, Gollen R, Nanavati C, He H. A simplified PBPK modeling approach for prediction of pharmacokinetics of four primarily renally excreted and CYP3A metabolized compounds during pregnancy. AAPS J. 2013;15(4):1012–24.
Pilari S, Preuße C, Huisinga W. Gestational influences on the pharmacokinetics of gestagenic drugs: a combined in silico, in vitro and in vivo analysis. Eur J Pharm Sci. 2011;42(4):318–31.
Gentry PR, Covington TR, Andersen ME, Clewell HJ III. Application of a physiologically based pharmacokinetic model for isopropanol in the derivation of a reference dose and reference concentration. Regul Toxicol Pharmacol. 2002;36(1):51–68.
Luecke RH, Wosilait WD, Pearce BA, Young JF. A computer model and program for xenobiotic disposition during pregnancy. Comput Methods Programs Biomed. 1997;53(3):201–24.
Yoon M, Schroeter JD, Nong A, Taylor MD, Dorman DC, Andersen ME, et al. Physiologically based pharmacokinetic modeling of fetal and neonatal manganese exposure in humans: describing manganese homeostasis during development. Toxicol Sci. 2011;122(2):297–316.
De Sousa Mendes M, Hirt D, Vinot C, Valade E, Lui G, Pressiat C, et al. Prediction of human fetal pharmacokinetics using ex-vivo human placenta perfusion studies and physiologically based models. Br J Clin Pharmacol. 2016;81(4):646–57.
Shintaku K, Hori S, Satoh H, Tsukimori K, Nakano H, Fujii T, et al. Prediction and evaluation of fetal toxicity induced by NSAIDs using transplacental kinetic parameters obtained from human placental perfusion studies. Br J Clin Pharmacol. 2012;73(2):248–56.
Abduljalil K, Furness P, Johnson TN, Rostami-Hodjegan A, Soltani H. Anatomical, physiological and metabolic changes with gestational age during normal pregnancy: a database for parameters required in physiologically based pharmacokinetic modelling. Clin Pharmacokinet. 2012;51(6):365–96.
Ke AB, Rostami-Hodjegan A, Zhao P, Unadkat JD. Pharmacometrics in pregnancy: an unmet need. Annu Rev Pharmacol Toxicol. 2014;54(1):53–69.
Isoherranen N, Thummel KE. Drug metabolism and transport during pregnancy: how does drug disposition change during pregnancy and what are the mechanisms that cause such changes? Drug Metab Dispos. 2013;41(2):256–62.
Chappuy H, Tréluyer J-M, Jullien V, Dimet J, Rey E, Fouché M, et al. Maternal-fetal transfer and amniotic fluid accumulation of nucleoside analogue reverse transcriptase inhibitors in human immunodeficiency virus-infected pregnant women. Antimicrob Agents Chemother. 2004;48(11):4332–6.
Mirochnick M, Taha T, Kreitchmann R, Nielsen-Saines K, Kumwenda N, Joao E, et al. Pharmacokinetics and safety of tenofovir in HIV-infected women during labor and their infants during the first week of life. J Acquir Immune Defic Syndr. 2014;65(1):33–41.
Hirt D, Urien S, Rey E, Arrivé E, Ekouévi DK, Coffié P, et al. Population pharmacokinetics of emtricitabine in human immunodeficiency virus type 1-infected pregnant women and their neonates. Antimicrob Agents Chemother. 2009;53(3):1067–73.
Hirt D, Urien S, Ekouévi DK, Rey E, Arrivé E, Blanche S, et al. Population pharmacokinetics of tenofovir in HIV-1-infected pregnant women and their neonates (ANRS 12109). Clin Pharmacol Ther. 2009;85(2):182–9.
Myllynen P, Vähäkangas K. Placental transfer and metabolism: an overview of the experimental models utilizing human placental tissue. Toxicol In Vitro. 2013;27(1):507–12.
Prouillac C, Lecoeur S. The role of the placenta in fetal exposure to xenobiotics: importance of membrane transporters and human models for transfer studies. Drug Metab Dispos. 2010;38(10):1623–35.
Price PS, Conolly RB, Chaisson CF, Gross EA, Young JS, Mathis ET, et al. Modeling Interindividual variation in physiological factors used in PBPK models of humans. Crit Rev Toxicol. 2003;33(5):469–503.
Rodgers T, Rowland M. Physiologically based pharmacokinetic modelling 2: predicting the tissue distribution of acids, very weak bases, neutrals and zwitterions. J Pharm Sci. 2006;95(6):1238–57.
Ibarra M, Vázquez M, Fagiolino P. Population pharmacokinetic model to analyze nevirapine multiple-peaks profile after a single oral dose. J Pharmacokinet Pharmacodyn. 2014;41(4):363–73.
