
Abstract
Background and Objectives
In the proximal tubule, basic drugs are transported from the renal cells to the tubule lumen through the concerted action of the H+/organic cation antiporters, multidrug and toxin extrusion (MATE) 1 and MATE2K. Dual inhibitors of the MATE transporters have been shown to have a clinically relevant effect on the pharmacokinetics of concomitantly administered basic drugs. However, the clinical impact of selective renal organic cation transport inhibition on the pharmacokinetics and pharmacodynamics of basic drugs, such as metformin, is unknown. This study sought to identify a selective MATE2K inhibitor in vitro and to determine its clinical impact on the pharmacokinetics and pharmacodynamics of metformin in healthy subjects.
Methods
Strategic cell-based screening of 71 US Food and Drug Administration (FDA)-approved medications was conducted to identify selective inhibitors of renal organic cation transporters that are capable of inhibiting at clinically relevant concentrations. From this screen, nizatidine was identified and predicted to be a clinically potent and selective inhibitor of MATE2K-mediated transport. The effect of nizatidine on the pharmacokinetics and pharmacodynamics of metformin was evaluated in 12 healthy volunteers in an open-label, randomized, two-phase crossover drug–drug interaction (DDI) study.
Results
In healthy volunteers, the MATE2K-selective inhibitor nizatidine significantly increased the apparent volume of distribution, half-life, and hypoglycemic activity of metformin. However, despite achieving unbound maximum concentrations greater than the in vitro inhibition potency (concentration of drug producing 50 % inhibition [IC50]) of MATE2K-mediated transport, nizatidine did not affect the renal clearance (CLR) or net secretory clearance of metformin.
Conclusion
This study demonstrates that a selective inhibition of MATE2K by nizatidine affected the apparent volume of distribution, tissue concentrations, and peripheral effects of metformin. However, nizatidine did not alter systemic concentrations or the CLR of metformin, suggesting that specific MATE2K inhibition may not be sufficient to cause renal DDIs with metformin.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Nizatidine was identified as a selective in vitro inhibitor of multidrug and toxin extrusion 2K (MATE2K)-mediated transport of metformin at clinically relevant concentrations. |
In healthy subjects, nizatidine increased the apparent volume of distribution and hypoglycemic activity of metformin. However, despite achieving unbound maximum concentrations that exceeded the in vitro inhibition potency of MATE2K-mediated transport, nizatidine did not alter the renal clearance or net secretory clearance of metformin. |
The data from this study demonstrate that very selective inhibitors may not be sufficient to cause measurable drug–drug interactions (DDIs) with organic cation transporters in the kidney. In addition, this study challenges current health authority guidelines that rely on in vitro predictions to inform the decision to conduct transporter-mediated clinical DDI studies. |
1 Introduction
In the proximal tubule of the kidney, basic drugs are transported from the blood to the lumen of the kidney by organic cation transporter 2 (OCT2) and are eliminated to the urine by the concerted action of the H+/organic cation antiporters, multidrug and toxin extrusion (MATE) 1 and MATE2K. Broadly selective inhibitors of multiple organic cation transporters (e.g., cimetidine for OCT2/MATE1/MATE2K, pyrimethamine for MATE1/MATE2K) have been shown to have a clinical impact on the pharmacokinetics of concomitantly administered organic cations (e.g., metformin, procainamide, ranitidine) through reduction in their renal clearance (CLR) [1–4]. However, the clinical impact of selective inhibition of a single organic cation transporter on the pharmacokinetics and pharmacodynamics of basic drugs is unknown.
MATE2K is believed to be an important renal transporter for many drugs. In comparison with MATE1, which is expressed in multiple tissues (e.g., kidney, liver, muscle), MATE2K is predominately expressed in the kidney [5], and at equivalent or higher levels than MATE1 (Yee, Chhibber, Kroetz, Giacomini, unpublished data). MATE2K also specifically transports some drugs (e.g., oxaliplatin), which do not appear to be substrates of MATE1 [6, 7]. Studies from our laboratory have shown that a common MATE2K promoter variant (g.−130G>A, rs12943590) is associated with poor response to the biguanide metformin in subjects with type 2 diabetes [8, 9]. Taken together, these data suggest that MATE2K is important for the renal elimination of many basic drugs including metformin.
As transporter-mediated drug–drug interactions (DDIs) occur in clinical situations and have an impact on pharmacokinetics and drug safety, regulatory agencies in the USA and EU have issued guidances that recommend using in vitro transporter studies to inform the decision of when to conduct a clinical DDI study. The US Food and Drug Administration (FDA) recommends that a clinical investigation of a transporter-mediated drug interaction should be conducted when the C max,u/IC50 ratio (maximum plasma concentration [C max] of the inhibitor that is not bound to plasma proteins [C max,u] divided by the concentration of drug producing 50 % inhibition in an in vitro assay [IC50]) of the new molecular entity is ≥0.1 [10]. The European Medicines Agency (EMA) guidance is more stringent with a clinical study initiation cut-off of ≥0.02 [11]. Although the current guidances focus primarily on the uptake transporters in the kidney (OCT2 and organic anion transporters 1 and 3 [OAT1 and OAT3]), the EMA and a recent publication from the International Transporter Consortium (ITC) recommend extending these guidelines to include MATE-mediated drug interactions [12].
The current decision trees within the FDA and EMA guidances focus on evaluating DDIs of individual transporters (e.g., OCT2, OAT1), rather than a transporter family (e.g., OATs, OCTs, MATEs). With the exception of cimetidine and pyrimethamine, few drugs have been identified as clinically potent inhibitors of renal organic ion transporters, and no drugs are selective inhibitors of individual renal organic cation transporters. In addition, because the inhibitors that have been identified are either promiscuous and inhibit multiple transporter families (e.g., quinidine) or are more potent inhibitors of OCT1, an organic cation transporter that is highly expressed in the liver (e.g., procainamide) [13–15], distinguishing the impact of renal organic cation transporters on the pharmacokinetics of the victim drug has been difficult.
