
Abstract
Background and Objective
Pazopanib is a multi-targeted anticancer tyrosine kinase inhibitor. This study was conducted to develop a population pharmacokinetic (popPK) model describing the complex pharmacokinetics of pazopanib in cancer patients.
Methods
Pharmacokinetic data were available from 96 patients from three clinical studies. A multi-compartment model including (i) a complex absorption profile, (ii) the potential non-linear dose–concentration relationship and (iii) the potential long-term decrease in exposure was developed.
Results
A two-compartment model best described pazopanib pharmacokinetics. The absorption phase was modelled by two first-order processes: 36 % (relative standard error [RSE] 34 %) of the administered dose was absorbed with a relatively fast rate (0.4 h−1 [RSE 31 %]); after a lag time of 1.0 h (RSE 6 %), the remaining dose was absorbed at a slower rate (0.1 h−1 [RSE 28 %]). The relative bioavailability (rF) at a dose of 200 mg was fixed to 1. With an increasing dose, the rF was strongly reduced, which was modelled with an E max (maximum effect) model (E max was fixed to 1, the dose at half of maximum effect was estimated as 480 mg [RSE 23 %]). Interestingly, the plasma exposure to pazopanib also decreased over time, modelled on rF with a maximum magnitude of 50 % (RSE 27 %) and a first-order decay constant of 0.15 day−1 (RSE 43 %). The inter-patient and intra-patient variability on rF were estimated as 36 % (RSE 16 %) and 75 % (RSE 22 %), respectively.
Conclusion
A popPK model for pazopanib was developed that illustrated the complex absorption process, the non-linear dose–concentration relationship, the high inter-patient and intra-patient variability, and the first-order decay of pazopanib concentration over time. The developed popPK model can be used in clinical practice to screen covariates and guide therapeutic drug monitoring.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
A population pharmacokinetic model for pazopanib was developed and evaluated. |
This model illustrated pharmacokinetic characteristics of pazopanib: the complex absorption process, the non-linear dose–concentration relationship, the high inter-patient and intra-patient variability, and the decreased exposure over time. |
It was suggested that pazopanib 400 mg twice daily could result in higher exposure than once-daily dosing of pazopanib 800 mg. |
1 Introduction
Pazopanib hydrochloride is a tyrosine kinase inhibitor that has been approved for the treatment of metastatic renal cell carcinoma (mRCC) and soft tissue sarcoma at an oral dose of 800 mg daily [1].
Pazopanib has a complex clinical pharmacological profile. Pazopanib is readily soluble in water at pH <4 but practically insoluble at higher pH [1, 2], which partly explains its low bioavailability (14–39 %) in humans [1–3]. In a phase I trial, the plasma exposure did not increase in a relevant way at doses exceeding 800 mg [4]. Accordingly, the bioavailability of pazopanib might be saturable. In addition, a recent study suggested that there might be a decrease in the plasma concentration of pazopanib during treatment [5]. Finally, pazopanib shows a large inter-patient variability in plasma concentrations (40–70 % coefficient of variation) [1, 4]. The intra-patient variability of pazopanib was found to be relatively large as well—nearly the same as the inter-patient variability [5].
A pazopanib trough plasma concentration (C trough) threshold of >20.5 mg/L has been identified, which was associated with the efficacy of treatment in patients with mRCC [6, 7]. With the reported large inter-patient variability in pharmacokinetics [1, 4], a subset of 30 % of patients are inadequately exposed to pazopanib when taking the fixed dose of 800 mg [6]. Therapeutic drug monitoring (TDM) may be implemented to reach plasma concentrations above this predefined target in all patients treated with pazopanib in order to optimise treatment outcomes.
The approach of modelling and simulation has proved its merits in understanding the pharmacokinetic characteristics of drugs [8], exploring complex drug–drug interactions or auto-induction/inhibition [9], capturing pharmacokinetic–pharmacogenetic relationships to partially explain the inter-patient variability [10, 11] and applying TDM by the extrapolation of randomly collected samples to C trough in the clinic [12]. Therefore, the aim of the current study was to systemically develop and evaluate a population pharmacokinetic (popPK) model of pazopanib to understand and quantify the complex pharmacokinetic characteristics of pazopanib.