Riska P, Lamson M, MacGregor T, Sabo J, Hattox S, Pav J, et al. Disposition and biotransformation of the antiretroviral drug nevirapine in humans. Drug Metab Dispos Biol Fate Chem. 1999;27(8):895–901.
Fan-Havard P, Liu Z, Chou M, Ling Y, Barrail-Tran A, Haas DW, et al. Pharmacokinetics of phase i nevirapine metabolites following a single dose and at steady state. Antimicrob Agents Chemother. 2013;57(5):2154–60.
Lamson M, MacGregor T, Riska P, Erickson D, Maxfield P, Rowland L, et al. Nevirapine induces both CYP3A4 and CYP2B6 metabolic pathways. Clin Pharmacol Ther. 1999;65(2):137.
Lamson MJ, Sabo JP, MacGregor TR, Pav JW, Rowland L, Hawi A, et al. Single dose pharmacokinetics and bioavailability of nevirapine in healthy volunteers. Biopharm Drug Dispos. 1999;20(6):285–91.
Schneider H, Panigel M, Dancis J. Transfer across the perfused human placenta of antipyrine, sodium and leucine. Am J Obstet Gynecol. 1972;114(6):822–8.
Forestier F, de Renty P, Peytavin G, Dohin E, Farinotti R, Mandelbrot L. Maternal-fetal transfer of saquinavir studied in the ex vivo placental perfusion model. Am J Obstet Gynecol. 2001;185(1):178–81.
Vinot C, Gavard L, Treluyer JM, Manceau S, Courbon E, Scherrmann JM, et al. Placental transfer of maraviroc in an ex vivo human cotyledon perfusion model and influence of ABC transporter expression. Antimicrob Agents Chemother. 2013;57(3):1415–20.
Kiserud T, Ebbing C, Kessler J, Rasmussen S. Fetal cardiac output, distribution to the placenta and impact of placental compromise. Ultrasound Obstet Gynecol. 2006;28(2):126–36.
Heller M, Burd L. Review of ethanol dispersion, distribution, and elimination from the fetal compartment. Birt Defects Res A Clin Mol Teratol. 2014;100(4):277–83.
Underwood MA, Gilbert WM, Sherman MP. Amniotic fluid: not just fetal urine anymore. J Perinatol. 2005;25(5):341–8.
Beall MH, van den Wijngaard JPHM, van Gemert MJC, Ross MG. Amniotic fluid water dynamics. Placenta. 2007;28(8–9):816–23.
Gilbert WM, Newman PS, Eby-Wilkens E, Brace RA. Technetium Tc 99m rapidly crosses the ovine placenta and intramembranous pathway. Am J Obstet Gynecol. 1996;175(6):1557–62.
Benaboud S, Ekouévi DK, Urien S, Rey E, Arrivé E, Blanche S, et al. Population pharmacokinetics of nevirapine in HIV-1-infected pregnant women and their neonates. Antimicrob Agents Chemother. 2011;55(1):331–7.
Capparelli EV, Aweeka F, Hitti J, Stek A, Hu C, Burchett SK, PACTG 1026S Study Team, PACTG P1022 Study Team, et al. Chronic administration of nevirapine during pregnancy: impact of pregnancy on pharmacokinetics. HIV Med. 2008;9(4):214–20.
Shintaku K, Arima Y, Dan Y, Takeda T, Kogushi K, Tsujimoto M, et al. Kinetic analysis of the transport of salicylic acid, a nonsteroidal anti-inflammatory drug, across human placenta. Drug Metab Dispos. 2007;35(5):772–8.
Yeh RF, Rezk NL, Kashuba ADM, Dumond JB, Tappouni HL, Tien H-C, et al. Genital tract, cord blood, and amniotic fluid exposures of seven antiretroviral drugs during and after pregnancy in human immunodeficiency virus type 1-infected women. Antimicrob Agents Chemother. 2009;53(6):2367–74.
Gingelmaier A, Kurowski M, Kästner R, Eberle J, Mylonas I, Belohradsky BH, et al. Placental transfer and pharmacokinetics of lopinavir and other protease inhibitors in combination with nevirapine at delivery. AIDS Lond Engl. 2006;20(13):1737–43.
Capparelli EV, Englund JA, Connor JD, Spector SA, McKinney RE, Palumbo P, et al. Population pharmacokinetics and pharmacodynamics of zidovudine in HIV-infected infants and children. J Clin Pharmacol. 2003;43(2):133–40.
Ivanovic J, Nicastri E, Anceschi MM, Ascenzi P, Signore F, Pisani G, Pregnancy and Newborn Clinical Outcome Group in HIV Infection (PANCOH), et al. Transplacental transfer of antiretroviral drugs and newborn birth weight in HIV-infected pregnant women. Curr HIV Res. 2009;7(6):620–5.
Betts S, Björkhem-Bergman L, Rane A, Ekström L. Expression of CYP3A4 and CYP3A7 in human foetal tissues and its correlation with nuclear receptors. Basic Clin Pharmacol Toxicol. 2015;117(4):261–6.