In this study, a strategic screen was implemented to identify probe inhibitors of renal organic cation transport that are selective and can inhibit transport at clinically relevant concentrations. Through in vitro assays we identified the histamine 2 antagonist nizatidine as a selective inhibitor of MATE2K-mediated transport with an I fu/IC50 ratio ≥0.1. To determine the clinical impact of MATE2K-selective inhibition on the pharmacokinetics and pharmacodynamics of a victim organic cation, an open-label, randomized, two-phase crossover DDI study was conducted using metformin as the victim drug and nizatidine as the perpetrator. Metformin is eliminated unchanged by renal mechanisms, and while the liver is important for the pharmacologic activity of metformin, biliary excretion of metformin is negligible [16, 17]. The hypotheses of this clinical DDI study were that (i) the co-administration of metformin and nizatidine will reduce the CLR of metformin, increase the metformin concentration in the kidney, and potentially lead to increased plasma concentrations; and (ii) the interaction will enhance the hypoglycemic activity of metformin.
2 Methods
2.1 Physicochemical Properties and Clinical Concentration of Test Inhibitors
Predicted charge at pH 7.4 was calculated using MarvinView version 5.3.6 (ChemAxon; http://www.chemaxon.com). C max values of test inhibitors in human subjects and percentage protein binding in human plasma were obtained from literature sources (Electronic Supplementary Material Table 1 [18, 19]). C max,u values were calculated by C max,u = C max·f u, where f u is the unbound fraction in human plasma.
2.2 In Vitro Identification of Renal Transporter Inhibitors
2.2.1 Cell Lines
Flp-In human embryonic kidney (HEK293-Flp-In) cells stably expressing human OCT1 (HEK-OCT1), OCT2 (HEK-OCT2), OCT3 (HEK-OCT3), MATE1 (HEK-MATE1), MATE2K (HEK-MATE2K), and the pcDNA5/FRT empty vector (HEK-EV) were previously established in our laboratory [8, 20–26]. Madin–Darby canine kidney type II (MDCK-II) cells stably expressing human plasma membrane monoamine transporter (PMAT) (MDCK-PMAT) and the pcDNA3.1(+) vector (MDCK-EV) were also established previously [27].
2.2.2 Cellular Uptake Study
Cells were suspended in cell culture medium, seeded on poly-d-lysine-coated 48-well plates (Greiner Bio-One, Monroe, NC, USA) and grown to ~90 % confluency (~48 h post seeding). Immediately prior to uptake, HEK-EV, HEK-OCT1, HEK-OCT2, and HEK-OCT3 cells were pre-incubated for 20 min with Hank’s balanced salt solution (HBSS). HEK-EV, HEK-MATE1, and HEK-MATE2K cells were pre-incubated with HBSS plus 30 mmol/L NH4Cl for 20 mins. MDCK-EV and MDCK-PMAT cells were pre-incubated with Krebs-Ringer-Henseleit (KRH) buffer for 20 min. Pre-incubation media was removed and uptake was initiated with the addition of uptake buffer (HBSS [HEK cell lines] or KRH [MDCK cell lines] containing [14C]-metformin [10 μmol/L] with and without a test inhibitor at its 10 × C max,u value) at 37 °C for a period of time for which linear uptake of metformin was observed (2–5 min). At the end of the uptake, cells were washed twice with ice-cold buffer (HBSS [HEK cell lines] or KRH [MDCK cell lines]) and lysed with 0.1 N NaOH/0.1 % sodium dodecyl sulfate (SDS). Intracellular radioactivity was determined by liquid scintillation counting and normalized per well of protein content as measured by bicinchoninic acid protein assay (Thermo Scientific, Rockford, IL, USA). Each test condition was conducted in triplicate. Compounds that were selective inhibitors of the renal organic cation transporters at their 10 × C max,u were subjected to experimental IC50 determination. Studies were conducted exactly as described above using increasing concentrations of the inhibitor (ranging from 0 to 40 × C max,u of the inhibitor for a total of eight concentrations).
2.2.3 Determination of the Concentration of Drug Producing 50 % Inhibition Values
After adjusting for protein quantity and subtracting non-specific transport of metformin (measured from empty vector cells), residual values were normalized to the rate of uptake in the absence of the inhibitor (set at 100 %). Dose-response curves and IC50 values were obtained using GraphPad Prism 4.0 (GraphPad Software, San Diego, CA, USA). Briefly, inhibitor concentrations were transformed to log scale and dose-response inhibition curves were fitted with the following equation (Eq. 1):
where bottom is the plateau of maximum inhibition observed.
Absolute IC50 values were calculated by interpolation of the fitted curve.
2.3 Clinical Drug–Drug Interaction Study in Healthy Volunteers
2.3.1 Study Design
This was an open-label, randomized, two-treatment crossover study conducted in healthy subjects (n = 12) at the University of California, San Francisco (UCSF) Clinical and Translational Science Institute Clinical Resources Services, San Francisco General Hospital (SFGH) clinical site (San Francisco, CA, USA). To be eligible for the study, subjects had to provide written informed consent, be between the ages of 18 and 45 years, be in good health (as evidenced by medical histories, physical examination, and routine clinical laboratory evaluations), not be on any medications other than oral contraceptives, and not have any known allergies to iodine. Once enrolled, volunteers were directed to follow a controlled carbohydrate diet (200–250 g/day) for 3 days prior to each inpatient visit. Subjects were admitted to the clinical facility the night before the first dose and remained onsite for the duration of the study (36 h). After an overnight fast (10 h), study participants received either (i) an 850 mg oral dose of metformin (Glucophage®); or (ii) a simultaneous 850 mg oral dose of metformin and a 600 mg oral dose of nizatidine (Axid®). Each study participant received both treatments separated by a minimum of 7 days. A 3-h oral glucose tolerance test (OGTT, 75 g) was conducted 2 h after metformin dosing (with or without nizatidine). Baseline OGTTs, without metformin, were obtained from the same subjects in a previous study [9]. Standardized meals were provided during inpatient visits after completion of the OGTT. Following the metformin dose, subjects were asked to drink 8 oz (~237 mL) of water every 2 h to maintain urine flow and pH. At 10 h after metformin dosing, 1500 mg of iohexol (Omnipaque®) was administered by slow intravenous push over 5 min.