2 Methods
2.1 Clinical Studies
Pharmacokinetic data were available from three clinical studies [5, 13, 14]. An overview of the pharmacokinetic data is given in Table 1 and is further explained in the following sections (sample numbers at different time points are listed in Electronic Supplementary Material table 1).
Study 1 evaluated the feasibility of pharmacokinetic-guided individualised dosing of pazopanib. In this phase I study, dense pharmacokinetic samples were collected during a dose interval in 13 patients at steady state on three occasions. For a detailed description of this study we refer the reader to de Wit et al. [5].
Study 2 explored the maximal tolerated dose of the combination of pazopanib and docetaxel in patients with advanced solid tumours. Co-treatment with docetaxel showed no influence on pazopanib pharmacokinetics. Pharmacokinetic data for pazopanib either with or without docetaxel were used for model development and were available on one occasion after 7 or 20 days of pazopanib treatment. A detailed description of Study 2 is given in Hamberg et al. [13].
Study 3 investigated the combination of pazopanib and ifosfamide in patients with advanced or metastatic solid tumours. Pazopanib pharmacokinetics were sampled extensively on 1 day with C trough measured on the following 2 days. These pharmacokinetic samples were taken after 7 or 13 days of pazopanib treatment. Pazopanib plasma concentrations were clearly influenced by the well-established enzyme induction by ifosfamide. Pharmacokinetic data of pazopanib either with or without ifosfamide were used in the model development. For a detailed description of Study 3 we refer the reader to Hamberg et al. [14].
2.2 Model Development
2.2.1 Structural Model Development
The starting point for model development was a one-compartment model with linear absorption and elimination [15]. As ifosfamide was co-administered in Study 3, induction of pazopanib clearance (CL) by ifosfamide was included a priori during model development. The pharmacokinetics of ifosfamide and pazopanib were modelled in a sequential manner [16]. Firstly, a metabolic enzyme turn-over model that has been developed to describe ifosfamide auto-induction [9] was used and fixed for ifosfamide pharmacokinetics in the current study. Secondly, individual parameter estimates of ifosfamide were used in the pazopanib popPK model with the amount of metabolic enzyme linearly correlated with pazopanib CL.
Subsequently, the popPK model for pazopanib was further developed. For this, different numbers of compartments, the optimal absorption model, potential saturable absorption and a long-term decrease in plasma exposure were systemically explored.
2.2.2 Statistical Model Development
Between-subject variability (BSV) and within-subject variability (WSV) were included on different structural pharmacokinetic parameters. The inclusion of BSV and WSV was screened according to the change of objective function value (OFV), clinical relevance, standard error and shrinkage. BSV and WSV were modelled according to Eq. 1:
where P i represents the individual parameter estimate for individual i, P represents the typical population parameter estimate and η i represents either the BSV or WSV effect distributed following N (0, ω 2).
Residual errors were described by a combined error model (Eq. 2):
where C obs,ij or C pred,ij represents, for the ith subject and the jth measurement, the observation or prediction, respectively. ε p,ij represents the proportional error that was assumed distributed following N (0, σ 2), and ε a,ij represents the additive error.
2.3 Model Evaluation
Model evaluation was performed by the inspection of multiple aspects: parameter precision, plausibility of parameter estimates and the correlation matrix. For hierarchical models, significant model improvement was defined according to the drop of OFV (dOFV) by a p value of <0.01 (degree of freedom [df] = 1, dOFV >6.63; df = 2, dOFV >9.21). Graphical evaluation was performed by model fit on each individual and goodness-of-fit (GOF) diagnostics. Besides, prediction-corrected visual predictive check (PcVPC) was used due to the high variability in data (n = 1000) [17].