Fanni D, Fanos V, Ambu R, Lai F, Gerosa C, Pampaloni P, et al. Overlapping between CYP3A4 and CYP3A7 expression in the fetal human liver during development. J Matern Fetal Neonatal Med. 2015;28(11):1291–5.
Shuster DL, Risler LJ, Prasad B, Calamia JC, Voellinger JL, Kelly EJ, et al. Identification of CYP3A7 for glyburide metabolism in human fetal livers. Biochem Pharmacol. 2014;92(4):690–700.
Vyhlidal CA, Pearce RE, Gaedigk R, Calamia JC, Shuster DL, Thummel KE, et al. Variability in expression of CYP3A5 in human fetal liver. Drug Metab Dispos Biol Fate Chem. 2015;43(8):1286–93.
Tracy TS, Venkataramanan R, Glover DD, Caritis SN, National Institute for Child Health and Human Development Network of Maternal-Fetal-Medicine Units. Temporal changes in drug metabolism (CYP1A2, CYP2D6 and CYP3A Activity) during pregnancy. Am J Obstet Gynecol. 2005;192(2):633–9.
Syme MR, Paxton JW, Keelan JA. Drug transfer and metabolism by the human placenta. Clin Pharmacokinet. 2004;43(8):487–514.
Cramer YS, Rosenkranz SL, Hall SD, Szczech LA, Amorosa V, Gupta SK. Hemodialysis does not significantly affect the pharmacokinetics of nevirapine in HIV-1-infected persons requiring hemodialysis: results from ACTG A5177. J Acquir Immune Defic Syndr. 2010;54(4):e7–9.
Erickson DA, Mather G, Trager WF, Levy RH, Keirns JJ. Characterization of the in vitro biotransformation of the HIV-1 reverse transcriptase inhibitor nevirapine by human hepatic cytochromes P-450. Drug Metab Dispos. 1999;27(12):1488–95.
Smith GCS, Cameron AD. Estimating human fetal blood volume on the basis of gestational age and fetal abdominal circumference. BJOG Int J Obstet Gynaecol. 2002;109(6):721–2.
Zanardo V, Gabrieli C, de Luca F, Trevisanuto D, De Santis M, Scambia G, et al. Head-to-body delivery by ‘two-step’ approach: effect on cord blood hematocrit. J Matern Fetal Neonatal Med. 2013;26(12):1234–8.
Chang Y-H, Yang S-H, Wang T-F, Lin T-Y, Yang K-L, Chen S-H. Complete blood count reference values of cord blood in Taiwan and the influence of gender and delivery route on them. Pediatr Neonatol. 2011;52(3):155–60.
Eskola M, Juutistenaho S, Aranko K, Sainio S, Kekomäki R. Association of cord blood platelet count and volume with hemoglobin in healthy term infants. J Perinatol. 2010;31(4):258–62.
Arant BS Jr. Developmental patterns of renal functional maturation compared in the human neonate. J Pediatr. 1978;92(5):705–12.
Moltó J, Valle M, Miranda C, Cedeño S, Miranda J, Santos JR, et al. Once- or twice-daily dosing of nevirapine in HIV-infected adults: a population pharmacokinetics approach. J Antimicrob Chemother. 2008;62(4):784–92.
Peters AM, Perry L, Hooker CA, Howard B, Neilly MDJ, Seshadri N, Sobnack R, Irwin A, Snelling H, Gruning T, Patel NH, Lawson RS, Shabo G, Williams N, Dave S, Barnfield MC. Extracellular fluid volume and glomerular filtration rate in 1878 healthy potential renal transplant donors: effects of age, gender, obesity and scaling. Nephrol Dial Transplant. 2012;27(4):1429–37.
Clapp JF, Stepanchak W, Tomaselli J, Kortan M, Faneslow S. Portal vein blood flow—Effects of pregnancy, gravity, and exercise. Am J Obstet Gynecol. 2000;183(1):167–72.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
Maïlys De Sousa Mendes, Gabrielle Lui, Yi Zheng, Claire Pressiat, Deborah Hirt, Elodie Valade, Naïm Bouazza, Frantz Foissac, Stephane Blanche, Jean-Marc Treluyer, SaïkUrien and Sihem Benaboud have no conflicts of interest to declare.
Funding
This work was funded by the University of Paris V.
Additional information
S. Urien and S. Benaboud contributed equally to this work as last authors.
Rights and permissions
About this article

Cite this article
De Sousa Mendes, M., Lui, G., Zheng, Y. et al. A Physiologically-Based Pharmacokinetic Model to Predict Human Fetal Exposure for a Drug Metabolized by Several CYP450 Pathways. Clin Pharmacokinet 56, 537–550 (2017). https://doi.org/10.1007/s40262-016-0457-5
Published:
Issue Date:
DOI: https://doi.org/10.1007/s40262-016-0457-5