Frequent timed blood samples were collected up to 24 h post dosing to determine plasma metformin and nizatidine concentrations. Additional blood samples were collected 10–14 h post metformin dosing to determine clearance of iohexol (a measure of glomerular filtration rate). For metformin pharmacodynamics, frequent blood samples were collected 0–180 min after glucose administration. An additional blood sample was collected at 12 h after the second dose of metformin to determine serum creatinine concentrations. Urine samples were collected 0–2, 2–4, 4–8, 8–12, and 12–24 h after metformin dosing to calculate metformin and creatinine CLR.
2.3.2 Bioanalytical Methods
Metformin concentrations in plasma and urine were assayed by a validated liquid chromatography–tandem mass spectrometry method [28]. Nizatidine plasma concentrations were measured using the transitions m/z 332.29–58.08. Both the intra-day and inter-day coefficients of analysis variation were <5 %. Iohexol concentrations in plasma were measured by University of Minnesota Physicians Outreach Labs (Minneapolis, MN, USA). Lactate and glucose concentrations in plasma were analyzed by ARUP Laboratories (Salt Lake City, UT, USA). Creatinine concentrations in plasma and urine were measured by the clinical laboratories of SFGH using standard methodologies.
2.3.3 Data Analysis
2.3.3.1 Clinical Pharmacokinetics
The concentration–time curves of metformin and nizatidine were plotted using GraphPad Prism 4.0. The pharmacokinetic parameters of metformin, nizatidine, and iohexol were determined by non-compartmental analysis (WinNonlin® 4.1, Pharsight Corporation, Mountain View, CA, USA).
2.3.3.2 Statistical Analysis
Using previously reported metformin pharmacokinetic data in healthy volunteers [29], a sample size of 12 was needed to detect a 30 % difference in the CLR of metformin between the two treatments with >80 % power. Data are presented as mean ± standard deviation unless indicated otherwise. Paired non-parametric Student’s t tests were used to analyze the differences in metformin pharmacokinetic and pharmacodynamic parameters using GraphPad Prism 4.0. A statistically significant result was defined as when p < 0.05.
3 Results
3.1 Identification of Drugs that Selectively Inhibit Renal Organic Cation Transporters at Clinically Relevant Concentrations
FDA-approved medications (n = 71, Electronic Supplementary Material Table 1) were selected to be screened as potential inhibitors of MATE1, MATE2K, and OCT2 if they met one or more of the following criteria: (i) C max in human subjects was >0.1 μmol/L; (ii) f u was >0.10; and (iii) they were predicted to have a positive or neutral charge at physiological pH (pH 7.4). These criteria were used to maximize the chance of identifying a clinically potent inhibitor of renal organic cation transporters. The selected drugs (Electronic Supplementary Material Table 1) span across various therapeutic classes (e.g., antibacterial, antiulcer, antiarrhythmic, antiemetic). Of the 71 drugs, which were screened at different concentrations reflecting their clinical concentrations, four (naloxone, quinine, procainamide, and praziquantel) were inhibitors of OCT2, ten (trimethoprim, cimetidine, ranitidine, moxifloxacin, chlorphenesin, quinine, clofazimine, abacavir, norfloxacin, and ondansetron) were inhibitors of MATE1, and eleven (trimethoprim, chlorphenesin, cimetidine, ranitidine, moxifloxacin, norfloxacin, procainamide, ondansetron, famciclovir, nizatidine, and quinine) were inhibitors of MATE2K, with ≥50 % inhibition of [14C]-metformin uptake at 10× C max,u (Electronic Supplementary Material Fig. 1a–c).
Based on the OCT2, MATE1, and MATE2K screening results, inhibitors were grouped as follows: (i) OCT2-selective (naloxone and praziquantel); (ii) MATE1-selective (abacavir and clofazimine); (iii) MATE2K-selective (famciclovir and nizatidine); (iv) MATE1/MATE2K dual inhibitors (chlorphenesin, cimetidine, moxifloxacin, norfloxacin, ondansetron, ranitidine, and trimethoprim); and (v) OCT2/apical inhibitors (procainamide and quinine). These compounds (n = 15) were then assessed for their potential to inhibit OCT1 and OCT3, which are not thought to play a significant role in renal drug elimination. Of the 15 compounds, moxifloxacin, norfloxacin, ondansetron, ranitidine, and nizatidine demonstrated minimal inhibition of OCT1- or OCT3-mediated metformin uptake at their 10× C max,u values (metformin uptake was >50 %) and were therefore designated as selective inhibitors of the renal organic cation transporters (Electronic Supplementary Material Fig. 2a–e).
Follow-up IC50 studies of moxifloxacin, norfloxacin, ondansetron, ranitidine, and nizatidine were conducted in cell lines expressing each of the known metformin transporters (OCT1, OCT2, OCT3, MATE1, MATE2K, and PMAT) (Fig. 1a–e; Table 1). Of note, moxifloxacin, norfloxacin, and ondansetron were identified as potential clinical inhibitors of both MATE1 (C max,u/IC50 = 0.74, 0.20, and 0.62, respectively) and MATE2K (C max,u/IC50 = 3.16, 0.84, and 0.39, respectively), but not of any other metformin transporters tested. In addition, nizatidine was identified as a potent and selective inhibitor of MATE2K (C max,u/IC50 = 0.37), but not of any other metformin transporters tested. Although ranitidine was a potent inhibitor of MATE1- and MATE2K-mediated transport, it did not demonstrate selective inhibition of the two MATE transporters. In all cases, the Hill coefficient was >1, suggesting positive cooperativity where more than one binding site is involved in the inhibition. However, additional kinetic analyses are warranted to confirm this observation.