2.4 Simulations
Simulations of the developed popPK model of pazopanib were performed at 800 mg once daily and 400 mg twice daily for a 6-week treatment period. The C trough over time and area under concentration–time curve (AUC) were compared between the two dose regimens. The changes in C trough through the treatment period were calculated. The published threshold of a C trough >20.5 mg/L was used as reference for the simulation [6].
2.5 Software
Model estimation, evaluation and simulation was performed by NONMEM® (version 7.3, ICON Development Solutions, Ellicott City, MD, USA) [18] together with a gfortran compiler and NONMEM® toolkit psn [19], with Piraña® used as the graphical interface [20]. The estimation method used in the model development was first-order conditional estimation with interaction. In addition, R (version 3.0.3) was used for pre-processing of the data and plotting [21] together with the R package Xpose [22].
3 Results
3.1 Model Development
The structure of the final popPK model of pazopanib is shown in Fig. 1 (control stream in Electronic Supplementary Material 1). In general, pazopanib pharmacokinetics were best described by a two-compartment model with a two-phase absorption model and linear elimination. The relative bioavailability (rF) was found to be correlated with the pazopanib dose and time on pazopanib treatment. The parameter estimates of the final popPK model are listed in Table 2. Details of the final model are explained in the following sections.

Schematic representation of the population pharmacokinetic model of pazopanib. The area within the grey square represents the previously developed population pharmacokinetic model of ifosfamide including (auto-)induction of metabolic enzymes [9]. ALAG lag time, CL clearance, fr fraction of pazopanib, Ifo ifosfamide, KaF fast absorption rate constant, KaS slow absorption rate constant, Paz pazopanib, Q between-compartmental clearance, Vc central volume of distribution, Vp peripheral volume of distribution
A first-order absorption process showed underestimation of the maximum concentration and misspecification at the elimination phase. This can be seen in Fig. 2, which shows the model fit with first-order absorption model in representative pharmacokinetic curves of pazopanib (dotted lines). As pazopanib is easily soluble at pH <4 but practically insoluble at higher pH, a sequential absorption process was considered. Ultimately, the absorption process was best described by two first-order processes: 36 % (relative standard error [RSE] 34 %) of pazopanib was absorbed with a relatively fast rate (fast absorption rate constant [KaF] 0.4 h−1 [RSE 31 %]); after a lag time of 1.0 h (RSE 6 %), the remaining part (64 %) of the pazopanib dose was absorbed with a slower rate (slow absorption rate constant [KaS] 0.1 h−1 [RSE 28 %]). The percentage of the two processes of absorption was estimated based on rF. Compared with a first-order absorption model, this two-phase absorption model dropped OFV by 34 points with a strong improvement in model fit (dashed lines in Fig. 2).

Representative concentration–time curves of pazopanib and the associated model fit of the first-order absorption model and two-phase absorption model. The triangles are the observed pharmacokinetic data and the lines are the individual model predictions: the dotted lines by first-order absorption model and the dashed lines by two-phase absorption model. The first-order absorption model shows underestimation of the maximum concentration or misspecification in elimination; the two-phase absorption model improves the model fit
A previous clinical trial suggested non-linearity between pazopanib dose and pharmacokinetics [4]. In Fig. 3, it is shown that not considering the effect of dose on the pharmacokinetic leads to underestimation at low doses and overestimation at high doses (dotted lines). To account for this effect, dose-related rF (rFd) was introduced in the model. With increasing dose, rFd decreased in an E max (maximum effect) manner. The rFd at a dose of 200 mg was fixed to 1; for the other doses the rFd was estimated according to Eq. 3:
where E max represents the maximum effect of dose concentrations on decrease of rF, and ED50 represents the dose level with half of the bioavailability at a dose of 200 mg.