Inhibitory potency of various drugs on metformin uptake in cell lines expressing organic cation transporters. Five compounds are shown that potently inhibited MATE2K in the screening studies at or below therapeutically relevant unbound concentrations. Increasing concentrations of a moxifloxacin, b norfloxacin, c ondansetron, d ranitidine, and e nizatidine were analyzed for their ability to inhibit metformin uptake in cells stably over-expressing OCT1, OCT2, OCT3, MATE1, MATE2K, and PMAT. IC50 values were determined for drugs/cell lines that display ≥50 % inhibition of metformin uptake (horizontal dashed line). The vertical gray and black lines are drawn at 1× and 10× the inhibitor’s C max,u, respectively. Values are presented as mean ± standard error of the mean (n = 3). C max,u maximum plasma concentration that is not bound to plasma proteins, IC 50 concentration of drug producing 50 % inhibition of active transport, MATE multidrug and toxin extrusion, OCT organic cation transporter, PMAT plasma membrane monoamine transporter
3.2 Determination of the Clinical Impact of MATE2K-Selective Inhibition on the Pharmacokinetics and Response of Metformin in Healthy Volunteers
An open-label, randomized, two-phase crossover DDI study was conducted in healthy volunteers (n = 12) to determine the effect of MATE2K-selective inhibition by nizatidine on the exposure and response of metformin. All subjects completed the study and no adverse events were reported. The demographic and baseline characteristics of the healthy volunteers are shown in Table 2.
To maximize the potential for a clinical impact of MATE2K inhibition by nizatidine, a 600 mg oral dose of nizatidine (the maximum recommended daily dose) was administered to healthy volunteers. To verify that this dose of nizatidine achieved sufficient concentrations predicted to alter MATE2K activity, the pharmacokinetics of nizatidine was determined (Fig. 2; Electronic Supplementary Material Table 2). Nizatidine reached a C max of 4.2 ± 0.3 μg/mL (12.7 μmol/L), and with 35 % protein binding [19] its calculated C max,u was 2.7 ± 0.2 μg/mL (8.2 μmol/L). This value is greater than the in vitro MATE2K inhibition potency (IC50 = 7.8 μmol/L) and above the recommended cut-off for conducting a clinical DDI study (C max,u/IC50 = 1.1) [11, 12].

Mean nizatidine plasma concentrations following administration of a single oral dose to 12 healthy volunteers. Nizatidine plasma concentrations were determined after a 600 mg single oral dose in combination with metformin. The dotted horizontal lines are drawn at C fu/IC50 ratios of 1, 0.1 (FDA cut-off), and 0.02 (EMA cut-off). Data represent mean ± standard error of the mean. C fu unbound concentration in plasma, EMA European Medicines Agency, FDA US Food and Drug Administration, IC 50 concentration of drug producing 50 % inhibition of active transport
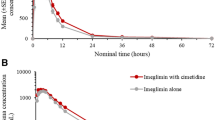
Plasma concentration–time profiles for metformin were similar following administration of metformin alone or with nizatidine (Fig. 3). When nizatidine was co-administered, the apparent volume of distribution (V d/F) and elimination half-life (t ½) of metformin increased slightly, but significantly, by 38 and 24 %, respectively (p < 0.05; Table 3; Fig. 4). Co-administration of nizatidine had no significant effect on the metformin time to C max (t max), C max, area under the plasma concentration–time curve [AUC] from time zero to 24 h (AUC24), AUC from time zero to infinity (AUC∞), and apparent oral clearance (CL/F) (Fig. 3; Table 3). Importantly, no effects of nizatidine on the CLR or CLSR (net CLR by secretion) of metformin were observed in the 11 subjects with complete urine collection.

Mean metformin plasma concentration–time curves after administration of metformin alone or with nizatidine to 12 healthy volunteers. Metformin plasma concentrations were determined after a single 850 mg dose alone (dotted line) or in combination with a 600 mg single oral dose of nizatidine (solid line). Data are given as mean ± standard error of the mean

Volume of distribution (a) and half-life (b) alterations between treatment groups. Symbols, the horizontal line, and brackets represent individual datapoints, the mean value of all subjects, and standard error of the mean, respectively. t ½ plasma terminal elimination half-life, V d /F apparent volume of distribution
A rare, but serious, adverse effect associated with metformin is lactic acidosis [30], where plasma lactate levels exceed 5 mmol/L. There was no significant difference in plasma lactate concentrations between treatment arms (data not shown) and observed C max values were below the 5 mmol/L toxicity threshold in both treatment groups (metformin alone, 1.7 ± 0.1 mmol/L; metformin with nizatidine, 1.6 ± 0.1 mmol/L).
The glucose-lowering effect of metformin was determined in healthy subjects by administering a 3-h OGTT (Electronic Supplementary Material Fig. 3). We observed similar glucose AUC values after the OGTT between pre-metformin and metformin alone treatment periods (pre-metformin, 3298 ± 145 mg·h/L; metformin alone, 3100 ± 121 mg·h/L; p > 0.1; Fig. 5). However, after metformin was co-administered with nizatidine, there was a significantly lower glucose AUC (greater response) than the pre-metformin values (pre-metformin, 3298 ± 145 mg·h/L; metformin with nizatidine, 3015 ± 74 mg·h/L; p = 0.03; Fig. 5).

The effect of nizatidine on plasma glucose concentrations after metformin treatment. The area under the plasma glucose concentration–time curve (AUC glucose) was calculated during the oral glucose tolerance test. Symbols, the horizontal line, and brackets represent individual datapoints, mean value of all subjects, and standard error of the mean, respectively. NS not significant
4 Discussion
Historically, DDIs were thought to occur primarily by interactions with drug-metabolizing enzymes. Current evidence suggests a role for drug transporters in mediating clinical DDIs [31, 32]; however, transporter-mediated DDIs have been less well-characterized. As noted previously, few inhibitors of renal organic cation transporters have been identified and, of these inhibitors, none has been shown to selectively inhibit a single transporter involved in the renal elimination of basic drugs. Further, previous clinical investigations of renal organic cation transporter-mediated DDIs have focused on drugs that are predicted to alter the activity of more than one transporter [1–4, 33–36]. Thus, there is a poor understanding of the mechanisms involved in the CLR of organic cations in general, and of metformin in particular. Through a strategic in vitro screen, nizatidine was identified as a clinically potent and selective inhibitor of MATE2K-mediated uptake of metformin. Thus, nizatidine represents a useful tool to understand the clinical significance of MATE2K in the renal elimination of metformin. In this study, nizatidine increased the V d/F and hypoglycemic activity of metformin. However, despite achieving C max,u values that exceeded the in vitro inhibition potency of MATE2K-mediated transport, nizatidine did not alter the CLR or net secretory clearance of metformin.