Pharmacokinetics of pazopanib at different dose levels and associated model fit with a linear and non-linear dose–concentration relationship. The triangles are the observed pharmacokinetic data and the lines are the population model predictions: the dotted lines by linear dose–concentration relationship and the dashed lines by non-linear dose–concentration relationship. The model with a linear dose-concentration relationship shows underestimation at low doses and overestimation at high doses. The model with a non-linear dose–concentration relationship improves the model fit
The implementation of rFd resulted in dOFV of 62 points (p < 0.01) together with strong improvement in model fit (dashed lines in Fig. 3). ED50 was estimated as 480 mg (RSE 23 %). The rFd at a dose of 400 mg was 59 % higher than that of 800 mg.
The potential decrease in pazopanib exposure over time [5] was investigated with several models. Firstly, an enzyme turn-over model, similar to that used to account for ifosfamide enzyme induction, was tested. However, this model proved to be over-parameterised. Ultimately, a model in which bioavailability decreased over time described the data best. In this model, time-related rF (rFt) was described as a first-order decay from 1 to a minimum level according to Eq. 4:
where A d represents the maximum decrease in rFt, and λ represents the first-order decay constant.
The inclusion of rFt dropped OFV by 16 points (p < 0.01). A d was estimated as 0.5 (RSE 27 %) and λ as 0.15 day−1 (RSE 43 %).
Combined, the rF of pazopanib was described using rFd and rFt according to Eq. 5:
Figure 4 shows the relationship between rF, dose and time. At pazopanib 800 mg once daily rF decreases from 44 % at start of treatment to 22 % after 8 weeks.

Relative bioavailability of pazopanib versus dose and time on treatment. The surface in the figure shows the relative bioavailability at different doses (200–1600 mg) and treatment periods (0–8 weeks). The relative bioavailability decreases with increments in the dose levels and treatment period. rF relative bioavailability
The variability was estimated to be very large. The BSV of KaF, CL, and peripheral volume of distribution were estimated as 140 % (RSE 20 %), 31 % (RSE 20 %) and 98 % (RSE 17 %), respectively. The BSV and WSV on rF were estimated as 36 % (RSE 16 %) and 74 % (RSE 22 %), respectively. In addition, a BSV was considered on the fraction of fast and slow absorption processes but was not included in the final model. This was because the estimate of this BSV was relatively small, with a drop in OFV of only 0.2 points.
The coadministration of docetaxel in Study 2 was estimated as a covariate on the parameters of rF, CL, KaF, KaS and the fraction of the fast and slow absorption processes, respectively. No significant difference was found on the parameter estimates by the coadministration of docetaxel (p > 0.01). In addition, to verify that the pazopanib pharmacokinetics in Study 3 were correctly estimated when combined with the ifosfamide pharmacokinetic model [9], the popPK models of pazopanib estimated with and without Study 3 were compared. It was found that the differences in the parameter estimates between these two models were all within an acceptable range (<25 %).
3.2 Model Evaluation
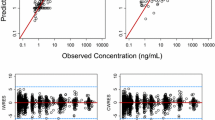
Individual model predictions of representative pazopanib pharmacokinetic curves shown in Fig. 2 (dashed lines) suggested the current model captured the highly variable absorption profiles of pazopanib pharmacokinetics well. Figure 5 shows the PcVPC stratified on three clinical studies. In general, the popPK model was sufficient to describe the pharmacokinetics in three studies; there was some underestimation in Study 1. The binning of three studies was not equally eligible due to different study sampling schedules. The GOF plots are presented in Electronic Supplementary Material figure 1, and the PcVPC stratified on treatment weeks 1, 2, 3, 4 and 6 are presented in Electronic Supplementary Material figure 2. A bootstrap analysis was considered but was found to be an unsuitable evaluation method due to the complex model structure and the variable sampling schedules between Study 1, 2 and 3.

Prediction-corrected visual predictive checks of the final population pharmacokinetic model of pazopanib stratified by the clinical studies (n = 1000). The shaded areas represent the 95 % confidence intervals, solid lines represent the observed values and dotted lines represent the simulated values of 10, 50, and 90 % percentiles
3.3 Simulations
Figure 6 shows the simulated pharmacokinetic profiles of pazopanib at the approved dose of 800 mg once daily compared with 400 mg twice daily. Pazopanib C trough first accumulates until day 10 and then decreases. From week 2 to week 4, both the C trough and AUC of pazopanib concentrations dropped by 11 %, with a further drop of 3 % from week 4 to week 6. After 6 weeks, the C trough of 400 mg twice daily was 75 % higher than that of 800 mg once daily (39 vs. 22 mg/L), and the AUC was 59 % higher (1056 vs. 665 mg·h/L).

Simulated pazopanib concentrations when administered at 800 mg once daily (approved dose) and 400 mg twice daily. Left panel pazopanib trough concentrations over 6 weeks; right panel pazopanib concentration–time curve over 24 h at steady-state concentrations (6 weeks of treatment). The dotted lines show the concentration level of 20.5 mg/L. 1dd once daily, 2dd twice daily
4 Discussion
In the current work, we systematically developed a popPK model for pazopanib based on data from 96 patients receiving a wide dose range (200–1200 mg). We studied the complex absorption process of pazopanib and identified and quantified a non-linear dose–concentration and time–concentration relationship that had not been revealed earlier. In addition, we quantified both inter-patient and intra-patient variability.
Previously, Imbs et al. [15] published a popPK model for pazopanib based on the pharmacokinetic data from the coadministration of pazopanib and bevacizumab in 25 patients at two dose concentrations (400 or 600 mg). This was a one-compartment model with first-order absorption with lag time and linear elimination. However, this model was based on a small study population and therefore did not evaluate essential pharmacokinetic factors such as the dose–concentration relationship of pazopanib, a decrease in exposure over time and intra-patient variability, which were studied in the current study. A similar model structure for pazopanib pharmacokinetics was also reported by the US Food and Drug Administration (FDA) [23] when they compared the bioavailability of a 400 mg dose and an 800 mg dose.
In our final model, the absorption profile of pazopanib was characterised by combined fast and slow processes. Pazopanib is water insoluble at pH >4 [2], which means that pazopanib is not absorbed in most parts of the human intestine. Meanwhile, the absorption of pazopanib is known to be limited by its dissolution rate [2]. Therefore, we speculated that pazopanib is first absorbed rapidly at the beginning of the intestine, the duodenum, where pazopanib might be still in solution. After transit through the duodenum (characterised by the lag time in our model), the second phase in the absorption of pazopanib is characterised by a fully dissolution rate-limited phase with a very low absorption rate. The estimated lag time of the slow absorption process (1 h) is approximately in agreement with the gastric emptying time [24].
The poor dissolution of pazopanib might also explain the dose-dependency in rF of pazopanib. A phase I trial of pazopanib found no obvious increase in plasma exposure at doses higher than 800 mg [4]. Based on the reported doses and corresponding AUCs in this trial, a further calculation of dose–normalised AUC (dAUC) at day 22 showed that the dAUC declined with increments of dose concentrations in an E max manner. The rF of 800 mg, calculated by dividing the dAUC of 800 mg by the dAUC of 200 mg, was 48 % (calculation procedure not shown), which was in agreement with our finding in the current study (44 %). The observed saturation in bioavailability suggests that a dose of pazopanib 400 mg twice daily may lead to higher exposure than an 800 mg once-daily dose (Fig. 6). Numerically, the rF of the 400 mg dose was estimated to be 59 % higher than that of the 800 mg dose in the current model, whereas the rF was found to be 40 % higher with the 400 mg dose in the FDA report [23]. Therefore, for the patients underexposed at 800 mg once daily, splitting the dose into twice-daily administration could potentially be effective both in treatment and costs. However, this has not yet been proven in a clinical study.