In our in vitro screen, several drugs appeared to stimulate metformin uptake in OCT2-, MATE1-, and MATE2K-transfected cells. This enhancement of in vitro activity has also been observed for a variety of solute carrier and adenosine triphosphate-binding cassette transporters including OAT1, MRP2, OATP1B1/1B3, and OCT2 [37–41]. Previous reports have observed the dependence of OCT2-mediated transport on inside-negative membrane potential [42, 43]. Thus, alterations in membrane potential by test compounds may explain an enhancement in metformin uptake. In our dataset, the enhancement in metformin uptake was more common in MATE-transfected cell lines, which are particularly affected by alterations in pH as they transport organic cations in exchange for a proton [5, 44, 45]. Therefore, it is possible that the test compounds could have altered the pH gradient, leading to an enhancement in metformin uptake by MATE-transfected cells. Other possibilities include positive cooperativity of compounds with the transporters as well as effects of compounds on potential difference across intracellular organelles (e.g., mitochondria).
Several reasons may be proposed to explain the lack of effect of nizatidine on metformin CLR. First, previous DDI studies examining the effect of perpetrator drugs on metformin CLR have used non-selective inhibitors of MATE1 and MATE2K. For example, in a previous clinical DDI study, there was a significant reduction in the CLR and CLSR of metformin after co-administration of a MATE1/MATE2K dual inhibitor, pyrimethamine [2]. However, in the current study, we did not observe a significant reduction in the CLR or CLSR of metformin when co-administered with nizatidine, a MATE2K-selective inhibitor. Compared to pyrimethamine, the t ½ of nizatidine is much shorter (pyrimethamine t ½, ~4 days [19]; nizatidine t ½, ~3 h). It is probable that the duration of inhibition is an important determinant of alterations in CLR, which is generally estimated over a 24-h period. However, we also examined the fractional CLR and CLSR (e.g., 0–2, 2–4, 0–4 h) of metformin, and found no differences between treatment arms (data not shown). Thus, the selective inhibition of MATE2K may be insufficient to have a measurable effect on the CLR and CLSR of metformin.
A second reason for the lack of reduction in metformin CLR by nizatidine is that metformin may cross the luminal membrane of the proximal tubule cell primarily by MATE1, and that MATE2K has a limited role. However, studies demonstrating an association between genetic variants of MATE2K and metformin CLR suggest that MATE2K does play a role in the renal elimination of metformin [8, 9, 46]. It is also possible that the in vitro methods used in this study incorrectly predicted the concentrations of nizatidine required to inhibit MATE2K in vivo. The IC50 for nizatidine on MATE2K-mediated transport was determined in vitro using a proton-driven influx of metformin. However, in vivo, MATE2K functions as an efflux pump. A previous study examining MATE1 transport of MPP+ (1-methyl-4-phenylpyridinium) demonstrated that there are symmetrical interactions of H+ with inward-facing and outward-facing MATE1 [47]. This has not been confirmed for MATE2K-mediated transport of metformin. However, it should be noted that for in vitro studies of both pyrimethamine and cimetidine, MATE1/MATE2K dual inhibitors of metformin transport, influx methods were used to predict IC50 values [2, 15].
Other possibilities are that concentrations of nizatidine in the renal tubule or tubule cells were not sufficient to inhibit the transporter. However, nizatidine had no effect on fractional CLR of metformin at earlier times, when nizatidine concentrations were higher. Notably, metformin concentrations were also higher at these earlier times; thus, nizatidine concentrations may not have been sufficient to reduce metformin CLR. Though speculative, it is also possible that nizatidine inhibited transporters involved in metformin reabsorption that counteracted its effects on CLSR. Because nizatidine is actively secreted in the kidney, its concentrations in the proximal tubule and proximal tubule cells should be higher than its plasma unbound concentrations [48].
These results suggest that further studies are needed to inform current guidelines provided by the EMA [11]. The EMA suggests that an I fu/IC50 ≥0.02 should trigger consideration of a clinical DDI study. However, our data suggest that very selective inhibitors may not be sufficient to cause measurable DDIs with organic cation transporters in the kidney, or that threshold ratios between I fu and IC50 for selective inhibitors should be higher to trigger consideration of a clinical study. More promiscuous inhibitors such as pyrimethamine and cimetidine clearly result in clinical DDIs and current ratios to trigger consideration of a clinical study, though conservative, are acceptable for these non-selective inhibitors. However, our data with nizatidine suggest that a MATE2K-selective inhibitor that achieved concentrations above its IC50 clearly did not result in a clinical DDI, and that different threshold ratios for triggering consideration of a clinical DDI may be warranted.
In this study, we observed a significant increase in metformin V d/F and a related increase in the metformin t ½. These results are consistent with previous reports that inhibitors of renal transporters have a direct effect on the volume of distribution of the victim drug [49]. As MATE2K is highly expressed in the kidney, an increase in metformin V d/F suggests that metformin may accumulate in the renal cell when its MATE2K-mediated efflux is blocked by nizatidine co-administration.
Interestingly, while we did not see any alterations in systemic concentrations of metformin, the hypoglycemic activity (as determined by the glucose AUC) was enhanced above pre-metformin values only when nizatidine was co-administered. Although not determined in the study, nizatidine is not known to alter glucose levels [50]. While the kidney is mostly known for its role in the reabsorption of glucose from the filtrate, it also has a significant impact on glucose uptake from blood (20 % of total uptake in the post-absorptive state) [51], gluconeogenesis (20–25 % of total glucose release in the post-absorptive state) [52–54], and glucose utilization (post-absorptive, 5–10 %; post-prandial, 10–15 % of total) [55, 56]. In the liver, metformin is known to enhance glucose uptake, decrease glucose production, and increase glucose utilization (for a review, see Woerle et al. [57]). Consistent with the increase in the metformin V d/F, our data suggest that nizatidine increases metformin concentrations in the kidney, where it is then able to enhance glucose uptake and utilization. Our data demonstrating that inhibition of MATE2K increases response to metformin in healthy subjects are consistent with previous studies from our laboratory demonstrating a reduced response to metformin in patients with type 2 diabetes who are carriers of an allele of MATE2K that is associated with increased transcription rates of the transporter [8, 9]. The concept that the pharmacologic effects of metformin are enhanced by increasing its concentrations in the liver or kidney through inhibition of MATEs needs further study. Similar concepts have been shown for renal toxicity to platinums [58], i.e., inhibition of MATEs enhances platinum accumulation in the kidney, resulting in increased nephrotoxicity [6, 7]. Further, mice with Mate1 deficiency exhibit increased metformin-induced lactic acidosis [59].