We characterised a decrease in pazopanib concentrations over time, especially within the first 4 weeks. The first mention of a possible decrease in the plasma concentration of pazopanib was in Study 1 [5]. Recently, Imbs et al. [25] described a lower concentration of pazopanib on day 15 as compared with day 8 (the day of cisplatin administration) in a phase I trial combining pazopanib and cisplatin [25]. In addition, a long-term decrease in concentrations has been found for other tyrosine kinase inhibitors, such as imatinib in patients with gastrointestinal stromal tumours [26] and sorafenib in patients with hepatocellular carcinoma [27]. However, the mechanism behind this decrease has not yet been elucidated. Pazopanib is a weak inhibitor for cytochrome P450 (CYP) 3A4 and CYP2D6 [28]. It has also been reported that pazopanib potentially induces human CYP3A4 via in vitro human pregnane X receptor [29]. However, whether the decrease in concentrations was due to the autoinduction of CYP3A4 has not been confirmed. The pharmacokinetic C trough target of >20.5 mg/L was identified based on week 4 pazopanib concentrations [6]. Therefore, attention should be paid for the TDM service applied within the first 4 weeks of treatment to the potential decrease in pazopanib exposure thereafter.
Both inter-patient and intra-patient variability were found to be high for pazopanib. One reason for this could be the variability in the absorption process. With the absorption model in mind, we speculate that gastric transit and absorption in the duodenum might be crucial in the absorption process of pazopanib and that these processes can be highly variable. The observed BSV strongly underlines the potential of TDM based on the predefined pharmacokinetic target (C trough >20.5 mg/L) for pazopanib, which has been explored by a clinical trial [30]. The actual reduction in variability by application of TDM will, however, be limited by the relative high WSV observed.
The presented popPK model has some limitations. Firstly, Study 1 was a TDM study in which the dose was adapted based on the observed AUC after the standard dose [5]. As previously shown, this might result in a biased model in favour of non-linear pharmacokinetics [31]. However, as complete pharmacokinetic profiles at the standard starting dose of 800 mg once daily were also available from each patient, this effect is likely to be minimal. Secondly, the exploration of concentration changes over time was limited by the relative short observation period of 6 weeks.
This popPK model can be useful in investigating clinically relevant questions. For example, it can be used to study drug–drug interactions and pharmacokinetics–toxicity and pharmacokinetics–efficacy relationships. Besides, various covariates, such as demographics, genotypes and phenotypes, can be systematically explored in order to further understand the variability and the decrease in concentrations in time. In addition, the model can assist in extrapolation of a random sampled concentration to C trough for the purpose of TDM and can also support the TDM service by comparing the outcome from different dose adjustments in patients.
5 Conclusions
We developed and evaluated a popPK model for pazopanib. This model illustrated multiple challenging aspects of the pharmacokinetic characteristics of pazopanib: the complex absorption process, the non-linear dose–concentration relationship, the high inter-patient and intra-patient variability, and the decreased exposure over time. Our results suggest that pazopanib 400 mg twice daily could result in a higher exposure than pazopanib 800 mg once daily.
References
Votrient 200 mg film-coated tablets. Summary of product characteristics. http://www.ema.europa.eu/docs/en_GB/document_library/EPAR_-_Product_Information/human/001141/WC500094272.pdf. Accessed 3 Sept 2015.
Center for Drug Evaluation and Research. Clinical pharmacology and biopharmaceutics review(s) of Votrient. http://www.accessdata.fda.gov/drugsatfda_docs/nda/2009/022465s000_ClinPharmR.pdf. Accessed 13 Jan 2016.
van Leeuwen RWF, van Gelder T, Mathijssen RHJ, Jansman FGA. Drug–drug interactions with tyrosine-kinase inhibitors: a clinical perspective. Lancet Oncol. 2014;15(8):e315–26.
Hurwitz HI, Dowlati A, Saini S, Savage S, Suttle AB, Gibson DM, et al. Phase I trial of pazopanib in patients with advanced cancer. Clin Cancer Res. 2009;15(12):4220–7.
de Wit D, van Erp NP, den Hartigh J, Wolterbeek R, den Hollander-van Deursen M, Labots M, et al. Therapeutic drug monitoring to individualize the dosing of pazopanib: a pharmacokinetic feasibility study. Ther Drug Monit. 2014;37(3):331–8.
Suttle AB, Ball HA, Molimard M, Hutson TE, Carpenter C, Rajagopalan D, et al. Relationships between pazopanib exposure and clinical safety and efficacy in patients with advanced renal cell carcinoma. Br J Cancer. 2014;111(10):1909–16.
Yu H, Steeghs N, Nijenhuis CM, Schellens JHM, Beijnen JH, Huitema ADR. Practical guidelines for therapeutic drug monitoring of anticancer tyrosine kinase inhibitors: focus on the pharmacokinetic targets. Clin Pharmacokinet. 2014;53(4):305–25.
Yu H, Steeghs N, Kloth JSL, de Wit D, van Hasselt JGC, van Erp NP, et al. Integrated semi-physiological pharmacokinetic model for both sunitinib and its active metabolite SU12662. Br J Clin Pharmacol. 2015;79(5):809–19.
Kerbusch T, Huitema ADR, Ouwerkerk J, Keizer HJ, Mathôt RA, Schellens JHM, et al. Evaluation of the autoinduction of ifosfamide metabolism by a population pharmacokinetic approach using NONMEM. Br J Clin Pharmacol. 2000;49(6):555–61.
Kloth JSL, Klümpen H-J, Yu H, Eechoute K, Samer CF, Kam BLR, et al. Predictive value of CYP3A and ABCB1 phenotyping probes for the pharmacokinetics of sunitinib: the ClearSun study. Clin Pharmacokinet. 2014;53(3):261–9.
Diekstra MHM, Klümpen HJ, Lolkema MPJK, Yu H, Kloth JSL, Gelderblom H, et al. Association analysis of genetic polymorphisms in genes related to sunitinib pharmacokinetics, specifically clearance of sunitinib and SU12662. Clin Pharmacol Ther. 2014;96(1):81–9.
Gotta V, Widmer N, Montemurro M, Leyvraz S, Haouala A, Decosterd LA, et al. Therapeutic drug monitoring of imatinib: Bayesian and alternative methods to predict trough levels. Clin Pharmacokinet. 2012;51(3):187–201.
Hamberg P, Mathijssen RHJ, de Bruijn P, Leonowens C, van der Biessen D, Eskens FA, et al. Impact of pazopanib on docetaxel exposure: results of a phase I combination study with two different docetaxel schedules. Cancer Chemother Pharmacol. 2014;75(2):365–71.
Hamberg P, Boers-Sonderen MJ, van der Graaf WTA, de Bruijn P, Suttle AB, Eskens FALM, et al. Pazopanib exposure decreases as a result of an ifosfamide-dependent drug-drug interaction: results of a phase I study. Br J Cancer. 2014;110(4):888–93.
Imbs D-C, Négrier S, Cassier P, Hollebecque A, Varga A, Blanc E, et al. Pharmacokinetics of pazopanib administered in combination with bevacizumab. Cancer Chemother Pharmacol. 2014;73(6):1189–96.
Zhang L, Beal SL, Sheiner LB. Simultaneous vs. sequential analysis for population PK/PD data I: best-case performance. J Pharmacokinet Pharmacodyn. 2003;30(6):387–404.
Bergstrand M, Hooker AC, Wallin JE, Karlsson MO. Prediction-corrected visual predictive checks for diagnosing nonlinear mixed-effects models. AAPS J. 2011;13(2):143–51.
Beal SL, Boeckman AJ, Sheiner LB, editors. NONMEM user guides. San Francisco: University of Califomia at San Francisco; 1988.
Lindbom L, Pihlgren P, Jonsson EN, Jonsson N. PsN-Toolkit–a collection of computer intensive statistical methods for non-linear mixed effect modeling using NONMEM. Comput Methods Programs Biomed. 2005;79(3):241–57.