This study raises questions about extrapolating in vitro inhibition IC50 data to clinical DDI studies, and suggests that further refinement of current FDA and EMA guidelines are needed. Those refinements may include recommendations related to measurements of intracellular drug concentrations, the relative t ½ of the inhibitor compared with the substrate, and selectivity of the inhibitor for renal transporters. Clearly, future studies are needed to define best practices and standardize in vitro methodologies that accurately predict clinical DDIs and to understand the effects of selective versus non-selective inhibitors on renal drug elimination.
5 Conclusions
In healthy volunteers, a MATE2K-selective inhibitor, nizatidine, increased the V d/F and hypoglycemic activity of metformin. However, despite achieving unbound C max values that were greater than the in vitro inhibition potency of MATE2K-mediated transport, nizatidine did not change the CLR or net secretory clearance of metformin. This study highlights that current guidelines, which rely on in vitro predictions to inform the decision to conduct transporter-mediated clinical DDI studies, need refinement [10–12]. Further, this study suggests that physiologically based pharmacokinetic models to predict intracellular and intra-tubular concentrations of drugs should be developed and used in combination with in vitro data to predict DDIs in the kidney. Finally, this study supports previous studies highlighting that inhibition of efflux transporters will have important effects on the V d/F, tissue concentrations, and peripheral effects of victim drugs [49].
References
Somogyi A, Stockley C, Keal J, Rolan P, Bochner F. Reduction of metformin renal tubular secretion by cimetidine in man. Br J Clin Pharmacol. 1987;23(5):545–51.
Kusuhara H, Ito S, Kumagai Y, Jiang M, Shiroshita T, Moriyama Y, et al. Effects of a MATE protein inhibitor, pyrimethamine, on the renal elimination of metformin at oral microdose and at therapeutic dose in healthy subjects. Clin Pharmacol Ther. 2011;89(6):837–44.
Somogyi A, McLean A, Heinzow B. Cimetidine-procainamide pharmacokinetic interaction in man: evidence of competition for tubular secretion of basic drugs. Eur J Clin Pharmacol. 1983;25(3):339–45.
van Crugten J, Bochner F, Keal J, Somogyi A. Selectivity of the cimetidine-induced alterations in the renal handling of organic substrates in humans. Studies with anionic, cationic and zwitterionic drugs. J Pharmacol Exp Ther. 1986;236(2):481–7.
Masuda S, Terada T, Yonezawa A, Tanihara Y, Kishimoto K, Katsura T, et al. Identification and functional characterization of a new human kidney-specific H+/organic cation antiporter, kidney-specific multidrug and toxin extrusion 2. J Am Soc Nephrol. 2006;17(8):2127–35.
Yokoo S, Yonezawa A, Masuda S, Fukatsu A, Katsura T, Inui K. Differential contribution of organic cation transporters, OCT2 and MATE1, in platinum agent-induced nephrotoxicity. Biochem Pharmacol. 2007;74(3):477–87.
Yonezawa A, Masuda S, Yokoo S, Katsura T, Inui K. Cisplatin and oxaliplatin, but not carboplatin and nedaplatin, are substrates for human organic cation transporters (SLC22A1-3 and multidrug and toxin extrusion family). J Pharmacol Exp Ther. 2006;319(2):879–86.
Choi JH, Yee SW, Ramirez AH, Morrissey KM, Jang GH, Joski PJ, et al. A common 5’-UTR variant in MATE2-K is associated with poor response to metformin. Clin Pharmacol Ther. 2011;90(5):674–84.
Stocker SL, Morrissey KM, Yee SW, Castro RA, Xu L, Dahlin A, et al. The effect of novel promoter variants in MATE1 and MATE2 on the pharmacokinetics and pharmacodynamics of metformin. Clin Pharmacol Ther. 2013;93(2):186–94.
US FDA. Guidance for industry (draft): drug interaction studies—study design, data analysis, implications for dosing, and labeling recommendations. http://www.fda.gov/Drugs/GuidanceComplianceRegulatoryInformation/Guidances/ucm064982.htm. Accessed May 2013.
European Medicines Agency’s guideline on the investigation of drug interactions. http://www.ema.europa.eu/docs/en_GB/document_library/Scientific_guideline/2012/07/WC500129606.pdf. Accessed May 2013.
Hillgren KM, Keppler D, Zur AA, Giacomini KM, Stieger B, Cass CE, et al. Emerging transporters of clinical importance: an update from the International Transporter Consortium. Clin Pharmacol Ther. 2013;94(1):52–63.
Bourdet DL, Pritchard JB, Thakker DR. Differential substrate and inhibitory activities of ranitidine and famotidine toward human organic cation transporter 1 (hOCT1; SLC22A1), hOCT2 (SLC22A2), and hOCT3 (SLC22A3). J Pharmacol Exp Ther. 2005;315(3):1288–97.
Minematsu T, Iwai M, Umehara K, Usui T, Kamimura H. Characterization of human organic cation transporter 1 (OCT1/SLC22A1)- and OCT2 (SLC22A2)-mediated transport of 1-(2-methoxyethyl)-2-methyl-4,9-dioxo-3-(pyrazin-2-ylmethyl)-4,9-dihydro-1H-naphtho[2,3-d]imidazolium bromide (YM155 monobromide), a novel small molecule survivin suppressant. Drug Metab Dispos. 2010;38(1):1–4.
Tsuda M, Terada T, Ueba M, Sato T, Masuda S, Katsura T, et al. Involvement of human multidrug and toxin extrusion 1 in the drug interaction between cimetidine and metformin in renal epithelial cells. J Pharmacol Exp Ther. 2009;329(1):185–91.
Ito S, Kusuhara H, Kuroiwa Y, Wu C, Moriyama Y, Inoue K, et al. Potent and specific inhibition of mMate1-mediated efflux of type I organic cations in the liver and kidney by pyrimethamine. J Pharmacol Exp Ther. 2010;333(1):341–50.
Tucker GT, Casey C, Phillips PJ, Connor H, Ward JD, Woods HF. Metformin kinetics in healthy subjects and in patients with diabetes mellitus. Br J Clin Pharmacol. 1981;12(2):235–46.
Goodman LS, Brunton LL, Chabner B, Knollmann BC. Goodman & Gilman’s pharmacological basis of therapeutics. 12th ed. New York: McGraw-Hill; 2011.
Moffat AC, Osselton MD, Widdop B, editors. Clarke’s analysis of drugs and poisons: in pharmaceuticals, body fluids and postmortem material. 3rd ed. London: Pharmaceutical Press; 2004.
Zhang S, Lovejoy KS, Shima JE, Lagpacan LL, Shu Y, Lapuk A, et al. Organic cation transporters are determinants of oxaliplatin cytotoxicity. Cancer Res. 2006;66(17):8847–57.
Chen Y, Zhang S, Sorani M, Giacomini KM. Transport of paraquat by human organic cation transporters and multidrug and toxic compound extrusion family. J Pharmacol Exp Ther. 2007;322(2):695–700.
Shu Y, Sheardown SA, Brown C, Owen RP, Zhang S, Castro RA, et al. Effect of genetic variation in the organic cation transporter 1 (OCT1) on metformin action. J Clin Invest. 2007;117(5):1422–31.
More SS, Li S, Yee SW, Chen L, Xu Z, Jablons DM, et al. Organic cation transporters modulate the uptake and cytotoxicity of picoplatin, a third-generation platinum analogue. Mol Cancer Ther. 2010;9(4):1058–69.
Kido Y, Matsson P, Giacomini KM. Profiling of a prescription drug library for potential renal drug-drug interactions mediated by the organic cation transporter 2. J Med Chem. 2011;54(13):4548–58.
Minematsu T, Giacomini KM. Interactions of tyrosine kinase inhibitors with organic cation transporters and multidrug and toxic compound extrusion proteins. Mol Cancer Ther. 2011;10(3):531–9.
Wittwer MB, Zur AA, Khuri N, Kido Y, Kosaka A, Zhang X, et al. Discovery of potent, selective multidrug and toxin extrusion transporter 1 (MATE1, SLC47A1) inhibitors through prescription drug profiling and computational modeling. J Med Chem. 2013;56(3):781–95.
Zhou M, Xia L, Wang J. Metformin transport by a newly cloned proton-stimulated organic cation transporter (plasma membrane monoamine transporter) expressed in human intestine. Drug Metab Dispos. 2007;35(10):1956–62.
Shu Y, Brown C, Castro RA, Shi RJ, Lin ET, Owen RP, et al. Effect of genetic variation in the organic cation transporter 1, OCT1, on metformin pharmacokinetics. Clin Pharmacol Ther. 2008;83(2):273–80.
Chen Y, Li S, Brown C, Cheatham S, Castro RA, Leabman MK, et al. Effect of genetic variation in the organic cation transporter 2 on the renal elimination of metformin. Pharmacogenet Genomics. 2009;19(7):497–504.
Howlett HC, Bailey CJ. A risk-benefit assessment of metformin in type 2 diabetes mellitus. Drug Saf. 1999;20(6):489–503.
Giacomini KM, Huang SM, Tweedie DJ, Benet LZ, Brouwer KL, Chu X, et al. Membrane transporters in drug development. Nat Rev Drug Discov. 2010;9(3):215–36.
Lau YY, Huang Y, Frassetto L, Benet LZ. effect of OATP1B transporter inhibition on the pharmacokinetics of atorvastatin in healthy volunteers. Clin Pharmacol Ther. 2007;81(2):194–204.
Abel S, Nichols DJ, Brearley CJ, Eve MD. Effect of cimetidine and ranitidine on pharmacokinetics and pharmacodynamics of a single dose of dofetilide. Br J Clin Pharmacol. 2000;49(1):64–71.
Feng B, Obach RS, Burstein AH, Clark DJ, de Morais SM, Faessel HM. Effect of human renal cationic transporter inhibition on the pharmacokinetics of varenicline, a new therapy for smoking cessation: an in vitro-in vivo study. Clin Pharmacol Ther. 2008;83(4):567–76.
Shiga T, Hashiguchi M, Urae A, Kasanuki H, Rikihisa T. Effect of cimetidine and probenecid on pilsicainide renal clearance in humans. Clin Pharmacol Ther. 2000;67(3):222–8.
Somogyi AA, Bochner F, Sallustio BC. Stereoselective inhibition of pindolol renal clearance by cimetidine in humans. Clin Pharmacol Ther. 1992;51(4):379–87.
Pedersen JM, Matsson P, Bergstrom CA, Norinder U, Hoogstraate J, Artursson P. Prediction and identification of drug interactions with the human ATP-binding cassette transporter multidrug-resistance associated protein 2 (MRP2; ABCC2). J Med Chem. 2008;51(11):3275–87.
Vanwert AL, Srimaroeng C, Sweet DH. Organic anion transporter 3 (oat3/slc22a8) interacts with carboxyfluoroquinolones, and deletion increases systemic exposure to ciprofloxacin. Mol Pharmacol. 2008;74(1):122–31.
Kindla J, Muller F, Mieth M, Fromm MF, Konig J. Influence of non-steroidal anti-inflammatory drugs on organic anion transporting polypeptide (OATP) 1B1- and OATP1B3-mediated drug transport. Drug Metab Dispos. 2011;39(6):1047–53.
Karlgren M, Vildhede A, Norinder U, Wisniewski JR, Kimoto E, Lai Y, et al. Classification of inhibitors of hepatic organic anion transporting polypeptides (OATPs): influence of protein expression on drug-drug interactions. J Med Chem. 2012;55(10):4740–63.