Keizer RJ, van Benten M, Beijnen JH, Schellens JHM, Huitema ADR. Piraña and PCluster: a modeling environment and cluster infrastructure for NONMEM. Comput Methods Programs Biomed. 2011;101(1):72–9.
R Development Core Team. R: a language and environment for statistical computing. Vienna: R Foundation for Statistical Computing; 2008.
Jonsson EN, Karlsson MO. Xpose–an S-PLUS based population pharmacokinetic/pharmacodynamic model building aid for NONMEM. Comput Methods Programs Biomed. 1999;58(1):51–64.
The FDA Clinical Pharmacology and Biopharmaceutics Review(s) (application number 22-465) of Pazopanib (Votrient) (patient population Advanced Renal Cell Carcinoma). http://www.accessdata.fda.gov/drugsatfda_docs/nda/2009/022465s000_ClinPharmR.pdf. Accessed 6 July 2016.
Rowland M, Tozer TN. Chapter 7: absorption. Clinical pharmacokinetics and pharmacodynamics, 4th ed. Baltimore: Lippincott Williams & Wilkins; 2011. pp. 183–216.
Imbs D-C, Diéras V, Bachelot T, Campone M, Isambert N, Joly F, et al. Pharmacokinetic interaction between pazopanib and cisplatin regimen. Cancer Chemother Pharmacol. 2016;77(2):385–92.
Eechoute K, Fransson MN, Reyners AK, De Jong FA, Sparreboom A, Van Der Graaf WTA, et al. A long-term prospective population pharmacokinetic study on imatinib plasma concentrations in GIST patients. Clin Cancer Res. 2012;18(20):5780–7.
Arrondeau J, Mir O, Boudou-Rouquette P, Coriat R, Ropert S, Dumas G, et al. Sorafenib exposure decreases over time in patients with hepatocellular carcinoma. Invest New Drugs. 2012;30(5):2046–9.
Goh BC, Reddy NJ, Dandamudi UB, Laubscher KH, Peckham T, Hodge JP, et al. An evaluation of the drug interaction potential of pazopanib, an oral vascular endothelial growth factor receptor tyrosine kinase inhibitor, using a modified Cooperstown 5 + 1 cocktail in patients with advanced solid tumors. Clin Pharmacol Ther. 2010;88(5):652–9.
Votrient: highlights of prescribing information. http://www.accessdata.fda.gov/drugsatfda_docs/label/2012/022465s-010S-012lbl.pdf. Accessed 3 Mar 2016.
Verheijen RB, Bins S, Mathijssen RH, Lolkema M, van Doorn L, Schellens JH, et al. Individualized pazopanib dosing: a prospective feasibility study in cancer patients. Clin Cancer Res. 2016. [Epub ahead of print]
Ahn JE, Birnbaum AK, Brundage RC. Inherent correlation between dose and clearance in therapeutic drug monitoring settings: possible misinterpretation in population pharmacokinetic analyses. J Pharmacokinet Pharmacodyn. 2005;32(5–6):703–18.
Acknowledgments
We would like to thank Prof. Dr. Hans Gelderblom who was the principal investigator of Study 1. We would like to thank Prof. Dr. Stefan Sleijfer and Dr. Paul Hamberg who were the principal investigators of Studies 2 and 3. In addition, we would like to thank Remy Verheijen for useful discussions.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Funding
No sources of funding were used.
Conflict of interest
Huixin Yu, Nielka van Erp, Sander Bins, Ron Mathijssen, Jan Schellens, Jos Beijnen, Neeltje Steeghs and Alwin Huitema declare no conflicts of interest.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
About this article

Cite this article
Yu, H., van Erp, N., Bins, S. et al. Development of a Pharmacokinetic Model to Describe the Complex Pharmacokinetics of Pazopanib in Cancer Patients. Clin Pharmacokinet 56, 293–303 (2017). https://doi.org/10.1007/s40262-016-0443-y
Published:
Issue Date:
DOI: https://doi.org/10.1007/s40262-016-0443-y