Mulgaonkar A, Venitz J, Grundemann D, Sweet DH. Human organic cation transporters 1 (SLC22A1), 2 (SLC22A2), and 3 (SLC22A3) as disposition pathways for fluoroquinolone antimicrobials. Antimicrob Agents Chemother. 2013;57(6):2705–11.
Busch AE, Karbach U, Miska D, Gorboulev V, Akhoundova A, Volk C, et al. Human neurons express the polyspecific cation transporter hOCT2, which translocates monoamine neurotransmitters, amantadine, and memantine. Mol Pharmacol. 1998;54(2):342–52.
Dudley AJ, Bleasby K, Brown CD. The organic cation transporter OCT2 mediates the uptake of beta-adrenoceptor antagonists across the apical membrane of renal LLC-PK1 cell monolayers. Br J Pharmacol. 2000;131(1):71–9.
Terada T, Masuda S, Asaka J, Tsuda M, Katsura T, Inui K. Molecular cloning, functional characterization and tissue distribution of rat H+/organic cation antiporter MATE1. Pharm Res. 2006;23(8):1696–701.
Tsuda M, Terada T, Asaka J, Ueba M, Katsura T, Inui K. Oppositely directed H + gradient functions as a driving force of rat H+/organic cation antiporter MATE1. Am J Physiol Renal Physiol. 2007;292(2):F593–8.
Chung JY, Cho SK, Kim TH, Kim KH, Jang GH, Kim CO, et al. Functional characterization of MATE2-K genetic variants and their effects on metformin pharmacokinetics. Pharmacogenet Genomics. 2013;23(7):365–73.
Dangprapai Y, Wright SH. Interaction of H+ with the extracellular and intracellular aspects of hMATE1. Am J Physiol Renal Physiol. 2011;301(3):F520–8.
Callaghan JT, Bergstrom RF, Rubin A, Chernish S, Crabtree R, Knadler MP, et al. A pharmacokinetic profile of nizatidine in man. Scand J Gastroenterol Suppl. 1987;136:9–17.
Grover A, Benet LZ. Effects of drug transporters on volume of distribution. AAPS J. 2009;11(2):250–61.
FDA. Highlights of prescribing information: nizatidine (Axid). http://www.accessdata.fda.gov/drugsatfda_docs/label/2005/21494s001lbl.pdf. Accessed July 2015.
Stumvoll M, Chintalapudi U, Perriello G, Welle S, Gutierrez O, Gerich J. Uptake and release of glucose by the human kidney. Postabsorptive rates and responses to epinephrine. J Clin Invest. 1995;96(5):2528–33.
Landau BR, Wahren J, Chandramouli V, Schumann WC, Ekberg K, Kalhan SC. Contributions of gluconeogenesis to glucose production in the fasted state. J Clin Invest. 1996;98(2):378–85.
Stumvoll M, Meyer C, Mitrakou A, Nadkarni V, Gerich JE. Renal glucose production and utilization: new aspects in humans. Diabetologia. 1997;40(7):749–57.
Gerich JE. Physiology of glucose homeostasis. Diabetes Obes Metab. 2000;2(6):345–50.
Gerich JE. Control of glycaemia. Baillieres Clin Endocrinol Metab. 1993;7(3):551–86.
Woerle HJ, Meyer C, Dostou JM, Gosmanov NR, Islam N, Popa E, et al. Pathways for glucose disposal after meal ingestion in humans. Am J Physiol Endocrinol Metab. 2003;284(4):E716–25.
Gong L, Goswami S, Giacomini KM, Altman RB, Klein TE. Metformin pathways: pharmacokinetics and pharmacodynamics. Pharmacogenet Genomics. 2012;22(11):820–7.
Rabik CA, Dolan ME. Molecular mechanisms of resistance and toxicity associated with platinating agents. Cancer Treat Rev. 2007;33(1):9–23.
Toyama K, Yonezawa A, Masuda S, Osawa R, Hosokawa M, Fujimoto S, et al. Loss of multidrug and toxin extrusion 1 (MATE1) is associated with metformin-induced lactic acidosis. Br J Pharmacol. 2012;166(3):1183–91.
Acknowledgments
We thank Jennifer Hibma for assistance in preparing the clinical study protocol. We thank Hector Vizoso and all the staff at the General Clinical Research Center (GCRC) at San Francisco General Hospital (SFGH) for their professionalism and enthusiastic assistance in our clinical study. We thank Yong Huang at the UCSF Drugs Services Unit for access to their bioanalytical facilities and Chav Doherty from UCSF SFGH Clinical Laboratory for excellent service.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Funding
This project was supported by National Institutes of Health/National Center for Research Resources (NIH/NCRR) University of California, San Francisco (UCSF)-Clinical & Translational Science Institute (CTSI) Grant UL1 RR024131 & NIH Grant GM61390. The authors are solely responsible for the contents and do not necessarily represent the official views of the NIH. This work was also made possible in part by core services provided by the General Clinical Research Center (GCRC) at San Francisco General Hospital (SFGH), funded by the NCRR (NIH MO1-RR00083-44). The authors also acknowledge the following funding sources for SLS. (Allergan) and ECC and KMM (NIH Training Grant T32 GM007175, FDA-CDER/ORISE).
Conflict of interest
The authors have no conflicts of interest that might be relevant to the content of this manuscript.
Ethical approval
The Committee on Human Research at the University of California, San Francisco (Institutional Review Board [IRB] 11-06968) approved the clinical drug–drug interaction study, and all subjects were recruited directly from the Study of Pharmacogenetics in Ethnically Diverse Populations (IRB 10-03167). Informed consent was obtained from all study participants.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
About this article

Cite this article
Morrissey, K.M., Stocker, S.L., Chen, E.C. et al. The Effect of Nizatidine, a MATE2K Selective Inhibitor, on the Pharmacokinetics and Pharmacodynamics of Metformin in Healthy Volunteers. Clin Pharmacokinet 55, 495–506 (2016). https://doi.org/10.1007/s40262-015-0332-9
Published:
Issue Date:
DOI: https://doi.org/10.1007/s40262-015-0332-9